

Abstract
Extracellular phosphate is toxic to the cell at high concentrations. When the phosphate level is increased in the blood by impaired urinary phosphate excretion, premature aging ensues. When the phosphate level is increased in the urine by dietary phosphate overload, this may lead to kidney damage (tubular injury and interstitial fibrosis). Extracellular phosphate exerts its cytotoxicity when it forms insoluble nanoparticles with calcium and fetuin-A, referred to as calciprotein particles (CPPs). CPPs are highly bioactive ligands that can induce various cellular responses, including osteogenic transformation of vascular smooth muscle cells and cell death in vascular endothelium and renal tubular epithelium. CPPs are detected in the blood of animal models and patients with chronic kidney disease (CKD) and associated with adaptation of the endocrine axes mediated by fibroblast growth factor-23 (FGF23) and Klotho that regulate mineral metabolism and aging. These observations have raised the possibility that CPPs may contribute to the pathophysiology of CKD. This notion, if validated, is expected to provide new diagnostic and therapeutic targets for CKD.
Keywords: calciprotein particle (CPP), fibroblast growth factor-23 (FGF23), Klotho, phosphate restriction
Phosphorus homeostasis is maintained primarily by balance between the intestinal absorption of phosphorus from diet and the renal excretion of blood phosphorus into urine. A normal 70 kg adult takes in ∼1.6 g phosphorus per day from food and a decent amount of phosphate from food additives and preservatives.1, 2 Approximately 40–60% of phosphorus in the food is absorbed from the intestine and the rest is excreted into feces. Phosphorus excretion into urine is ∼1.0 g/day and regulated to match the intestinal absorption of phosphorus in non-growing adults in the steady state, thereby maintaining a net zero balance.3
Absorption and excretion of phosphorus and its distribution within the body is coordinately regulated by three hormones, parathyroid hormone (PTH), 1,25-dihydroxyvitamin D3 (calcitriol), and fibroblast growth factor-23 (FGF23).4 FGF23 functions as a phosphaturic hormone as well as a counter-regulatory hormone for calcitriol.5, 6 FGF23 is secreted from the bone upon dietary phosphate load through mechanisms yet to be completely understood. FGF23 acts on the kidney to suppress phosphate reabsorption and calcitriol synthesis, thereby inducing a negative phosphate balance. FGF23 also acts on the parathyroid to lower PTH.7 A critical feature of FGF23 is the fact that it requires Klotho, a single-pass transmembrane protein, as an obligate co-receptor.8
Identification of the FGF23–Klotho endocrine axis has significantly advanced our knowledge on the phosphate metabolism and has transformed our view on phosphate: phosphate is not merely a mineral stored in the bone, but should be recognized as a critical nutrient and bioactive ligand that has tremendous impacts on our health. This review discusses new insights into the pathophysiology of chronic kidney disease (CKD) and a novel mechanism of aging prompted by the discovery of the FGF–Klotho endocrine system.
FGF23 AND KLOTHO
Klotho was originally identified as a gene inactivated in a mutant mouse that inherited a syndrome resembling human aging in an autosomal recessive manner.9 This strain is a transgenic mouse line carrying transgene insertion at the 5′ flanking region of the klotho gene. The transgene insertion caused extensive hypermethylation around the insertion site and silenced expression of the transgene and the neighboring klotho gene.10 As a result, mice homozygous for the transgene (kl/kl mice) lack Klotho expression and develop complex phenotypes resembling human aging around 4 weeks of age, including growth arrest, cognition impairment,11 hearing loss,12 hypogonadotropic hypogonadism,13 vascular calcification, cardiac hypertrophy,14 emphysematous lung,15 osteopenia,16 ectopic calcification in soft tissues, atrophy of thymus, fat, skin, and skeletal muscle, resulting in premature death around 9 weeks of age.9 They also exhibit a marked increase in blood phosphate, calcium, and calcitriol levels.17 The klotho gene encodes a single-pass transmembrane protein and is expressed primarily in distal tubules in the kidney, chief cells in the parathyroid,7 and the choroid plexus in the brain.9 Klotho protein forms constitutive binary complexes with FGF receptors (FGFR1c, 3c, and 4) and increases their affinity selectively to FGF23.8 Without Klotho, FGF23 cannot bind to and activate its cognate FGFRs at physiological concentrations. This explains the fact that kl/kl mice and Fgf23−/− mice develop identical aging-like phenotypes and abnormal mineral metabolism.18 The tissue-specific expression of Klotho explains why FGF23 can identify the kidney and parathyroid as its target organs among many other tissues expressing FGFRs. However, it remains to be determined what Klotho does in the choroid plexus and why FGF23 can regulate function of renal proximal tubules (i.e., phosphate reabsorption and calcitriol synthesis) despite its obligate co-receptor Klotho being expressed primarily in distal tubules.19, 20
HYPERPHOSPHATEMIA AND AGING
Why do defects in the FGF23–Klotho endocrine axis lead to a premature-aging syndrome? Specifically, is the premature-aging syndrome in mice lacking Klotho or FGF23 due to disturbed mineral metabolism? To address this question, several laboratories ablated calcitriol activity in kl/kl mice and Fgf23−/− mice. The premature-aging syndrome in these mutants was ameliorated by feeding a vitamin D-deficient diet17, 21 or by disrupting the vitamin D receptor gene22 or the Cyp27b1 gene that encodes 25-hydroxyvitamin D-1α-hydroxylase essential for calcitriol synthesis.23, 24 These findings indicated that vitamin D intoxication might be the cause of the aging-like phenotypes. However, these dietary and genetic interventions lowered blood calcium and phosphate levels simultaneously and were unable to specify which was guilty, calcitriol, calcium, or phosphate. Subsequently, it was reported that deletion of the type-IIa sodium-dependent phosphate co-transporter (Npt2a) gene mitigated the premature-aging syndrome in kl/kl mice.25 Npt2a mediates renal phosphate reabsorption in proximal tubules and, when ablated, induces phosphate wasting into urine and hypophosphatemia.26 Importantly, mice lacking Npt2a increase calcitriol synthesis in an attempt to increase intestinal phosphate absorption and compensate for the renal phosphate loss. This causes hypercalcemia, because calcitriol increases intestinal calcium absorption and renal calcium reabsorption. Thus, deletion of Npt2a lowers the blood phosphate levels but further increases the already elevated blood calcium and calcitriol levels in kl/kl mice.27 Nonetheless, Npt2a gene knockout rescued the aging-like phenotypes, clearly indicating that it is phosphate, and not calcium or calcitriol, which is responsible for the premature-aging syndrome. This notion was further confirmed by the facts that (1) low phosphate diet lowered blood phosphate levels and rescued Fgf23−/− mice21 and that (2) mice lacking both Klotho and Npt2a regained aging-like phenotypes after dietary phosphate overload.25
CYTOTOXIC POTENTIAL OF EXTRACELLULAR PHOSPHATE
Although the mechanism by which hyperphosphatemia induces premature aging remains elusive, the fact that high extracellular phosphate is directly toxic to vascular endothelial cells may partly explain vascular senescence and calcification in CKD patients. Regular cell culture medium contains approximately 1 mM phosphate, which is equivalent to the normal human blood phosphate level. When cultured in high phosphate medium containing 1.4–2.8 mM phosphate (similar to blood phosphate levels in end-stage renal disease patients), vascular endothelial cells increase production of reactive oxygen species and eventually undergo apoptosis.28, 29, 30 Endothelial cell death can expose underlying smooth muscle cells to the high ambient phosphate. Vascular smooth muscle cells cultured in high phosphate medium increase expression of BMP-2 (a potent osteogenic cytokine), Runx2 (a master transcription factor for osteoblast differentiation), and osteopontin, followed by phenotypic transition to osteoblast-like cells.31, 32 This cascade of events may contribute to vascular calcification associated with hyperphosphatemia.33
When phosphate concentrations are increased by 1–2 mM in regular cell culture medium, phosphate forms insoluble crystals with calcium.34 This fact raised the possibility that insoluble calcium–phosphate crystals, rather than soluble phosphate, may be toxic to the cell. Several lines of evidence support this notion: first, calcium–phosphate crystals induced cellular responses mentioned above without increasing soluble phosphate concentrations. Second, these cellular responses did not occur in the high phosphate medium when calcium–phosphate crystals were removed by centrifugation or when formation of calcium–phosphate crystals were inhibited by pyrophosphate or phosphonoformic acid.34, 35, 36 It was reported that stimulation of vascular smooth muscle cells with calcium–phosphate crystals rapidly increased the intracellular calcium concentration and induced apoptosis, which was attenuated by pre-treatment of the cells with an inhibitor of the lysosomal proton pump. These observations suggest that calcium–phosphate crystals are endocytosed and targeted to the lysosome, where they dissolve under the acidic environment, leak as free calcium into the cytosol, and induce apoptosis.36
CALCIPROTEIN PARTICLES (CPPs)
Formation of calcium–phosphate crystals is a physicochemical process that occurs when concentrations of calcium and phosphate exceed the solubility limit (or the formation product). This process is referred to as nucleation. After nucleation, calcium–phosphate crystals undergo phase transformation among CaHPO4 (monetite), CaHPO4·2H2O (brushite), Ca4H(PO4)3 (octacalcium phosphate or OCP), Ca9(PO4)6 (amorphous calcium phosphate or ACP), and Ca10(PO4)6(OH)2 (hydroxyapatite or HA).37 The phase transformation is affected by various factors besides calcium and phosphate concentrations, including ionic strength, pH, temperature, and incubation time. Under biologically relevant conditions, the initial phase is monetite and brushite, which grow in size to become OCP, ACP, and HA over time and eventually precipitate.38
Serum strongly inhibits growth (but not nucleation) of calcium–phosphate crystals. Immediately after nucleation, mineral-binding proteins in the serum such as fetuin-A sequester the crystals while they are still very small (<1 nm in diameter) and inhibit their growth.39 The calcium–phosphate crystal-laden fetuin-A molecules aggregate to form nanoparticles (50–100 nm in diameter), which are called primary CPPs. Primary CPPs are subject to a topological rearrangement to turn into a stable structure, in which a densely packed fetuin-A monolayer covers a mineral core, thereby preventing further crystal growth.40, 41 These particles are referred to secondary CPPs (100–200 nm in diameter). CPPs exist as colloids and do not precipitate spontaneously.42 Thus, formation of CPPs can be regarded as a defense mechanism against precipitation of solid-phase calcium–phosphate in the blood, urine, and soft tissues. In fact, fetuin-A knockout mice suffer from severe ectopic calcifications all over the body.43
CPPs are generated in high phosphate medium containing serum and responsible for the phenotypic transition of vascular smooth muscle cells to osteoblast-like cells.34 It is possible that many other reported effects of high phosphate medium on various types of cells may actually be attributed not to soluble phosphate but to CPPs. Indeed, Khoshniat et al.44 reported that induction of mineralization-associated gene expression (e.g., osteopontin, matrix gla protein) in cultured osteoblasts by high phosphate medium never occurred in the absence of extracellular calcium and in the presence of an inhibitor for calcium–phosphate crystal formation (phosphocitrate). They observed calcium–phosphate nanoparticles (most likely CPPs) in the high phosphate medium but not in the calcium-free or phosphocitrate-containing high phosphate medium. However, CPPs did not need to enter the cell but required intact lipid rafts on the cell surface to induce expression of osteopontin. These observations suggest that, unlike inorganic calcium–phosphate crystals, CPPs may not necessarily enter the cell but can transmit signal through machinery localized in the lipid raft on the cell surface to induce the cellular responses. More recently, it was reported that synthesized CPPs, when injected into mice, were cleared from the blood by Kupffer cells in the liver through phagocytosis via class A scavenger receptors (SR-A),45 indicating that SR-A can function as a clearance receptor for CPPs. It remains to be determined whether SR-A also functions as a signaling receptor for CPPs.
CPPS IN CKD
Are CPPs merely an experimental artifact generated in high phosphate medium or indeed present in vivo? Recent studies have demonstrated that CPPs circulate in the blood of CKD patients and animals but not in the blood of normal subjects.46, 47, 48 These studies isolated CPPs from the blood by sequential centrifugation: first, serum was prepared from clotted blood by a standard condition (centrifugation at 3000 g for 10 min). As CPPs are nanoparticles existing as colloids, they never precipitated under this condition and stayed in the serum. Next, the serum was centrifuged at a higher speed (16,000–22,000 g) for a longer time (2 h) to precipitate CPPs. CPPs in the pellet were dissolved with EDTA or acid and then subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis for quantification of fetuin-A. Alternatively, serum CPP levels were measured as the difference between the serum fetuin-A levels before and after the high-speed centrifugation using enzyme-linked immunosorbent assay.
Serum CPP levels increased with CKD progression.46 Furthermore, serum CPP levels were independently correlated with serum phosphate, C-reactive protein (systemic inflammation), oxidized low-density lipoprotein, and BMP-2/7 ratio, and inversely correlated with estimated glomerular filtration rate (eGFR).48 Oxidized low-density lipoprotein and the BMP-2/7 ratio are thought to represent propensity for vascular calcification, because oxidized low-density lipoprotein induces expression of BMP-2 that promotes osteogenic transition in vascular smooth muscle cells, whereas BMP-7 inhibits it. Indeed, vascular stiffness and calcification as determined by aortic pulse wave velocity and coronary artery calcification scores, respectively, were positively correlated with serum CPP levels.46, 48 In addition, appearance of CPPs precedes increase in the aortic calcium content and vascular calcification in a rat CKD model.47 Taken together, the hypothesis that CPPs has a pathogenic role in vascular calcification in CKD should be further evaluated.
DIETARY PHOSPHATE OVERLOAD AND KIDNEY DAMAGE
It has been known that oral phosphate overload can induce de novo CKD in normal animals49 and humans.50 The degree of kidney damage depends not only on the amount of phosphate load but also the nephron number. Specifically, by combining partial nephrectomy and diets containing different amounts of phosphate, Haut et al.51 changed phosphate excretion per nephron as an independent variable and observed a positive correlation with the severity of histological kidney damage, which was characterized by tubulointerstitial lesions including interstitial fibrosis, interstitial cell infiltration, interstitial edema, tubular atrophy, and tubular dilatation, but no glomerular changes. Namely,
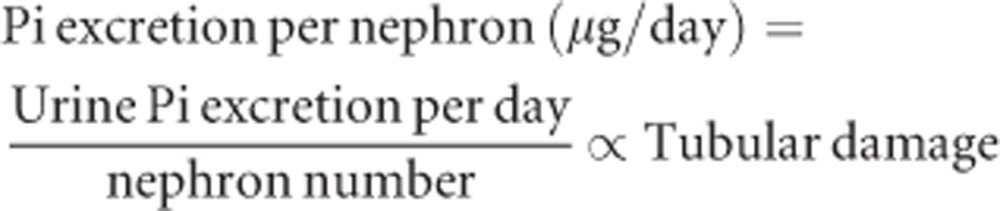 |
The phosphate excretion per nephron is increased when the numerator (urine phosphate excretion per day) is increased and/or the denominator (the nephron number) is decreased. Normal rats placed on diets containing 0.5%, 1.0%, or 2.0% phosphate excreted approximately 30, 70, or 150 mg/day of phosphate into urine, respectively.51 Assuming 3 × 104 nephrons per kidney, phosphate excretion per nephron is estimated at 0.5, 1.2, or 2.5 μg/day, respectively. The linear correlation between the dietary phosphate content and the urinary phosphate excretion indicates that no effective regulatory mechanism exists by which excess phosphate intake suppresses intestinal phosphate absorption. In other words, the phosphate balance upon dietary phosphate overload is mainly maintained not at the level of intestinal absorption but at the level of renal excretion, which justifies the assumption that the urinary phosphate excretion correlates with the dietary phosphate intake. Thus, dietary phosphate overload increases phosphate excretion per nephron and can induce tubular damage. It is likely that such tubular damage may eventually reduce the nephron number, thereby forming a vicious cycle leading to progressive tubular damage unless phosphate intake is reduced. The equation above also predicts that individuals with reduced nephron number (e.g., CKD patients and the aged) should reduce phosphate intake accordingly to prevent tubular damage. Notably, there was no correlation between the tubular damage and the blood phosphate level: normal rats placed on high phosphate diet developed tubular damage without a significant increase in the blood phosphate level.51
Unless renal function is critically impaired, dietary phosphate overload never causes persistent hyperphosphatemia, because the kidney excretes excess phosphate into urine and maintains the phosphate balance. Thus, dietary phosphate overload increases phosphate levels not in the blood but in the urine (more precisely, in the tubular luminal fluid). Measurement of the tubular luminal fluid composition by micropuncture in rats indicated that calcium and phosphate concentrations likely exceeded the formation product in the proximal tubule upon dietary phosphate overload,52, 53 suggesting that nucleation and possibly CPP formation could occur in the proximal tubular lumen.53 In cultured renal epithelial cells, exposure to calcium–phosphate crystals can induce oxidative stress and cellular damage.54 Thus, formation of cytotoxic CPPs in the tubular lumen may contribute to the tubular damage. In further support of this notion, administration of a bisphosphonate (etidronate) alleviated the tubular damage induced by phosphate load in rats.51 Bisphosphonates have been used for the treatment of osteoporosis because of their activity that kills osteoclasts.55 Besides this biological activity, bisphosphonates have a chemical activity as pyrophosphate analogs that inhibit nucleation of calcium–phosphate crystals at very low concentrations.56 As bisphosphonates are not metabolized and excreted into urine unchanged, etidronate may inhibit CPP formation in the tubular lumen.
When phosphate excretion per nephron exceeds ∼1 μg/day, tubular damage occurs in rats.51 Although it remains to be determined whether this threshold is applicable to other species, it is informative to estimate phosphate excretion per nephron in humans. A normal adult excretes ∼1 g/day of phosphate into urine.3 Assuming 1 × 106 nephrons per kidney,57 phosphate excretion per nephron is estimated at 0.5 μg/day. Thus, a 50% decrease in nephron number by CKD and/or during the normal aging process is sufficient to increase phosphate excretion per nephron beyond 1 μg/day, which potentially causes tubular damage and interstitial fibrosis. In fact, renal fibrosis is universally observed not only in CKD patients but also in the aged without overt CKD.58 On the other hand, living kidney donors, whose phosphate excretion per nephron should be around 1 μg/day, show no increase in mortality or end-stage renal disease in the long term59 despite significant decrease in GFR60 and increase in blood FGF23 levels.61 Although no substantial data on renal histology of living kidney donors is available, it is possible that the threshold of phosphate excretion per nephron for tubular damage may be higher than 1 μg/day in humans. This may be partly because normal adults have lower calcium excretion per nephron than the normal rats (∼0.075 μg/day62 vs. ∼0.105 μg/day51).
PHOSPHATE OVERLOAD AND THE FGF23–KLOTHO SYSTEM
There are two possible ways to increase phosphate excretion into urine: one is to increase the volume of ultrafiltered plasma by increasing GFR (hyperfiltration). The other is to increase the fractional excretion of phosphate (FEp). Unlike healthy people, CKD patients are not always capable of increasing GFR but can increase FEp to maintain phosphate balance.63 The increase in FEp is primarily attributed to increased serum FGF23 in early stage CKD. PTH also has phosphaturic activity and is increased in CKD, however, PTH may not contribute substantially to the increase in FEp, at least in early stage CKD. This notion is based on the following findings: first, epidemiological studies showed that the increase in FGF23 and FEp preceded the increase in PTH during CKD progression.64 Second, animal experiments showed that the high PTH in CKD was secondary to low calcitriol caused by high FGF23.65 These experiments used CKD rats that exhibited high FEp associated with high FGF23, high PTH, low calcitriol, and normal phosphate in the blood. In these rats with moderate CKD, FGF23 activity was blocked by injection of an anti-FGF23 neutralizing antibody. The inhibition of FGF23 activity reduced FEp and increased blood phosphate and calcitriol levels within 24 h, whereas PTH remained high for 24 h and then decreased thereafter. These observations indicate that high FEp in early stage CKD is likely due to high FGF23 but not due to high PTH and that the high PTH is a feedback response to low calcitriol caused by high FGF23.
FGF23 is a hormone that increases phosphate excretion per nephron by increasing FEp. Provided that FGF23 is primarily responsible for the maintenance of phosphate homeostasis in early stage CKD, it is conceivable that the blood FGF23 level may correlate with, and therefore, serve as a surrogate marker for phosphate excretion per nephron. This notion is supported by the facts that (1) blood FGF23 levels increase when dietary phosphate intake is increased but not when the blood phosphate level is increased by bolus intravenous phosphate injection66, 67, 68 and that (2) the blood FGF23 level increases as the nephron number decreases with age and CKD progression.64, 69 However, the correlation between FGF23 and phosphate excretion per nephron may be blunted among healthy people whose nephrons are not operating with full throttle but still holding the spare capacity to increase single nephron GFR in response to the demand to increase phosphate excretion per nephron. In such population, phosphate excretion per nephron can be increased not only by increasing FEp with FGF23 but also by increasing GFR (hyperfiltration), which may explain why some clinical studies failed to detect a significant correlation between FGF23 and phosphate intake in healthy people.70 Thus, the equation can be modified as follows, which may be applicable to patients with decreased GFR (CKD stages 2 and higher):
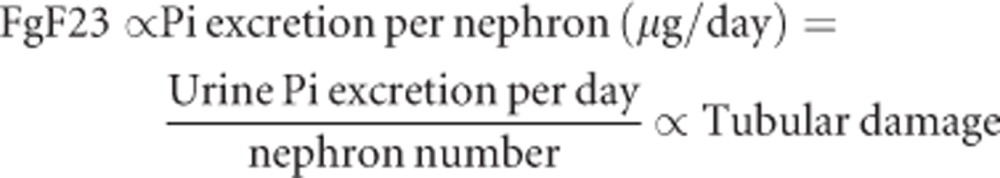 |
A NEW PARADIGM FOR PHOSPHATE RESTRICTION
The rationale of the phosphate-lowering therapy has been the theory that hyperphosphatemia causes vascular calcification and increases cardiovascular events. This theory justifies the use of phosphate binders only for CKD patients with hyperphosphatemia, which account for only ∼0.3% of the total CKD patient population.71, 72 Naturally, the therapeutic goal is to lower blood phosphate levels.73 This current paradigm may be challenged, provided that the equation above is proven applicable not only to rodents but also to humans. The new paradigm for phosphate restriction would be as follows: regardless of the blood phosphate level, phosphate restriction should be started when the blood FGF23 level begins to rise, because this is a sign of excess phosphate intake relative to the residual nephron number and increased risk for CPP formation in the tubular lumen. The primary purpose of phosphate restriction at this stage is to prevent tubular damage and delay CKD progression but not to prevent vascular calcification per se. The therapeutic goal is not to lower the blood phosphate level but to lower the blood FGF23 level, a surrogate marker for phosphate excretion per nephron. This new paradigm justifies phosphate restriction for CKD patients with high blood FGF23 levels, which are observed as early as stage 2[ref. [64]] and account for ∼35% of all CKD patients together (Table 1).71
Table 1. Comparison between the current and the proposed new paradigm of phosphate restriction.
| Current paradigm | New paradigm | |
|---|---|---|
| Rationale | High blood phosphate promotes vascular calcification and cardiovascular events | High urine phosphate injures renal tubules and induces renal fibrosis |
| Goal | To lower blood phosphate | To lower blood FGF23 |
| Indication | ESRD with hyperphosphatemia (∼0.3% of the total CKD patients) | CKD stages 2–5 with high blood FGF23 levels (∼35% of all CKD stages 2–5 patients) |
Abbreviations: CKD, chronic kidney disease; ESRD, end-stage renal disease; FGF23, fibroblast growth factor-23.
The equation above, if proven, also makes it possible to monitor CKD progression not by GFR but by the residual functional nephron number: measure the blood FGF23 level regularly. If FGF23 is increased, measure the 24-h urine phosphate. If it is also increased, excess phosphate intake is suspected, but not decrease in the nephron number. If the 24-h urine phosphate is not increased, however, the increase in FGF23 means decrease in the nephron number or progression of CKD, even if GFR may not be decreased. In either case, phosphate restriction should be enhanced by dietary counseling and/or by increasing the dose of phosphate binders.
New designs are needed for clinical studies to verify these new paradigms, because the existing clinical studies on dietary phosphate intake and blood FGF23 have not measured histological kidney damage as an outcome. In addition, there is a fundamental limitation in the estimation of dietary phosphate intake. The daily phosphate intake can be most accurately estimated by measuring the 24-h urine phosphate. However, it has rarely been used in large-scale epidemiological studies because of practical difficulty. Instead, the ‘24-h recall' method has been used, which is unacceptably inaccurate because of at least three reasons: first, this method estimates dietary phosphorus intake based on interview with the participants for what they ate in the past 24 h, which inevitably entails intentional and unintentional errors. Second, a large amount of phosphate added to processed foods as additives and preservatives is not included, which potentially increases phosphorus intake by as much as 1.0 g/day2. This is because current regulations do not require the amount of phosphate-containing food additives to be listed in the Nutrition Facts label. Finally, dietary phosphorus intake does not necessarily correlate with the amount of phosphate actually absorbed from the intestine. Phosphorus in natural foods exists mainly as various forms of organic phosphate and their bioavailability varies from 0 to 60%.74 Of note, phosphorus in plants is not absorbed as effectively as that in meats, which explains why in CKD patients vegetarian diet reduces blood FGF23 levels more effectively than meat diet with equivalent phosphorus content.75 In contrast, phosphorus in food additives is in the form of inorganic phosphate and 100% is absorbable. Thus, dietary phosphorus intake does not tell us how much phosphate is actually absorbed from the intestine. Considering these fundamental limitations, any conclusions drawn from the phosphorus intake data using the 24-h recall method should be carefully interpreted and re-evaluated.
CONCLUDING REMARKS
This review has focused on the hypothesis that excess phosphate intake relative to the residual functional nephron number leads to FGF23 increase and may be a trigger of CKD initiation and progression. FGF23 reduces serum calcitriol and can result in two adverse outcomes: increase in PTH (secondary hyperparathyroidism) and decrease in Klotho expression.4 A decrease in Klotho can induce FGF23 resistance and further increase FGF23, thereby forming a self-aggravating deteriorating spiral of decreased Klotho and increased FGF23. To interrupt this vicious cycle, stimulation of Klotho expression using calcitriol and/or proliferator-activated receptor-γ agonists may be of therapeutic potential besides phosphate restriction.
In the 1980s, the smallest life form called ‘nanobacteria' was reported as self-propagating particles residing in serum, urine, and tissues of animals and humans.76, 77 Nanobacteria were thought blood-born infectious agents causing vascular calcification, kidney stones, and renal tubular damages.78 However, recent studies have provided unequivocal evidence showing that nanobacteria are not living entities but nanoparticles composed of calcium–phosphate crystals and fetuin-A,79, 80, 81, 82 which are identical with CPPs. On the other hand, the fact that dietary phosphate overload induces renal tubular injury and interstitial fibrosis has been pointed out since the 1930s.49 By revisiting these decade-old pioneering observations in the light of the FGF23–Klotho system, novel hypotheses on the pathophysiology of CKD and new paradigms for the diagnosis and treatment of CKD patients have emerged. Clinical studies to verify these paradigms require non-invasive assessment of tubular injury and interstitial fibrosis. It was recently reported that diffusion-weighted magnetic resonance imaging was useful for detecting renal fibrosis in a sensitive and quantitative manner in mice.83, 84 If validated in humans as well, diffusion-weighted magnetic resonance imaging, which has been widely used for detecting brain damage, may be applied for detecting kidney damage to complement renal biopsy.
Acknowledgments
This supplement was supported by a grant from the 58th Annual Meeting of the Japanese Society for Dialysis Therapy.
MK has received lecture fees from Chugai Pharmaceuticals and Bayer Japan.
References
- Hiza H, Bente L, Fungwe T.Nutrient content of the US food supply, 2005 U.S. Department of Agriculture, Center for Nutritional Policy and PromotionHome Economics Research Report No. 58,2008
- Uribarri J. Phosphorus additives in food and their effect in dialysis patients. Clin J Am Soc Nephrol. 2009;4:1290–1292. doi: 10.2215/CJN.03950609. [DOI] [PubMed] [Google Scholar]
- Schiavi SC, Kumar R. The phosphatonin pathway: new insights in phosphate homeostasis. Kidney Int. 2004;65:1–14. doi: 10.1111/j.1523-1755.2004.00355.x. [DOI] [PubMed] [Google Scholar]
- Kuro-o M. Phosphate and Klotho. Kidney Int. 2011;79:S20–S23. doi: 10.1038/ki.2011.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada T, Hasegawa H, Yamazaki Y, et al. FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J Bone Miner Res. 2004;19:429–435. doi: 10.1359/JBMR.0301264. [DOI] [PubMed] [Google Scholar]
- White KE, Carn G, Lorenz-Depiereux B, et al. Autosomal-dominant hypophosphatemic rickets (ADHR) mutations stabilize FGF-23. Kidney Int. 2001;60:2079–2086. doi: 10.1046/j.1523-1755.2001.00064.x. [DOI] [PubMed] [Google Scholar]
- Ben-Dov IZ, Galitzer H, Lavi-Moshayoff V, et al. The parathyroid is a target organ for FGF23 in rats. J Clin Invest. 2007;117:4003–4008. doi: 10.1172/JCI32409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurosu H, Ogawa Y, Miyoshi M, et al. Regulation of fibroblast growth factor-23 signaling by Klotho. J Biol Chem. 2006;281:6120–6613. doi: 10.1074/jbc.C500457200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuro-o M, Matsumura Y, Aizawa H, et al. Mutation of the mouse Klotho gene leads to a syndrome resembling ageing. Nature. 1997;390:45–51. doi: 10.1038/36285. [DOI] [PubMed] [Google Scholar]
- Azuma M, Koyama D, Kikuchi J, et al. Promoter methylation confers kidney-specific expression of the Klotho gene. FASEB J. 2012;26:4264–4274. doi: 10.1096/fj.12-211631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai T, Yamada K, Kim HC, et al. Cognition impairment in the genetic model of aging Klotho gene mutant mice: a role of oxidative stress. FASEB J. 2003;17:50–52. doi: 10.1096/fj.02-0448fje. [DOI] [PubMed] [Google Scholar]
- Kamemori M, Ohyama Y, Kurabayashi M, et al. Expression of Klotho protein in the inner ear. Hear Res. 2002;171:103–110. doi: 10.1016/s0378-5955(02)00483-5. [DOI] [PubMed] [Google Scholar]
- Toyama R, Fujimori T, Nabeshima Y, et al. Impaired regulation of gonadotropins leads to the atrophy of the female reproductive system in Klotho-deficient mice. Endocrinology. 2006;147:120–129. doi: 10.1210/en.2005-0429. [DOI] [PubMed] [Google Scholar]
- Faul C, Amaral AP, Oskouei B, et al. FGF23 induces left ventricular hypertrophy. J Clin Invest. 2011;121:4393–4408. doi: 10.1172/JCI46122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suga T, Kurabayashi M, Sando Y, et al. Disruption of the Klotho gene causes pulmonary emphysema in mice. Defect in maintenance of pulmonary integrity during postnatal life. Am J Respir Cell Mol Biol. 2000;22:26–33. doi: 10.1165/ajrcmb.22.1.3554. [DOI] [PubMed] [Google Scholar]
- Kawaguchi H, Manabe N, Miyaura C, et al. Independent impairment of osteoblast and osteoclast differentiation in Klotho mouse exhibiting low-turnover osteopenia. J Clin Invest. 1999;104:229–237. doi: 10.1172/JCI5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujikawa H, Kurotaki Y, Fujimori T, et al. Klotho, a gene related to a syndrome resembling human premature aging, functions in a negative regulatory circuit of vitamin D endocrine system. Mol Endocrinol. 2003;17:2393–2403. doi: 10.1210/me.2003-0048. [DOI] [PubMed] [Google Scholar]
- Shimada T, Kakitani M, Yamazaki Y, et al. Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J Clin Invest. 2004;113:561–568. doi: 10.1172/JCI19081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrow EG, Davis SI, Summers LJ, et al. Initial FGF23-mediated signaling occurs in the distal convoluted tubule. J Am Soc Nephrol. 2009;20:955–960. doi: 10.1681/ASN.2008070783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olauson H, Lindberg K, Amin R, et al. Targeted deletion of Klotho in kidney distal tubule disrupts mineral metabolism. J Am Soc Nephrol. 2012;23:1641–1651. doi: 10.1681/ASN.2012010048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stubbs JR, Liu S, Tang W, et al. Role of hyperphosphatemia and 1,25-dihydroxyvitamin D in vascular calcification and mortality in fibroblastic growth factor 23 null mice. J Am Soc Nephrol. 2007;18:2116–2124. doi: 10.1681/ASN.2006121385. [DOI] [PubMed] [Google Scholar]
- Hesse M, Frohlich LF, Zeitz U, et al. Ablation of vitamin D signaling rescues bone, mineral, and glucose homeostasis in Fgf-23 deficient mice. Matrix Biol. 2007;26:75–84. doi: 10.1016/j.matbio.2006.10.003. [DOI] [PubMed] [Google Scholar]
- Razzaque MS, Sitara D, Taguchi T, et al. Premature aging-like phenotype in fibroblast growth factor 23 null mice is a vitamin D-mediated process. FASEB J. 2006;20:720–722. doi: 10.1096/fj.05-5432fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohnishi M, Nakatani T, Lanske B, et al. Reversal of mineral ion homeostasis and soft-tissue calcification of Klotho knockout mice by deletion of vitamin D 1alpha-hydroxylase. Kidney Int. 2009;75:1166–1172. doi: 10.1038/ki.2009.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohnishi M, Razzaque MS. Dietary and genetic evidence for phosphate toxicity accelerating mammalian aging. FASEB J. 2010;24:3562–3571. doi: 10.1096/fj.09-152488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck L, Karaplis AC, Amizuka N, et al. Targeted inactivation of Npt2 in mice leads to severe renal phosphate wasting, hypercalciuria, and skeletal abnormalities. Proc Natl Acad Sci USA. 1998;95:5372–5377. doi: 10.1073/pnas.95.9.5372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohnishi M, Nakatani T, Lanske B, et al. In vivo genetic evidence for suppressing vascular and soft-tissue calcification through the reduction of serum phosphate levels, even in the presence of high serum calcium and 1,25-dihydroxyvitamin d levels. Circ Cardiovasc Genet. 2009;2:583–590. doi: 10.1161/CIRCGENETICS.108.847814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Marco GS, Hausberg M, Hillebrand U, et al. Increased inorganic phosphate induces human endothelial cell apoptosis in vitro. Am J Physiol Renal Physiol. 2008;294:F1381–F1387. doi: 10.1152/ajprenal.00003.2008. [DOI] [PubMed] [Google Scholar]
- Shuto E, Taketani Y, Tanaka R, et al. Dietary phosphorus acutely impairs endothelial function. J Am Soc Nephrol. 2009;20:1504–1512. doi: 10.1681/ASN.2008101106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Six I, Maizel J, Barreto FC, et al. Effects of phosphate on vascular function under normal conditions and influence of the uraemic state. Cardiovasc Res. 2012;96:130–139. doi: 10.1093/cvr/cvs240. [DOI] [PubMed] [Google Scholar]
- Steitz SA, Speer MY, Curinga G, et al. Smooth muscle cell phenotypic transition associated with calcification: upregulation of Cbfa1 and downregulation of smooth muscle lineage markers. Circ Res. 2001;89:1147–1154. doi: 10.1161/hh2401.101070. [DOI] [PubMed] [Google Scholar]
- Jono S, McKee MD, Murry CE, et al. Phosphate regulation of vascular smooth muscle cell calcification. Circ Res. 2000;87:E10–E17. doi: 10.1161/01.res.87.7.e10. [DOI] [PubMed] [Google Scholar]
- Reynolds JL, Joannides AJ, Skepper JN, et al. Human vascular smooth muscle cells undergo vesicle-mediated calcification in response to changes in extracellular calcium and phosphate concentrations: a potential mechanism for accelerated vascular calcification in ESRD. J Am Soc Nephrol. 2004;15:2857–2867. doi: 10.1097/01.ASN.0000141960.01035.28. [DOI] [PubMed] [Google Scholar]
- Sage AP, Lu J, Tintut Y, et al. Hyperphosphatemia-induced nanocrystals upregulate the expression of bone morphogenetic protein-2 and osteopontin genes in mouse smooth muscle cells in vitro. Kidney Int. 2011;79:414–422. doi: 10.1038/ki.2010.390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villa-Bellosta R, Sorribas V. Phosphonoformic acid prevents vascular smooth muscle cell calcification by inhibiting calcium-phosphate deposition. Arterioscler Thromb Vasc Biol. 2009;29:761–766. doi: 10.1161/ATVBAHA.108.183384. [DOI] [PubMed] [Google Scholar]
- Ewence AE, Bootman M, Roderick HL, et al. Calcium phosphate crystals induce cell death in human vascular smooth muscle cells: a potential mechanism in atherosclerotic plaque destabilization. Circ Res. 2008;103:e28–e34. doi: 10.1161/CIRCRESAHA.108.181305. [DOI] [PubMed] [Google Scholar]
- Nancollas GH, LoRe M, Perez L, et al. Mineral phases of calcium phosphate. Anat Rec. 1989;224:234–241. doi: 10.1002/ar.1092240213. [DOI] [PubMed] [Google Scholar]
- Pak CYC, Eanes ED, Ruskin B. Spontaneous precipitation of brushite in urine: evidence that brushite is the nidus of renal stones originating as calcium phosphate. Proc Natl Acad Sci USA. 1971;68:1456–1460. doi: 10.1073/pnas.68.7.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiss A, Pipich V, Jahnen-Dechent W, et al. Fetuin-A is a mineral carrier protein: small angle neutron scattering provides new insight on fetuin-a controlled calcification inhibition. Biophys J. 2010;99:3986–3995. doi: 10.1016/j.bpj.2010.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rochette CN, Rosenfeldt S, Heiss A, et al. A shielding topology stabilizes the early stage protein-mineral complexes of fetuin-A and calcium phosphate: a time-resolved small-angle X-ray study. Chembiochem. 2009;10:735–740. doi: 10.1002/cbic.200800719. [DOI] [PubMed] [Google Scholar]
- Heiss A, Jahnen-Dechent W, Endo H, et al. Structural dynamics of a colloidal protein-mineral complex bestowing on calcium phosphate a high solubility in biological fluids. Biointerphases. 2007;2:16–20. doi: 10.1116/1.2714924. [DOI] [PubMed] [Google Scholar]
- Heiss A, DuChesne A, Denecke B, et al. Structural basis of calcification inhibition by alpha 2-HS glycoprotein/fetuin-A. Formation of colloidal calciprotein particles. J Biol Chem. 2003;278:13333–13341. doi: 10.1074/jbc.M210868200. [DOI] [PubMed] [Google Scholar]
- Schafer C, Heiss A, Schwarz A, et al. The serum protein alpha 2-Heremans-Schmid glycoprotein/fetuin-A is a systemically acting inhibitor of ectopic calcification. J Clin Invest. 2003;112:357–366. doi: 10.1172/JCI17202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoshniat S, Bourgine A, Julien M, et al. Phosphate-dependent stimulation of MGP and OPN expression in osteoblasts via the ERK1/2 pathway is modulated by calcium. Bone. 2010;48:894–902. doi: 10.1016/j.bone.2010.12.002. [DOI] [PubMed] [Google Scholar]
- Herrmann M, Schäfer C, Heiss A, et al. Clearance of fetuin-A--containing calciprotein particles is mediated by scavenger receptor-A. Circ Res. 2012;111:575–584. doi: 10.1161/CIRCRESAHA.111.261479. [DOI] [PubMed] [Google Scholar]
- Hamano T, Matsui I, Mikami S, et al. Fetuin-mineral complex reflects extraosseous calcification stress in CKD. J Am Soc Nephrol. 2010;21:1998–2007. doi: 10.1681/ASN.2009090944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui I, Hamano T, Mikami S, et al. Fully phosphorylated fetuin-A forms a mineral complex in the serum of rats with adenine-induced renal failure. Kidney Int. 2009;75:915–928. doi: 10.1038/ki.2008.700. [DOI] [PubMed] [Google Scholar]
- Smith ER, Ford ML, Tomlinson LA, et al. Phosphorylated fetuin-A-containing calciprotein particles are associated with aortic stiffness and a procalcific milieu in patients with pre-dialysis CKD. Nephrol Dial Transplant. 2012;27:1957–1966. doi: 10.1093/ndt/gfr609. [DOI] [PubMed] [Google Scholar]
- Mackay EM, Oliver J. Renal damage following the ingestion of a diet containing an excess of inorganic phosphate. J Exp Med. 1935;61:319–334. doi: 10.1084/jem.61.3.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ori Y, Herman M, Tobar A, et al. Acute phosphate nephropathy-an emerging threat. Am J Med Sci. 2008;336:309–314. doi: 10.1097/MAJ.0b013e318167410c. [DOI] [PubMed] [Google Scholar]
- Haut LL, Alfrey AC, Guggenheim S, et al. Renal toxicity of phosphate in rats. Kidney Int. 1980;17:722–731. doi: 10.1038/ki.1980.85. [DOI] [PubMed] [Google Scholar]
- Bank N, Su WS, Aynedjian HS. A micropuncture study of renal phosphate transport in rats with chronic renal failure and secondary hyperparathyroidism. J Clin Invest. 1978;61:884–894. doi: 10.1172/JCI109014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau K. Phosphate excess and progressive renal failure: the precipitation-calcification hypothesis. Kidney Int. 1989;36:918–937. doi: 10.1038/ki.1989.281. [DOI] [PubMed] [Google Scholar]
- Aihara K, Byer KJ, Khan SR. Calcium phosphate-induced renal epithelial injury and stone formation: involvement of reactive oxygen species. Kidney Int. 2003;64:1283–1291. doi: 10.1046/j.1523-1755.2003.00226.x. [DOI] [PubMed] [Google Scholar]
- Miller PD. The kidney and bisphosphonates. Bone. 2011;49:77–81. doi: 10.1016/j.bone.2010.12.024. [DOI] [PubMed] [Google Scholar]
- Francis MD, Russell RG, Fleisch H. Diphosphonates inhibit formation of calcium phosphate crystals in vitro and pathological calcification in vivo. Science. 1969;165:1264–1266. doi: 10.1126/science.165.3899.1264. [DOI] [PubMed] [Google Scholar]
- Bertram JF, Douglas-Denton RN, Diouf B, et al. Human nephron number: implications for health and disease. Pediatr Nephrol. 2011. [DOI] [PubMed]
- Zhou XJ, Rakheja D, Yu X, et al. The aging kidney. Kidney Int. 2008;74:710–772. doi: 10.1038/ki.2008.319. [DOI] [PubMed] [Google Scholar]
- Ibrahim HN, Foley R, Tan LT, et al. Long-term consequences of kidney donation. N Engl J Med. 2009;360:459–469. doi: 10.1056/NEJMoa0804883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan JC, Busque S, Workeneh B, et al. Effects of aging on glomerular function and number in living kidney donors. Kidney Int. 2010;78:686–692. doi: 10.1038/ki.2010.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young A, Hodsman AB, Boudville N, et al. Bone and mineral metabolism and fibroblast growth factor 23 levels after kidney donation. Am J Kidney Dis. 2011;59:761–769. doi: 10.1053/j.ajkd.2011.09.019. [DOI] [PubMed] [Google Scholar]
- Robertson WG, Morgan DB. The distribution of urinary calcium excretions in normal persons and stone-formers. Clin Chim Acta. 1972;37:503–508. doi: 10.1016/0009-8981(72)90475-5. [DOI] [PubMed] [Google Scholar]
- Gutierrez O, Isakova T, Rhee E, et al. Fibroblast growth factor-23 mitigates hyperphosphatemia but accentuates calcitriol deficiency in chronic kidney disease. J Am Soc Nephrol. 2005;16:2205–2215. doi: 10.1681/ASN.2005010052. [DOI] [PubMed] [Google Scholar]
- Isakova T, Wahl P, Vargas GS, et al. Fibroblast growth factor 23 is elevated before parathyroid hormone and phosphate in chronic kidney disease. Kidney Int. 2011;79:1370–1378. doi: 10.1038/ki.2011.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa H, Nagano N, Urakawa I, et al. Direct evidence for a causative role of FGF23 in the abnormal renal phosphate handling and vitamin D metabolism in rats with early-stage chronic kidney disease. Kidney Int. 2010;78:975–980. doi: 10.1038/ki.2010.313. [DOI] [PubMed] [Google Scholar]
- Vervloet MG, van Ittersum FJ, Büttler RM, et al. Effects of dietary phosphate and calcium intake on fibroblast growth factor-23. Clin J Am Soc Nephrol. 2010;6:383–389. doi: 10.2215/CJN.04730510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antoniucci DM, Yamashita T, Portale AA. Dietary phosphorus regulates serum fibroblast growth factor-23 concentrations in healthy men. J Clin Endocrinol Metab. 2006;91:3144–3149. doi: 10.1210/jc.2006-0021. [DOI] [PubMed] [Google Scholar]
- Ito N, Fukumoto S, Takeuchi Y, et al. Effect of acute changes of serum phosphate on fibroblast growth factor (FGF)23 levels in humans. J Bone Miner Metab. 2007;25:419–422. doi: 10.1007/s00774-007-0779-3. [DOI] [PubMed] [Google Scholar]
- Gutierrez OM, Wolf M, Taylor EN. Fibroblast growth factor 23, cardiovascular disease risk factors, and phosphorus intake in the health professionals follow-up study. Clin J Am Soc Nephrol. 2011;6:2871–2878. doi: 10.2215/CJN.02740311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson T, Nisbeth U, Ljunggren O, et al. Circulating concentration of FGF-23 increases as renal function declines in patients with chronic kidney disease, but does not change in response to variation in phosphate intake in healthy volunteers. Kidney Int. 2003;64:2272–2279. doi: 10.1046/j.1523-1755.2003.00328.x. [DOI] [PubMed] [Google Scholar]
- Coresh J, Selvin E, Stevens LA, et al. Prevalence of chronic kidney disease in the United States. JAMA. 2007;298:2038–2047. doi: 10.1001/jama.298.17.2038. [DOI] [PubMed] [Google Scholar]
- Levin A, Bakris GL, Molitch M, et al. Prevalence of abnormal serum vitamin D, PTH, calcium, and phosphorus in patients with chronic kidney disease: results of the study to evaluate early kidney disease. Kidney Int. 2007;71:31–38. doi: 10.1038/sj.ki.5002009. [DOI] [PubMed] [Google Scholar]
- Nelson RG, Tuttle KR. The new KDOQI clinical practice guidelines and clinical practice recommendations for diabetes and CKD. Blood Purif. 2007;25:112–114. doi: 10.1159/000096407. [DOI] [PubMed] [Google Scholar]
- Gutierrez OM, Wolf M. Dietary phosphorus restriction in advanced chronic kidney disease: merits, challenges, and emerging strategies. Semin Dial. 2010;23:401–406. doi: 10.1111/j.1525-139X.2010.00750.x. [DOI] [PubMed] [Google Scholar]
- Moe SM, Zidehsarai MP, Chambers MA, et al. Vegetarian compared with meat dietary protein source and phosphorus homeostasis in chronic kidney disease. Clin J Am Soc Nephrol. 2011;6:257–264. doi: 10.2215/CJN.05040610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajander EO, Ciftcioglu N. Nanobacteria: an alternative mechanism for pathogenic intra- and extracellular calcification and stone formation. Proc Natl Acad Sci USA. 1998;95:8274–8279. doi: 10.1073/pnas.95.14.8274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajander EO. Nanobacteria--propagating calcifying nanoparticles. Lett Appl Microbiol. 2006;42:549–552. doi: 10.1111/j.1472-765X.2006.01945.x. [DOI] [PubMed] [Google Scholar]
- Kajander EO, Ciftcioglu N, Miller-Hjelle MA, et al. Nanobacteria: controversial pathogens in nephrolithiasis and polycystic kidney disease. Curr Opin Nephrol Hypertens. 2001;10:445–452. doi: 10.1097/00041552-200105000-00023. [DOI] [PubMed] [Google Scholar]
- Young JD, Martel J, Young D, et al. Characterization of granulations of calcium and apatite in serum as pleomorphic mineralo-protein complexes and as precursors of putative nanobacteria. PLoS One. 2009;4:e5421. doi: 10.1371/journal.pone.0005421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JD, Martel J, Young L, et al. Putative nanobacteria represent physiological remnants and culture by-products of normal calcium homeostasis. PLoS One. 2009;4:e4417. doi: 10.1371/journal.pone.0004417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CY, Martel J, Young D, et al. Fetuin-A/albumin-mineral complexes resembling serum calcium granules and putative nanobacteria: demonstration of a dual inhibition-seeding concept. PLoS One. 2009;4:e8058. doi: 10.1371/journal.pone.0008058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raoult D, Drancourt M, Azza S, et al. Nanobacteria are mineralo fetuin complexes. PLoS Pathog. 2008;4:e41. doi: 10.1371/journal.ppat.0040041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Togao O, Doi S, Kuro-o M, et al. Assessment of renal fibrosis with diffusion-weighted MR imaging: study with murine model of unilateral ureteral obstruction. Radiology. 2010;255:772–780. doi: 10.1148/radiol.10091735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doi S, Zou Y, Togao O, et al. Klotho inhibits transforming growth factor-β1 (TGF-β1) signaling and suppresses renal fibrosis and cancer metastasis in mice. J Biol Chem. 2011;286:8655–8665. doi: 10.1074/jbc.M110.174037. [DOI] [PMC free article] [PubMed] [Google Scholar]