

Abstract
Nanoparticle-based therapeutics with local delivery and external electromagnetic field modulation holds extraordinary promise for soft-tissue cancers such as breast cancer; however, knowledge of the distribution and fate of nanoparticles in vivo is crucial for clinical translation. Here we demonstrate that multiple diagnostic capabilities can be introduced in photothermal therapeutic nanocomplexes by simultaneously enhancing both near-infrared fluorescence and magnetic resonance imaging (MRI). We track nanocomplexes in vivo, examining the influence of HER2 antibody targeting on nanocomplex distribution over 72 h. This approach provides valuable, detailed information regarding the distribution and fate of complex nanoparticles designed for specific diagnostic and therapeutic functions.
Keywords: Multimodal imaging in vivo, nanoshells, fluorescence optical imaging, magnetic resonance imaging, biodistribution
Nanoparticle-based imaging probes and therapeutic carriers are rapidly emerging as an alternative to current, conventional diagnostics and therapeutics for cancer, offering new approaches for earlier diagnosis and minimally invasive treatment.1–6 One advantage of this approach is that diagnostic and therapeutic function can easily be designed into the same nanoscale complex, with the potential for ultimately combining diagnosis and treatment strategies in clinical applications. Nanoparticle complexes that combine contrast agents for imaging with photothermal heating to induce tumor cell ablation are an example of this multifunctional, “theranostic” approach.7–10
Gold nanoshells are spherical silica nanoparticles wrapped in a nanometer-scale gold shell. They induce a strong photothermal response under near-infrared illumination which shows extraordinary promise for cancer therapy, demonstrating 100% efficacy in the remission of tumors11–15 The surfaces of gold nanoshells can be used to bind targeting, diagnostic, and therapeutic functionalities to the nanoparticle, forming a multifunctional nanocomplex. While gold nanoshells have outstanding therapeutic potential, the ability to effectively image them in vivo has mostly been limited to tissue depths of ∼1 mm.16,17 Additional moieties are needed to accurately determine the localization of the nanoshells and to evaluate their biodistribution in vivo.
Nanoshells can dramatically enhance the fluorescence of near-infrared (NIR) fluorophores, changing the quantum yield of dyes such as indocyanine green (ICG) or IR800 from typically only a few percent to above 80%.18,19 This is accomplished by growing a thin dielectric layer around the nanoshell as a spacer, upon which the molecules are then adsorbed. When dressed with these enhanced fluorophores, the bright, beaconlike nanoshells can be used as contrast agents for fluorescence optical imaging (FOI). Iron oxide (Fe3O4) nanoparticles can be incorporated into the thin dielectric layer, facilitating magnetic resonance imaging (MRI) contrast with high spatial resolution.20 These bimodal (FOI–MRI) nanoshell-based contrast agents combine the advantages of these complementary imaging modalities and extend diagnostic imaging capabilities to tissue depths of many centimeters. These multifunctional markers can serve as simultaneous reporter–actuators, useful for image guided interventions ranging from completely noninvasive treatment planning to intraoperative tumor margin detection.21,22 With addition of antibody conjugation, these nanocomplexes can be actively targeted and delivered specifically to tumors, providing tracking capabilities before, during, and after treatment. As nanoshell-based photothermal cancer therapy transitions to clinical trials and applications,13 these multifunctional nanoshells can provide additional information valuable for the next critical phase of cancer research: transitioning this potentially revolutionary technology into clinical practice.
Recently, we illustrated the design and theranostic capabilities of multifunctional magnetic–fluorescent gold nanoshells (nanocomplexes) for diagnosis and photothermal therapy of breast cancer cells in vitro.20 Here we perform studies in vivo, utilizing both the FOI and MRI contrast enhancement properties of the nanocomplexes to track the nanocomplexes in a live animal study over 72 h. Nanocomplexes conjugated with antibodies specific for targeting human epidermal growth factor receptor 2 (HER2) overexpressing breast cancer tumors could then be identified using both the FOI and MRI enhancements of the nanocomplex. As the antibody-conjugated nanocomplexes are tracked throughout the body, we observe clear differences in tumor uptake quantities between HER2 overexpressing and HER2 low expressing tumors. To our best knowledge, this study is the first to show that remarkably small concentrations (650 nM) of ICG molecules, when enhanced in this complex, can provide sufficient fluorescence contrast to visualize tumors in vivo and that combined FOI and MRI can reveal a detailed picture of geographic distribution of nanoparticles in tumors and internal organs. This work also shows that modification of the nanoparticle surface with appropriate functional groups can favorably accumulate nanocomplexes in the tumor relative to other organs, even for nanoparticles larger than 150 nm which are expected to trap in tumors primarily by enhanced permeation and retention (EPR) effect.
The multifunctional nanocomplexes were fabricated by following a similar procedure described in our in vitro studies.20 The diagnostic capabilities of the nanocomplexes were visualized in vivo by targeting HER2 expressing breast cancer tumors in animal models. Molecular targeting was achieved by assembling anti-HER2 antibodies on silica surface via streptavidin-biotin binding procedure. This conjugation protocol yields ∼300–350 antibodies per nanocomplex as quantified by enzyme-linked immunosorbent assay.20 Poly(ethylene glycol) (PEG) molecules were then conjugated to nanocomplexes to reduce nonspecific binding in vivo, provide steric stability, improve circulation time, reduce immunogenicity, and, in combination with antibodies, increase nanoparticle accumulation in tumor.23 PEG, a nontoxic U.S. Food and Drug Administration (FDA) approved biopolymer, is known to promote solubility in aqueous media and provide steric repulsion between colloidal nanoparticles reducing aggregation in tissue.24 A schematic diagram describing the antibody and PEG conjugation procedure is illustrated in Figure 1a. The silica epilayer encapsulating the Fe3O4 nanoparticle coated nanoshells also serves multiple functions: (i) it provides an effective spacer between the Au nanoshell surface and ICG molecules, maximizing fluorescence enhancement;18,19 (ii) it reduces photobleaching of the ICG molecules;25 (iii) it provides access for H2O within its pores, enhancing the interaction between Fe3O4 and the protons of water molecules and thus improving the proton relaxation rate and providing high T2 relaxivity and significant MR contrast;20,26 (iv) it facilitates antibodies and PEG binding via simple conjugation chemistry.
Figure 1.
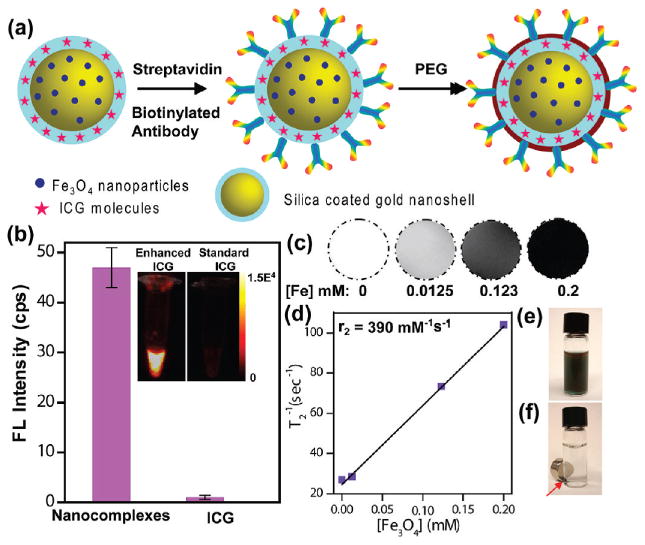
Characterization of multifunctional nanocomplexes. (a) Schematic representation of antibody and PEG conjugation to nanocomplexes. (b) Fluorescence (FL) intensity of nanocomplexes at 830 nm compared with standard unenhanced ICG showing ∼50-fold enhancement by nanocomplexes. Inset: Near-IR FL image of vials with enhanced ICG (nanocomplexes) and standard ICG of equivalent concentration. (c) T2-weighted MR images of nanocomplexes in aqueous media at various concentrations. The [Fe] concentration in each sample is provided at the bottom of the respective images. (d) Spin-spin relaxation rate (T2−1) as a function of [Fe] of the nanocomplexes. Optical image of nanocomplexes (e) dispersed in aqueous media and (f) with magnet (shown with arrow).
The nanocomplexes demonstrated intense fluorescence enhancement of the ICG molecules doped within the silica epilayer. A comparison of the fluorescence intensity of nanocomplexes dispersed in aqueous media with standard unenhanced aqueous ICG solution of equivalent concentration at 830 nm (Figure 1b and inset) clearly shows a ∼50-fold enhancement of ICG molecules in the presence of the nanoshells. Nearly 650 nM of ICG is doped in a particle concentration of ∼9 × 109 particles/mL. This concentration of ICG would be entirely inadequate to provide image enhancement in vivo, even for shallow, subcutaneous imaging. Other fluorophore-encapsulated nanoparticles which have been examined in vivo utilized a significantly higher concentration of doped-fluorophore (>10 μM)27–29 for image analysis, signifying that nanoshell-mediated fluorescence enhancement provides a route to tumor diagnosis at significant tissue depths with substantially low doses of fluorophore. The nanocomplexes also exhibited excellent magnetic characteristics as shown in the T2-weighted MR images (Figure 1c) with a high T2 relaxivity, r2 = 390 mM−1 s−1 (Figure 1d). Upon placement of a magnet next to a vial of nanocomplexes, they accumulate near the magnet within 2 h, resulting in a clear solution (Figure 1f).
The in vivo experiments were executed by first subcutaneously injecting HER2 overexpressing human breast cancer cells, BT474AZ, and control, HER2 low expressing breast cancer cells, MDAMB231, at the dorsal flank of female nude mice (20 ± 3 g) near the fourth mammary chain. Care was taken to inject the cells in the mammary fat pad. The tumors were allowed to grow to 7–8 mm before nanocomplex injection and imaging. Nanocomplexes at a concentration of 9 × 109 particles/mL were injected systemically via the tail vein of mice, ∼10 μL/g body weight, and animals were imaged immediately. This was equivalent to a dose of approximately 5 μg of ICG per kg body weight, which is 400 times lower than the maximum FDA approved clinical dose of 2 mg of ICG/kg body weight.30–32 NIR fluorescence images of mice with MDAMB231 xenografts (top) and BT474AZ xenografts (bottom) are shown (Figure 2a) at 0.3, 2, 4, 24, 48, and 72 h postinjection of nanocomplexes. The NIR images were obtained using a 100 mW, 785 nm diode laser with the beam expanded uniformly over the entire mouse body.
Figure 2.
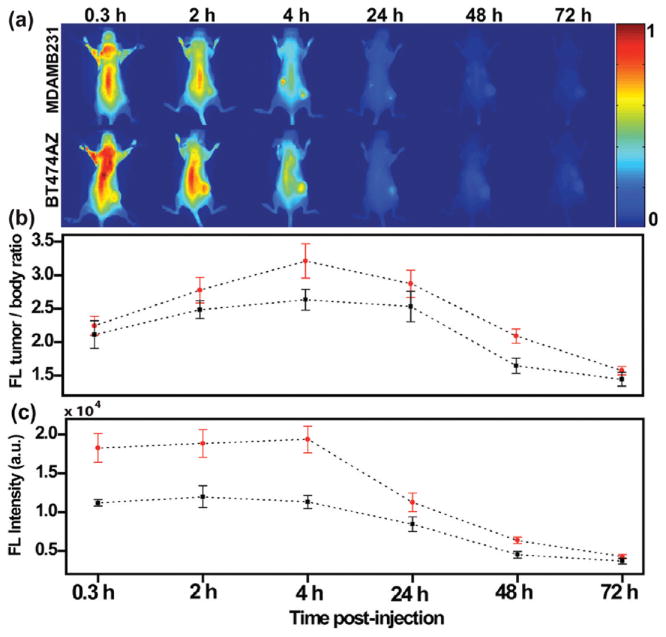
Nanocomplexes delivery in vivo imaged via NIR-FOI (a) NIR images of mice with HER2 low expressing MDAMB231 xenografts (top) and HER2 overexpressing BT474AZ xenografts (bottom) at 0.3, 2, 4, 24, 48, and 72 h postinjection of nanocomplexes. (b) Fluorescence (FL) intensity of tumor-to-body ratio at different time points of mice with BT474AZ xenografts (n = 6) and MDAMB231 xenografts (n = 3) and showing maximum fluorescence at 4 h. Significant variation across tumor types, p = 0.007, determined by ANOVA is observed. (c) Fluorescence intensity comparison of tumors only between BT474AZ (n = 6) and MDAMB231 (n = 3) showing 71.5% increase in signal at 4 h in BT474AZ tumors compared to MDAMB231 tumors, p = 0.003 across tumor types. The dotted lines have been provided as guide to the eye.
The fluorescence intensity of the MDAMB231 xenografts (n = 3) and the BT474AZ xenografts (n = 6), determined as tumor-to-body ratio, was analyzed at a series of time points, showing maximum contrast at 4 h postinjection (Figure 2b). Fluorescence intensities determined as tumor-to-body ratio represent the normalized signal, taking into account the total number of nanocomplexes injected into the body. Significant variation across tumor types (p = 0.007) and across time points (p = 3e−10) was observed. The considerable fluorescence signal observed at 0.3 and 2 h is possibly due to nanocomplex circulation in the body. It is noticeable, however, that within this short time a significant difference in nanocomplexes uptake between MDAMB231 and BT474AZ tumors are already observable. Over time, a portion of the nanocomplexes are cleared from the liver and spleen, most likely by mononuclear phagocytes, and some nanocomplexes would either accumulate in the peripheral vessels of the kidneys or be excreted. This is the likely explanation for the fluorescence signal observed from the body of the mice in the region of the liver, spleen, and kidneys at 4 and 24 h. Within 72 h, nanocomplexes were cleared from the body, indicated by the decrease in florescence intensity to levels equal to the background noise. Due to a higher binding affinity of anti-HER2 conjugated nanocomplexes to the BT474AZ xenografts, more nanocomplexes were accumulated and retained in the BT474AZ tumors. To verify the specificity and sensitivity of nanocomplex–antibody conjugates in targeting HER2 overexpressing tumors, a comparison of the fluorescence intensities of the tumors only was also evaluated at different time points (Figure 2c). This comparison reveals a 71.5% increase in the BT474AZ tumor signal at 4 h compared to the signal from the MDAMB231 tumors. This significant variation, across tumor types (p = 0.003) and across time points (p = 1e−11), indicates that the antibody–nanocomplex conjugates accumulate in the tumor more effectively by specifically targeting cell surface markers on the tumor, as well as by the EPR effect.33
The Fe3O4 nanoparticles incorporated within the porous silica epilayer of nanocomplexes provide significant MR contrast enabling tumor diagnosis at considerable depths. The T2-weighted MR images of mice with BT474AZ xenografts (Figure 3a top) and MDAMB231 xenografts (Figure 3a bottom) preinjection, 0 h, and 4, 24, 48, and 72 h postinjection of nanocomplexes are shown in Figure 3. The tumor area is shown in the red circle. As the nanocomplexes accumulate in the tumor, higher T2 contrast is observed in the tumors, making the tumors appear darker (intensity decreases). Within 72 h, as nanocomplexes clear from the tumor, they regain their original, brighter appearance (intensity increases).
Figure 3.
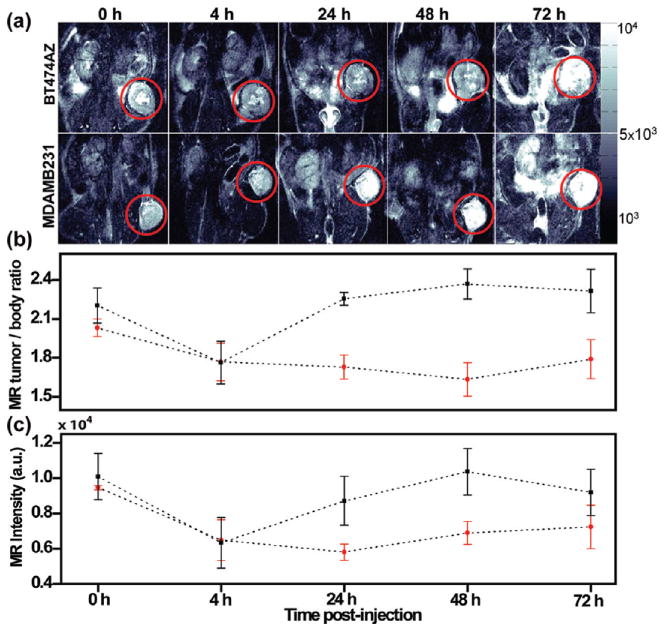
Monitoring uptake of nanocomplexes in vivo via MRI. (a) T2-weighted MR images of mice with HER2 overexpressing, BT474AZ, xenografts (top) and HER2 low expressing, MDAMB231, xenografts (bottom) preinjection, 0 h, and 4, 24, 48, and 72 h postinjection of nanocomplexes. The tumor is shown in red circle. (b) MR image intensity of tumor-to-body ratio at different time points of mice with BT474AZ xenografts (n = 3) and MDAMB231 xenografts (n = 3) and showing T2-weighted contrast for BT474AZ even at 72 h. Significant variation across tumor types, p = 0.002, determined by ANOVA is observed. (c) MR image intensity comparison of tumors only between BT474AZ (n = 3) and MDAMB231 (n = 3) showing 50.5% darker contrast at 24 h in BT474AZ tumors compared to MDAMB231 tumors, p = 0.038.
The MR contrast over time was evaluated by analyzing the T2 values of tumor-to-body ratio averaged over all slices, from which tumor cross sections were manually segmented. This accounts for variation in the performance of the MRI instrument over time as well as distribution of nanocomplexes in the tumor relative to the body. The MR image intensity of tumor-to-body ratio at different time points of mice with BT474AZ xenografts (n = 3) and MDAMB231 xenografts (n = 3) (Figure 3b) shows T2-weighted contrast for BT474AZ tumors even at 72 h. Significant variations are observed across tumor types (p = 0.002) but not across time points (p = 0.360). Analogous to fluorescence analysis, comparison of the MR intensities of tumors only at different time points (Figure 3c) validates the specificity of the nanocomplexes in targeting HER2 overexpressing BT474AZ xenografts compared to HER2 low expressing MDAMB231 xenografts. The BT474AZ tumors are ∼50.5% darker at 24 h compared to MDAMB231 tumors demonstrating maximum accumulation at 24 h. Significant variation is observed among tumor types, (p = 0.038), but not across time points (p = 0.118). The low sensitivity of MRI and low signal-to-noise ratio can explain p > 0.05 across time points, which explains in part the discrepancy between the results of MR and fluorescence imaging. In addition, some of the tumor cores were necrotic (as observed in the BT474AZ tumor) indicating the vasculature is unable to deliver sufficient oxygen and glucose. This would result in poor delivery of the nanocomplexes to the center of the tumor and hence low MR intensity.
The discrepancy between MR and fluorescence analysis in vivo not only is due to low sensitivity of MRI but also can be attributed to the surface-weighted characteristics of fluorescence imaging. With fluorescence imaging, nanocomplexes accumulated near the tumor surface are preferentially visualized, but with MRI, complete cross sections of the tumor are imaged. It is not surprising that nanocomplexes take longer to distribute within the tumor core compared to accumulation in peripheral vasculature, which explains the difference between MRI and fluorescence analysis. The discrepancy in the results between the two diagnostic modalities, however, essentially signifies the intrinsic differences between the two techniques. Nanocomplexes take longer to diffuse into the tumor interior relative to the peripheral vasculature, as reflected in the difference between MRI and fluorescence analysis. MRI provides high spatial resolution and the capability to obtain detailed anatomical structure information at significant probe depths, but lacks sensitivity relative to fluorescence imaging. Fluorescence imaging offers exceptional detection sensitivity, to the single photon level, but is limited by photon penetration depth. Combining these two complementary imaging modalities by means of a single contrast agent nanocomplex is therefore quite advantageous. The nanocomplexes both facilitate highly sensitive time-dependent fluorescence distribution in vivo and also provide detailed spatial profiling of the tumors in addition to contrast agent location. This enables a detailed map of the tumor interior, including the distribution of nanocomplexes within the tumor, at specific time points. This detailed information will be highly useful in developing and optimizing nanoparticle-based cancer therapies.22
The individual tissues of both BT474AZ and MDAMB231 mice were analyzed to determine the biodistribution of these nanocomplexes in different organs and their bioelimination. NIR fluorescence images of individual tissues (Figure 4a), retrieved from MDAMB231 (n = 3) and BT474AZ (n = 5) mice sacrificed 72 h postinjection of nanocomplexes, show clearly that the maximum accumulation is in the tumor relative to the other tissues sampled. Since a higher concentration of nanocomplexes accumulated in the tumor of BT474AZ mice (Figure 2 and Figure 3), consequently, more nanocomplexes are observed to be eliminated from the body into liver and spleen. This explains the higher fluorescence intensity observed in these organs of BT474AZ mice relative to MDAMB231 mice (Figure 4a). A surface averaged fluorescence intensity analysis, where the fluorescence intensity was divided by the surface area of each tissue (Figure 4b) demonstrate that tumors have maximum nanocomplex uptake, for both tumor types, followed by liver, kidneys, spleen, lungs, heart, and brain. Significant variation, (p = 0.005), is observed in fluorescence intensity across tumor types. Nanocomplex accumulation observed in MDAMB231 xenografts may be due to some (low) HER2 expression in this cell line as well as the EPR effect. In both tumor types, it is surprising that kidneys and liver have similar surface averaged fluorescence intensities since nanoparticles >10 nm are known to accumulate in liver. This may be attributed to the surface weighted characteristics of optical images, and since liver is larger and highly absorbing in the NIR region, nanocomplexes buried deeper into liver may not be observable by fluorescence. While FOI is an effective technique, a method that analyzes the atomic Au content directly and accurately is required to verify the nanocomplex distribution in all tissues. The Au content in each tissue was measured using inductively coupled plasma mass spectrometry (ICP-MS). The Au distribution (μg) per mass of tissue (g) for each tissue type, for both BT474AZ and MDAMB231 mice (Figure 4c), correlates well with the measured fluorescence intensities (Figure 4b) with minor variations. In both tumor types, the tumors and liver have significant Au content followed by kidneys, spleen, lungs, heart, and brain. Significant variation is observed across tumor types (p = 0.019). Tissues of BT474AZ have a higher nanocomplex accumulation relative to MDAMB231; it is also noted that BT474AZ liver has a higher distribution of gold than the kidneys, as expected. The Au levels in the lungs and heart may be due to some small amounts of residual blood left behind in these highly perfused tissues. Both fluorescence and ICP-MS measurements show low Au concentration in the brain, indicating that the nanocomplexes have low permeation through the blood–brain barrier.
Figure 4.
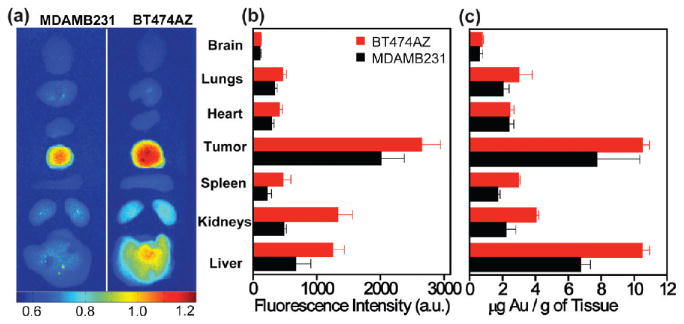
Nanocomplexes biodistribution in vivo. (a) NIR fluorescence images of mice tissues harvested from BT474AZ (left) and MDAMB231 (right) 72 h postinjection of nanocomplexes. (b) Surface averaged fluorescence intensity analysis of mice organs of BT474AZ (red, n = 5) and MDAMB231 (black, n = 3) showing maximum fluorescence in tumors, p = 0.005. (c) Gold distribution per mass of tissue obtained from ICP-MS in various mice organs of BT474AZ (red, n = 5) and MDAMB231 (black, n = 3) showing significant Au content in tumors, p = 0.019.
The differences in nanoparticle uptake in BT474AZ tumors may arise due to differences in vascular densities irrespective of the contrast in HER2 expression levels. To determine the differences in vascular density, xenografted tumors were grown in two mice per cell line in locations identical to animals treated with nanoshell injections. The tumors in these four animals were allowed to grow to ∼1 cm, and the animals were then sacrificed and tumors harvested. Tumor sections were stained with CD31 antibody to delineate endothelial cells. A clinical pathologist quantified the microvessel density from CD31 stained images. The measurements were made at eight randomly selected sites for each tumor type. We determined the microvessel density to be 43 ± 1.77 (standard error) vessels/mm2 for BT474 AZ tumors and 43.25 ± 2.29 (standard error) vessels/mm2 for the MDA-MB-231 tumors. These results are illustrated in Figure 5. Since, the vascular density was found to be the similar in two tumors, the differences in NIR/MR signal intensity were most probably due to differences in HER2 expression levels of the two tumors.
Figure 5.
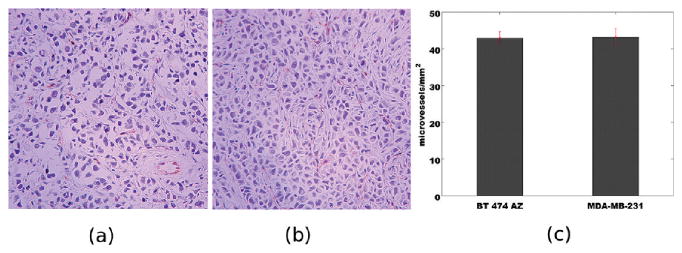
Immunohistochemistry based analysis of microvessel density via CD31 antibody staining for (a) BT474AZ and (b) MDA-MB-231 xenografts. Images are depicted at 200× magnification. The endothelial cells are stained in red; cell nuclei are stained in purple. (c) Vessel density counts for two tumor sections. Error bars are drawn with standard error of mean over eight random locations per tumor type.
After in vivo administration of nanoparticles, their bio-distribution is largely determined by particle size, surface properties such as surface charge, hydrophobicity, and moieties bound to the surface.34 The mononuclear phagocytes of the reticulo-endothelial system (RES) are known to rapidly scavenge nanoparticles from the bloodstream. Larger nanoparticles hence accumulate in the liver and spleen, and smaller nanoparticles are eliminated through the kidneys.35,36 However, PEG conjugated nanoparticles have been shown to minimize the recognition by the RES, prolonging blood circulation time.37,38 The stealth character of PEG grafted surfaces can be attributed to the low interfacial free energy of PEG in aqueous media and steric repulsion arising from a loss of conformational entropy of PEG chains in close proximity to foreign materials.24 While PEG-mediated passive targeting is effective for nanoparticle accumulation in the tumor by EPR effect, active targeting via antibodies has been shown to have higher specificity due to the direct recognition of the cell surface receptors.1,39 Antibody targeted nanocarriers have also demonstrated enhanced cellular internalization in vivo relative to nontargeted agents.40,41 The nanocomplexes utilized in this study simultaneously promote active and passive targeting via the attached PEG chains and the anti-HER2 antibodies. The high targeting capability of the nanocomplexes is attributable to their substantial accumulation in tumor.
The localization of nanocomplexes in the liver is anticipated since nanoparticles >10 nm are scavenged by the macrophages of the RES and typically accumulate in the liver and spleen.35,42–44 However, in this study, it is noteworthy that although livers have ∼3–4 times higher weight than tumors, the Au concentration is equivalent in both tissues. Furthermore, the final antibody concentration injected in vivo is ≤0.1 μg/g of body weight, which is ∼100× less dose of anti-HER2 administered intravenously in Herceptin based therapies (10 mg/kg body weight)45 which exemplify the extraordinary targeting efficacy of these multifunctional nanocomplexes to breast cancer in vivo. The significant uptake of nanocomplexes in tumor remarkably demonstrates the “smart” design of the nanocomplexes as well as efficient antibody and PEG conjugation on the nanocomplexes surface. The presence of Au in kidneys was surprising since nanoparticles >6 nm are less likely to have renal clearance via glomerular filtration36 in mice. Although, 250 nm ferrofluid particles have been shown to accumulate in kidneys of slightly larger animals, for example rabbits46 and nearly 6 μm particles have been shown to localize in human kidneys.47 In this study, the presence of nanocomplexes in the mice kidneys can be attributed to the streptavidin layer, which was used to bind the antibodies to the silica surface of the nanocomplexes. Streptavidin is known to have unusually high affinity to localize and retain in the kidneys due to the presence of the RYD motifs (Arginine-Tyrosine-Aspartic acid) which promotes adhesion to integrin receptors present in the kidneys.48–51
Tumor sections retrieved from BT474AZ mice 72 h postinjection of nanocomplexes were examined with transmission electron microscopy (TEM) to verify the structural integrity of the nanocomplexes in vivo and tumor internalization (Figure 6). Low-resolution image (Figure 6a) and corresponding high-resolution image show a nanocomplex bound to the cell surface (Figure 6b) and nanocomplexes internalized in the cell (Figure 6c). In Figure 6c, a few nanocomplexes are embedded in different planes of the tumor section appearing as dark gray spots in the image. A different area of the tumor section shows another nanocomplex internalized in the cell (Figure 6d). Although, the SiO2 epilayer and the tissue background have similar contrast in TEM, the high-resolution images (Figure 6, panel b, c, and d insets) clearly show the intact SiO2 layer encapsulating the darker Au shell. This indicates that the nanocomplexes retained their topology and structural integrity in vivo.
Figure 6.
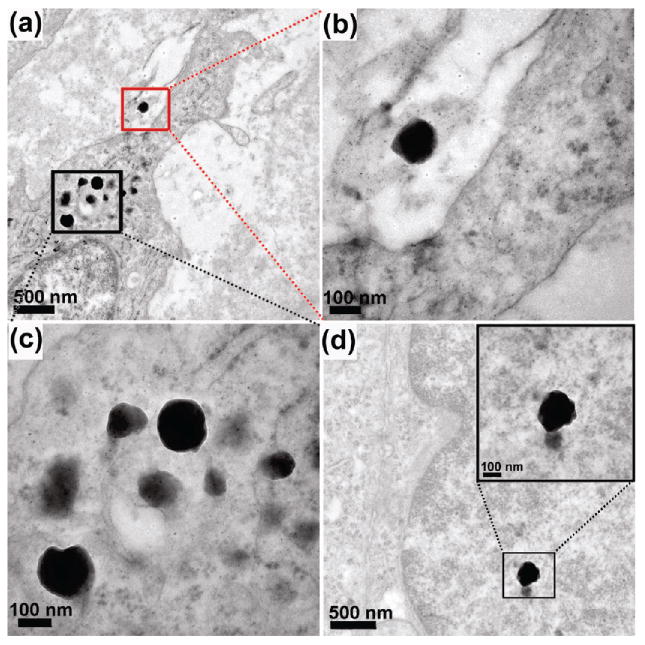
TEM images of BT474AZ tumor sections retrieved 72 h postinjection. (a) Low-resolution image showing nanocomplex bound to the cell surface (red) and nanocomplexes internalized in the cytoplasm (black). (b) High-resolution image of the nanocomplex shown within red box in part a. (c) High-resolution image of the nanocomplexes shown within black box in part a. (d) Low-resolution image of a different area of the tumor section showing nanocomplex in cytoplasm. High-resolution image of the nanocomplex provided as inset.
In summary, we have demonstrated the efficacy of multifunctional nanocomplexes which simultaneously enhance NIR fluorescence and MR contrast. In combination with PEG molecules and antibodies, the nanocomplexes actively target breast carcinoma tumors in vivo, which then can be diagnosed via MRI and FOI. We have examined the biodistribution of nanocomplexes 72 h postinjection, observing the accumulation of nanocomplexes primarily in the tumor. The structural integrity of the nanocomplexes was verified in vivo. For effective clinical translation, nanoparticle based diagnostic and therapeutic agents should essentially (i) be composed of well-characterized nontoxic materials, (ii) be hydrophilic, (iii) have long blood circulation time, (iv) exhibit high targeting and uptake efficiency in diseased cells, tissues, and/or tumors, (v) be less prone to aggregation in vivo and retain the structural integrity for increased therapeutic benefits, and (vi) have surface properties to maximize tumor accumulation while evading the macrophages of the RES.1,21,52 The nanocomplexes used in this study effectively combine these qualities. Ultimately these nanocomplexes will be capable of providing far more information than single modality conventional imaging probes. Following exhaustive biodistribution studies and photothermal cancer therapy in vivo these nanocomplexes could potentially revolutionize early diagnosis of cancer followed by rapid treatment, merging, and improving both detection and therapy for cancer.
Supplementary Material
Acknowledgments
We gratefully acknowledge funding support provided by the Baylor College of Medicine Faculty Seed Grant No. 2680150801, Caroline M Weiss Junior Faculty seed award, and NIH Grants R42 CA115028-02, and R01 CA151962-01 to A.J. R.B. and N.J.H, were supported by DoD NSSEFF, Air Force Office of Scientific Research (FA9550-06-1-0021), and Robert A. Welch Foundation (C-1220). C.P.T. and R.P. were supported by NIH Grant P30DK079638. R.M.W, M.B., and R.S. were supported by Breast Cancer Specialized Program of Research Excellence grant (SPORE) P50 CA058183 from the National Cancer Institute. We thank J. T. Mayo and E. Day for assistance with ICP-MS, S. Mukherjee, C. Ayala, and Dr. X. Liu for valuable discussions, B. Ji for help with animal experiments, Dr. M. Mancini and D. Townley from Integrated Microscopy Core, Baylor College of Medicine for assistance with TEM.
Footnotes
Supporting Information Available. Multifunctional nanocomplexes fabrication, xenograft preparation, in vivo fluorescence and MR imaging methodology, ICPMS sample preparation, tissue preparation for TEM, and statistical analysis details. This material is available free of charge via the Internet at http://pubs.acs.org.
References and Notes
- 1.Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nat Nanotechnol. 2007;2(12):751–760. doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]
- 2.Hearty S, Leonard P, O'Kennedy R. Nat Nanotechnol. 2010;5:9–10. doi: 10.1038/nnano.2009.459. [DOI] [PubMed] [Google Scholar]
- 3.Horcajada P, Chalati T, Serre C, Gillet B, Sebrie C, Baati T, Eubank JF, Heurtaux D, Clayette P, Kreuz C, Chang JS, Hwang YK, Marsaud V, Bories PN, Cynober L, Gil S, Férey G, Couvreur P, Gref R. Nat Mater. 2010;9:172–178. doi: 10.1038/nmat2608. [DOI] [PubMed] [Google Scholar]
- 4.Choi HS, Liu W, Liu F, Nasr K, Misra P, Bawendi MG, Frangioni JV. Nat Nanotechnol. 2010;5:42–47. doi: 10.1038/nnano.2009.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weissleder R. Science. 2006;312:1168–1171. doi: 10.1126/science.1125949. [DOI] [PubMed] [Google Scholar]
- 6.Bruns OT, Ittrich H, Peldschus K, Kaul MG, Tromsdorf UI, Lauterwasser J, Nikolic MS, Mollwitz B, Merkel M, Bigall NC, Sapra S, Reimer R, Hohenberg H, Weller H, Eychmuller A, Adam G, Beisiegel U, Heeren J. Nat Nanotechnol. 2009;4:193–201. doi: 10.1038/nnano.2008.405. [DOI] [PubMed] [Google Scholar]
- 7.McCarthy JR. Nanomedicine (London, U K) 2009;4:693–695. doi: 10.2217/nnm.09.58. [DOI] [PubMed] [Google Scholar]
- 8.Davis ME, Chen ZG, Shin DM. Nat Rev Drug Discovery. 2008;7:771–782. doi: 10.1038/nrd2614. [DOI] [PubMed] [Google Scholar]
- 9.McCarthy JR, Jaffer FA, Weissleder R. Small. 2006;2:983–987. doi: 10.1002/smll.200600139. [DOI] [PubMed] [Google Scholar]
- 10.Huang XH, Jain PK, El-Sayed IH, El-Sayed MA. Nanomedicine. 2007;2:681–693. doi: 10.2217/17435889.2.5.681. [DOI] [PubMed] [Google Scholar]
- 11.Choi MR, Stanton-Maxey KJ, Stanley JK, Levin CS, Bardhan R, Akin D, Badve S, Sturgis J, Robinson JP, Bashir R, Halas NJ, Clare SE. Nano Lett. 2007;7:3759–3765. doi: 10.1021/nl072209h. [DOI] [PubMed] [Google Scholar]
- 12.Hirsch LR, Stafford RJ, Bankson JA, Sershen SR, Rivera B, Price RE, Hazle JD, Halas NJ, West JL. Proc Natl Acad Sci U S A. 2003;100:13549–13554. doi: 10.1073/pnas.2232479100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lal S, Clare SE, Halas NJ. Acc Chem Res. 2008;41:1842–1851. doi: 10.1021/ar800150g. [DOI] [PubMed] [Google Scholar]
- 14.O'Neal DP, Hirsch LR, Halas NJ, Payne JD, West JL. Cancer Lett. 2004;209:171–176. doi: 10.1016/j.canlet.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 15.Stern JM, Stanfield J, Kabbani W, Hsieh JT, Cadeddu JA. J Urol. 2008;179:748–753. doi: 10.1016/j.juro.2007.09.018. [DOI] [PubMed] [Google Scholar]
- 16.Gobin AM, Lee MH, Halas NJ, James WD, Drezek RA, West JL. Nano Lett. 2007;7:1929–1934. doi: 10.1021/nl070610y. [DOI] [PubMed] [Google Scholar]
- 17.Loo C, Lowery A, Halas NJ, West JL, Drezek RA. Nano Lett. 2005;5:709–711. doi: 10.1021/nl050127s. [DOI] [PubMed] [Google Scholar]
- 18.Bardhan R, Grady NK, Cole J, Joshi A, Halas NJ. ACS Nano. 2009;3:744–752. doi: 10.1021/nn900001q. [DOI] [PubMed] [Google Scholar]
- 19.Bardhan R, Grady NK, Halas NJ. Small. 2008;4:1716–1722. doi: 10.1002/smll.200800405. [DOI] [PubMed] [Google Scholar]
- 20.Bardhan R, Chen W, Perez-Torres C, Bartels M, Huschka RM, Zhao LL, Morosan E, Pautler RG, Joshi A, Halas NJ. Adv Funct Mater. 2009;19(24):3901–3909. [Google Scholar]
- 21.Cheon J, Lee JH. Acc Chem Res. 2008;41:1630–1640. doi: 10.1021/ar800045c. [DOI] [PubMed] [Google Scholar]
- 22.Kircher MF, Mahmood U, King RS, Weissleder R, Josephson L. Cancer Res. 2003;63:8122–8125. [PubMed] [Google Scholar]
- 23.Akiyama Y, Mori T, Katayama Y, Niidome T. J Controlled Release. 2009;139:81–84. doi: 10.1016/j.jconrel.2009.06.006. [DOI] [PubMed] [Google Scholar]
- 24.Otsuka H, Nagasaki Y, Kataoka K. Adv Drug Delivery Rev. 2003;55:403–419. doi: 10.1016/s0169-409x(02)00226-0. [DOI] [PubMed] [Google Scholar]
- 25.Burns A, Ow H, Wiesner U. Chem Soc Rev. 2006;35:1028–1042. doi: 10.1039/b600562b. [DOI] [PubMed] [Google Scholar]
- 26.Tovbina ZM, Kuts VS, Strelko VV. Teor Eksp Khim. 1969;5:848–850. [Google Scholar]
- 27.Altýnoglu EI, Russin TJ, Kaiser JM, Barth BM, Eklund PC, Kester M, Adair JH. ACS Nano. 2008;2(10):2075–2084. doi: 10.1021/nn800448r. [DOI] [PubMed] [Google Scholar]
- 28.Texier I, Goutayer M, Silva AD, Guyon L, Djaker N, Josserand V, Neumann E, Bibette J, Vinet F. J Biomed Opt. 2009;14:054005. doi: 10.1117/1.3213606. [DOI] [PubMed] [Google Scholar]
- 29.Bumb A, Regino CAS, Perkins MR, Bernardo M, Ogawa M, Fugger L, Choyke PL, Dobson PJ, Brechbiel MW. Nanotechnology. 2010;21:175704. doi: 10.1088/0957-4484/21/17/175704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.IC Green. http://www.drugs.com/pro/ic-green.html.
- 31.Costa RA, Farah ME, Freymüller E, Morales PH, Smith R, Cardillo JA. Am J Ophthalmol. 2001;132(4):557–565. doi: 10.1016/s0002-9394(01)01138-2. [DOI] [PubMed] [Google Scholar]
- 32.Gandorfer A, Haritoglou C, Gandorfer A, Kampik A. Invest Ophthalmol Visual Sci. 2003;44:316–323. doi: 10.1167/iovs.02-0545. [DOI] [PubMed] [Google Scholar]
- 33.McDonald DM, Baluk P. Cancer Res. 2002;62:5381–5385. [PubMed] [Google Scholar]
- 34.Sonavane G, Tomoda K, Makino K. Colloids and Surf, B. 2008;66:274–280. doi: 10.1016/j.colsurfb.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 35.James WD, Hirsch LR, West JL, O'Neal PD, Payne JD. J Radioanal Nucl Chem. 2007;271(2):455–459. [Google Scholar]
- 36.Longmire M, Choyke PL, Kobayashi H. Nanomedicine. 2008;3(5):703–17. doi: 10.2217/17435889.3.5.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Akerman ME, Chan WCW, Laakkonen P, Bhatia SN, Ruoslahti E. Proc Natl Acad Sci U S A. 2002;99(20):12617–12621. doi: 10.1073/pnas.152463399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Prencipe G, Tabakman SM, Welsher K, Liu Z, Goodwin AP, Zhang L, Henry J, Dai H. J Am Chem Soc. 2009;131:4783–4787. doi: 10.1021/ja809086q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang L, Mao H, Wang YA, Cao Z, Peng X, Wang X, Duan H, Ni C, Yuan Q, Adams G, Smith MQ, Wood WC, Gao X, Nie S. Small. 2009;5(2):235–243. doi: 10.1002/smll.200800714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kirpotin DB, Drummond DC, Shao Y, Shalaby MR, Hong K, Nielsen U, Marks JD, Benz CC, Park JW. Cancer Res. 2006;66:6732–6740. doi: 10.1158/0008-5472.CAN-05-4199. [DOI] [PubMed] [Google Scholar]
- 41.Lammers T, Hennink WE, Storm G. Br J Cancer. 2008;99:392–397. doi: 10.1038/sj.bjc.6604483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen J, Glaus C, Laforest R, Zhang Q, Yang M, Gidding M, Welch MJ, Xia Y. Small. 2010;6(7):811–817. doi: 10.1002/smll.200902216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Niidome T, Yamagata M, Okamoto Y, Akiyama Y, Takahashi H, Kawano T, Katayama Y, Niidome Y. J Controlled Release. 2006;114:343–347. doi: 10.1016/j.jconrel.2006.06.017. [DOI] [PubMed] [Google Scholar]
- 44.Terentyuk GS, Maslyakova GN, Suleymanova LV, Khlebtsov BN, Kogan BY, Akchurin GG, Shantrocha AV, Maksimova IL, Khlebtsov NG, Tuchin VV. J Biophotonics. 2009;2(5):292–302. doi: 10.1002/jbio.200910005. [DOI] [PubMed] [Google Scholar]
- 45.Arpino G, Gutierrez C, Weiss H, Rimawi M, Massarweh S, Bharwani L, Placido SD, Osborne CK, Schiff R. J Natl Cancer Inst. 2007;99:694–705. doi: 10.1093/jnci/djk151. [DOI] [PubMed] [Google Scholar]
- 46.Wiegand S, Heinen T, Ramaswamy A, Sesterhenn AM, Bergemann C, Werner JA, Lübbe AS. J Drug Targeting. 2009;17(3):194–199. doi: 10.1080/10611860802582467. [DOI] [PubMed] [Google Scholar]
- 47.Gattia AM, Rivasi F. Biomaterials. 2002;23:2381–2387. doi: 10.1016/s0142-9612(01)00374-x. [DOI] [PubMed] [Google Scholar]
- 48.Wilbur DS, Hamlin DK, Sanderson J, Lin Y. Bioconjugate Chem. 2004;15:1454–1463. doi: 10.1021/bc049869n. [DOI] [PubMed] [Google Scholar]
- 49.Murray S, Maraveyas A, Dougan T, Chu AC. Biochim Biophys Acta. 2002;1570:81–88. doi: 10.1016/s0304-4165(02)00157-5. [DOI] [PubMed] [Google Scholar]
- 50.Schechter B, Arnon R, Colas C, Burakova T, Wilchek M. Kidney Int. 1995;47:1327–1335. doi: 10.1038/ki.1995.188. [DOI] [PubMed] [Google Scholar]
- 51.Wilbur DS, Hamlin DK, Buhler KR, Pathare PM, Vessella RL, Stayton PS, To R. Bioconjugate Chem. 1998;9:322–330. doi: 10.1021/bc970182v. [DOI] [PubMed] [Google Scholar]
- 52.Couvreur P, Vauthier C. Pharm Res. 2006;23(7):1417–50. doi: 10.1007/s11095-006-0284-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.