

Abstract
Bryonolic acid (BA) (1) is a naturally occurring triterpenoid with pleiotropic properties. This study characterizes the mechanisms mediating the anti-inflammatory and antioxidant activities of BA and validates the utility of BA as a tool to explore the relationships between triterpenoid structure and activity. BA reduces the inflammatory mediator NO by suppressing the expression of the inflammatory enzyme inducible nitric oxide synthase (iNOS) in LPS-activated RAW 264.7 macrophage cells. In addition, BA robustly induces the antioxidant protein heme oxygenase-1 (HO-1) in vitro and in vivo in an Nrf2-dependent manner. Further analyses of Nrf2 target genes reveal selectivity for the timing and level of gene induction by BA in treated macrophages with distinct patterns for Nrf2-regulated antioxidant genes. Additionally, the distinct expression profile of BA on Nrf2 target genes relative to oleanolic acid suggests the importance of the triterpenoid scaffold in dictating the pleiotropic effects exerted by these molecules.
Triterpenoids are one of the most functionally and structurally diverse class of secondary metabolites ubiquitous in the plant kingdom. Triterpenoids are cyclized from oxidosqualene to form approximately 200 chemically diverse triterpene skeletons. More than 20,000 triterpenoids have been documented, with new structures continually being identified and studied for their biological activity. In addition to the impressive skeletal diversity of these molecules, they also possess a variety of biological activities including anti-inflammatory, hepatoprotective, analgesic, antimicrobial, antimycotic, virostatic, immunomodulatory, and tonic effects.1 One of the best-studied triterpenoids, oleanolic acid (OA) (2), served as a platform for the discovery of the semi-synthetic triterpenoid 2-cyano-3,12-dioxooleana-1,9(11)-dien-28-oic acid (CDDO).2,3 CDDO and its methyl ester derviative, CDDO-Me, are potent anti-inflammatory3,4 and chemopreventive agents5-8 that have advanced to Phase III clinical trials for renal sparing effects in diabetic nephropathy.9,10
We became interested in bryonolic acid (BA) (1) in part because of its unique chemical attributes within the triterpenoid family (namely the unsaturated B-C ring fusion) and partly due to its interesting pleiotropic profile of biological activity. The activities reported for BA (1) include anti-allergic properties, inhibition of homologous passive cutaneous anaphylaxis in rats, delayed hypersensitivity in mice,11,12 anti-tumor activity13 and cytotoxicity towards various tumor cell lines.14,15 Although reports have shown BA (1) to be a promising natural anti-inflammatory agent, the mechanism of action mediating these effects has yet to be identified. We hypothesized that the previously observed BA (1) phenotypes could be explained by the activation of the transcription factor Nrf2.
The NF-E2-related factor 2 (Nrf2) was first isolated as a DNA binding protein to tandem repeats in the β-globin locus region and had been targeted for prevention of chemical carcinogenesis even before its complete characterization.16 Nrf2 acts as an electrophilic and oxidative damage sensor and induces a battery of cytoprotective genes that detoxify reactive electrophiles and oxidants. Among the hundreds of genes regulated by Nrf2, the most studied include heme oxygenase-1 (HO-1), NAD(P)H dehydrogenase, quinone 1 (NQO1), catalase (CAT), glutamate-cysteine ligase catalytic subunit GCLC), and glutathione reductase (GR). Several classes of endogenous and exogenous ligands induce Nrf2, with triterpenoids being one of the most promising and clinically relevant examples. Upon induction, Nrf2 dissociates from Keap1, the principal cytoplasmic inhibitor of Nrf2 function.17 Nrf2 subsequently escapes ubiquitination and proteosomal degredation, translocates to the nucleus and effectively upregulates the expression of cytoprotective and antioxidant genes.18
Here we provide the first demonstration of the molecular mechanisms contributing to the anti-inflammatory/anti-allergic effects of BA (1). Through in vitro analysis of BA (1) activity in mouse macrophages and in vivo studies following systemic administration of BA (1) in mice, we show potent suppression of iNOS expression and a robust induction of HO-1 by BA (1) in an Nrf2-dependent manner. BA (1) induces other antioxidant and cytoprotective genes and triggers a unique expression profile for Nrf2 target genes when compared to a structurally similar triterpenoid, OA (2). The observed differential regulation by BA (1) of genes in this pathway is characterized by a rapid and more potent induction of HO-1 and NQO1, while other Nrf2 target genes such as CAT, GR, and GCLC respond with a more gradual and modest increase in expression. This newly discovered ability of BA to regulate expression of inflammatory and antioxidant enzymes validates the utility of BA (1) as a platform to explore the importance of the triterpenoid scaffold in defining the anti-inflammatory and chemopreventive properties of triterpenoids. In addition, BA (1) may potentially uncover unique mechanism(s) in regulating the inflammatory and antioxidant pathway in comparison to the oleanane triterpenoids due to its different regulation of antioxidant genes. More specifically, these studies set the stage for an effort combining the application of synthetic chemistry and chemical biology screens that has the potential to yield diverse triterpenoid structures with selective therapeutic properties.
Results and Discussion
Bryonolic Acid Decreases NO Levels and iNOS Expression in LPS-activated RAW 264.7 Cells
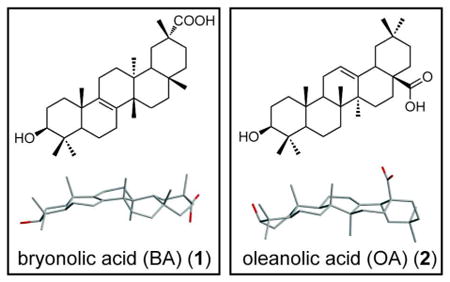
Initial studies of BA (1) have shown in vivo anti-inflammatory properties in rats and mice by inhibiting the allergic response.11,12,19 However, the mechanisms mediating these effects have not been explored. In order to elucidate the anti-inflammatory effects of BA (1), we used an established in vitro model of LPS-activated RAW 264.7 leukemic mouse macrophage cells (RAW). Upon LPS activation of RAW cells, NO is produced which spontaneously oxidizes to nitrite. In an initial experiment, LPS-activated RAW cells were treated with BA (1) and nitrite levels were measured from cell culture supernatants. Treatment with BA (1) reduced nitrite levels, demonstrating an IC50 value of 53.3 μM ± 3 μM after a 24-hour treatment (Figure 1A). RAW cells remained viable in BA (1) concentrations as high as 300 μM (gray shaded area), but cytotoxicity was apparent at higher concentrations as measured by the MTT assay (Figure 1B). This decrease in viability may be attributed to higher DMSO exposure at this dose range (1.5% at a concentration of 300 μM BA (1) from the maximum soluble stock of 20 mM). Due to the solubility of BA (1) and possible toxicity from DMSO, the maximum concentration included in the calculated IC50 value and the subsequent cell culture experiments was 100 μM at 0.5% DMSO.
Figure 1.
Bryonolic acid (BA) (1) decreases NO levels and inhibits iNOS expression in RAW 264.7 cells in a dose dependent and time dependent manner. RAW 264.7 cells were activated with 5 ng/mL LPS and treated with varying concentrations of BA for 24 hours (A-D) or varying time points (E). (A) Nitrite levels were measured via Griess assay in LPS-activated cells treated with BA for 24 hours. (B) RAW 264.7 cells were treated with varying concentrations of BA for 24 hours and the viability measured by the MTT assay. (C) iNOS protein levels were quantified through immunoblot analysis in LPS-activated cells treated with varying concentrations of BA for 24 hours. (D) iNOS mRNA levels were measured by RT-PCR. (E) iNOS protein levels were quantified through immunoblot analysis in LPS-activated cells treated with 100 μM BA or DMSO control at various time points.
To determine the mechanism through which BA (1) suppresses the production of NO following LPS exposure, we examined the expression profile of iNOS through immunoblot analysis in LPS-activated RAW cells. Treatment with 50 μM BA (1) significantly reduced iNOS protein levels after a 24-hour treatment compared to the DMSO control (Figure 1C). At 100 μM BA (1), iNOS protein levels are no longer detectable. The iNOS mRNA levels in LPS-activated RAW cells were also reduced in the presence of 50 μM and 100 μM BA (1) after a 24-hour treatment (Figure 1D). Thus, the decrease of NO production is mediated by the suppression of iNOS expression, as shown by the decrease in both protein and mRNA levels in the presence of BA (1). To further assess this effect, RAW cells were treated at various time points with 100 μM BA (1). LPS induced iNOS expression at 4 hours and BA (1) suppressed iNOS levels at this early time point (Figure 1E).
Bryonolic Acid Induces the Antioxidant Heme Oxygenase-1 Expression in RAW 264.7 Cells
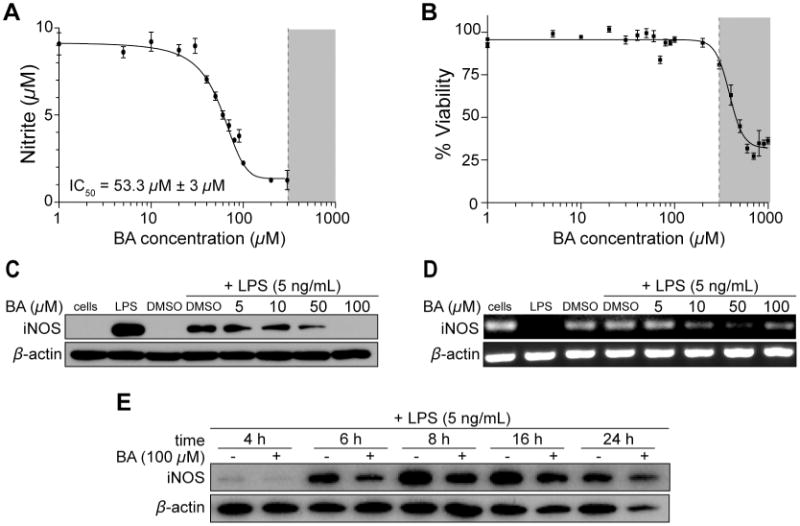
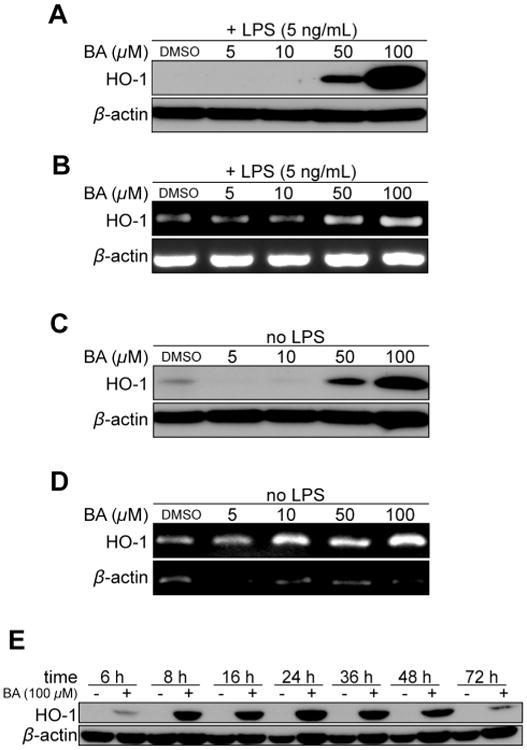
Previous data have shown robust induction of HO-1 by other triterpenoids.20,21 Therefore, we next determined whether BA (1) might also induce HO-1. We observed a dose-dependent induction of HO-1 by BA (1), with HO-1 protein and mRNA levels induced at 50 μM and 100 μM after 24 hours (Figure 2A and 2B) in LPS-activated RAW cells. Since LPS is known to induce HO-1, we next determined whether LPS is a requirement for HO-1 induction by BA (1). RAW cells were treated with BA (1) in the absence of LPS and probed for HO-1. As seen in Figure 2C, HO-1 was induced in the presence of 50 μM and 100 μM BA (1) in the absence of LPS. HO-1 mRNA levels were induced in the presence of 10 μM BA (1) with increasing expression at 100 μM BA (1) (Figure 2D). To investigate the time course of HO-1 induction, RAW cells treated with 100 μM BA (1) were harvested at different time points and probed for HO-1. BA (1) induced HO-1 protein levels as early as 6 hours and expression peaked approximately 24 hours after treatment. The induction extends beyond 48 hours and is diminished by 72 hours (Figure 2E). This profile for HO-1 induction illustrates a long-term induction by BA (1) amounting to a total of approximately 66 hours. HO-1 mRNA levels were also induced at 4 hours with a 4.1-fold change compared to control at time = 0 (Figure 3). HO-1 mRNA levels continued to increase up to 20 hours, at which time the peak induction demonstrated a 11.3-fold change in mRNA levels compared to control.
Figure 2.
Bryonolic acid (BA) (1) induces HO-1 expression in RAW 264.7 cells in an LPS independent manner. RAW 264.7 cells were either activated with 5 ng/mL LPS (A-B) or without (C-D) with varying concentrations of BA for 24 hours (A-D) or varying time points (E). (A, C and E) Immunoblot analysis of HO-1 protein levels. (B and D) mRNA level measurement of HO-1 by RT-PCR.
Figure 3.
Bryonolic acid (BA) (1) induces Nrf2 and its target genes. RAW 264.7 cells were treated with BA at different time points. qRT-PCR was performed probing for Nrf2 and Nrf2 target genes including catalase (CAT), glutamate-cysteine ligase catalytic subunit (GCLC), glutathione reductase (GR), heme oxygenase-1 (HO-1) and NAD(P)H dehydrogenase, quinone 1 (NQO1) at varying time points in cells treated with 100 μM BA.
Bryonolic Acid Induces Nrf2 Target Genes
The induction of HO-1 by BA (1) led us to question whether BA (1) could also induce other phase 2 genes. Previous data showed an inverse correlation between the expression of the inflammatory gene iNOS and phase 2 genes in triterpenoid treated cells.21 Several phase 2 genes were probed including HO-1, NQO1, CAT, GR, and GCLC at various time points in BA (1) treated RAW cells (Figure 3). Since Nrf2 also controls its own expression, we initially probed for Nrf2 mRNA levels. Nrf2 was induced 1.8-fold at 20 hours in 100 μM treated cells compared to control. Significant induction in expression for all of the genes occurred at the 20-hour time point. NQO1 expression was significantly induced, demonstrating an 11.7-fold increase, similar in magnitude to the 11.3-fold increase in HO-1 mRNA levels. CAT was induced 1.7-fold at 16 hours and almost doubled to 3.5-fold induction at 20 hours. GCLC was induced 1.8-fold at 20 hours and decreased to basal levels by 24 hours. This pattern of induction is also similar for GR which was induced 1.5-fold at 20 hours and decreased to basal levels by 24 hours.
Unique Dose Response Profiles for Bryonolic Acid and Oleanolic Acid for NO Suppression and HO-1 Induction
The numerous biological activities of triterpenoids led us to compare BA (1) with the well-studied and structurally similar triterpenoid OA (2). We compared the ability of OA (2) and BA (1) to suppress NO production in RAW cells. Nitrite levels decreased 85% from control in OA (2) treated cells, and 72% in BA (1) treated cells (Figure 4A). Studies of OA (2) have led to the discovery of several potent synthetic triterpenoids, and selected OA (2) derivatives are now in late phase clinical trials.22,23 However, when we compared the ability of both naturally occurring triterpenoids to induce HO-1, we found that BA (1) is considerably more effective than OA (2) (Figure 4B). At 50 μM, there is greater HO-1 induction in BA (1) treated cells compared to OA (2) treated cells after 24 hours. BA's (1) potency for inducing HO-1 is more apparent at the higher concentration of 100 μM. In addition, we compared the potency of HO-1 induction by BA (1) with the structurally similar triterpenoids ursolic acid (UA) (3), betulinic acid (4), boswellic acid (5), and glycyrrhetinic acid (6) (Figure S1, Supporting Information). We found that BA (1) induces HO-1 more potently compared to ursolic acid (3) and betulinic acid (4), but similary toboswellic acid (5) and glycyrhetinic acid (6) after 8 hours of treatment. Interestingly, BA (1) is morepotent at inducing HO-1 mRNA levels at the earlier 4 hour time point compared to all of thesetriterpenoids (2-6). We noted that unlike BA (1) and OA (2), UA (3), betulinic acid (4) and glycrrhetinicacids (6) are all toxic to cells as early as 8 hours after treatment.
Figure 4.
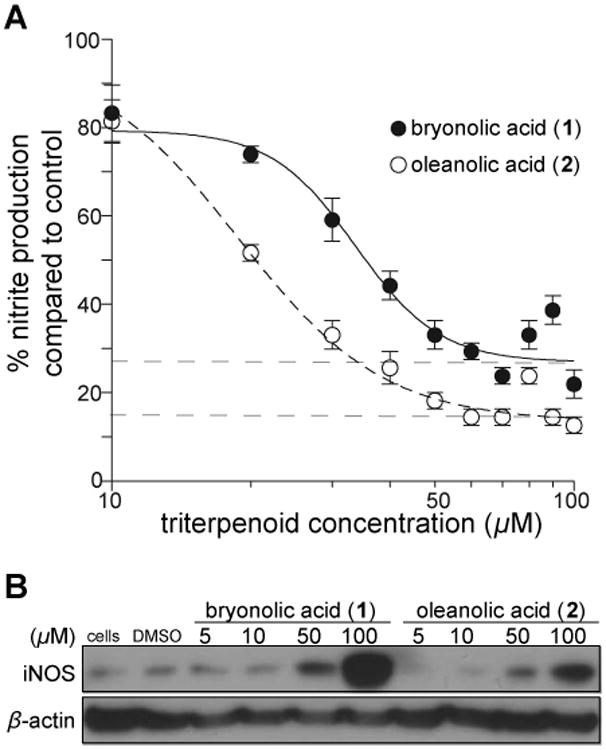
Activity comparison of bryonolic acid (BA) (1) versus oleanolic acid (OA) (2) at reducing nitrite levels and inducing HO-1. RAW 264.7 cells were treated with various concentrations of BA (1) or OA (2) at 24 hours. (A) Nitrite levels were measured via the Griess assay. (B) Immunoblot analysis of HO-1 in the whole cell lysates of triterpenoid treated cells.
Bryonolic Acid Induces Translocation of Nrf2 into the Nucleus
The induction of HO-1 is regulated by the transcription factor Nrf2. In the presence of an inducer, Nrf2 is released from its cytoplasmic inhibitor Keap1 and is translocated into the nucleus where it then binds to genes containing antioxidant response element (ARE) sites and induces transcription of the antioxidant phase 2 enzymes. In order to determine the mechanism of HO-1 induction by BA (1), we examined whether BA (1) is able to translocate Nrf2 into the nucleus. We treated RAW cells with 50 μM and 100 μM BA (1) at various time points and probed both cytoplasmic and nuclear fractions for Nrf2. We observed that exposure to BA (1) decreased cytoplasmic Nrf2 in treated cells as early as 1 hour (Figure S2, Supporting Information), and this decrease was more evident in the presence of 100 μM BA (1). This reduction in cytoplasmic Nrf2 was more evident in BA (1) treated cells from 1 to 4 hours after treatment. Nrf2 accumulates in the nucleus in both BA (1) and OA (2) treated cells and remained nuclear throughout the time course, whereas no accumulation was observed in the control. These results show that BA (1) is more potent at inducing Nrf2 nuclear translocation when compared to OA (2).
Differential Regulation of Nrf2 Target Genes by Bryonolic Acid
We next determined whether the observed enhanced BA-induced (1) Nrf2 activation relative to OA (2) correlates with distinct expression profiles for Nrf2 target genes. Quantitative RT-PCR analysesrevealed peak HO-1 induction in OA (2) treated cells beginning 2-4 hours after treatment, while peakHO-1 induction in BA-treated (1) cells occurred 16-20 hours after treatment (Figure 5A). HO-1 expression levels in OA (2) treated cells decreased to basal levels while HO-1 expression levels in BA (1) treated cells remained elevated at later time points as compared to OA (2). In addition, another prototypical target of Nrf2, NQO1 was induced in a similar manner as HO-1, (Figure 5B), while the expression profiles of CAT and GR were disparate in BA- (1) and OA-treated (2) cells. Whereas OA-treated (2) cells showed a peak induction followed by a steep decline in gene expression, BA-treated cells (1) demonstrated only a gradual increase in expression of these genes over the entire time course (Figures 5C and E). This differential regulation exerted by BA (1) does not affect non-Nrf2 regulated genes, β-actin or GAPDH (Figure S3, Supporting Information). Taken together, these data show that subtle structural differences between triterpenoids with a similar carbocyclic skeleton affects the ability of these small molecules to modulate expression of the Nrf2 target genes in a different manner.
Figure 5.
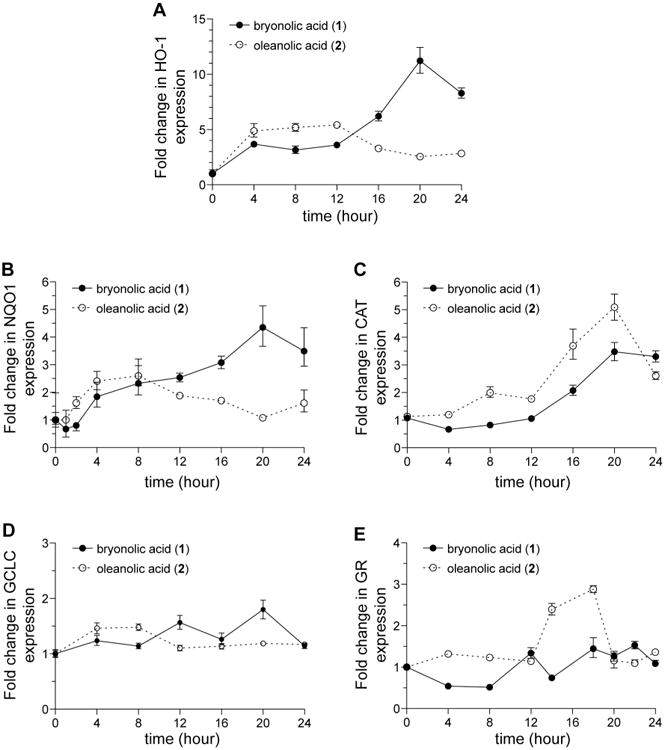
Bryonolic acid (BA) (1) differentially induces Nrf2 target genes compared with oleanolic acid (2). RAW 264.7 cells were treated with 100 μM BA (1) or OA (2) at different time points. qRT-PCR was performed to measure HO-1 (A), NQO1 (B), CAT (C), GCLC (D), and GR (E) expression at various time points.
Bryonolic Acid Activity in Primary Macrophage is Dependent on the Nrf2-Keap1 Pathway
In order to demonstrate the requirement for Nrf2 in the BA-induction (1) of HO-1, we performed a series of experiments in primary peritoneal macrophages and probed for HO-1. Consistent with the observed activity in the RAW cell line, BA (1) induced HO-1 expression in a dose-dependent manner in primary peritoneal macrophages, and it did so with greater potency (induction observed at 10 μM BA (1) in primary macrophages in comparison to 50 μM in the RAW cells; Figure 6A). However, when we treated primary macrophages from Nrf2 deficient mice (Nrf2-/-), BA (1) no longer induced HO-1. Thus, the induction of HO-1 by BA (1) is dependent on the Nrf2-Keap1 pathway.
Figure 6.
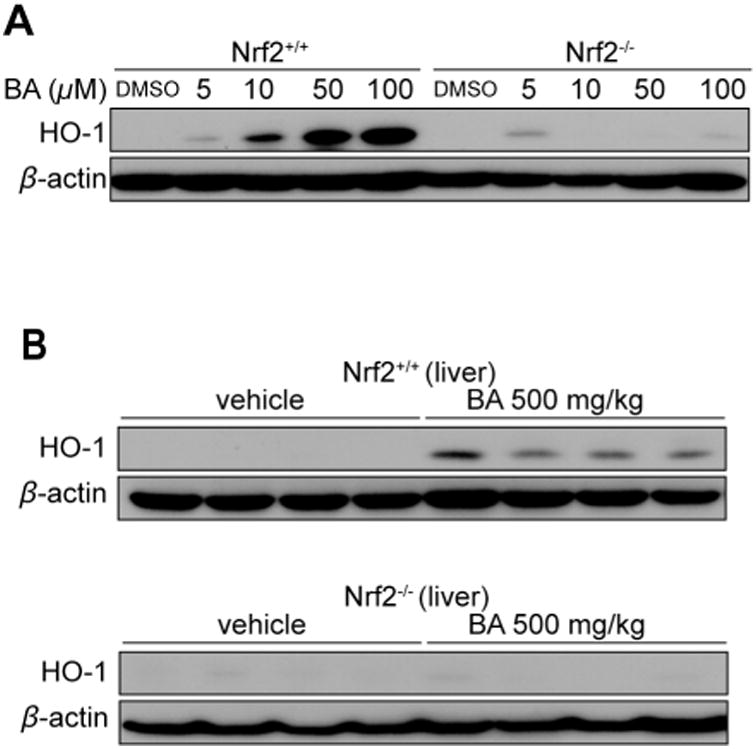
Bryonolic acid (BA) (1) induces HO-1 in primary peritoneal macrophage and liver and is dependent on the Nrf2 pathway. (A) Primary peritoneal macrophages harvested from Nrf2 wild-type or Nrf2 knockout (C57BL6/J background) mice were treated with varying concentration of BA (1) for 48 hours. Immunoblot analysis of HO-1 protein levels in BA (1) treated primary macrophages. (B) immunoblot analysis of HO-1 protein levels in liver tissues of Nrf2 wild-type (upper panel) or Nrf2 knockout (lower panel) mice were treated with 500 mg/kg BA or vehicle for 8 hours by i.p.
Bryonolic Acid Induces HO-1 In Vivo in an Nrf2-dependent Manner
In order to determine whether this demonstrated capacity of BA (1) to induce HO-1 can be exerted following systemic exposure to BA (1) in vivo, wild-type mice were treated with 500 mg/kg BA (1) by i.p. injection, and sacrificed 8 hours after treatment. Mouse livers were then harvested as hepatocytes have been reported to exhibit highly inducible HO-1 expression.24,25 Mouse livers which had been homogenized and probed for HO-1 after 8 hours of BA (1) treatment showed a significant induction of HO-1 as determined by immunoblot analysis (Figure 6B). The same experiment performed in Nrf2-/- mice similarly demonstrated that in the absence of an intact Nrf2-Keap1 pathway, BA (1) was unable to induce HO-1 in vivo. Taken together, these data show that BA (1) potently induces HO-1 in a manner dependent on the Nrf2-Keap1 pathway.
Our goal in this study was to understand the molecular mechanism of the anti-inflammatory activity of BA (1) and to validate the use of BA (1) as platform for studies designed to explore the relationship of structure to the pleiotropic effects of triterpenoids. This is the first definitive report demonstrating a molecular mechanism through which BA (1) exerts potent anti-inflammatory activity, by reducing NO levels via suppression of iNOS expression as shown in LPS-activated macrophages, in a dose and time-dependent manner. We previously reported that BA (1) induces HO-1 in vitro in LPS-activated RAW cells26 and herein report the induction of HO-1 expression in a dose-dependent and time-dependent manner independent of LPS. In addition, we show that BA (1) induces HO-1 in an Nrf2-dependent manner in primary mouse macrophages and in hepatocytes in vivo following systemic administration of BA (1).
These data, together with the observed induction of antioxidant genes, provide a mechanism to explain how BA (1) exerts the previously reported anti-allergic and anti-inflammatory properties observed in preclinical models in mice and rats.11,12 More importantly, the observed effects of BA (1) on the production of iNOS and HO-1 are consistent with the reported role of these molecules in allergy and inflammation as highlighted in several studies. For example, iNOS expression is significantly increased after an allergen challenge in preclinical anaphylaxis mouse model,27 and is highly expressed in several forms of dermatoses in humans.28 Furthermore, induction of HO-1 inhibits allergic inflammation in mice29 and in humans30 and it is intriguing that the anti-allergic properties of several molecules have been attributed to an effect on HO-1 expression or activity.31,32 Although the anti-inflammatory activity of HO-1 is established, further studies are required to determine how HO-1 contributes to the anti-inflammatory activity of BA (1).33-35
Surprisingly, BA (1) exhibits a unique expression profile compared to OA (2), inducing the characteristic robust HO-1 expression while yielding a different expression profile of the Nrf2-target genes from the structurally similar OA (2). In comparison to other structurally similar triterpenoids – ursolic acid (3), betulinic acid (4), boswellic acid (5), and glycyrrhetinic acid (6), BA (1) induces HO-1 mRNA at the earlier time point (Figure S3, Supporting Information). In the absence of Nrf2, BA (1) failed to induce HO-1, suggesting that BA (1) acts primarily through this pathway. This is further corroborated by an analysis of Nrf2 nuclear translocation in which we observed a marked decrease in cytoplasmic Nrf2 and an increase in nuclear accumulation of Nrf2 in BA (1) treated cells. The chemopreventive and chemotherapeutic effects of the oleanane-derived semi-synthetic triterpenoids (CDDO and CDDO derivatives) have been attributed to the potent induction of HO-1 and the Nrf2-depdendent genes.24,36-43 The synthetic effort leading to the discovery of CDDO originated from improvement upon the weak anti-inflammatory activity of OA (2). Since BA (1) is more potent in comparison to OA (2) at inducing HO-1, combined with the extended gradual induction of other Nrf2-dependent genes, we can anticipate BA (1) to be an excellent platform for the development of potent anti-inflammatory and chemopreventive agents.
While one might expect that targeting the Nrf2 pathway with any triterpenoid would result in similar upregulation of antioxidant and cytoprotective genes,25,44,45 our studies indicate that the minimal structural difference between the BA (1) and OA (2) triterpenoids influence their capacity to modulate a similar set of target genes. There are multiple interpretations to the observed HO-1 phenotype and the unique expression profile of BA (1). One possibility is that BA (1) is targeting a pathway different from Nrf2, which results in the difference of gene expression between BA (1) and OA (2). However, the mechanistic studies in which we explored BA (1) effects in primary mouse macrophages in vitro and in hepatocytes in vivo show that BA (1) failed to induce HO-1 without an intact Nrf2 pathway. These data support the conclusion that the observed BA (1) phenotype is mediated through the Nrf2 pathway. A second interpretation is that the primary target of BA (1) is Nrf2, but there exists structural specificity that dictates the differential regulation of Nrf2 target genes. There is precedence for this interpretation, as several ligands including OA (2) have been shown to bind to the farnesoid x receptor (FXR) and selectively modulate the expression of specific target genes important for bile acid regulation.46 Our results suggest that BA (1) may be acting in a similar fashion while targeting the Nrf2 pathway. In addition, although Nrf2 is a key transcription factor for the induction of antioxidant genes, several studies have shown differences in the regulation of these genes. Small Maf accessory proteins which heterodimer with Nrf2 have been shown to play a role in the differential regulation of HO-1 and NQO1.47,48,48 However, further studies are required to identify the cognate binding partner of BA (1), and to determine the molecular underpinnings of the observed expression profile.
It has been suggested that inducers of the phase 2 response are also suppressors of inflammation. Triterpenoids have been shown to coordinately regulate markers of inflammation and phase 2 response and that inducer potency is a reliable predictor of anti-inflammatory activity.21,49 However, it is interesting to note that this is not the case for the observed BA (1) phenotype, where HO-1 induction is more potent and suppression of iNOS is weaker in BA (1) treated cells compared to OA (2). Further mechanistic studies on BA (1) will hopefully uncover novel regulation of both the phase 2 and inflammatory pathway by triterpenoids.
Translationally, the capacity of BA (1) to induce HO-1 and other target genes could be leveraged through the use of BA (1) as a platform for developing novel selective modulators of Nrf2–dependent gene expression. By fully exploring the triterpenoid scaffold, there is potential to unlock the full potential of triterpenoids as selective inflammatory regulators. Such an effort could lead to the design of synthetic triterpenoids that may modulate expression of specific genes in a selected disease context. For example, the HO-1 phenotype observed in vivo has clear translational implications in the context of malaria, caused by infection of the Plasmodium genus of parasites. Many of the clinical manifestations of infection by Plasmodium are directly linked to the hemolysis of red blood cells and release of hemoglobin and the effects of hemoglobin degradation products. Hemoglobin released from red blood cells is oxidized by reactive oxygen species (ROS)50 during inflammation resulting in free heme, which serves as an amino acid source for the parasite.51,52 HO-1 plays an important role in modulating the inflammatory response by breaking down the deleterious heme and produces anti-inflammatory byproducts, effectively protecting the host from developing cerebral and non-cerebral forms of malaria.53,54 The importance of this mechanism is supported by the observation that Plasmodium infection leads to rapid hepatic failure and lethality in mice with a targeted disruption of the HO-1 gene.55 Thus, host survival in this context is dependent on the capacity to upregulate the HO-1 enzyme.56 Ultimately, further studies will set a foundation for in depth analyses of the triterpenoid scaffold and how this may be manipulated to generate potent and selective modulators of inflammation.
Experimental Section
Materials
C57BL/6 mice (wild-type) were purchased from Jackson Laboratory (Bar Harbor, ME) and the Nrf2-knockout (Nrf2-/-) mice on C57BL/6 background were purchased from RIKEN BioResource Center (Tsukuba, Japan). BA (1) was isolated from the roots of Cucurbita pepo L. as previously reported26 and OA (2) was purchased from Tokyo Chemical Industry (Tokyo, Japan). Stocks were made fresh in DMSO. Ursolic acid (3), betulinic acid (4), boswellic acid (5), glycyrrhetinic acid (6), DMSO, Cremophore EL and LPS from E. coli were purchased from Sigma (St. Louis, MO). All the primary antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA) and secondary antibodies from Southern Biotech (Birmingham, AL). DMEM and RPMI media were purchased from GIBCO (Grand Island, NY) and then supplemented with low endotoxin FBS (<0.06 EU), obtained from Thermo Scientific (Logan, UT). The Tryphan Blue Exclusion, Penicillin/streptomycin, Griess assay reagents, TRIZol® reagent, PureLink™ RNA Mini Kit, SuperScript One-Step RT-PCR, SuperScript® III First-Strand Synthesis System for RT-PCR, RIPA Buffer, 0.2 μm PVDF membrane, Novex® 4-20% tris glycine gels, and running and transfer buffers were all purchased from Invitrogen (Grand Island, NY). The Protease cocktail inhibitor tablet was purchased from Roche (Indianapolis, IN) and MTT cell proliferation assay kit from ATCC (Manassas, VA). TaqMan® Fast Universal PCR Master Mix was obtained from Applied Biosystems (Foster City, CA). Thioglycollate for primary macrophage stimulation was purchased from Becton Dickinson (Sparks, MD) and ECL plus from Amersham (Buckinghamshire, UK). PBS was obtained from Cellgro by Mediatech, Inc. (Manassas, VA). Autoradiography film was purchased from MidSci (St. Louis, MO).
Cell Culture
The leukemic mouse macrophage cells (RAW 264.7) were obtained as a gift from Dr. Michael Sporn (Dartmouth College, NH), and cultured in DMEM media supplemented with 10% FBS and 1% Penicillin/streptomycin and kept in culture at 37°C in a 5% CO2 environment. Cells were kept in culture for no longer than a month and routinely checked for LPS responsiveness every few passages via detection of NO production measured using the Griess assay.
NO Measurement
RAW 264.7 cells were plated (1 × 105 cells/well) in a 96-well plate and allowed to attach for several hours before activation with 5 ng/mL LPS. LPS-activated cells were treated with varying concentrations of BA (1) (1-1000 μM) for 24 hours. NO levels were measured via Griess assay using 100 μL Griess reagent with 100 μL cell culture supernatant. Absorbance was read at 550 nm using the Sunrise plate reader by TECAN (Mannedorf, Switzerland). IC50 values were calculated by fitting a non-linear sigmoidal variant slope curve to the data using Prism 5.0 software by Graphpad Inc.
Toxicity Measurement
The MTT cell proliferation assay kit was used according to manufacturer's specifications to measure toxicity of BA (1) treated RAW 264.7 cells after 24 hours. Tryphan blue exclusion test was used in Figure S3 (Supporting Information).
RAW 264.7 Cell Treatment
RAW 264.7 cells were plated at 4 × 106 cells/60 mm plate and allowed to attach for 2 hours. Cells were activated with 5 ng/mL LPS and immediately treated with varying concentrations of BA (1) (maintaining 0.5% DMSO), BA (1) alone without LPS activation or DMSO control. Cells were harvested for immunoblotting analysis after a 24-hour treatment.
Primary Peritoneal Macrophage Isolation and Treatment
Prior to primary peritoneal macrophage harvest, 6-8 week old C57BL/6 and Nrf2-/- (C57BL/6 background) mice were injected with 2 mL 4% thioglycollate via i.p. Primary peritoneal macrophages were collected with PBS, plated in 60 mm plates (3 × 106 cells/plate) in RPMI medium supplemented with 10% FBS and 1% penicillin/streptomycin, and allowed to attach for 2-3 hours. Prior to LPS activation and BA (1) treatment, cells were washed with PBS 3-4 times to remove non-macrophage cells. Cells were treated with varying concentrations of BA (1) (5, 10, 50, or 100 μM), or DMSO control. Cells were harvested for immunoblotting analysis after a 48-hour treatment.
Immunoblotting Analysis
Cells and homogenized tissues were lysed with RIPA buffer containing protease inhibitors. Lysates were probed for HO-1 and iNOS using a 1:1000 primary antibody dilution for RAW 264.7 cells and primary macrophage cells and a 1:500 HO-1 antibody dilution for tissue lysates. Secondary antibodies (1:5000 dilution) were detected using ECL plus with autoradiography.
RT-PCR Analysis
Total RNA from the treated cells was extracted and purified using TRIZol reagent. cDNA was synthesized and PCR reactions were performed using Superscript One-Step RT-PCR. Primer sequences for RT-PCR analysis for iNOS and HO-157 and catalase, GCLC, GR, NQO1, β-actin were adopted from a previous publication.58 The conditions were used accordingly: 55 °C for 30 min for reverse transcription, 94 °C for 2 min for pre-denaturation, followed by 30 cycles of 94 °C for 30 sec for denaturing, 55 °C for 30 sec for annealing, and 72 °C for 1 min for extension, and followed by 1 cycle of 72 °C for 10 min for final extension.
Quantitative Real-time PCR Analysis
Total RNA from treated cells was isolated with the PureLink RNA Mini Kit and converted to cDNA using the SuperScript III First-Strand Synthesis System for RT-PCR. TaqMan Fast Universal PCR Master Mix was used for real time RT-PCR with mouse-specific Taqman gene expression assay from Applied Biosystems. The TaqMan PCR primers and probes used were as follows: iNOS (Mm01309902_m1), HO-1 (Mm00516007_m1), NQO1 (Mm00500821_m1), catalase (Mm00437992_m1), GCLC (Mm00802655_m1), GR (Mm00833903_m1), and 18S rRNA (Hs99999901_s1) as a control. Amplification was performed using the 7500 Fast Real-Time PCR system and the 7500 Fast System SDS Software-Sequence Detection Software version 1.3.1.21 by Applied Biosystems. The assay used for the study was the relative quantification assay (ΔΔCt) using the Run mode Fast 7500 profile (95 °C for 20 sec, followed by 40 cycles of 95 °C for 3 sec, and 60 °C for 30 sec).
In Vivo BA Administration
Both the C57BL/6 and the Nrf2-/- mice were treated with either a single dose of 500 mg/kg BA (1) (dissolved in 80% PBS/10% DMSO/10% Cremophore) or vehicle control administered by intraperitoneal (i.p.) injection. Mice were sacrificed and liver tissue harvested 8 hours following BA (1) administration. All experiments were performed in accordance with an approved protocol by the Institutional Animal Care and Use Committee at Case Western Reserve University.
Supplementary Material
Acknowledgments
The work was supported by grants from the NIH, R01CA157735 and R03CA132168, and Reuter Foundation to G.P.T. and J. J. T., Landon Foundation INNOVATOR award from AACR to G.P.T., and an NIH fellowship, F31CA134211 to T.N.G.
Footnotes
Supporting Information. Comparative analysis of HO-1 induction by triterpenoids (Figure S1), immunoblot analysis of cytoplasmic and nuclear Nrf2 in bryonolic acid and oleanolic acid treated RAW 264.7 cells (Figure S2), positive and negative controls for BA (1) treated RAW 264.7 cells (Figure S3), and selective regulation of Nrf2 target genes by BA (1) (Figure S4) is presented. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Dzubak P, Hajduch M, Vydra D, Hustova A, Kvasnica M, Biedermann D, Markova L, Urban M, Sarek J. Nat Prod Rep. 2006;23:394–411. doi: 10.1039/b515312n. [DOI] [PubMed] [Google Scholar]
- 2.Honda T, Rounds BV, Gribble GW, Suh N, Wang Y, Sporn MB. Bioorg Med Chem Lett. 1998;8:2711–2714. doi: 10.1016/s0960-894x(98)00479-x. [DOI] [PubMed] [Google Scholar]
- 3.Suh N, Honda T, Finlay HJ, Barchowsky A, Williams C, Benoit NE, Xie QW, Nathan C, Gribble GW, Sporn MB. Cancer Res. 1998;58:717–723. [PubMed] [Google Scholar]
- 4.Honda T, Rounds BV, Bore L, Finlay HJ, Favaloro FG, Jr, Suh N, Wang Y, Sporn MB, Gribble GW. J Med Chem. 2000;43:4233–4246. doi: 10.1021/jm0002230. [DOI] [PubMed] [Google Scholar]
- 5.Suh N, Wang Y, Honda T, Gribble GW, Dmitrovsky E, Hickey WF, Maue RA, Place AE, Porter DM, Spinella MJ, Williams CR, Wu G, Dannenberg AJ, Flanders KC, Letterio JJ, Mangelsdorf DJ, Nathan CF, Nguyen L, Porter WW, Ren RF, Roberts AB, Roche NS, Subbaramaiah K, Sporn MB. Cancer Res. 1999;59:336–341. [PubMed] [Google Scholar]
- 6.Deeb D, Gao X, Dulchavsky SA, Gautam SC. J Exp Ther Oncol. 2008;7:31–39. [PubMed] [Google Scholar]
- 7.Sogno I, Vannini N, Lorusso G, Cammarota R, Noonan DM, Generoso L, Sporn MB, Albini A. Recent Results Cancer Res. 2009;181:209–212. doi: 10.1007/978-3-540-69297-3_19. [DOI] [PubMed] [Google Scholar]
- 8.Kim EH, Deng C, Sporn MB, Royce DB, Risingsong R, Williams CR, Liby KT. Cancer Prev Res. 2012;5:89–97. doi: 10.1158/1940-6207.CAPR-11-0359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Petronelli A, Pelosi E, Santoro S, Saulle E, Cerio AM, Mariani G, Labbaye C, Testa U. Leuk Res. 2011;35:534–544. doi: 10.1016/j.leukres.2010.09.013. [DOI] [PubMed] [Google Scholar]
- 10.Pergola PE, Raskin P, Toto RD, Meyer CJ, Huff JW, Grossman EB, Krauth M, Ruiz S, Audhya P, Christ-Schmidt H, Wittes J, Warnock DG. N Engl J Med. 2011;365:327–336. doi: 10.1056/NEJMoa1105351. [DOI] [PubMed] [Google Scholar]
- 11.Tanaka S, Uno C, Akimoto M, Tabata M, Honda C, Kamisako W. Planta Med. 1991;57:527–530. doi: 10.1055/s-2006-960199. [DOI] [PubMed] [Google Scholar]
- 12.Tabata M, Tanaka S, Cho HJ, Uno C, Shimakura J, Ito M, Kamisako W, Honda C. J Nat Prod. 1993;56:165–174. doi: 10.1021/np50092a001. [DOI] [PubMed] [Google Scholar]
- 13.Kondo T, Inoue M, Mizukami H, Ogihara Y. Biol Pharm Bull. 1995;18:726–729. doi: 10.1248/bpb.18.726. [DOI] [PubMed] [Google Scholar]
- 14.Takeda T, Kondo T, Mizukami H, Ogihara Y. Chem Pharm Bull. 1994;42:730–732. doi: 10.1248/cpb.42.730. [DOI] [PubMed] [Google Scholar]
- 15.Kongtun S, Jiratchariyakul W, Kummalue T, Tan-ariya P, Kunnachak S, Frahm AW. Planta Med. 2009;75:839–842. doi: 10.1055/s-0029-1185455. [DOI] [PubMed] [Google Scholar]
- 16.Moi P, Chan K, Asunis I, Cao A, Kan YW. Proc Natl Acad Sci U S A. 1994;91:9926–9930. doi: 10.1073/pnas.91.21.9926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Itoh K, Wakabayashi N, Katoh Y, Ishii T, O'Connor T, Yamamoto M. Genes Cells. 2003;8:379–391. doi: 10.1046/j.1365-2443.2003.00640.x. [DOI] [PubMed] [Google Scholar]
- 18.Itoh K, Tong KI, Yamamoto M. Free Radical Biol Med. 2004;36:1208–1213. doi: 10.1016/j.freeradbiomed.2004.02.075. [DOI] [PubMed] [Google Scholar]
- 19.Rios JL, Bas E, Recio MC. Curr Med Chem: Anti-Inflammatory Anti-Allergy Agents. 2005;4:65–80. [Google Scholar]
- 20.Thimmulappa RK, Scollick C, Traore K, Yates M, Trush MA, Liby KT, Sporn MB, Yamamoto M, Kensler TW, Biswal S. Biochem Biophys Res Commun. 2006;351:883–889. doi: 10.1016/j.bbrc.2006.10.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dinkova-Kostova AT, Liby KT, Stephenson KK, Holtzclaw WD, Gao X, Suh N, Williams C, Risingsong R, Honda T, Gribble GW, Sporn MB, Talalay P. Proc Natl Acad Sci U S A. 2005;102:4584–4589. doi: 10.1073/pnas.0500815102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koschmieder S, D'Alo F, Radomska H, Schoneich C, Chang JS, Konopleva M, Kobayashi S, Levantini E, Suh N, Di Ruscio A, Voso MT, Watt JC, Santhanam R, Sargin B, Kantarjian H, Andreeff M, Sporn MB, Perrotti D, Berdel WE, Muller-Tidow C, Serve H, Tenen DG. Blood. 2007;110:3695–3705. doi: 10.1182/blood-2006-11-058941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tsao T, Kornblau S, Safe S, Watt JC, Ruvolo V, Chen W, Qiu Y, Coombes KR, Ju Z, Abdelrahim M, Schober W, Ling X, Kardassis D, Meyer C, Schimmer A, Kantarjian H, Andreeff M, Konopleva M. Cancer Res. 2010;70:4949–4960. doi: 10.1158/0008-5472.CAN-09-1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reisman SA, Buckley DB, Tanaka Y, Klaassen CD. Toxicol Appl Pharmacol. 2009;236:109–114. doi: 10.1016/j.taap.2008.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reisman SA, Aleksunes LM, Klaassen CD. Biochem Pharmacol. 2009;77:1273–1282. doi: 10.1016/j.bcp.2008.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barker EC, Gatbonton-Schwager TN, Han Y, Clay JE, Letterio JJ, Tochtrop GP. J Nat Prod. 2010;73:1064–1068. doi: 10.1021/np1000076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sade K, Schwartz IF, Etkin S, Schwartzenberg S, Levo Y, Kivity S. J Invest Allergol Clin Immunol. 2007;17:379–385. [PubMed] [Google Scholar]
- 28.Rowe A, Farrell AM, Bunker CB. Br J Dermatol. 1997;136:18–23. [PubMed] [Google Scholar]
- 29.Listopad J, Asadullah K, Sievers C, Ritter T, Meisel C, Sabat R, Docke WD. Exp Dermatol. 2007;16:661–670. doi: 10.1111/j.1600-0625.2007.00581.x. [DOI] [PubMed] [Google Scholar]
- 30.Kirino M, Kirino Y, Takeno M, Nagashima Y, Takahashi K, Kobayashi M, Murakami S, Hirasawa T, Ueda A, Aihara M, Ikezawa Z, Ishigatsubo Y. J Allergy Clin Immunol. 2008;122:290–297. doi: 10.1016/j.jaci.2008.05.031. [DOI] [PubMed] [Google Scholar]
- 31.Matsushima M, Takagi K, Ogawa M, Hirose E, Ota Y, Abe F, Baba K, Hasegawa T, Hasegawa Y, Kawabe T. Inflammation Res. 2009;58:705–715. doi: 10.1007/s00011-009-0039-1. [DOI] [PubMed] [Google Scholar]
- 32.Matsushima H, Tanaka H, Mizumoto N, Takashima A. Blood. 2009;114:64–73. doi: 10.1182/blood-2009-02-204297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim HP, Wang X, Zhang J, Suh GY, Benjamin IJ, Ryter SW, Choi AM. J Immunol. 2005;175:2622–2629. doi: 10.4049/jimmunol.175.4.2622. [DOI] [PubMed] [Google Scholar]
- 34.Otterbein LE, Bach FH, Alam J, Soares M, Tao Lu H, Wysk M, Davis RJ, Flavell RA, Choi AM. Nat Med (N Y, NY, U S) 2000;6:422–428. doi: 10.1038/74680. [DOI] [PubMed] [Google Scholar]
- 35.Rocuts F, Ma Y, Zhang X, Gao W, Yue Y, Vartanian T, Wang H. J Immunol. 2010;185:2134–2139. doi: 10.4049/jimmunol.0902782. [DOI] [PubMed] [Google Scholar]
- 36.Thimmulappa RK, Scollick C, Traore K, Yates M, Trush MA, Liby KT, Sporn MB, Yamamoto M, Kensler TW, Biswal S. Biochem Biophys Res Commun. 2006;351:883–889. doi: 10.1016/j.bbrc.2006.10.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thimmulappa RK, Fuchs RJ, Malhotra D, Scollick C, Traore K, Bream JH, Trush MA, Liby KT, Sporn MB, Kensler TW, Biswal S. Antioxid Redox Signaling. 2007;9:1963–1970. doi: 10.1089/ars.2007.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ferguson HE, Thatcher TH, Olsen KC, Garcia-Bates TM, Baglole CJ, Kottmann RM, Strong ER, Phipps RP, Sime PJ. Am J Physiol: Lung Cell Mol Physiol. 2009;297:L912–L919. doi: 10.1152/ajplung.00148.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ichikawa T, Li J, Meyer CJ, Janicki JS, Hannink M, Cui T. PLoS One. 2009;4:e8391. doi: 10.1371/journal.pone.0008391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aleksunes LM, Goedken MJ, Rockwell CE, Thomale J, Manautou JE, Klaassen CD. J Pharmacol Exp Ther. 2010;335:2–12. doi: 10.1124/jpet.110.170084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Auletta JJ, Alabran JL, Kim BG, Meyer CJ, Letterio JJ. J Interferon Cytokine Res. 2010;30:497–508. doi: 10.1089/jir.2009.0100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stack C, Ho D, Wille E, Calingasan NY, Williams C, Liby K, Sporn M, Dumont M, Beal MF. Free Radicals Biol Med. 2010;49:147–158. doi: 10.1016/j.freeradbiomed.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Neymotin A, Calingasan NY, Wille E, Naseri N, Petri S, Damiano M, Liby KT, Risingsong R, Sporn M, Beal MF, Kiaei M. Free Radicals Biol Med. 2011;51:88–96. doi: 10.1016/j.freeradbiomed.2011.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yates MS, Tauchi M, Katsuoka F, Flanders KC, Liby KT, Honda T, Gribble GW, Johnson DA, Johnson JA, Burton NC, Guilarte TR, Yamamoto M, Sporn MB, Kensler TW. Mol Cancer Ther. 2007;6:154–162. doi: 10.1158/1535-7163.MCT-06-0516. [DOI] [PubMed] [Google Scholar]
- 45.Wang X, Ye XL, Liu R, Chen HL, Bai H, Liang X, Zhang XD, Wang Z, Li WL, Hai CX. Chem-Biol Interact. 2010;184:328–337. doi: 10.1016/j.cbi.2010.01.034. [DOI] [PubMed] [Google Scholar]
- 46.Liu W, Wong C. Phytother Res. 2010;24:369–373. doi: 10.1002/ptr.2948. [DOI] [PubMed] [Google Scholar]
- 47.Yeligar SM, Machida K, Kalra VK. J Biol Chem. 2010;285:35359–35373. doi: 10.1074/jbc.M110.138636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sun J, Hoshino H, Takaku K, Nakajima O, Muto A, Suzuki H, Tashiro S, Takahashi S, Shibahara S, Alam J, Taketo MM, Yamamoto M, Igarashi K. EMBO J. 2002;21:5216–5224. doi: 10.1093/emboj/cdf516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu H, Dinkova-Kostova AT, Talalay P. Proc Natl Acad Sci U S A. 2008;105:15926–15931. doi: 10.1073/pnas.0808346105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Francis SE, Sullivan DJ, Jr, Goldberg DE. Annu Rev Microbiol. 1997;51:97–123. doi: 10.1146/annurev.micro.51.1.97. [DOI] [PubMed] [Google Scholar]
- 51.Goldberg DE, Slater AF, Cerami A, Henderson GB. Proc Natl Acad Sci U S A. 1990;87:2931–2935. doi: 10.1073/pnas.87.8.2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sherman IW, Ruble JA, Tanigoshi L. Mil Med. 1969;134:954–961. [PubMed] [Google Scholar]
- 53.Ferreira A, Marguti I, Bechmann I, Jeney V, Chora A, Palha NR, Rebelo S, Henri A, Beuzard Y, Soares MP. Cell. 2011;145:398–409. doi: 10.1016/j.cell.2011.03.049. [DOI] [PubMed] [Google Scholar]
- 54.Pamplona A, Ferreira A, Balla J, Jeney V, Balla G, Epiphanio S, Chora A, Rodrigues CD, Gregoire IP, Cunha-Rodrigues M, Portugal S, Soares MP, Mota MM. Nat Med (N Y, NY, U S) 2007;13:703–710. doi: 10.1038/nm1586. [DOI] [PubMed] [Google Scholar]
- 55.Seixas E, Gozzelino R, Chora A, Ferreira A, Silva G, Larsen R, Rebelo S, Penido C, Smith NR, Coutinho A, Soares MP. Proc Natl Acad Sci U S A. 2009;106:15837–15842. doi: 10.1073/pnas.0903419106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ferreira A, Balla J, Jeney V, Balla G, Soares MP. J Mol Med (Heidelberg, Ger) 2008;86:1097–1111. doi: 10.1007/s00109-008-0368-5. [DOI] [PubMed] [Google Scholar]
- 57.Khor TO, Huang MT, Kwon KH, Chan JY, Reddy BS, Kong AN. Cancer Res. 2006;66:11580–11584. doi: 10.1158/0008-5472.CAN-06-3562. [DOI] [PubMed] [Google Scholar]
- 58.Zhu H, Jia Z, Zhang L, Yamamoto M, Misra HP, Trush MA, Li Y. Exp Biol Med (Maywood, NJ, U S) 2008;233:463–474. doi: 10.3181/0711-RM-304. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.