

Abstract
The intracellular protozoan parasite Trypanosoma cruzi causes Chagas disease, a serious disorder that affects millions of people in Latin America. Cell invasion by T. cruzi and its intracellular replication are essential to the parasite's life cycle and for the development of Chagas disease. Here, we present evidence suggesting the involvement of the host's cyclooxygenase (COX) enzyme during T. cruzi invasion. Pharmacological antagonist for COX-1, aspirin (ASA), caused marked inhibition of T. cruzi infection when peritoneal macrophages were pretreated with ASA for 30 min at 37°C before inoculation. This inhibition was associated with increased production of IL-1β and nitric oxide (NO∙) by macrophages. The treatment of macrophages with either NOS inhibitors or prostaglandin E2 (PGE2) restored the invasive action of T. cruzi in macrophages previously treated with ASA. Lipoxin ALX-receptor antagonist Boc2 reversed the inhibitory effect of ASA on trypomastigote invasion. Our results indicate that PGE2, NO∙, and lipoxins are involved in the regulation of anti-T. cruzi activity by macrophages, providing a better understanding of the role of prostaglandins in innate inflammatory response to T. cruzi infection as well as adding a new perspective to specific immune interventions.
1. Introduction
Trypanosoma cruzi is an intracellular protozoan parasite causing Chagas disease, which affects millions of people in Latin America. During the acute inflammatory phase of the T. cruzi infection, high-level expression of inducible nitric oxide synthase (iNOS) [1], proinflammatory cytokines [2], and arachidonic acid- (AA-) derived lipids such as leukotrienes, lipoxins (LXs), H (P) ETEs, prostaglandins, and thromboxane is prevalent [3, 4]. In the early T. cruzi infection, nitric oxide (NO∙) and arachidonic acid metabolites could be attributed to resistance, but later on to tissue damage [4].
Prostaglandins (PGs) are oxygenated lipid mediators formed from the ω6 essential fatty acid, arachidonic acid (AA). The committed step in PG biosynthesis is the conversion of AA to PG H2 (PGH2), catalyzed by either PG endoperoxide H synthase-1 or -2, commonly known as cyclooxygenase-1 (COX-1) and cyclooxygenase-2 (COX-2), respectively [5, 6]. Both COX-1 and COX-2 are nonselectively inhibited by nonsteroidal anti-inflammatory drugs (NSAIDs) such as aspirin and ibuprofen, whereas COX-2 activity is selectively blocked by COX-2 inhibitors called coxibs (e.g., celecoxib) [7, 8]. The relevance of these enzymes and the bioactive lipids that they produce are not well understood in parasitic disease, although the role of eicosanoids in the pathogenesis of Chagas disease is becoming more defined [3]. Pharmacological antagonists of COX-1 (aspirin, ASA), COX-2 (celecoxib), or both (indomethacin) have been found to increase mortality and parasitemia (parasite load in peripheral blood and cardiac tissue) regardless of which mouse or T. cruzi strains were used [9–13]. Moreover, evidence suggests that administration of NSAIDs may enhance mortality in chagasic patients [12]. Conversely, others have found that inhibition of PG synthesis/release reduces parasitemia and extends survival of mice infected with T. cruzi [14–17]. This was often associated with a decrease in the levels of circulating inflammatory cytokines (such as TNF-α, IFN-γ, and IL-10) [16]. More recently, treatment with ASA during chronic infection was found to be beneficial with no increase in mortality and substantial improvement in cardiac function [13]. Additionally, the protective effect of ASA could be mediated by the synthesis of 15-epi-lipoxin A4 (15-epi-LXA4) [18].
Given the increasing interest in the role of eicosanoids in T. cruzi infection, we decided to investigate the effect of prostaglandin synthesis inhibition with ASA on inflammatory response and macrophage invasion by T. cruzi.
2. Material and Methods
2.1. Animals
Six- to eight-week-old BALB/c female and male mice were supplied by the Multi-Institutional Center for Biological Investigation, State University of Campinas, Brazil. Mice were maintained under standard conditions in the animal house of the Department of Pathological Sciences, Center for Biological Sciences, State University of Londrina. Commercial rodent diet (Nuvilab-CR1, Quimtia-Nuvital, Colombo, Brazil) and sterilized water were available ad libitum.
All animal procedures were performed in accordance with the guidelines of the Brazilian Code for the Use of Laboratory Animals. The protocols were approved by the Internal Scientific Commission and the Ethics in Animal Experimentation Committee of Londrina State University (Approval Number: CEEA 5492.2012.22).
2.2. Parasites
T. cruzi Y [19] was maintained by weekly intraperitoneal inoculation of Swiss mice with 2 × 105 trypomastigotes. To conduct our experiments, blood from previously infected mice was obtained by cardiac puncture without anticoagulant. The blood was centrifuged at 1,500 ×g for 1 min and allowed to stand at 37°C for 60 min. The supernatant serum containing most of the T. cruzi was centrifuged at 1,200 ×g for 15 min. The sediment was resuspended in 1 mL of RPMI 1640 medium (GIBCO, Gran Island, NY) containing 10% inactivated fetal bovine serum (FBS), 100 units of penicillin, and 100 μg streptomycin (GIBCO, Gran Island, NY).
Trypomastigotes were derived from the supernatant of T. cruzi-infected LLC-Mk2 culture cells (ATCC CCL-7; American Type Culture Collection, Rockville, MD) grown in RPMI 1640 medium containing 10% inactivated fetal bovine serum (FBS), 40 μg mL−1 gentamicin, 100 units of penicillin, and 100 μg streptomycin (GIBCO, Gran Island, Y). Subconfluent cultures of LLC-Mk2 were infected with 5 × 106 trypomastigotes. Free parasites were removed after 24 h and cultures were maintained in 10% FBS-RPMI 1640. Five days postinfection, free trypomastigote forms could be found in the cell supernatants.
2.3. Macrophage Culture
Mice were inoculated intraperitoneally with 2 mL of 5% thioglycollate and, 4 days later, the elicited cells from the peritoneal exudates were harvested in cold PBS. Mouse peritoneum was washed with 5 mL ice-cold, serum-free RPMI. Peritoneal cells from 3–6 mice were pooled and left to adhere in complete medium (RPMI, 2 mM glutamine, 1 mM sodium pyruvate, 40 μg mL−1 gentamicin, and 10 mM HEPES) for 24 h in 24-well plates at 2 × 105 cells/well. Each suspension of pooled peritoneal cells was plated in triplicate wells. Then, nonadherent cells were washed away and adherent cells received complete medium. The macrophages were plated onto 13 mm round glass coverslips and washed in warm phosphate-buffered saline (PBS) before the interaction assays. In addition, 2.0 × 105 macrophages were plated onto 96-well dishes. One set of plates was used to quantify IL-1β and the other set for NO∙ detection.
2.4. Treatment of Macrophages with Drugs and Macrophage Invasion Assay
Before the experiments, peritoneal macrophages previously washed were incubated for 30 min at 37°C in a 5% CO2 atmosphere in the presence of different concentrations of ASA (2.5 mM, 1.25 mM, and 0.625 mM) to test its effect on internalization of the parasite into the host cell. After incubation, the medium containing ASA was removed, and macrophages were allowed to interact with trypomastigote forms added in a ratio of 5 parasites per cell. The interaction was allowed to proceed for 2 h, at 37°C in a 5% CO2 atmosphere. The cells were then washed three times, fixed with Bouin's fixative, stained with Giemsa (Merck) stain, and observed with a light microscope at 1000x magnification. Other treatments included incubation with aminoguanidine (1 mM) or L-NAME (1.0 mM) for 60 min at 37°C with or without ASA.
The internalization index was calculated by multiplying the percentage of infected cells by the mean number of parasites per infected cell [20]. All internalization indices were normalized. Experiments were performed in triplicate, and six independent experiments were completed. All experiments included untreated, infected peritoneal macrophages as controls. The quantification was carried out using light microscopy where a total of 500 cells were randomly counted. The viability of the cells obtained from the cultures before and after incubation experiments was determined using MTT (Sigma-Aldrich) assay, showing the mitochondrial activity of living cells. The culture medium was aspirated, and MTT (0.5 mg mL−1) was added to the cells prior to incubation at 37°C for 4 h. The supernatant was aspirated and dimethyl sulfoxide (Sigma-Aldrich) was added to the wells. Insoluble crystals were dissolved by mixing and the plates were read using a BioRad multiplate reader (Hercules, CA), at a test wavelength of 570 nm and a reference wavelength of 630 nm.
2.5. Nitrite Measurements
Production of nitric oxide (NO∙) was determined by measuring the level of accumulated nitrite, a metabolite of NO∙ in the culture supernatant using Griess reagent (Sigma-Aldrich). After 24 h of treatment with ASA (0.625 mM), the culture supernatants were collected and mixed with an equal volume of Griess reagent in 96-well culture plates and incubated at room temperature for 10 min. The absorbance was measured at 540 nm and nitrite concentrations were calculated by reference to a standard curve generated by known concentrations of sodium nitrite.
2.6. Immunocytochemistry Labeling for iNOS
Immunocytochemistry for iNOS was performed on coverslip-adherent cells using the labeled streptavidin biotin method with a LSAB KIT (DAKO Japan, Kyoto, Japan) without microwave accentuation. The coverslips were incubated with 10% Triton X-100 solution for 1 h, washed 3 times in PBS, and treated for 40 min at room temperature with 10% BSA. The coverslips were then incubated overnight at 4°C with the primary antibody (anti-iNOS rabbit monoclonal antibody diluted 1 : 200, BD Biosciences, catalog number 610599), followed by secondary antibody treatment for 2 h at room temperature. Horseradish peroxidase activity was visualized by treatment with H2O2 and 3,3′-diaminobenzidine (DAB) for 5 min. At the last step, the sections were weakly counterstained with Harry's hematoxylin (Merck). For each case, negative controls were prepared by omitting the primary antibody. Intensity and localization of immune reaction against primary antibody used were examined on all coverslips using a photomicroscope (Olympus BX41, Olympus Optical Co., Ltd., Tokyo, Japan). For the image analysis study, photomicroscopic color slides of representative areas (magnification, 40x) were digitally acquired. After conversion of the images into grey scale (Adobe Photoshop) iNOS-positive pixels and total pixels became thresholds and were processed by Image J program. Positive immunostained area was calculated as the proportion of positive pixels to total pixels (%).
2.7. ELISA for IL-1β
Culture supernatants from peritoneal macrophages in 96-well plates treated with ASA (0.625 mM) or untreated, either infected or not infected with T. cruzi, were incubated for 24 h. Levels of IL-1β in 100 μL medium were measured by commercial ELISA kits (Ready-SET-Go! eBioscience, San Diego, CA), according to the manufacturer's instructions.
2.8. Statistical Analysis
The statistical analysis was conducted using one way ANOVA with Bonferroni's multiple comparison test. Values are presented as ± standard error of mean. The results were considered significant when P < 0.05. Statistical analysis was performed using the GraphPad Prism 5.0 computer software application (GraphPad Software, San Diego, CA, USA).
3. Results
3.1. Aspirin Inhibits T. cruzi Entry into Macrophages
To determine whether COX-derived mediators are involved in T. cruzi entry into host cells, cells were treated with increasing amounts of ASA for 30 min and after treatment, the medium containing ASA was removed before macrophage invasion assay in order to guarantee that ASA affected only the host cell and not the parasites. After 2 h of incubation with parasites, which provides sufficient time for them to enter into macrophages, the free parasites were removed. Aspirin irreversibly inhibits COX-1 by acetylation of a single serine residue on the enzyme [21] and this inactivation persists, that is, ≥24 hours. In some cases, the medium with increasing ASA concentrations was added every 24 hours until the end of the T. cruzi infection period (7 days, Figure 4(d)). Figure 1(a) shows that ASA markedly inhibited the internalization of trypomastigote by macrophages at all concentrations tested (P < 0.0001). Thus, PGE2 synthesis inhibition using ASA improves macrophage response against T. cruzi infection. The cytotoxicity of ASA in macrophages was evaluated by MTT assay (Figure 1(a), insert). ASA did not induce cell death, as the concentrations of ASA used in all experiments reported were too low to cause cytotoxicity [22]. Light microscopy observations confirm T. cruzi inhibition invasion of ASA treated cells (Figure 1(c)).
Figure 4.
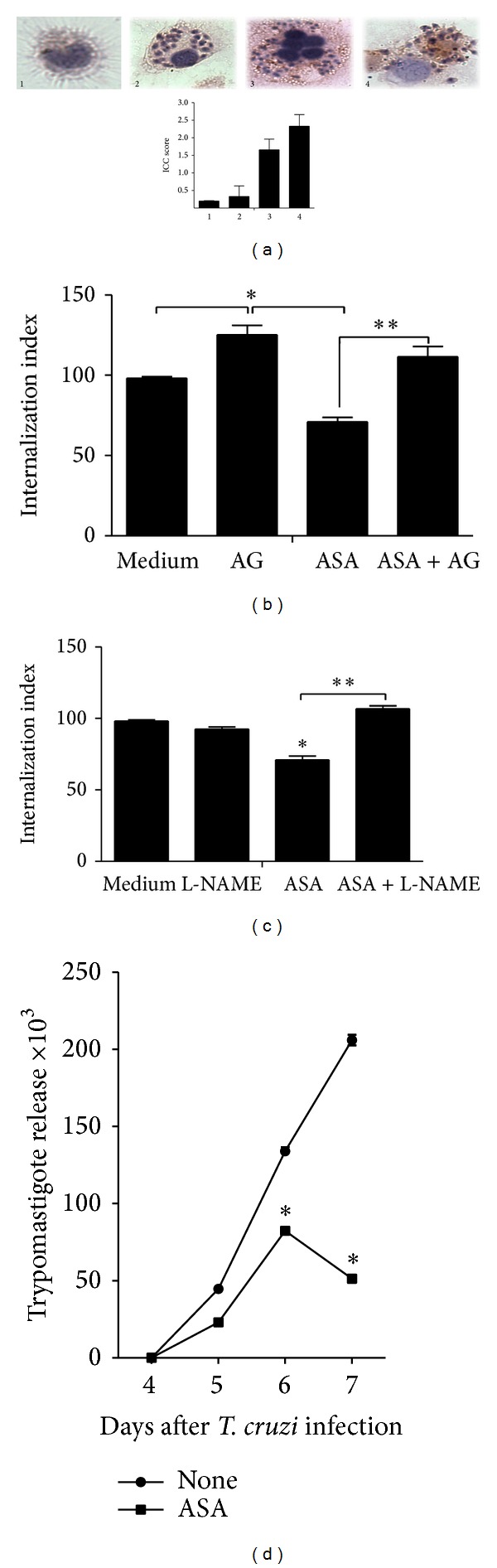
Effect of ASA on macrophage activity depends on NO∙ production. (a) Aspirin treatment stimulated iNOs expression in T. cruzi-infected macrophages. Immunocytochemistry for iNOS was performed on coverslip-adherent cells using the labeled streptavidin biotin method with a LSAB KIT (DAKO Japan, Kyoto, Japan) without microwave accentuation. (1) Intracellular iNOS protein cannot be detected by immunocytochemistry in uninfected (control) macrophages, (2) T. cruzi-infected cell, (3) ASA (0.625 mM) was an effective inducer of iNOS expression in peritoneal macrophages, and (4) the addition of PGE2 (3.52 ng mL−1) to culture media increased iNOS mRNA expression in macrophages infected. Macrophages were treated for 30 minutes separately with 0.625 μM ASA. After treatment, macrophages were washed and incubated with aminoguanidine (AG, 1.0 mM) (b) or L-NAME (c) at 1.0 mM for 1 h at 37°C. After treatment, macrophages were washed again and interacted with 5 : 1 trypomastigotes for 2 hours at 37°C, after which they are washed, fixed with Bouin's fixative, and stained with Giemsa. Quantification was carried out under a light microscope where the number of intracellular parasites was counted in a total of at least 500 cells. Results are the mean ± standard error for triplicate determinations and are representative of two independent experiments. (d) Effect of aspirin upon trypomastigote release in aspirin-treated T. cruzi-infected macrophages. Cells were infected with T. cruzi trypomastigotes and treated daily with ASA at 0.625 mM; after 4 days of treatment, trypomastigotes release to supernatants was found and was measured until day 7 after infection. Results are the mean ± standard error for triplicate determinations and are representative of two independent experiments. *P < 0.01 for a comparison with infected cells cultured in medium alone. **P < 0.01 for a comparison with infected cells cultured treated with ASA.
Figure 1.
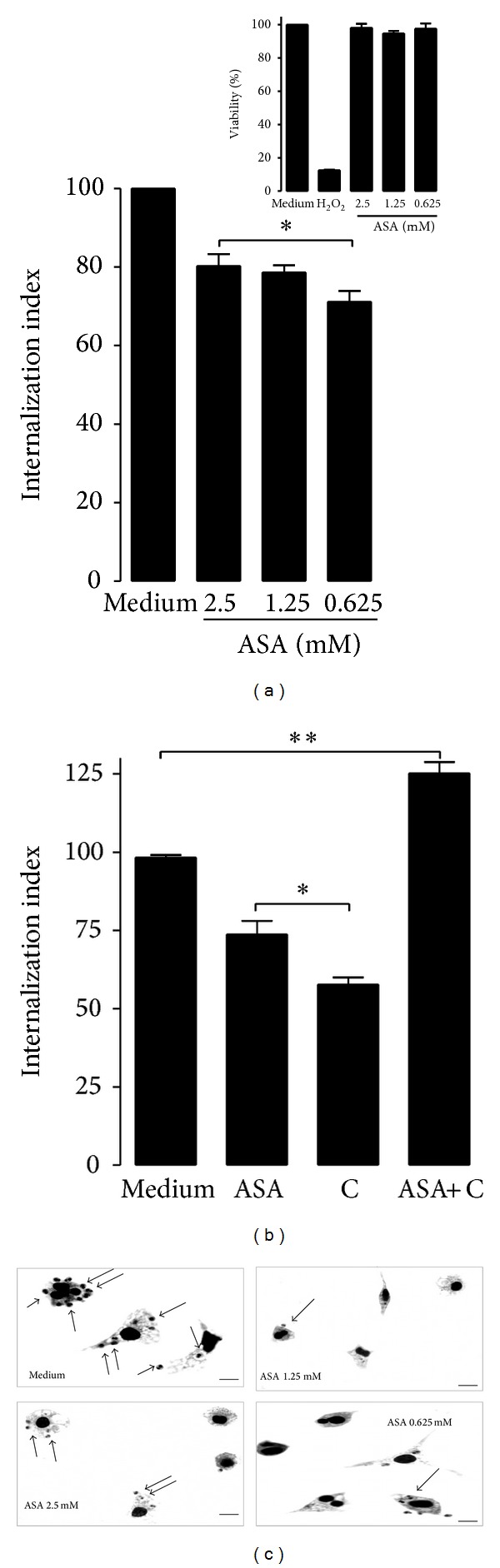
Aspirin (ASA) impairs Trypanosoma cruzi internalization by peritoneal macrophages. (a) Internalization index of the interaction process between macrophages treated for 30 minutes with increasing concentrations of ASA (0.625, 1.25, and 2.5 mM) and exposed to T. cruzi (Y strain). After treatment with ASA, peritoneal macrophages interacted with 5 : 1 trypomastigotes for 2 hours, after which they are washed, fixed with Bouin's fixative, and stained with Giemsa. Quantification was carried out under a light microscope where the number of intracellular parasites was counted in a total of at least 500 cells. MTT assay to measure cell viability in macrophages after treatment with ASA at 0.625 to 2.5 mM concentrations. H2O2 (1000 μM) was used as negative control (insert). Values are the means ± standard error of mean of 10 experiments or two experiments (MTT assay). *P < 0.0001 for a comparison with infected cells cultured in medium alone. (b) The combined effect of celecoxib and aspirin on the entry of T. cruzi into phagocytic cells. Macrophages were treated for 30 minutes with celecoxib (0.625 mM) and ASA (0.625 mM) exposed to T. cruzi as describe above. Results are the mean ± standard error for triplicate determinations and are representative of three independent experiments. *P < 0.0001, **P < 0.001 for a comparison with infected cells cultured in medium alone. (c) Light microscopy observations confirm T. cruzi inhibition invasion of ASA treated cells. Observation after Giemsa staining by light microscopy of the interaction process between peritoneal macrophages treated (or not) with different concentration of ASA and exposed to trypomastigotes forms of T. cruzi. The black arrows indicate internalized parasites. Bars = 10 μm.
3.2. COX-2 Inhibition Together with Aspirin Restores the Infectivity of Trypomastigotes in Peritoneal Macrophages
We examined the combined effect of celecoxib (COX-2 selective inhibitor) and ASA on the entry of T. cruzi into phagocytic cells. Peritoneal macrophages were incubated separately, with or without drugs (ASA and celecoxib) either alone or in combination. Figure 1(b) shows that the drugs in combination significantly (P < 0.01) restored the invasive capacity of trypomastigotes. None of the drugs (ASA, celecoxib, or their combination) showed cytotoxicity against uninfected peritoneal macrophages (data not shown). These results indicate that the enzyme activities of COX-1 and COX-2 favor invasion, but simultaneous inhibition of both activities also favors parasite invasion, probably by allowing a new panorama of eicosanoids production.
3.3. Aspirin Decreases Trypomastigotes Release to Culture Supernatants from T. cruzi-Infected Macrophages
Four days postinfection, macrophages began releasing trypomastigotes into the supernatant (Figure 4(d)). Trypomastigotes release to culture supernatants from T. cruzi-infected macrophages was diminished by ASA (0.625 mM). This result is in agreement with previous reports, showing that aspirin and other COX inhibitors decrease T. cruzi infection in vitro [14–16, 23].
3.4. Aspirin Inhibits T. cruzi Entry into Macrophages via ALX
Lipoxins (LXs) are endogenous lipid mediators with potent anti-inflammatory and proresolving actions [24, 25]. Native LXs and their stable analogues exert their biological effects by binding to a G-protein-coupled receptor, denoted as ALX [26, 27]. The effect of ASA has been associated, in part, to a switch to the AA pathway linked to the acetylation of the COX-2 isoenzyme. This reaction enables COX-2 to synthesize LXs as 15-epi-LXs (15-epi-LXA4) [22]. To evaluate the effect of ASA-triggered LXs on the entry of T. cruzi into phagocytic cells, peritoneal macrophages were incubated separately either with or without drugs (ASA or Boc-2, a specific antagonist of the 15-epi-LXs) either alone or in combination. The inhibitory effect of ASA on the entry of T. cruzi into macrophages was prevented by Boc-2 demonstrating that the effect of ASA on the entry of T. cruzi into macrophages could be mediated by the synthesis of 15-epi-LXA4. Furthermore, Boc-2 did not have any effect on the entry of T. cruzi into macrophages when used alone (Figure 2(a)).
Figure 2.
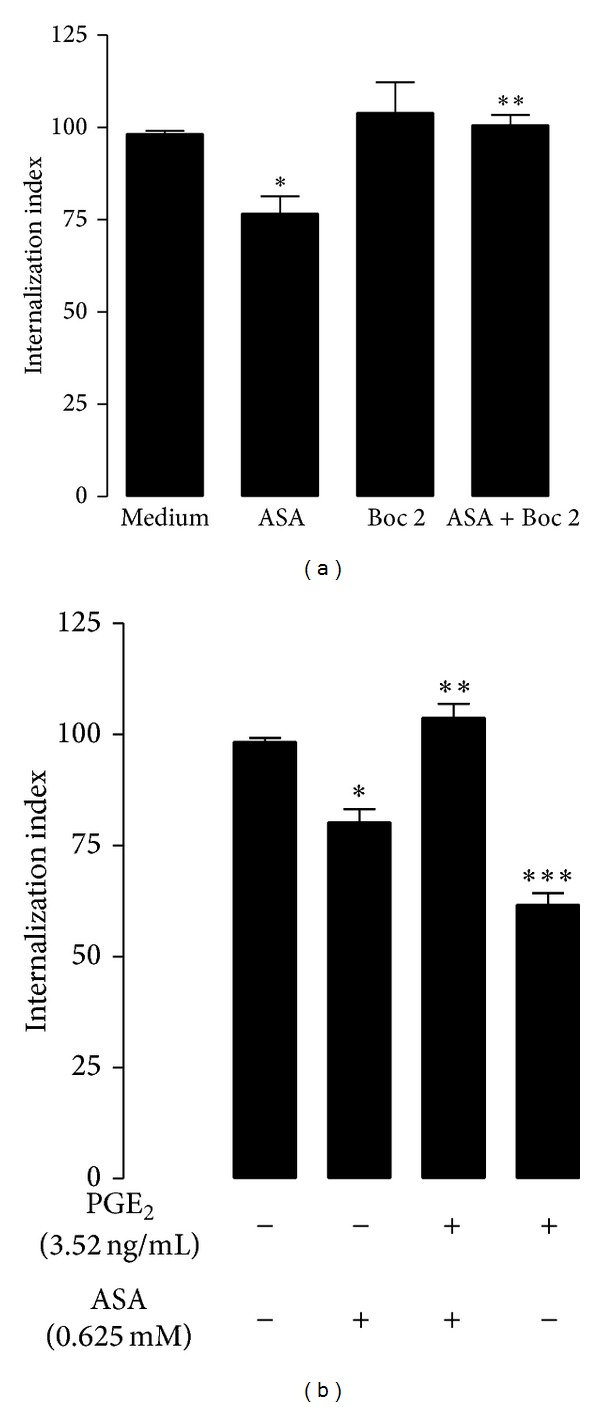
The effect of ASA on the entry of T. cruzi into macrophages was prevented by Boc-2. (a) Internalization index of the interaction process between macrophages treated for 30 minutes separately either with or without drugs (0.625 μM ASA or 100 μM Boc-2). Results are the mean ± standard error for triplicate determinations and are representative of two independent experiments. *P < 0.01, for a comparison with infected cells cultured in medium alone. **P < 0.001, for a comparison with infected cell treated with ASA. (b) PGE2 restores aspirin effect on T. cruzi entry into macrophages. Macrophages were treated for 30 minutes separately either with or without PGE2 (35.2, 3.52, or 0.35 ng mL−1) alone or in combination (ASA 0.625 mM + PGE2 3.52 ng mL−1). Results are the mean ± standard error and are representative of two independent experiments. *P < 0.001 for a comparison with infected cells cultured in medium alone. **P < 0.05 for comparison with infected cell treated with ASA or PGE2 alone. ***P < 0.05 for a comparison with infected cells cultured in medium alone.
3.5. PGE2 Restores Aspirin Effect on T. cruzi Entry into Macrophages
Figure 2(b) (insert) shows that treatment of macrophages with high concentrations of PGE2 causes inhibition of T. cruzi entry into macrophages. When PGE2 (3.52 ng mL−1) was added alone or in combination with ASA, the effect of ASA was inhibited, indicating that PGE2 is involved in the internalization of T. cruzi trypomastigotes into peritoneal macrophages.
3.6. Aspirin Modulates Innate Inflammatory Response of Macrophages Infected with T. cruzi
The internalization of T. cruzi into macrophages stimulated the release of IL-1β while ASA increased IL-1β production by infected macrophages (Figure 3(a)). The effect of ASA on NO∙ production was evaluated by detection of nitrite in T. cruzi-infected macrophage supernatants using Griess reaction (Figure 3(b)). NO∙ production in macrophages was stimulated by T. cruzi and was also increased by prior treatment of macrophages with ASA.
Figure 3.
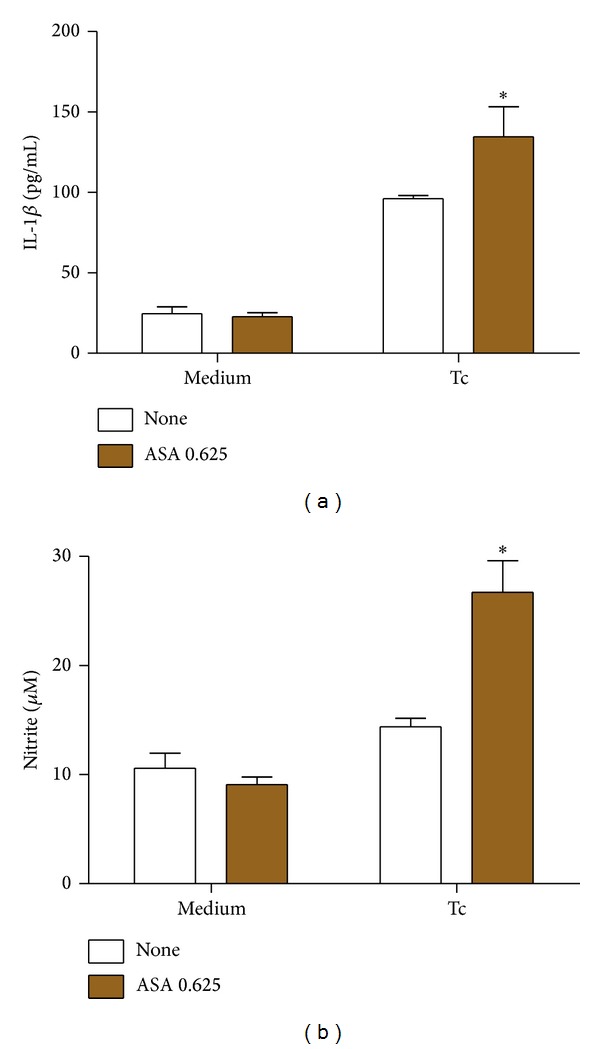
Effects of ASA upon IL-1β and nitrite production in T. cruzi-infected macrophages. Macrophages were treated for 30 minutes with ASA (0.625 mM) and exposed to T. cruzi (Y strain). After treatment with ASA, cells interacted with 5 : 1 trypomastigotes for 2 hours, after which they are cultured at 37°C in 5% CO2 during 24 h. Afterwards, IL-1β (a) and nitrite (b) levels in supernatant were measured with a specific enzyme-linked immunosorbent assay and by Griess reaction, respectively. Results are the mean ± standard error for duplicate determinations and are representative of four independent experiments. *P < 0.05 for a comparison with cell culture in medium alone.
In addition, we observed that ASA treatment stimulated iNOS expression in T. cruzi-infected macrophages (Figure 4(a)). To confirm that the effect of ASA on macrophage activity depends on NO∙ levels, we assessed the entry of trypomastigotes into macrophages incubated with aminoguanidine (AG, iNOS inhibitor) or with L-NAME (c-NOS inhibitor). We found that both inhibitors reversed the effects of ASA (Figures 4(b) and 4(c)).
4. Discussion
Previous studies have shown that the release of eicosanoids during infection with T. cruzi regulates host responses and controls disease progression [3, 10, 11, 13, 14, 28–31]. PGs, together with NO∙ and TNF-α, participate in a complex circuit that controls lymphoproliferative and cytokine responses in T. cruzi infection [10]. However, the involvement of COX-mediated PG production in the entry of T. cruzi into macrophages is largely unexplored. The data shown herein demonstrates that the treatment of macrophages with ASA significantly inhibits internalization of T. cruzi trypomastigotes and strongly supports the idea that the COX pathway plays a fundamental role in the process of parasite invasion. In fact, PGE2 production increases significantly in T. cruzi-infected macrophages as compared to uninfected macrophages [32] and synergistically enhances the activity of nifurtimox and benznidazole on infected RAW 264.7 cells [23].
Ours results suggest that the actions of ASA depend on COX-2-derived biosynthesis of products. This is demonstrated by the observation that celecoxib reversed the aspirin-induced inhibition of entry of T. cruzi into macrophages. The problem of NSAIDs coadministration is actively discussed in literature in the context of uncertainty of the resulting therapeutic and side effects arising from such combinations. The mechanism of such a suppression of aspirin inhibitory effect on COX-1 by other NSAIDs has been difficult to satisfactorily explain. This could be due to celecoxib binding strongly to a monomer of COX-1 without affecting AA oxygenation [7]. In fact, a related study has shown that celecoxib prevented ASA inhibition in a dog model of thrombosis [33]. However, the application of our approach to investigate the combined effect of aspirin and celecoxib on COX-1 during coadministration confirmed the ability of celecoxib to suppress the aspirin-mediated inhibition of COX-1 in vitro conditions [7].
In addition, the effects of ASA on T. cruzi infection have been associated in part, to a switch to the AA pathway linked to the acetylation of the COX-2 isoenzyme [18]. Such modification promotes the synthesis of the 15-R-HETE intermediate, which can be transformed by 5-lipooxygenase to 15-epi-LXA4, a lipid involved in the resolution of inflammation [34]. Accordingly, we assessed the effect of Boc-2 (a specific antagonist of the 15-epi-LXs) on low doses of ASA, indicating that 15-epi-LXA4 is probably involved in the inhibitory effect exerted by ASA on the internalization of T. cruzi by macrophages.
Interestingly, when macrophages were treated with PGE2 concentration as high as 3.52 ng mL−1, we observed a reduction in the entry of parasites, but when we used low concentration of PGE2 (0.35 ng mL−1), the entry of T. cruzi was similar to that observed in untreated macrophages. In addition, we found reversal of ASA effect, even when PGE2 concentration as low as 3.52 ng mL−1 was used, indicating that PGE2 has an important role in ASA effect. Inhibition of COX activity may increase NO∙ levels, thus restoring the antiparasitic activity of macrophages [23]. Our results are in agreement with this hypothesis. To confirm that the effect of ASA on macrophage activity depends upon restoring NO∙ levels, we assessed the invasive capacity of T. cruzi when cells were incubated with aminoguanidine (AG), an iNOS inhibitor. We found that 1.0 mM AG reverses ASA effect totally. In addition, we showed that iNOS expression in macrophages was increased with ASA treatment, suggesting that iNOS-dependent NO∙ production is responsible for ASA effects. We did find reversal of ASA effect with L-NAME (1.0 mM), indicating the role of cNOS in ASA activity. This can be explained as high concentrations of L-NAME may interfere in the selectivity for cNOS and can inhibit other isoforms of NOS, such as nNOS and iNOS. So, NO∙ deficiency induced by L-NAME could be explaining our results.
Additionally, polyamines seem to be crucial for trypomastigote internalization process in, at least, some cellular types and infection progression [35]. In T. cruzi-infected macrophages, COX is related to the increase of ornithine decarboxylase (ODC) activity [15], which might increase the polyamine content in macrophages. Since T. cruzi uses these polyamines to synthesize trypanothione (an enzyme that participates in the hydroperoxide detoxification of T. cruzi), the inhibition of COX by ASA probably resulted in a reduction in polyamine levels caused by inhibition of ODC, indirectly contributing to decrease trypanothione synthesis in T. cruzi, as suggested by López-Muñoz and collaborators [23].
Finally, in T. cruzi-infected macrophages, COX inhibition by ASA was related to the increase of IL-1β, which also might explain the increase of antiparasitic activity of macrophages treated with ASA. In fact, IL-1β is critical for the restriction of Leishmania amazonensis infection [36] and it recently was demonstrated that macrophages treated with IL-1β released fewer trypomastigotes than untreated macrophages and IL-1β triggered NO∙ release by T. cruzi-infected macrophages in a dose dependent manner [37].
5. Conclusion
In conclusion, this is the first report, to our knowledge, showing the in vitro effect of aspirin on T. cruzi entry into peritoneal macrophages and the influence of COX pathway on innate inflammatory response to T. cruzi infection, adding a new perspective to immune interventions against Chagas disease.
Acknowledgments
This work was supported by the Conselho Nacional de Desenvolvimento Científico e Tecnológico, Brazil (CNPq, 302097/2010-474792/2011-0), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), and the Fundação Araucaria (Convênio 419/2009). LVR, WAVJ, MCMP, and PPF received research fellowships from CNPq.
Conflict of Interests
The authors declare no conflict of interests.
Authors' Contribution
P. Pinge-Filho, S. F. Yamada-Ogatta, M. C. Martins-Pinge, and L. V. Rizzo participated in research design. A. D. Malvezi, C. Panis, R. Valeriano da Silva, M. I. Lovo-Martins, N. G. Zanluqui, and V. L. H. Tatakihara conducted experiments. W. A. Verri Jr., and L. M. Yamauchi contributed to the new reagents or analytical tools. A. D. Malvezi, C. Panis, R. Valeriano da Silva, M. I. Lovo-Martins, and P. Pinge-Filho performed data analysis. A. D. Malvezi, C. Panis, and P. Pinge-Filho contributed to the writing of the paper.
References
- 1.Rottenberg ME, Castaños-Velez E, de Mesquita R, Laguardia OG, Biberfeld P, Örn A. Intracellular co-localization of Trypanosoma cruzi and inducible nitric oxide synthase (iNOS): evidence for dual pathway of iNOS induction. European Journal of Immunology. 1996;26(12):3203–3213. doi: 10.1002/eji.1830261254. [DOI] [PubMed] [Google Scholar]
- 2.Gomes JA, Molica AM, Keesen TS, et al. Inflammatory mediators from monocytes down-regulate cellular proliferation and enhance cytokines production in patients with polar clinical forms of Chagas disease. Human Immunology. 2014;75(1):20–28. doi: 10.1016/j.humimm.2013.09.009. [DOI] [PubMed] [Google Scholar]
- 3.Machado FS, Mukherjee S, Weiss LM, Tanowitz HB, Ashton AW. Bioactive lipids in Trypanosoma cruzi infection. Advances in Parasitology. 2011;76:1–31. doi: 10.1016/B978-0-12-385895-5.00001-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cardoni RL, Antúnez MI. Circulating levels of cyclooxygenase metabolites in experimental Trypanosoma cruzi infections. Mediators of Inflammation. 2004;13(4):235–240. doi: 10.1080/09637480400003022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rocca B, FitzGerald GA. Cyclooxygenases and prostaglandins: shaping up the immune response. International Immunopharmacology. 2002;2(5):603–630. doi: 10.1016/s1567-5769(01)00204-1. [DOI] [PubMed] [Google Scholar]
- 6.Park JY, Pillinger MH, Abramson SB. Prostaglandin E2 synthesis and secretion: the role of PGE2 synthases. Clinical Immunology. 2006;119(3):229–240. doi: 10.1016/j.clim.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 7.Rimon G, Sidhu RS, Lauver DA, et al. Coxibs interfere with the action of aspirin by binding tightly to one monomer of cyclooxygenase-1. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(1):28–33. doi: 10.1073/pnas.0909765106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pountos I, Georgouli T, Bird H, Giannoudis PV. Nonsteroidal anti-inflammatory drugs: prostaglandins, indications, and side effects. International Journal of Interferon, Cytokine and Mediator Research. 2011;3(1):19–27. [Google Scholar]
- 9.Celentano AM, Gorelik G, Solana ME, Sterin-Borda L, Borda E, González Cappa SM. PGE2 involvement in experimental infection with Trypanosoma cruzi subpopulations. Prostaglandins. 1995;49(3):141–153. doi: 10.1016/0090-6980(95)00002-r. [DOI] [PubMed] [Google Scholar]
- 10.Pinge-Filho P, Tadokoro CE, Abrahamsohn IDA. Prostaglandins mediate suppression of lymphocyte proliferation and cytokine synthesis in acute Trypanosoma cruzi infection. Cellular Immunology. 1999;193(1):90–98. doi: 10.1006/cimm.1999.1463. [DOI] [PubMed] [Google Scholar]
- 11.Hideko Tatakihara VL, Cecchini R, Borges CL, et al. Effects of cyclooxygenase inhibitors on parasite burden, anemia and oxidative stress in murine Trypanosoma cruzi infection. FEMS Immunology and Medical Microbiology. 2008;52(1):47–58. doi: 10.1111/j.1574-695X.2007.00340.x. [DOI] [PubMed] [Google Scholar]
- 12.Sterin-Borda L, Gorelik G, Goren N, Gonzalez Cappa S, Celentano AM, Borda E. Lymphocyte muscarinic cholinergic activity and PGE2 involvement in experimental Trypanosoma cruzi infection. Clinical Immunology and Immunopathology. 1996;81(2):122–128. doi: 10.1006/clin.1996.0167. [DOI] [PubMed] [Google Scholar]
- 13.Mukherjee S, Machado FS, Huang H, et al. Aspirin treatment of mice infected with Trypanosoma cruzi and implications for the pathogenesis of chagas disease. PLoS ONE. 2011;6(2) doi: 10.1371/journal.pone.0016959.e16959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abdalla GK, Faria GEL, Silva KT, Castro ECC, Reis MA, Michelin MA. Trypanosoma cruzi: the role of PGE2 in immune response during the acute phase of experimental infection. Experimental Parasitology. 2008;118(4):514–521. doi: 10.1016/j.exppara.2007.11.003. [DOI] [PubMed] [Google Scholar]
- 15.Freire-de-Lima CG, Nascimento DO, Soares MBP, et al. Erratum: uptake of apoptotic cells drives the growth of a pathogenic trypanosome in macrophages (Nature (2000) 403 (199-203)) Nature. 2000;404(6780):p. 904. doi: 10.1038/35003208. [DOI] [PubMed] [Google Scholar]
- 16.Michelin MA, Silva JS, Cunha FQC. Inducible cyclooxygenase released prostaglandin mediates immunosuppression in acute phase of experimental Trypanosoma cruzi infection. Experimental Parasitology. 2005;111(2):71–79. doi: 10.1016/j.exppara.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 17.Paiva CN, Arras RH, Lessa LP, et al. Unraveling the lethal synergism between Trypanosoma cruzi infection and LPS: a role for increased macrophage reactivity. European Journal of Immunology. 2007;37(5):1355–1364. doi: 10.1002/eji.200636705. [DOI] [PubMed] [Google Scholar]
- 18.Molina-Berríos A, Campos-Estrada C, Henriquez N, et al. Protective role of acetylsalicylic acid in experimental Trypanosoma cruzi infection: evidence of a 15-epi-lipoxin A4-mediated effect. PLoS Neglected Tropical Diseases. 2013;7(4) doi: 10.1371/journal.pntd.0002173.e2173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Silva LHP, Nussenzweig V. Sobre uma cepa de Trypanosoma cruzi altamente virulenta para o camundongo branco. Folia Clinica et Biológica. 1953;20:191–203. [Google Scholar]
- 20.Barrias ES, Reignault LC, de Souza W, Carvalho TMU. Dynasore, a dynamin inhibitor, inhibits Trypanosoma cruzi entry into peritoneal macrophages. PLoS ONE. 2010;5(1) doi: 10.1371/journal.pone.0007764.e7764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smith WL. Nutritionally essential fatty acids and biologically indispensable cyclooxygenases. Trends in Biochemical Sciences. 2008;33(1):27–37. doi: 10.1016/j.tibs.2007.09.013. [DOI] [PubMed] [Google Scholar]
- 22.Cezar-De-Mello PFT, Vieira AM, Nascimento-Silva V, Villela CG, Barja-Fidalgo C, Fierro IM. ATL-1, an analogue of aspirin-triggered lipoxin A 4, is a potent inhibitor of several steps in angiogenesis induced by vascular endothelial growth factor. British Journal of Pharmacology. 2008;153(5):956–965. doi: 10.1038/sj.bjp.0707650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.López-Muñoz R, Faúndez M, Klein S. Trypanosoma cruzi: in vitro effect of aspirin with nifurtimox and benznidazole. Experimental Parasitology. 2010;124(2):167–171. doi: 10.1016/j.exppara.2009.09.005. [DOI] [PubMed] [Google Scholar]
- 24.Serhan CN, Chiang N, van Dyke TE. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nature Reviews Immunology. 2008;8(5):349–361. doi: 10.1038/nri2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Russell CD, Schwarze J. The role of pro-resolution lipid mediators in infectious disease. Immunology. 2014;141(2):166–173. doi: 10.1111/imm.12206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fiore S, Antico G, Aloman M, Sodin-Semrl S. Lipoxin A4 biology in the human synovium. Role of the ALX signaling pathways in modulation of inflammatory arthritis. Prostaglandins Leukotrienes and Essential Fatty Acids. 2005;73(3-4):189–196. doi: 10.1016/j.plefa.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 27.Chiang N, Serhan CN, Dahlén S-E, et al. The lipoxin receptor ALX: potent ligand-specific and stereoselective actions in vivo. Pharmacological Reviews. 2006;58(3):463–487. doi: 10.1124/pr.58.3.4. [DOI] [PubMed] [Google Scholar]
- 28.Clària J, Lee MH, Serhan CN. Aspirin-triggered lipoxins (15-epi-LX) are generated by the human lung adenocarcinoma cell line (A549)-neutrophil interactions and are potent inhibitors of cell proliferation. Molecular Medicine. 1996;2(5):583–596. [PMC free article] [PubMed] [Google Scholar]
- 29.Ashton AW, Mukherjee S, Nagajyothi FNU, et al. Thromboxane A2 is a key regulator of pathogenesis during Trypanosoma cruzi infection. Journal of Experimental Medicine. 2007;204(4):929–940. doi: 10.1084/jem.20062432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Borges CL, Cecchini R, Tatakihara VLH, et al. 5-Lipoxygenase plays a role in the control of parasite burden and contributes to oxidative damage of erythrocytes in murine Chagas’ disease. Immunology Letters. 2009;123(1):38–45. doi: 10.1016/j.imlet.2009.02.002. [DOI] [PubMed] [Google Scholar]
- 31.Panis C, Mazzuco TL, Costa CZF, et al. Trypanosoma cruzi: effect of the absence of 5-lipoxygenase (5-LO)-derived leukotrienes on levels of cytokines, nitric oxide and iNOS expression in cardiac tissue in the acute phase of infection in mice. Experimental Parasitology. 2011;127(1):58–65. doi: 10.1016/j.exppara.2010.06.030. [DOI] [PubMed] [Google Scholar]
- 32.Borges MM, Kloetzel JK, Andrade HF, Jr., Tadokoro CE, Pinge-Filho P, Abrahamsohn I. Prostaglandin and nitric oxide regulate TNF-α production during Trypanosoma cruzi infection. Immunology Letters. 1998;63(1):1–8. doi: 10.1016/s0165-2478(98)00034-0. [DOI] [PubMed] [Google Scholar]
- 33.Hennan JK, Huang J, Barrett TD, et al. Effects of selective cyclooxygenase-2 inhibition on vascular responses and thrombosis in canine coronary arteries. Circulation. 2001;104(7):820–825. doi: 10.1161/hc3301.092790. [DOI] [PubMed] [Google Scholar]
- 34.Wang Y-P, Wu Y, Li L-Y, et al. Aspirin-triggered lipoxin A4attenuates LPS-induced pro-inflammatory responses by inhibiting activation of NF-κB and MAPKs in BV-2 microglial cells. Journal of Neuroinflammation. 2011;8, article no. 95 doi: 10.1186/1742-2094-8-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kierszenbaum F, Wirth JJ, McCann PP, Sjoerdsma A. Impairment of macrophage function by inhibitors of ornithine decarboxylase activity. Infection and Immunity. 1987;55(10):2461–2464. doi: 10.1128/iai.55.10.2461-2464.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lima-Junior DS, Costa DL, Carregaro V, et al. Inflammasome-derived IL-1β production induces nitric oxide-mediated resistance to Leishmania. Nature Medicine. 2013;19(7):909–915. doi: 10.1038/nm.3221. [DOI] [PubMed] [Google Scholar]
- 37.Silva GK, Costa RS, Silveira TN, Caetano BC. Apoptosis-associated speck-like protein containing a caspase recruitment domain inflammasomes mediate IL-1β response and host resistance to Trypanosoma cruzi infection. Journal of Immunology. 2013;191:3373–3383. doi: 10.4049/jimmunol.1203293. [DOI] [PubMed] [Google Scholar]