

Abstract
Microorganisms live in fluctuating environments, requiring stress response pathways to resist environmental insults and stress. These pathways dynamically monitor cellular status, and mediate adaptive changes by remodeling the proteome, largely accomplished by remodeling transcriptional networks and protein degradation. The complementarity of fast, specific proteolytic degradation and slower, broad transcriptomic changes gives cells the mechanistic repertoire to dynamically adjust cellular processes and optimize response behavior. Together, this enables cells to minimize the “cost” of the response while maximizing the ability to survive environmental stress. Here we highlight recent progress in the understanding of transcriptional networks and proteolysis that illustrates design principles used by bacteria to generate the complex behaviors required to resist stress.
1. Introduction
Bacteria and other single-celled organisms have evolved to survive in variable and at times extreme conditions, and must sense and mount effective responses to environmental challenges as diverse as heat, oxidative damage, anti-microbial agents, and nutritional limitation. While bacteria have a number of programs that they can use to combat these environmental challenges, mounting a costly response in the absence of stress is detrimental, as resources that could be utilized for growth are wastefully funneled into unneeded adaptations [1]. Since bacteria are in constant competition with other species in their environment, organisms with more efficient stress responses have a competitive advantage. Thus, stress responses are carefully regulated so that they are activated only when required and to the extent necessary.
This review will describe emerging stories in bacterial stress responses that highlight design principles used by bacteria to mount stress responses that are fast, accurate, cost efficient, and successful. We focus on two complementary mechanisms that remodel the proteome to oppose stress: rewiring the transcriptome and modulating proteolysis. While transcription can activate broad swathes of genes in concert, proteolysis is best suited to quickly adjust the availability of specific cellular proteins to favor required processes. Together, these mechanisms allow cells to maintain a dynamic equilibrium, continually re-optimizing processes in response to changing environmental cues.
2. Transcriptional remodeling in response to stress
The first step in a transcriptional response is to convert the signals from the environment into transcriptional change, leading to production of new proteins and adaptation. Regulators can sense stress through two general mechanisms: 1) Consequence sensing (e.g., sensing heat by the accumulation of unfolded proteins); 2) Direct sensing (e.g., a regulatory RNA whose structure is melted by heat), also called “feed-forward” sensing [2] (note that this is distinct from the “feed-forward loop” regulatory motif [3, 4]). Notably, these stress signals often control transcription factors post-transcriptionally (e.g., by protein degradation or regulation of activity). This decreases the lag time of transcriptional responses, enabling both a rapid initial response and rapid adaptation. As stresses are alleviated, the activity of stress-responsive transcription factors then decreases to reach a new homeostasis.
In this section, we review emerging stories about bacterial stress-responsive transcription factors, focusing on two large families, two-component systems and alternative sigma factors (σs). Two-component systems are comprised of a sensor histidine kinase and a cognate response regulator [5, 6]. When activated, a histidine kinase auto-phosphorylates and then transfers the phosphate group to the response regulator, which modulates gene expression [5–11]. σs are subunits of RNA polymerase holoenzyme that mediate promoter recognition; alternative, non-housekeeping σs are widely used in stress responsive signal-transduction pathways [12–14]. Typically, every bacterial species contains multiple members of each of these families. We discuss how these transcription factors sense and relieve the deleterious effects of stress as quickly and accurately as possible, and how stress systems limit spurious cross-activation between pathways to ensure an accurate and specific response.
Stress sensory domains in two-component systems
How do two-component systems sense stress signals? For histidine kinases, which auto-phosphorylate on a specific histidine residue, the current model is that ligand binding induces conformational changes that properly position the catalytic domain and facilitate phosphorylation of the target histidine, activating the response [10, 15–21]. Indeed, this is the mechanism proposed for the Escherichia coli histidine kinase EnvZ, which regulates the membrane porins OmpC and OmpF with its response regulator, OmpR [22–24]. EnvZ crosses the inner membrane and monitors a variety of signals (osmolarity, pH, temperature, procaine), though the location of the primary signal (periplasm vs cytoplasm) is unknown [22, 23].
Recent work has demonstrated that high osmolarity directly alters the conformation of the cytoplasmic fragment of EnvZ (EnvZ-C) [24], an example of feed-forward sensing. High osmolarity drives EnvZ-C to adopt a more compact structure, properly positioning the catalytic and auto-phosphorylation sites, and activating OmpR [24]. While EnvZ-C may be sufficient for osmo-sensing [24, 25], the other domains of EnvZ may still play a role in response, as substitutions in the EnvZ transmembrane domains are known to affect EnvZ activity [26]. Like many histidine kinases, EnvZ contains inner membrane proximal HAMP domains, which mediate transduction of periplasmic or transmembrane stimuli into conformational changes in the cytoplasm [20, 21, 27, 28]. Thus, the periplasmic portions of EnvZ may sense other types of signals, play a role in EnvZ dimerization, or sense osmolarity in a concerted fashion with EnvZ-C by mediating conformational change of EnvZ-C [22]. Lastly, the periplasmic portion of EnvZ interacts with MzrA, which modulates EnvZ activity, but does not preclude EnvZ signal sensing [29]. While the direct signals that modulate MzrA activity are unknown, MzrA may be regulated by both CpxA/CpxR and σE, two sensors of membrane status [29, 30]. While EnvZ-C may be a feed-forward sensor for osmolarity, the EnvZ periplasmic domains may have a role in sensing other EnvZ signals and properly modulating the activity of EnvZ.
Other two-component systems often contain Per-ARNT-Sim (PAS) domains, a structural motif found across all kingdoms of life that can be feed-forward sensors for signals as varied as light, redox potential, and metabolites, though the mechanisms that activate most PAS domains remain unknown [31–33]. As PAS domains are highly modular, they can be exchanged or conjugated to alternative proteins to reprogram signaling and response [34–38]. This allows development of various genetic tools; for example, a light activated histine kinase, by switching the oxygen sensitive PAS domain of Bradyrhizobium japonicum FixL for the light sensitive PAS domain of Bacillus subtilis YtvA [36], or a chimeric histidine kinase that cooperatively responds to both light and oxygen, by fusing the YtvA light sensitive PAS domain to the FixL oxygen sensing domain [35].
Unfortunately, the activating signals and the mechanism of activation are unknown for most signaling pathways. We have excellent tools (e.g., microarrays, proteomics, ChIP) to identify the downstream targets of regulatory systems, but the methods for identifying the signals that activate the relevant regulators and the mechanisms for this activation have yet to mature. This is an important area of investigation, as identification of these signals is critical for both understanding the organism and systems biology.
How σ32 maintains protein-folding homeostasis
Maintaining protein-folding homeostasis is a critical task for all cells. It is especially important for cells living in environments with variable temperature, as heat alters protein folding. The highly regulated universal heat shock response controls expression of a core set of chaperones in all organisms, as well as many additional organism-specific proteins, including a set of conserved proteases in bacteria [39–42]. In E. coli and other proteobacteria, the HSR is controlled by σ32, the master regulator of ~100 genes [42, 43]. Recent progress in understanding E. coli σ32 illustrates the complexity of control that allows σ32 to monitor protein folding in the cytoplasm and inner membrane.
σ32 is controlled by two mechanisms that enable a rapid response: σ32 translation is regulated by a feed-forward sensing mechanism, as heat directly melts an inhibitory mRNA structure that dampens σ32 translation [44, 45]; σ32 activity and stability is controlled by two feedback loops that sense protein folding status [42]. σ32 activity is regulated by the cytoplasmic chaperones (e.g. DnaK/DnaJ), which bind directly to σ32 to inhibit its activity, and σ32 protein level is mediated by the inner membrane localized FtsH protease, which degrades σ32 (Fig. 1) [46–49]. When stresses induce protein unfolding, the chaperones and proteases are titrated away from σ32, activating the heat shock response (Fig. 1) [42]. These regulators are also themselves transcriptionally activated by σ32, forming a negative feedback loop [42]. Thus, regulation of σ32 is responsive both to heat and to cellular protein folding status.
Figure 1. Regulation of σ32 at the inner membrane.

(Left) σ32 inhibition. Membrane localized σ32 is inhibited via degradation by FtsH and inactivation by the cytoplasmic chaperones. Unfolded proteins relieve inhibition by competing for FtsH and titrating chaperones away from σ32. (Right) σ32 membrane localization. σ32 is brought to the membrane by the signal recognition particle (SRP), which also traffics inner membrane proteins (inner membranePs) to the membrane. When stress stalls or prevents proper SRP-dependent inner membraneP insertion, this may prevent σ32 from being trafficked to the membrane for inactivation.
Despite this complexity, the known circuitry could not explain two key features of σ32 response: a) mutations in a small region of σ32 (a “homeostatic control region”) disrupt inhibition of σ32 by chaperones and FtsH in vivo, leading to hyperactive σ32, but do not alter σ32 regulation by these factors in vitro [50–53]; b) σ32 is thought to monitor the folding status of inner membrane proteins, but the mechanism for this was unknown [42, 54]. These observations suggest that σ32 may monitor the inner membrane through a key regulator that had not been found.
This missing regulator was recently identified as the Signal Recognition Particle (SRP; Ffh + 4.5S RNA) [54]. SRP is part of the co-translational membrane trafficking system that mediates inner membrane protein biogenesis. SRP binds to and targets ribosomes with nascent proteins that contain hydrophobic N-terminal signal sequences to the inner membrane for cotranslational insertion and folding (Fig. 1) [55–57]. Surprisingly, although σ32 does not have a signal sequence, SRP also traffics σ32 to the inner membrane; membrane localization of σ32 is essential for proper regulation by chaperones and FtsH (Fig. 1) [54]. In fact, mutants in the σ32 homeostatic control region are hyperactive because they reduce binding to SRP, therefore they are not membrane localized by SRP and cannot be inhibited by chaperones and FtsH [54]. Thus, membrane localization is vital for proper σ32 regulation.
SRP allows σ32 to sense the protein folding status of the inner membrane. Since SRP is substoichiometric relative to ribosome (~1:100 SRP:ribosome), free SRP levels depend on efficient SRP recycling [58, 59]. As almost all inner membrane proteins are trafficked by SRP, defects in trafficking may alter SRP recycling or lead to accumulation of ribosomes with signal sequence proteins, preventing SRP from interacting with σ32 or localizing σ32 to the membrane [54, 55]. Thus, the amount and activity of σ32 will dynamically adjust in response to flux of proteins through the inner membrane (Fig. 1).
Why would σ32 sense inner membrane protein folding? The σ32 regulon contains SRP, FtsH, and is additionally enriched in proteins that are involved in or reside in the inner membrane [42, 43]. Furthermore, FtsH not only degrades σ32 but also is the main protease that mediates quality control of membrane proteins [60, 61]. Active σ32 will reduce inner membrane dysfunction by increasing levels of SRP (to ameliorate trafficking) and FtsH (to reduce unfolded protein load). As σ32 activity is further regulated by cytoplasmic chaperones, this allows σ32 to integrate the folding status of both inner membrane and cytosolic proteins, a significant advantage as inner membrane proteins comprise 20–30% of total cellular protein [55, 62].
How σE maintains homeostasis of the outer membrane
The first line of defense for gram-negative bacteria is the outer membrane, which presents a formidable permeability barrier to protect against antibiotics and other stresses [63, 64]. The outer membrane is an asymmetric lipid bilayer: its outer leaflet is composed of lipopolysaccharides (LPS) and its inner leaflet of phospholipids [63, 65]. The outer membrane additionally contains proteins, including the outer membrane proteins (OMPs) that allow access to selected solutes [63, 64, 66, 67]. Both LPS and OMPs rely on complex machines for their transport and assembly into the outer membrane [65, 68–72]. As outer membrane integrity depends on proper balance of its components [63, 64, 71], maintaining appropriate levels of assembly machines and substrates is vital.
To monitor stress in this compartment, E. coli and other γ-proteobacteria employ σE, which regulates genes required for assembly of all major components of the outer membrane [73, 74]. The major challenge for the σE system is how to convey the information about the status of the outer membrane into the cytoplasm to mediate transcriptional change. We discuss the current model for how each σE regulator senses assembly of a different outer membrane component to generate an integrated portrait of envelope status (Fig. 2).
Figure 2. Two signals are required for σE activation.
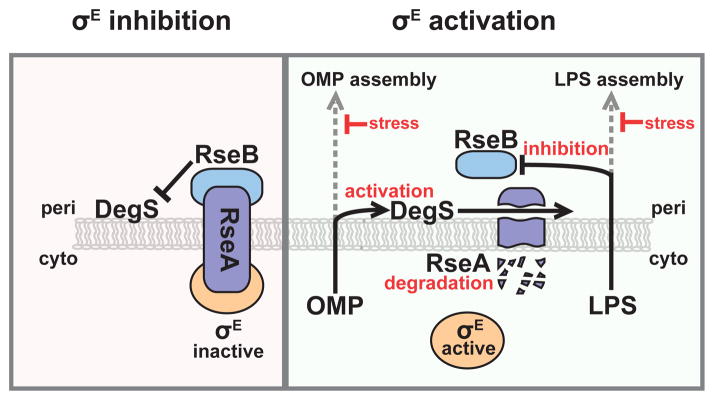
(Left) σE inhibition. σE is held inactive by RseA in the inner membrane. DegS, a protease, can cleave RseA if activated, but RseA cleavage is prevented by RseB. (Right) σE activation. When concomitant defects in OMP assembly and LPS assembly occur, σE is activated. Periplasmic LPS dissociates RseB from RseA, and periplasmic OMPs activate DegS to cleave RseA. This leads to a proteolytic cascade that degrades RseA, releasing and activating σE.
σE monitors outer membrane protein folding through the rate of cleavage of its negative regulator RseA [75–77]. RseA, an inner membrane protein that sequesters σE in an inactive conformation, can be cleaved by the protease DegS, which permits secondary cleavage by RseP and subsequent degradation of RseA, freeing σE to activate transcription [78–83]. DegS is activated only when it binds to unfolded OMP C-termini in the periplasm [77, 84–86]. As these unfolded species are thought to accumulate when OMPs are inefficiently assembled into the outer membrane, activation of DegS is a reflection of outer membrane dysfunction [87, 88].
How is the status of outer membrane LPS sensed? σE has a second negative regulator, RseB, which binds to RseA and protects it from cleavage by DegS [89–92]. Recent studies have shown that LPS can bind to and dissociate RseB from RseA [93]. In vitro, RseA degradation in the presence of RseB requires both OMPs and LPS: OMPs activate DegS, and LPS dissociates the RseA/RseB complex [93]. Similarly, in vivo, perturbations that lead to accumulation of off-pathway LPS (e.g., mutations that partially inactivate the LPS assembly machinery or alter LPS structure), in combination with activated DegS, lead to dramatic activation of σE [93, 94]. Thus, maximal activation of σE in vivo requires two signals of outer membrane stress (Fig. 2).
Why do cells integrate these two signals of outer membrane assembly? OMPs and LPS are the major unique components of the bacterial outer membrane, and thus excellent indicators of outer membrane status [65]. Requiring concomitant defects in the assembly of both OMPs and LPS reduces the chances for spurious activation, ensuring that a large and costly response is not provoked by normal variation in the flux of proteins or LPS through the periplasm. For this mechanism to be an effective response, sustained defects in either OMP or LPS assembly must provoke defects in assembly of the other, ensuring σE activation. Indeed, certain LPS species have been shown to reduce the efficiency of OMP assembly, as the altered outer membrane environment may be less conducive to proper OMP assembly [63, 95, 96]. Similarly, the major component of the LPS assembly machine is inserted into the outer membrane by the same mechanism as is used for other OMPs, thus defects in OMP assembly will eventually lead to defects in LPS assembly [97, 98]. Thus, sensing assembly intermediates for multiple outer membrane components allows bacteria to monitor outer membrane status more accurately and comprehensively.
OMPs are among the most abundant proteins in the cell and there is tremendous OMP flux to the outer membrane [63]. For this reason, increasing the production of OMP chaperones, proteases and assembly factors may be insufficient to rapidly restore proper folding. Thus, σE also reduces OMP synthesis by inducing two small RNAs (sRNAs), MicA and RybB, that target OMP mRNA for degradation, thereby dramatically decreasing the flow of OMP precursors to the envelope [99–103]. The vital role of these sRNAs is demonstrated by the fact that overexpression of either sRNA can protect the cell from the deleterious effects of depleting σE, which normally leads to lysis and cell death [104, 105]. Interestingly, the strategy employed by bacteria to address OMP folding is reminiscent of the intercompartmental eukaryotic unfolded protein response (UPR). Upon sensing stress in the endoplasmic reticulum, the UPR opposes folding stress both by upregulating folding factors and by downregulating the flow of precursors to the endoplasmic reticulum [106–108].
Dynamic responses in σB activation
To optimize stress responses, cells must tailor the timing, amplitude, and dynamics of the response to each stress. Indeed, many responses contain entwined positive and negative feedback loops that can generate distinct, sophisticated behaviors like bistability or oscillation [4, 109]. Furthermore, while many systems have regulators that suppress stochastic fluctuations (noise) to prevent spurious activation (e.g., σE system, see section 2.3), noise can also be utilized to generate sophisticated response behaviors [4, 92, 110–113]. Indeed, recent studies have demonstrated that noise is used in the Bacillus subtilis σB system to generate vastly different dynamic behaviors depending on the inducing stress (Fig. 3) [114, 115].
Figure 3. Activation of σB by different stresses leads to distinct responses.
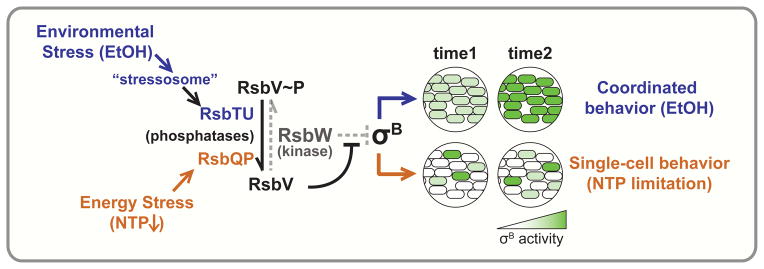
Activation of σB by environmental stress (e.g., EtOH, blue) and energy stress (decreased NTP, orange) lead to two different σB behaviors: EtOH induces a coordinated response, while nucleotide limitation induces stochastic pulses (same maximum amplitude) on a single-cell level. σB is inhibited by RsbW, which is in turn inhibited by RsbV. RsbW, a kinase, phosphorylates RsbV to relieve its own inhibition. Countering this, stress-specific phosphatases such as RsbTU (activated by EtOH, blue) and RsbQP (activated by NTP limitation, orange) dephosphorylate RsbV~P, allowing RsbV to bind to and inhibit RsbW and thereby promoting release and activation of σB.
In B. subtilis and related gram-positives, σB is a “general stress factor”, which induces a core stress regulon that is expressed in concert with stress-specific responses [116, 117]. In steady state, σB is bound and inhibited by its anti-σ, RsbW [117–119]. Under stress, the antagonist RsbV binds RsbW and frees σB, activating response [119–121]. This partner-switching mechanism is regulated by the phosphorylation state of RsbV: unphosphorylated RsbV binds RsbW, but RsbV~P cannot [119, 120]. Notably, RsbW is the kinase that phosphorylates RsbV [119]. Therefore RsbW keeps σB activity in check both by binding σB and phosphorylating RsbV [119, 120]. To activate σB, two different phosphatase systems can dephosphorylate RsbV~P: 1) RsbP, activated by nutrient limitation (e.g., limiting NTPs), and 2) RsbU, activated by environmental stress (e.g., ethanol). Environmental stress induces the highly conserved 1.8 MDa supermolecular “stressosome” complex to release RsbT, which, in turn, activates RsbU phosphatase activity (Fig. 3) [121–125].
Although both energy and environmental stress modulate σB activity through RsbV dephosphorylation, each stress leads to different dynamics in the σB response [114, 115]. Nucleotide limitation (via mycophenoic acid treatment) leads to continuous stochastic pulses of σB activity that vary in timing but not in intensity from cell-to-cell (Fig. 3) [114, 115]. In contrast, ethanol induces a single pulse of σB activity that is synchronous across the population (Fig. 3) [114, 115]. How does the circuitry governing σB generate these diverse responses? Opposing kinase (RsbW) and phosphatase (RsbP or RsbU) activities leads to an ultrasensitive response, so that σB is activated in a sharp, switch-like manner [114]. Cell-to-cell variability in either the initial level phosphatase or RsbW would mean that different cells would require different levels of phosphatase to oppose RsbW and cross the threshold of activation. Thus, during energy stress, small fluctuations (noise) in RsbW/RsbP ratio per cell could lead single-cell variability in the timing of σB activation [114]. In contrast, environmental stress induces mass release of RsbT from the stressosome, enabling RsbT to activate RsbU and overwhelm inhibition by RsbW, thus activating σB in a synchronous manner in all cells [115].
What could be the advantage in responding differently to these stresses? This is an important question that requires investigation. As Bacillus devotes up to 40% of its translational capacity to the σB regulon during stress, misregulation of σB is an enormous metabolic cost [126]. One posibility is that different patterns of σB activity are optimizations that minimize the cost of response for each stress. Another possibility could be that σB pulsing is a bet-hedging mechanism, as the cell may anticipate that nutrient limitation is a precursor for other stresses that may require σB [115]. This is supported by the fact that σB pulses indefinitely during nucleotide limitation, suggesting that σB activity does not lead to adaptation in this condition [114]. Testing these types of hypotheses is a difficult but important challenge, as these different dynamic response behaviors will be present in other systems, particularly those that sense multiple types of stress. These studies suggest that single-cell analysis of stress systems will continue to reveal novel behaviors not previously appreciated in bulk studies.
Cross-talk in signaling systems
Most bacteria have dozens if not hundreds of paralogous two-component systems, each recognizing their own signals [127, 128]. Since these systems evolved and proliferated via genomic duplication events, they often share considerable sequence and structural similarity, creating significant potential for spurious cross-activation, or “cross-talk” [128, 129]. Indeed, histidine kinases have been observed to activate non-cognate response regulators when their cognate regulator is lost [130, 131]. As cross-talk may activate non-beneficial responses, networks that are prone to cross-talk will evolve mechanisms to insulate responses [128, 129].
The current model is that the co-evolution of residues in the interaction surfaces of histidine kinases and their cognate response regulators is a major molecular basis for preventing cross-talk in two-component systems [128, 132–134]. As these residues (called “specificity residues”) determine the histidine kinase/response regulator interaction, amino acid substitutions on either the histidine kinase or response regulator can lead to recognition of non-cognate histidine kinases or response regulators [132, 134–136]. For example, a single amino acid change to the specificity residues of the histidine kinase EnvZ is sufficient to allow phosphorylation of the non-cognate response regulator RstA [132]. Global approaches have demonstrated that orthogonality (lack of cross-talk) is the norm for nearly all histidine kinases within a genome, ensuring proper insulation of signaling [133, 137].
How is orthogonality maintained during genome evolution? This was examined for the broadly conserved PhoB/PhoR two-component system, which is present in α, β, and γ-proteobacteria. Interestingly, while the specificity residues of PhoR are highly similar in β- and γ-proteobacteria, there is lower conservation between γ- and α-proteobacteria [133]. Analysis revealed this to be an evolutionary adaptation in α–proteobacteria to insulate PhoB/R from an α-specific paralog, NtrY/NtrX, preventing cross-talk between these two systems. Indeed, E. coli PhoR (γ) can phosphorylate both Caulobacter PhoB and NtrX (α), whereas Caulobacter PhoR is specific for PhoB [133]. Furthermore, a mutant PhoR that cross-activates NtrX is detrimental to growth under PhoR-inducing conditions; this growth defect is almost fully suppressed by deletion of ntrX [133]. Thus, cross-talk can produce selective pressure that drives newly acquired signaling pathways to diverge and insulate themselves against paralogous systems.
These principles are also observed for the extra-cytoplasmic σ-factors, a highly diverse group of alternative σs, which comprise 43 phyolgenetically distinct subgroups [14, 138]. Recent work with 40 σs from 20 different subgroups indicates that, in general, σs are inhibited only by their cognate anti-σ, and recognize only promoters within their subgroup [139]. However, questions remain as to whether σs within a subgroup are as well insulated. For example, the soil bacterium Streptomyces coelicolor contains an astonishing 63 σs, with 4 σs derived from subgroup 39 [138]. Do these σs initiate cooperative response, or are they well-insulated from each other? Furthermore, the evolutionary trajectories that mediate σ orthogonality are not well understood. Such analyses are key to understanding the design of signaling systems and the selective pressures that drive their evolution.
3. Regulatory proteolysis in stress response
As a counterpoint to transcriptional remodeling, regulatory proteolysis represents an alternative way of altering the protein content of the cell in response to stress. In all organisms, failure to degrade proteins that are unfolded or damaged by stress leads to protein aggregation and deleterious consequences such as cell death in bacteria and disease and aging in eukaryotes [39, 40, 140]. Proteolytic control is particularly important in bacteria, as most proteins are otherwise stable and diluted only by cell division [141]. Recently, the role of proteolytic machines in regulating transcriptional response as well as being direct sensors and effectors for stress has emerged. We have already described how proteolysis controls the amount of σ32 (see section 2.2), and the activity of σE (see section 2.3). In this section, we focus on recent stories about how the major cytoplasmic proteases ClpXP and Lon directly sense stress and modulate their proteolytic activity in response.
The AAA+ proteases ClpXP and Lon, are members of a large, well-conserved family of proteins that assemble into heptameric or hexameric rings [142–144]. Proteolysis occurs in a central pore that acts as a degradation chamber [142, 143]. Cycles of ATP hydrolysis drive conformational changes that promote target protein unfolding and translocation into this chamber [142]. It is estimated that together, Clp and Lon are responsible for ~75% of ATP-dependent proteolysis in bacteria [141, 145]. Importantly, as degradation is irreversible, these proteases utilize adapter proteins to specifically recognize intended targets and thus avoid spurious degradation [142, 143, 146].
The intimate role of proteolysis in controlling the general stress response
In E. coli and related gram-negatives, the “general stress response” is mediated by σS, which is induced by many different conditions, including DNA damage, low Mg2+ or PO4, and low nutrients/stationary phase [147]. While σS is controlled in every possible way, σS protein level is regulated by proteolysis [147, 148]. In unstressed cells, the adaptor protein RssB targets σS to ClpXP for degradation [149–151]. In appropriately stressed cells, σS is stabilized, activating its regulon. Recent studies demonstrate that σS is stabilized by two discrete mechanisms (Fig. 4).
Figure 4. ClpXP degradation of σS is regulated via two mechanisms.

The adaptor RssB targets σS to ClpXP for degradation. Stress prevents σS degradation via two mechanisms: (1) Specific stresses induce expression of corresponding anti-adaptor proteins (low PO4, IraP; low Mg2+, IraM; stationary phase/DNA damage, IraD), which prevent RssB from interacting with σS. (2) ClpXP degradation of σS is particularly sensitive to ATP levels; low ATP (nutrient limitation) thus specifically prevents ClpXP degradation of σS.
Firstly, a suite of stress responsive “anti-adaptor” proteins (IraD, IraM, IraP) bind to RssB and prevent it from targeting σS to ClpXP for degradation [147, 152, 153]. Each Ira is induced by a different stress condition (in E. coli: IraD – nutrient limitation/stationary phase or DNA-damage, IraM – low Mg2+ or Ca2+, IraP – low PO4) [147, 152–155], thereby communicating each discrete stress to ClpXP by interfering with RssB function (Fig. 4). Although Ira proteins all bind to RssB, they are not members of the same protein family, do not have sequence similarity, and interact with different residues of RssB [147, 156], indicating that they have arisen independently to tune σS proteolysis. How Ira proteins are themselves inhibited to turn off the σS response remains unclear. An additional question is how σS responds to stresses that activate multiple Ira proteins. Most laboratory experiments focus on examining effects of a single stress, but in the environment, multiple stresses may occur simultaneously. These stresses may have combinatorial effects, encouraging bacteria to evolve systems that process information from multiple stresses in an integrated way.
Secondly, ClpXP tunes its own proteolytic capacity to alter σS degradation in response to ATP limitation [157]. ClpXP is ATP-dependent, creating the potential for ATP availability to affect rates of substrate degradation [158]. ClpXP degradation of σS is in fact exceptionally sensitive to intracellular ATP concentration: at low levels of ATP, many canonical ClpXP substrates are degraded normally, but degradation of σS is blocked (Fig. 4) [157]. Though the mechanism for this ATP dependence is unknown, it is thought that reducing ATP levels slows ClpXP translocation and may cause accumulation of partially folded substrates that interfere with further unfolding or degradation [157–159]. Interestingly, since low ATP is an indicator of nutrient stress, nutrient limitation regulates ClpXP degradation of σS both directly by ATP and indirectly by the nutrient responsive anti-adaptor IraD [155]. What differentiates these two mechanisms? Direct ATP control of σS proteolysis may be a feed-forward response that couples σS activity directly and dynamically to cellular metabolism. In contrast, while accumulation of IraD during transition to stationary phase may be slower, once made, IraD can constitutively block σS degradation, since it is not degraded with σS [153]. These complementary mechanisms may allow σS to be highly responsive to nutrient state, leading to both rapid and sustained activation of σS.
Proteolysis can generate alternative forms of proteins required during stress
The proteome can also be altered by programmed ribosomal frameshifting that generates alternative forms of proteins, such as the two forms of DnaX in E. coli. DnaX is a subunit of the complex that loads the DNA replication sliding clamp, which is required for processive replication in all organisms [160]. Both forms of DnaX protein (shorter γ and full-length τ) are present in the cell, with full-length τ generated by a frameshifting event [161–163]. However, the significance of these two forms of DnaX and whether and how bacteria other than E. coli and Salmonella produced these forms was unclear.
Recent work has shown that Caulobacter produces the shorter γ form of DnaX from full-length τ by ClpXP proteolysis, rather than by ribosomal frameshifting [164]. Caulobacter DnaX contains a glycine-rich, “slippery” tract adjacent to a stably folded domain that promotes release of partially degraded DnaX γ from ClpXP [164]. In vitro, ClpXP had been observed to release degradation intermediates of specific artificial substrates, but native substrates with this property had not been previously identified [165–168].
Both long and short form of DnaX are required for growth in Caulobacter [164]. Importantly, processing of τ DnaX to the γ form is required for proper recovery from DNA damage, as cells that constitutively express γ and a form of τ that cannot be processed are sensitive to DNA damaging agents [164]. Processing to γ may be required for efficient exchange to alternative, mutagenic DNA polymerases, which are employed during DNA damage [164]. Indeed, loss of τ processing leads to a reduced level of UV-induced mutagenesis, suggesting that proper usage of the alternative DNA polymerases has been inhibited [164]. This suggests that there is a stress-related rationale for generating two variants of the clamp loader. Interestingly, there are several known eukaryotic examples of partial proteolysis by the ubiquitin-proteosome system [169–171]. Ci, a regulator of hedgehog signaling, and NFκB, a mammalian transcription factor involved in inflammatory response, are both released when the proteosome encounters a low complexity sequence (e.g., glycine tracts) adjacent to a stably folded domain [169–171]. As this is the same mechanism that causes relese of DnaX, this conservation suggests that there are likely more examples of partial proteolysis in other organisms.
Proteome Remodeling by Lon
Lon, the first protease to be discovered, is thought to be the most widely conserved of all energy-dependent proteases [172]. Its housekeeping function is degradation of unfolded and abnormally folded proteins [144, 172]. This model is supported by the recent realization that the recognition tags for Lon comprise aromatic and hydrophobic residues that are buried in folded proteins [142, 173, 174]. Like ClpXP, Lon also participates in regulating stress responses. Indeed, the first phenotype determined for a deletion of lon was extreme UV-sensitivity [144, 172]. New studies highlight two further activities for Lon and additionally suggest that Lon may target additional proteins when stimulated by stress.
Recently, it was realized that Lon could act as a chaperone as well as a protease (Fig. 5). Lon variants that neither hydrolyze ATP nor degrade substrates can suppress severe unfolded protein stress, by binding to target proteins [175]. This chaperone activity is proposed to arise from ATP-independent conformational changes that are coupled to protein remodeling [175, 176]. Thus, chaperone activity may be a dominant function when ATP is limiting. Lon can also use its ATPase activity to inactivate the cell-division inhibitor, SulA (Fig. 5) [177–179]. As Lon mutants that are defective in chaperone activity can still inhibit SulA, this suggests that chaperone activity and SulA inhibition are distinct mechanisms for Lon [175].
Figure 5. Many cellular functions of Lon.

(1) Lon is responsible for degradation of many cellular proteins. (2) Lon can act as a chaperone to prevent protein aggregation. (3) Lon inactivates the cell division inhibitor SulA. (4) Lon is allosterically activated by unfolded proteins to degrade the initiation protein DnaA. (5) Lon exists as a hexamer and a dodecamer. Large proteins are thought to be excluded from entering the pore of the dodecamer and being degraded by Lon. It is not known if the dodecamer may also have chaperone activity, mediate SulA inhibition, or degrade DnaA.
Additionally, Lon can remodel its substrate specificity by altering its quaternary structure. Normally hexameric, Lon can also exist in a dodecameric state that closes off the entryway to its degradation chamber (Fig. 5) [180]. This may gate this chamber so that large substrates (>12–25 kDa) can no longer enter and be proteolyzed [180]. Importantly, the cellular concentration of Lon is high enough to support dodecamer formation, and constitutively dodecameric Lon mutants can complement many lon deletion phenotypes in vivo [180, 181]. As dodecamers cannot recognize large protein aggregates, dodecamer formation can realign the powerful degradation capacity of Lon to focus on important small regulatory proteins during times of high protein unfolding and aggregation [180].
Although it is an open question whether specific conditions or stresses promote chaperone activity or dodecamer formation, it is already known that heat can alter the substrate specificity of Lon. Under conditions of severe heat shock (shift to 45°C), replication is arrested in C. crescentus as a result of degradation of the DNA replication initiation protein DnaA [182, 183]. DnaA is stable in rich media, but rapidly depleted during severe temperature upshift and in several other stress conditions, leading to growth arrest [183–186]. This effect was recently traced to Lon-mediated degradation of DnaA [187].
Intriguingly, while DnaA is not a normal substrate for Lon in vitro, addition of a model unfolded protein substrate stimulated specific, robust degradation of DnaA (Fig. 5) [187]. Folded substrates of Lon could not stimulate degradation of DnaA, nor did unfolded protein significantly increase the degradation rate of other known Lon substrates [187]. In normal in vivo conditions, unfolded proteins are continually removed by cytoplasmic chaperones (e.g., Hsp70), and thus unavailable to activate Lon degradation of DnaA. However, following sudden onset of severe stress (shift to 45°C), unfolded pr oteins exceed the capacity of the protein refolding machinery, activating DnaA degradation and arresting replication [187]. As Lon and the chaperone machinery are widely distributed among bacteria, regulated DnaA degradation by Lon may be a broad mechanism for inducing growth arrest during stress. Intriguingly, there is an additional example of Lon targeting proliferation proteins for degradation: In E. coli that have lost the Hsp70 chaperone machine (ΔdnaKJ), Hsp33 (HslO) can interact with the ribosomal elongation factor Tu (Ef-Tu) and target it for degradation by Lon, thereby inhibiting translation of proteins and leading to growth arrest [188].
Why would cells want to target proliferation factors for degradation? During times of severe stress, if cells cannot maintain genome integrity or ensure survival during growth, it may become better for them to assume a nonproliferative (persister) state [189–193]. In fact, persister cells are highly resistant to stresses and antibiotics [189–192]. By stopping replication and reducing protein synthesis, the cell can focus on stress response while waiting for a more opportune condition to resume growth. Such behavior would be a form of bet-hedging, trading current fitness for future benefits [194, 195]. These may be examples of general mechanism, whereby stress exposes vital proliferation factors as proteolytic targets to induce growth arrest.
4.0 Perspective
Stress responses are not disconnected pathways, but are closely integrated into bacterial physiology. As there is no limit in the variety of ways that stress can alter cellular pathways, responses have evolved to be equally complex, monitoring and maintaining every cellular process. Since stress responses are so intimately connected to cellular state, studying them has provided an elegant window into the mechanisms that regulate the homeostasis of the cell. We have started to develop an understanding of the mechanisms that sense stress, the molecular tools that comprise responses, the logic of how responses are constructed and linked, and the dynamic outcomes that can result. However, many questions remain. For many responses, we still do not know the inducing signal, all the players, how the players fit together, or the behaviors that can result. As there are only a finite number of sensors and regulators to face an infinite variety of stresses, not all responses may be perfectly adaptive [196]. Additionally, we do not know how bacteria integrate the combinatorial stresses they are likely to have faced in the environment. This is especially important for pathogens, as they experience a characteristic set of stresses in a defined temporal order, and responses may be optimized to reflect this [197, 198]. Lastly, we are only beginning to grasp at the variability that may occur on the single-cell level. How pervasive are these behaviors in stress? Why have particular response behaviors been selected over others? Answering these and other questions will be crucial for understanding bacterial physiology and engineering.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Scott M, Gunderson CW, Mateescu EM, Zhang Z, Hwa T. Interdependence of cell growth and gene expression: origins and consequences. Science. 2010;330:1099–1102. doi: 10.1126/science.1192588. [DOI] [PubMed] [Google Scholar]
- 2.Kortmann J, Narberhaus F. Bacterial RNA thermometers: molecular zippers and switches. Nat Rev Microbiol. 2012;10:255–265. doi: 10.1038/nrmicro2730. [DOI] [PubMed] [Google Scholar]
- 3.Mangan S, Alon U. Structure and function of the feed-forward loop network motif. Proc Natl Acad Sci USA. 2003;100:11980–11985. doi: 10.1073/pnas.2133841100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Silva-Rocha R, De Lorenzo V. Noise and robustness in prokaryotic regulatory networks. Annu Rev Microbiol. 2010;64:257–275. doi: 10.1146/annurev.micro.091208.073229. [DOI] [PubMed] [Google Scholar]
- 5.Stock AM, Robinson VL, Goudreau PN. Two-component signal transduction. Annu Rev Biochem. 2000;69:183–215. doi: 10.1146/annurev.biochem.69.1.183. [DOI] [PubMed] [Google Scholar]
- 6.Inouye M, Dutta R. Histidine kinases in signal transduction. San Diego, California: Elsevier Science; 2002. [Google Scholar]
- 7.Utsumi R. Bacterial Signal Transduction: Networks and Drug Targets. Vol. 631. Austin, TX: Landes Bioscience; 2008. [PubMed] [Google Scholar]
- 8.Galperin MY. Diversity of structure and function of response regulator output domains. Curr Opin Microbiol. 2010;13:150–159. doi: 10.1016/j.mib.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stewart RC. Protein histidine kinases: assembly of active sites and their regulation in signaling pathways. Curr Opin Microbiol. 2010;13:133–141. doi: 10.1016/j.mib.2009.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jung K, Fried L, Behr S, Heermann R. Histidine kinases and response regulators in networks. Curr Opin Microbiol. 2012;15:118–124. doi: 10.1016/j.mib.2011.11.009. [DOI] [PubMed] [Google Scholar]
- 11.Gopel Y, Gorke B. Rewiring two-component signal transduction with small RNAs. Curr Opin Microbiol. 2012;15:132–139. doi: 10.1016/j.mib.2011.12.001. [DOI] [PubMed] [Google Scholar]
- 12.Gruber TM, Gross CA. Multiple sigma subunits and the partitioning of bacterial transcription space. Annu Rev Microbiol. 2003;57:441–466. doi: 10.1146/annurev.micro.57.030502.090913. [DOI] [PubMed] [Google Scholar]
- 13.Osterberg S, del Peso-Santos T, Shingler V. Regulation of alternative sigma factor use. Annu Rev Microbiol. 2011;65:37–55. doi: 10.1146/annurev.micro.112408.134219. [DOI] [PubMed] [Google Scholar]
- 14.Mascher T. Signaling diversity and evolution of extracytoplasmic function (ECF) σ factors. Curr Opin Microbiol. 2013;16:148–155. doi: 10.1016/j.mib.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 15.Cho US, Bader MW, Amaya MF, Daley ME, Klevit RE, Miller SI, Xu W. Metal bridges between the PhoQ sensor domain and the membrane regulate transmembrane signaling. J Mol Biol. 2006;356:1193–1206. doi: 10.1016/j.jmb.2005.12.032. [DOI] [PubMed] [Google Scholar]
- 16.Parkinson JS. Signaling mechanisms of HAMP domains in chemoreceptors and sensor kinases. Annu Rev Microbiol. 2010;64:101–122. doi: 10.1146/annurev.micro.112408.134215. [DOI] [PubMed] [Google Scholar]
- 17.Cheung J, Hendrickson WA. Sensor domains of two-component regulatory systems. Curr Opin Microbiol. 2010;13:116–123. doi: 10.1016/j.mib.2010.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krell T, Lacal J, Busch A, Silva-Jimenez H, Guazzaroni ME, Ramos JL. Bacterial sensor kinases: diversity in the recognition of environmental signals. Annu Rev Microbiol. 2010;64:539–559. doi: 10.1146/annurev.micro.112408.134054. [DOI] [PubMed] [Google Scholar]
- 19.Ferris HU, Dunin-Horkawicz S, Mondejar LG, Hulko M, Hantke K, Martin J, Schultz JE, Zeth K, Lupas AN, Coles M. The mechanisms of HAMP-mediated signaling in transmembrane receptors. Structure. 2011;19:378–385. doi: 10.1016/j.str.2011.01.006. [DOI] [PubMed] [Google Scholar]
- 20.Ferris HU, Dunin-Horkawicz S, Hornig N, Hulko M, Martin J, Schultz JE, Zeth K, Lupas AN, Coles M. Mechanism of regulation of receptor histidine kinases. Structure. 2012;20:56–66. doi: 10.1016/j.str.2011.11.014. [DOI] [PubMed] [Google Scholar]
- 21.Wang C, Sang J, Wang J, Su M, Downey JS, Wu Q, Wang S, Cai Y, Xu X, Wu J, et al. Mechanistic insights revealed by the crystal structure of a histidine kinase with signal transducer and sensor domains. PLoS Biol. 2013;11:e1001493. doi: 10.1371/journal.pbio.1001493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Egger LA, Park H, Inouye M. Signal transduction via the histidyl-aspartyl phosphorelay. Genes Cells. 1997;2:167–184. doi: 10.1046/j.1365-2443.1997.d01-311.x. [DOI] [PubMed] [Google Scholar]
- 23.Pratt LA, Hsing W, Gibson KE, Silhavy TJ. From acids to osmZ: multiple factors influence synthesis of the OmpF and OmpC porins in Escherichia coli. Mol Microbiol. 1996;20:911–917. doi: 10.1111/j.1365-2958.1996.tb02532.x. [DOI] [PubMed] [Google Scholar]
- 24.Wang LC, Morgan LK, Godakumbura P, Kenney LJ, Anand GS. The inner membrane histidine kinase EnvZ senses osmolality via helix-coil transitions in the cytoplasm. EMBO J. 2012;31:2648–2659. doi: 10.1038/emboj.2012.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Park H, Saha SK, Inouye M. Two-domain reconstitution of a functional protein histidine kinase. Proc Natl Acad Sci USA. 1998;95:6728–6732. doi: 10.1073/pnas.95.12.6728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tokishita S, Kojima A, Mizuno T. Transmembrane signal transduction and osmoregulation in Escherichia coli: functional importance of the transmembrane regions of membrane-located protein kinase, EnvZ. J Biochem. 1992;111:707–713. doi: 10.1093/oxfordjournals.jbchem.a123823. [DOI] [PubMed] [Google Scholar]
- 27.Hulko M, Berndt F, Gruber M, Linder JU, Truffault V, Schultz A, Martin J, Schultz JE, Lupas AN, Coles M. The HAMP domain structure implies helix rotation in transmembrane signaling. Cell. 2006;126:929–940. doi: 10.1016/j.cell.2006.06.058. [DOI] [PubMed] [Google Scholar]
- 28.Airola MV, Huh D, Sukomon N, Widom J, Sircar R, Borbat PP, Freed JH, Watts KJ, Crane BR. Architecture of the soluble receptor Aer2 indicates an in-line mechanism for PAS and HAMP domain signaling. J Mol Biol. 2013;425:886–901. doi: 10.1016/j.jmb.2012.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gerken H, Charlson ES, Cicirelli EM, Kenney LJ, Misra R. MzrA: a novel modulator of the EnvZ/OmpR two-component regulon. Mol Microbiol. 2009;72:1408–1422. doi: 10.1111/j.1365-2958.2009.06728.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gerken H, Misra R. MzrA-EnvZ interactions in the periplasm influence the EnvZ/OmpR two-component regulon. J Bacteriol. 2010;192:6271–6278. doi: 10.1128/JB.00855-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Taylor BL, Zhulin IB. PAS domains: internal sensors of oxygen, redox potential, and light. Microbiol Mol Biol Rev. 1999;63:479–506. doi: 10.1128/mmbr.63.2.479-506.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moglich A, Ayers RA, Moffat K. Structure and signaling mechanism of Per-ARNT-Sim domains. Structure. 2009;17:1282–1294. doi: 10.1016/j.str.2009.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Henry JT, Crosson S. Ligand-binding PAS domains in a genomic, cellular, and structural context. Annu Rev Microbiol. 2011;65:261–286. doi: 10.1146/annurev-micro-121809-151631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zelzer E, Wappner P, Shilo BZ. The PAS domain confers target gene specificity of Drosophila bHLH/PAS proteins. Genes Dev. 1997;11:2079–2089. doi: 10.1101/gad.11.16.2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moglich A, Ayers RA, Moffat K. Addition at the molecular level: signal integration in designed Per-ARNT-Sim receptor proteins. J Mol Biol. 2010;400:477–486. doi: 10.1016/j.jmb.2010.05.019. [DOI] [PubMed] [Google Scholar]
- 36.Moglich A, Ayers RA, Moffat K. Design and signaling mechanism of light-regulated histidine kinases. J Mol Biol. 2009;385:1433–1444. doi: 10.1016/j.jmb.2008.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu YI, Frey D, Lungu OI, Jaehrig A, Schlichting I, Kuhlman B, Hahn KM. A genetically encoded photoactivatable Rac controls the motility of living cells. Nature. 2009;461:104–108. doi: 10.1038/nature08241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lungu OI, Hallett RA, Choi EJ, Aiken MJ, Hahn KM, Kuhlman B. Designing photoswitchable peptides using the AsLOV2 domain. Chem Biol. 2012;19:507–517. doi: 10.1016/j.chembiol.2012.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morimoto RI. The heat shock response: systems biology of proteotoxic stress in aging and disease. Cold Spring Harbor Symp Quant Biol. 2011;76:91–99. doi: 10.1101/sqb.2012.76.010637. [DOI] [PubMed] [Google Scholar]
- 40.Mogk A, Huber D, Bukau B. Cold Spring Harbor Perspectives in Biology. 2011. Integrating protein homeostasis strategies in prokaryotes; p. 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Anckar J, Sistonen L. Regulation of HSF1 function in the heat stress response: implications in aging and disease. Annu Rev Biochem. 2011;80:1089–1115. doi: 10.1146/annurev-biochem-060809-095203. [DOI] [PubMed] [Google Scholar]
- 42.Guisbert E, Yura T, Rhodius VA, Gross CA. Convergence of molecular, modeling, and systems approaches for an understanding of the Escherichia coli heat shock response. Microbiol Mol Biol Rev. 2008;72:545–554. doi: 10.1128/MMBR.00007-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nonaka G, Blankschien M, Herman C, Gross CA, Rhodius VA. Regulon and promoter analysis of the E. coli heat-shock factor, sigma32, reveals a multifaceted cellular response to heat stress. Genes Dev. 2006;20:1776–1789. doi: 10.1101/gad.1428206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Morita M, Kanemori M, Yanagi H, Yura T. Heat-induced synthesis of sigma32 in Escherichia coli: structural and functional dissection of rpoH mRNA secondary structure. J Bacteriol. 1999;181:401–410. doi: 10.1128/jb.181.2.401-410.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Morita MT, Tanaka Y, Kodama TS, Kyogoku Y, Yanagi H, Yura T. Translational induction of heat shock transcription factor sigma32: evidence for a built-in RNA thermosensor. Genes Dev. 1999;13:655–665. doi: 10.1101/gad.13.6.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guisbert E, Herman C, Lu CZ, Gross CA. A chaperone network controls the heat shock response in E. coli. Genes Dev. 2004;18:2812–2821. doi: 10.1101/gad.1219204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gamer J, Multhaup G, Tomoyasu T, McCarty JS, Rudiger S, Schonfeld HJ, Schirra C, Bujard H, Bukau B. A cycle of binding and release of the DnaK, DnaJ and GrpE chaperones regulates activity of the Escherichia coli heat shock transcription factor sigma32. EMBO J. 1996;15:607–617. [PMC free article] [PubMed] [Google Scholar]
- 48.Herman C, Thevenet D, D’Ari R, Bouloc P. Degradation of sigma 32, the heat shock regulator in Escherichia coli, is governed by HflB. Proc Natl Acad Sci USA. 1995;92:3516–3520. doi: 10.1073/pnas.92.8.3516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tomoyasu T, Gamer J, Bukau B, Kanemori M, Mori H, Rutman AJ, Oppenheim AB, Yura T, Yamanaka K, Niki H. Escherichia coli FtsH is a membrane-bound, ATP-dependent protease which degrades the heat-shock transcription factor sigma 32. EMBO J. 1995;14:2551–2560. doi: 10.1002/j.1460-2075.1995.tb07253.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Horikoshi M, Yura T, Tsuchimoto S, Fukumori Y, Kanemori M. Conserved region 2.1 of Escherichia coli heat shock transcription factor sigma32 is required for modulating both metabolic stability and transcriptional activity. J Bacteriol. 2004;186:7474–7480. doi: 10.1128/JB.186.22.7474-7480.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Obrist M, Narberhaus F. Identification of a turnover element in region 2.1 of Escherichia coli sigma32 by a bacterial one-hybrid approach. J Bacteriol. 2005;187:3807–3813. doi: 10.1128/JB.187.11.3807-3813.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yura T, Guisbert E, Poritz M, Lu CZ, Campbell E, Gross CA. Analysis of sigma32 mutants defective in chaperone-mediated feedback control reveals unexpected complexity of the heat shock response. Proc Natl Acad Sci USA. 2007;104:17638–17643. doi: 10.1073/pnas.0708819104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Suzuki H, Ikeda A, Tsuchimoto S, Adachi K-i, Noguchi A, Fukumori Y, Kanemori M. Synergistic binding of DnaJ and DnaK chaperones to heat shock transcription factor σ32 ensures its characteristic high metabolic instability: implications for heat shock protein 70 (Hsp70)-Hsp40 mode of function. J Biol Chem. 2012;287:19275–19283. doi: 10.1074/jbc.M111.331470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lim B, Miyazaki R, Neher S, Siegele DA, Ito K, Walter P, Akiyama Y, Yura T, Gross CA. Heat Shock Transcription Factor σ(32) Co-opts the Signal Recognition Particle to Regulate Protein Homeostasis in E. coli. PLoS Biol. 2013;11:e1001735. doi: 10.1371/journal.pbio.1001735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Driessen AJM, Nouwen N. Protein translocation across the bacterial cytoplasmic membrane. Annu Rev Biochem. 2008;77:643–667. doi: 10.1146/annurev.biochem.77.061606.160747. [DOI] [PubMed] [Google Scholar]
- 56.Bibi E. Early targeting events during membrane protein biogenesis in Escherichia coli. Biochim Biophys Acta. 2011;1808:841–850. doi: 10.1016/j.bbamem.2010.07.025. [DOI] [PubMed] [Google Scholar]
- 57.Saraogi I, Shan S-o. Co-translational protein targeting to the bacterial membrane. Biochim Biophys Acta. 2013 doi: 10.1016/j.bbamcr.2013.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jensen CG, Pedersen S. Concentrations of 4.5S RNA and Ffh protein in Escherichia coli: the stability of Ffh protein is dependent on the concentration of 4.5S RNA. J Bacteriol. 1994;176:7148–7154. doi: 10.1128/jb.176.23.7148-7154.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Holtkamp W, Lee S, Bornemann T, Senyushkina T, Rodnina MV, Wintermeyer W. Dynamic switch of the signal recognition particle from scanning to targeting. Nat Struct Mol Biol. 2012;19:1332–1337. doi: 10.1038/nsmb.2421. [DOI] [PubMed] [Google Scholar]
- 60.Okuno T, Ogura T. FtsH Protease-Mediated Regulation of Various Cellular Functions. Subcell Biochem. 2013;66:53–69. doi: 10.1007/978-94-007-5940-4_3. [DOI] [PubMed] [Google Scholar]
- 61.Ito K, Akiyama Y. Cellular functions, mechanism of action, and regulation of FtsH protease. Annu Rev Microbiol. 2005;59:211–231. doi: 10.1146/annurev.micro.59.030804.121316. [DOI] [PubMed] [Google Scholar]
- 62.Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 63.Nikaido H. Molecular basis of bacterial outer membrane permeability revisited. Microbiol Mol Biol Rev. 2003;67:593–656. doi: 10.1128/MMBR.67.4.593-656.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Delcour AH. Outer membrane permeability and antibiotic resistance. Biochim Biophys Acta. 2009;1794:808–816. doi: 10.1016/j.bbapap.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Silhavy TJ, Kahne D, Walker S. The bacterial cell envelope. Cold Spring Harbor Perspectives in Biology. 2010;2:a000414. doi: 10.1101/cshperspect.a000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tokuda H. Biogenesis of outer membranes in Gram-negative bacteria. Bioscience, Biotechnology, and Biochemistry. 2009;73:465–473. doi: 10.1271/bbb.80778. [DOI] [PubMed] [Google Scholar]
- 67.Okuda S, Tokuda H. Lipoprotein sorting in bacteria. Annu Rev Microbiol. 2011;65:239–259. doi: 10.1146/annurev-micro-090110-102859. [DOI] [PubMed] [Google Scholar]
- 68.Sperandeo P, Deho G, Polissi A. The lipopolysaccharide transport system of Gram-negative bacteria. Biochim Biophys Acta. 2009;1791:594–602. doi: 10.1016/j.bbalip.2009.01.011. [DOI] [PubMed] [Google Scholar]
- 69.Ricci DP, Silhavy TJ. The Bam machine: a molecular cooper. Biochim Biophys Acta. 2012;1818:1067–1084. doi: 10.1016/j.bbamem.2011.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Raetz CRH, Guan Z, Ingram BO, Six DA, Song F, Wang X, Zhao J. Discovery of new biosynthetic pathways: the lipid A story. The Journal of Lipid Research. 2009;50(Suppl):S103–108. doi: 10.1194/jlr.R800060-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang G, Meredith TC, Kahne D. On the essentiality of lipopolysaccharide to Gram-negative bacteria. Curr Opin Microbiol. 2013;16:779–785. doi: 10.1016/j.mib.2013.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hagan CL, Silhavy TJ, Kahne D. β-Barrel membrane protein assembly by the Bam complex. Annu Rev Biochem. 2011;80:189–210. doi: 10.1146/annurev-biochem-061408-144611. [DOI] [PubMed] [Google Scholar]
- 73.Rhodius VA, Suh WC, Nonaka G, West J, Gross CA. Conserved and variable functions of the sigmaE stress response in related genomes. PLoS Biol. 2006;4:e2. doi: 10.1371/journal.pbio.0040002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Barchinger SE, Ades SE. Regulated Proteolysis: Control of the Escherichia coli σ(E)-Dependent Cell Envelope Stress Response. Subcell Biochem. 2013;66:129–160. doi: 10.1007/978-94-007-5940-4_6. [DOI] [PubMed] [Google Scholar]
- 75.Missiakas D, Mayer MP, Lemaire M, Georgopoulos C, Raina S. Modulation of the Escherichia coli sigmaE (RpoE) heat-shock transcription-factor activity by the RseA, RseB and RseC proteins. Mol Microbiol. 1997;24:355–371. doi: 10.1046/j.1365-2958.1997.3601713.x. [DOI] [PubMed] [Google Scholar]
- 76.De Las Penas A, Connolly L, Gross CA. The sigmaE-mediated response to extracytoplasmic stress in Escherichia coli is transduced by RseA and RseB, two negative regulators of sigmaE. Mol Microbiol. 1997;24:373–385. doi: 10.1046/j.1365-2958.1997.3611718.x. [DOI] [PubMed] [Google Scholar]
- 77.Walsh NP, Alba BM, Bose B, Gross CA, Sauer RT. OMP peptide signals initiate the envelope-stress response by activating DegS protease via relief of inhibition mediated by its PDZ domain. Cell. 2003;113:61–71. doi: 10.1016/s0092-8674(03)00203-4. [DOI] [PubMed] [Google Scholar]
- 78.Ades SE, Connolly LE, Alba BM, Gross CA. The Escherichia coli sigma(E)-dependent extracytoplasmic stress response is controlled by the regulated proteolysis of an anti-sigma factor. Genes Dev. 1999;13:2449–2461. doi: 10.1101/gad.13.18.2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Alba BM, Leeds JA, Onufryk C, Lu CZ, Gross CA. DegS and YaeL participate sequentially in the cleavage of RseA to activate the sigma(E)-dependent extracytoplasmic stress response. Genes Dev. 2002;16:2156–2168. doi: 10.1101/gad.1008902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Campbell EA, Tupy JL, Gruber TM, Wang S, Sharp MM, Gross CA, Darst SA. Crystal structure of Escherichia coli sigmaE with the cytoplasmic domain of its anti-sigma RseA. Mol Cell. 2003;11:1067–1078. doi: 10.1016/s1097-2765(03)00148-5. [DOI] [PubMed] [Google Scholar]
- 81.Kanehara K, Ito K, Akiyama Y. YaeL proteolysis of RseA is controlled by the PDZ domain of YaeL and a Gln-rich region of RseA. EMBO J. 2003;22:6389–6398. doi: 10.1093/emboj/cdg602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bohn C, Collier J, Bouloc P. Dispensable PDZ domain of Escherichia coli YaeL essential protease. Mol Microbiol. 2004;52:427–435. doi: 10.1111/j.1365-2958.2004.03985.x. [DOI] [PubMed] [Google Scholar]
- 83.Grigorova IL, Chaba R, Zhong HJ, Alba BM, Rhodius V, Herman C, Gross CA. Fine-tuning of the Escherichia coli sigmaE envelope stress response relies on multiple mechanisms to inhibit signal-independent proteolysis of the transmembrane anti-sigma factor, RseA. Genes Dev. 2004;18:2686–2697. doi: 10.1101/gad.1238604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sohn J, Grant RA, Sauer RT. Allosteric activation of DegS, a stress sensor PDZ protease. Cell. 2007;131:572–583. doi: 10.1016/j.cell.2007.08.044. [DOI] [PubMed] [Google Scholar]
- 85.Sohn J, Sauer RT. OMP peptides modulate the activity of DegS protease by differential binding to active and inactive conformations. Mol Cell. 2009;33:64–74. doi: 10.1016/j.molcel.2008.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sohn J, Grant RA, Sauer RT. OMP peptides activate the DegS stress-sensor protease by a relief of inhibition mechanism. Structure. 2009;17:1411–1421. doi: 10.1016/j.str.2009.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mecsas J, Rouviere PE, Erickson JW, Donohue TJ, Gross CA. The activity of sigma E, an Escherichia coli heat-inducible sigma-factor, is modulated by expression of outer membrane proteins. Genes Dev. 1993;7:2618–2628. doi: 10.1101/gad.7.12b.2618. [DOI] [PubMed] [Google Scholar]
- 88.Alba BM, Gross CA. Regulation of the Escherichia coli sigma-dependent envelope stress response. Mol Microbiol. 2004;52:613–619. doi: 10.1111/j.1365-2958.2003.03982.x. [DOI] [PubMed] [Google Scholar]
- 89.Collinet B, Yuzawa H, Chen T, Herrera C, Missiakas D. RseB binding to the periplasmic domain of RseA modulates the RseA:sigmaE interaction in the cytoplasm and the availability of sigmaE:RNA polymerase. J Biol Chem. 2000;275:33898–33904. doi: 10.1074/jbc.m006214200. [DOI] [PubMed] [Google Scholar]
- 90.Cezairliyan BO, Sauer RT. Inhibition of regulated proteolysis by RseB. Proc Natl Acad Sci USA. 2007;104:3771–3776. doi: 10.1073/pnas.0611567104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kim DY, Kwon E, Choi J, Hwang HY, Kim KK. Structural basis for the negative regulation of bacterial stress response by RseB. Protein Sci. 2010;19:1258–1263. doi: 10.1002/pro.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chaba R, Alba BM, Guo MS, Sohn J, Ahuja N, Sauer RT, Gross CA. Signal integration by DegS and RseB governs the σ E-mediated envelope stress response in Escherichia coli. Proc Natl Acad Sci USA. 2011;108:2106–2111. doi: 10.1073/pnas.1019277108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lima S, Guo MS, Chaba R, Gross CA, Sauer RT. Dual molecular signals mediate the bacterial response to outer-membrane stress. Science. 2013;340:837–841. doi: 10.1126/science.1235358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tam C, Missiakas D. Changes in lipopolysaccharide structure induce the sigma(E)-dependent response of Escherichia coli. Mol Microbiol. 2005;55:1403–1412. doi: 10.1111/j.1365-2958.2005.04497.x. [DOI] [PubMed] [Google Scholar]
- 95.Ames GF, Spudich EN, Nikaido H. Protein composition of the outer membrane of Salmonella typhimurium: effect of lipopolysaccharide mutations. J Bacteriol. 1974;117:406–416. doi: 10.1128/jb.117.2.406-416.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Austin EA, Graves JF, Hite LA, Parker CT, Schnaitman CA. Genetic analysis of lipopolysaccharide core biosynthesis by Escherichia coli K-12: insertion mutagenesis of the rfa locus. J Bacteriol. 1990;172:5312–5325. doi: 10.1128/jb.172.9.5312-5325.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ruiz N, Chng S-S, Hiniker A, Kahne D, Silhavy TJ. Nonconsecutive disulfide bond formation in an essential integral outer membrane protein. Proc Natl Acad Sci USA. 2010 doi: 10.1073/pnas.1007319107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Vertommen D, Ruiz N, Leverrier P, Silhavy TJ, Collet JF. Characterization of the role of the Escherichia coli periplasmic chaperone SurA using differential proteomics. Proteomics. 2009;9:2432–2443. doi: 10.1002/pmic.200800794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Udekwu KI, Darfeuille F, Vogel J, Reimegard J, Holmqvist E, Wagner EGH. Hfq-dependent regulation of OmpA synthesis is mediated by an antisense RNA. Genes Dev. 2005;19:2355–2366. doi: 10.1101/gad.354405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Rasmussen AA, Eriksen M, Gilany K, Udesen C, Franch T, Petersen C, Valentin-Hansen P. Regulation of ompA mRNA stability: the role of a small regulatory RNA in growth phase-dependent control. Mol Microbiol. 2005;58:1421–1429. doi: 10.1111/j.1365-2958.2005.04911.x. [DOI] [PubMed] [Google Scholar]
- 101.Johansen J, Rasmussen AA, Overgaard M, Valentin-Hansen P. Conserved small non-coding RNAs that belong to the sigmaE regulon: role in down-regulation of outer membrane proteins. J Mol Biol. 2006;364:1–8. doi: 10.1016/j.jmb.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 102.Papenfort K, Pfeiffer V, Mika F, Lucchini S, Hinton JCD, Vogel J. SigmaE-dependent small RNAs of Salmonella respond to membrane stress by accelerating global omp mRNA decay. Mol Microbiol. 2006;62:1674–1688. doi: 10.1111/j.1365-2958.2006.05524.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Thompson KM, Rhodius VA, Gottesman S. SigmaE regulates and is regulated by a small RNA in Escherichia coli. J Bacteriol. 2007;189:4243–4256. doi: 10.1128/JB.00020-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hayden JD, Ades SE. The extracytoplasmic stress factor, sigmaE, is required to maintain cell envelope integrity in Escherichia coli. PLoS ONE. 2008;3:e1573. doi: 10.1371/journal.pone.0001573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gogol EB, Rhodius VA, Papenfort K, Vogel J, Gross CA. Small RNAs endow a transcriptional activator with essential repressor functions for single-tier control of a global stress regulon. Proc Natl Acad Sci USA. 2011;108:12875–12880. doi: 10.1073/pnas.1109379108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- 107.Pavitt GD, Ron D. New insights into translational regulation in the endoplasmic reticulum unfolded protein response. Cold Spring Harbor Perspectives in Biology. 2012:4. doi: 10.1101/cshperspect.a012278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Maurel M, Chevet E. Endoplasmic reticulum stress signaling: the microRNA connection. Am J Physiol Cell Physiol. 2013;304:C1117–1126. doi: 10.1152/ajpcell.00061.2013. [DOI] [PubMed] [Google Scholar]
- 109.Alon U. Network motifs: theory and experimental approaches. Nat Rev Genet. 2007;8:450–461. doi: 10.1038/nrg2102. [DOI] [PubMed] [Google Scholar]
- 110.Thattai M, Van Oudenaarden A. Stochastic gene expression in fluctuating environments. Genetics. 2004;167:523–530. doi: 10.1534/genetics.167.1.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.El-Samad H, Khammash M. Regulated degradation is a mechanism for suppressing stochastic fluctuations in gene regulatory networks. Biophys J. 2006;90:3749–3761. doi: 10.1529/biophysj.105.060491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Eldar A, Elowitz MB. Functional roles for noise in genetic circuits. Nature. 2010;467:167–173. doi: 10.1038/nature09326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Cağatay T, Turcotte M, Elowitz MB, Garcia-Ojalvo J, Suel GM. Architecture-dependent noise discriminates functionally analogous differentiation circuits. Cell. 2009;139:512–522. doi: 10.1016/j.cell.2009.07.046. [DOI] [PubMed] [Google Scholar]
- 114.Locke JCW, Young JW, Fontes M, Hernandez Jimenez MJ, Elowitz MB. Stochastic pulse regulation in bacterial stress response. Science. 2011;334:366–369. doi: 10.1126/science.1208144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Young JW, Locke JCW, Elowitz MB. Rate of environmental change determines stress response specificity. Proc Natl Acad Sci USA. 2013;110:4140–4145. doi: 10.1073/pnas.1213060110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hecker M, Volker U. Non-specific, general and multiple stress resistance of growth-restricted Bacillus subtilis cells by the expression of the sigmaB regulon. Mol Microbiol. 1998;29:1129–1136. doi: 10.1046/j.1365-2958.1998.00977.x. [DOI] [PubMed] [Google Scholar]
- 117.Hecker M, Pane-Farre J, Volker U. SigB-dependent general stress response in Bacillus subtilis and related gram-positive bacteria. Annu Rev Microbiol. 2007;61:215–236. doi: 10.1146/annurev.micro.61.080706.093445. [DOI] [PubMed] [Google Scholar]
- 118.Benson AK, Haldenwang WG. Bacillus subtilis sigma B is regulated by a binding protein (RsbW) that blocks its association with core RNA polymerase. Proc Natl Acad Sci USA. 1993;90:2330–2334. doi: 10.1073/pnas.90.6.2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Alper S, Dufour A, Garsin DA, Duncan L, Losick R. Role of adenosine nucleotides in the regulation of a stress-response transcription factor in Bacillus subtilis. J Mol Biol. 1996;260:165–177. doi: 10.1006/jmbi.1996.0390. [DOI] [PubMed] [Google Scholar]
- 120.Dufour A, Haldenwang WG. Interactions between a Bacillus subtilis anti-sigma factor (RsbW) and its antagonist (RsbV) J Bacteriol. 1994;176:1813–1820. doi: 10.1128/jb.176.7.1813-1820.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Voelker U, Voelker A, Maul B, Hecker M, Dufour A, Haldenwang WG. Separate mechanisms activate sigma B of Bacillus subtilis in response to environmental and metabolic stresses. J Bacteriol. 1995;177:3771–3780. doi: 10.1128/jb.177.13.3771-3780.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Yang X, Kang CM, Brody MS, Price CW. Opposing pairs of serine protein kinases and phosphatases transmit signals of environmental stress to activate a bacterial transcription factor. Genes Dev. 1996;10:2265–2275. doi: 10.1101/gad.10.18.2265. [DOI] [PubMed] [Google Scholar]
- 123.Vijay K, Brody MS, Fredlund E, Price CW. A PP2C phosphatase containing a PAS domain is required to convey signals of energy stress to the sigmaB transcription factor of Bacillus subtilis. Mol Microbiol. 2000;35:180–188. doi: 10.1046/j.1365-2958.2000.01697.x. [DOI] [PubMed] [Google Scholar]
- 124.Kim TJ, Gaidenko TA, Price CW. In vivo phosphorylation of partner switching regulators correlates with stress transmission in the environmental signaling pathway of Bacillus subtilis. J Bacteriol. 2004;186:6124–6132. doi: 10.1128/JB.186.18.6124-6132.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Marles-Wright J, Lewis RJ. The stressosome: molecular architecture of a signalling hub. Biochem Soc Trans. 2010;38:928–933. doi: 10.1042/BST0380928. [DOI] [PubMed] [Google Scholar]
- 126.Bernhardt J, Volker U, Volker A, Antelmann H, Schmid R, Mach H, Hecker M. Specific and general stress proteins in Bacillus subtilis--a two-deimensional protein electrophoresis study. Microbiology. 1997;143:999–1017. doi: 10.1099/00221287-143-3-999. [DOI] [PubMed] [Google Scholar]
- 127.Szurmant H, Hoch JA. Interaction fidelity in two-component signaling. Curr Opin Microbiol. 2010;13:190–197. doi: 10.1016/j.mib.2010.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Podgornaia AI, Laub MT. Determinants of specificity in two-component signal transduction. Curr Opin Microbiol. 2013;16:156–162. doi: 10.1016/j.mib.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 129.Laub MT, Goulian M. Specificity in two-component signal transduction pathways. Annu Rev Genet. 2007;41:121–145. doi: 10.1146/annurev.genet.41.042007.170548. [DOI] [PubMed] [Google Scholar]
- 130.Siryaporn A, Goulian M. Cross-talk suppression between the CpxA-CpxR and EnvZ-OmpR two-component systems in E. coli. Mol Microbiol. 2008;70:494–506. doi: 10.1111/j.1365-2958.2008.06426.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Groban ES, Clarke EJ, Salis HM, Miller SM, Voigt CA. Kinetic buffering of cross talk between bacterial two-component sensors. J Mol Biol. 2009;390:380–393. doi: 10.1016/j.jmb.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Capra EJ, Perchuk BS, Lubin EA, Ashenberg O, Skerker JM, Laub MT. Systematic dissection and trajectory-scanning mutagenesis of the molecular interface that ensures specificity of two-component signaling pathways. PLoS Genet. 2010;6:e1001220. doi: 10.1371/journal.pgen.1001220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Capra EJ, Perchuk BS, Skerker JM, Laub MT. Adaptive mutations that prevent crosstalk enable the expansion of paralogous signaling protein families. Cell. 2012;150:222–232. doi: 10.1016/j.cell.2012.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Podgornaia AI, Casino P, Marina A, Laub MT. Structural basis of a rationally rewired protein-protein interface critical to bacterial signaling. Structure. 2013;21:1636–1647. doi: 10.1016/j.str.2013.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Skerker JM, Perchuk BS, Siryaporn A, Lubin EA, Ashenberg O, Goulian M, Laub MT. Rewiring the specificity of two-component signal transduction systems. Cell. 2008;133:1043–1054. doi: 10.1016/j.cell.2008.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Siryaporn A, Perchuk BS, Laub MT, Goulian M. Evolving a robust signal transduction pathway from weak cross-talk. Mol Syst Biol. 2010;6:452. doi: 10.1038/msb.2010.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Willett JW, Tiwari N, Muller S, Hummels KR, Houtman JCD, Fuentes EJ, Kirby JR. Specificity residues determine binding affinity for two-component signal transduction systems. mBio. 2013;4:e00420–00413. doi: 10.1128/mBio.00420-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Staroń A, Sofia H, Dietrich S, Ulrich L, Liesegang H, Mascher T. The third pillar of bacterial signal transduction: classification of the extracytoplasmic function (ECF) sigma factor protein family. Mol Microbiol. 2009 doi: 10.1111/j.1365-2958.2009.06870.x. [DOI] [PubMed] [Google Scholar]
- 139.Rhodius VA, Segall-Shapiro TH, Sharon BD, Ghodasara A, Orlova E, Tabakh H, Burkhardt DH, Clancy K, Peterson TC, Gross CA, et al. Design of orthogonal genetic switches based on a crosstalk map of σs, anti-σs, and promoters. Mol Syst Biol. 2013;9:702. doi: 10.1038/msb.2013.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Gur E, Biran D, Ron EZ. Regulated proteolysis in Gramnegative bacteria--how and when? Nat Rev Microbiol. 2011;9:839–848. doi: 10.1038/nrmicro2669. [DOI] [PubMed] [Google Scholar]
- 141.Maurizi MR. Proteases and protein degradation in Escherichia coli. Experientia. 1992;48:178–201. doi: 10.1007/BF01923511. [DOI] [PubMed] [Google Scholar]
- 142.Sauer RT, Baker TA. AAA+ proteases: ATP-fueled machines of protein destruction. Annu Rev Biochem. 2011;80:587–612. doi: 10.1146/annurev-biochem-060408-172623. [DOI] [PubMed] [Google Scholar]
- 143.Gur E, Ottofueling R, Dougan DA. Machines of Destruction - AAA+ Proteases and the Adaptors That Control Them. Subcell Biochem. 2013;66:3–33. doi: 10.1007/978-94-007-5940-4_1. [DOI] [PubMed] [Google Scholar]
- 144.Gur E. The Lon AAA+ Protease. Subcell Biochem. 2013;66:35–51. doi: 10.1007/978-94-007-5940-4_2. [DOI] [PubMed] [Google Scholar]
- 145.Rosen R, Biran D, Gur E, Becher D, Hecker M, Ron EZ. Protein aggregation in Escherichia coli: role of proteases. FEMS Microbiol Lett. 2002;207:9–12. doi: 10.1111/j.1574-6968.2002.tb11020.x. [DOI] [PubMed] [Google Scholar]
- 146.Gottesman S. Proteolysis in bacterial regulatory circuits. Annu Rev Cell Dev Biol. 2003;19:565–587. doi: 10.1146/annurev.cellbio.19.110701.153228. [DOI] [PubMed] [Google Scholar]
- 147.Battesti A, Majdalani N, Gottesman S. The RpoS-mediated general stress response in Escherichia coli. Annu Rev Microbiol. 2011;65:189–213. doi: 10.1146/annurev-micro-090110-102946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Hengge R. Proteolysis of sigmaS (RpoS) and the general stress response in Escherichia coli. Res Microbiol. 2009;160:667–676. doi: 10.1016/j.resmic.2009.08.014. [DOI] [PubMed] [Google Scholar]
- 149.Muffler A, Fischer D, Altuvia S, Storz G, Hengge-Aronis R. The response regulator RssB controls stability of the sigma(S) subunit of RNA polymerase in Escherichia coli. EMBO J. 1996;15:1333–1339. [PMC free article] [PubMed] [Google Scholar]
- 150.Pratt LA, Silhavy TJ. The response regulator SprE controls the stability of RpoS. Proc Natl Acad Sci USA. 1996;93:2488–2492. doi: 10.1073/pnas.93.6.2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Zhou Y, Gottesman S, Hoskins JR, Maurizi MR, Wickner S. The RssB response regulator directly targets sigma(S) for degradation by ClpXP. Genes Dev. 2001;15:627–637. doi: 10.1101/gad.864401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Bougdour A, Wickner S, Gottesman S. Modulating RssB activity: IraP, a novel regulator of sigma(S) stability in Escherichia coli. Genes Dev. 2006;20:884–897. doi: 10.1101/gad.1400306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Bougdour A, Cunning C, Baptiste PJ, Elliott T, Gottesman S. Multiple pathways for regulation of sigmaS (RpoS) stability in Escherichia coli via the action of multiple anti-adaptors. Mol Microbiol. 2008;68:298–313. doi: 10.1111/j.1365-2958.2008.06146.x. [DOI] [PubMed] [Google Scholar]
- 154.Merrikh H, Ferrazzoli AE, Bougdour A, Olivier-Mason A, Lovett ST. A DNA damage response in Escherichia coli involving the alternative sigma factor, RpoS. Proc Natl Acad Sci USA. 2009;106:611–616. doi: 10.1073/pnas.0803665106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Merrikh H, Ferrazzoli AE, Lovett ST. Growth phase and (p)ppGpp control of IraD, a regulator of RpoS stability, in Escherichia coli. J Bacteriol. 2009;191:7436–7446. doi: 10.1128/JB.00412-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Battesti A, Hoskins JR, Tong S, Milanesio P, Mann JM, Kravats A, Tsegaye YM, Bougdour A, Wickner S, Gottesman S. Anti-adaptors provide multiple modes for regulation of the RssB adaptor protein. Genes Dev. 2013;27:2722–2735. doi: 10.1101/gad.229617.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Peterson CN, Levchenko I, Rabinowitz JD, Baker TA, Silhavy TJ. RpoS proteolysis is controlled directly by ATP levels in Escherichia coli. Genes Dev. 2012;26:548–553. doi: 10.1101/gad.183517.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Martin A, Baker TA, Sauer RT. Protein unfolding by a AAA+ protease is dependent on ATP-hydrolysis rates and substrate energy landscapes. Nat Struct Mol Biol. 2008;15:139–145. doi: 10.1038/nsmb.1380. [DOI] [PubMed] [Google Scholar]
- 159.Nager AR, Baker TA, Sauer RT. Stepwise unfolding of a β barrel protein by the AAA+ ClpXP protease. J Mol Biol. 2011;413:4–16. doi: 10.1016/j.jmb.2011.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Johnson A, O’donnell M. Cellular DNA replicases: components and dynamics at the replication fork. Annu Rev Biochem. 2005;74:283–315. doi: 10.1146/annurev.biochem.73.011303.073859. [DOI] [PubMed] [Google Scholar]
- 161.Tsuchihashi Z, Kornberg A. Translational frameshifting generates the gamma subunit of DNA polymerase III holoenzyme. Proc Natl Acad Sci USA. 1990;87:2516–2520. doi: 10.1073/pnas.87.7.2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Blinkowa AL, Walker JR. Programmed ribosomal frameshifting generates the Escherichia coli DNA polymerase III gamma subunit from within the tau subunit reading frame. Nucleic Acids Res. 1990;18:1725–1729. doi: 10.1093/nar/18.7.1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Flower AM, McHenry CS. The gamma subunit of DNA polymerase III holoenzyme of Escherichia coli is produced by ribosomal frameshifting. Proc Natl Acad Sci USA. 1990;87:3713–3717. doi: 10.1073/pnas.87.10.3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Vass RH, Chien P. Critical clamp loader processing by an essential AAA+ protease in Caulobacter crescentus. Proc Natl Acad Sci USA. 2013;110:18138–18143. doi: 10.1073/pnas.1311302110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Kenniston JA, Baker TA, Sauer RT. Partitioning between unfolding and release of native domains during ClpXP degradation determines substrate selectivity and partial processing. Proc Natl Acad Sci USA. 2005;102:1390–1395. doi: 10.1073/pnas.0409634102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Aubin-Tam ME, Olivares AO, Sauer RT, Baker TA, Lang MJ. Single-molecule protein unfolding and translocation by an ATP-fueled proteolytic machine. Cell. 2011;145:257–267. doi: 10.1016/j.cell.2011.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Maillard RA, Chistol G, Sen M, Righini M, Tan J, Kaiser CM, Hodges C, Martin A, Bustamante C. ClpX(P) Generates Mechanical Force to Unfold and Translocate Its Protein Substrates. Cell. 2011;145:459–469. doi: 10.1016/j.cell.2011.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Too PHM, Erales J, Simen JD, Marjanovic A, Coffino P. Slippery substrates impair function of a bacterial protease ATPase by unbalancing translocation versus exit. J Biol Chem. 2013;288:13243–13257. doi: 10.1074/jbc.M113.452524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Tian L, Holmgren RA, Matouschek A. A conserved processing mechanism regulates the activity of transcription factors Cubitus interruptus and NF-kappaB. Nat Struct Mol Biol. 2005;12:1045–1053. doi: 10.1038/nsmb1018. [DOI] [PubMed] [Google Scholar]
- 170.Palombella VJ, Rando OJ, Goldberg AL, Maniatis T. The ubiquitin-proteasome pathway is required for processing the NF-kappa B1 precursor protein and the activation of NF-kappa B. Cell. 1994;78:773–785. doi: 10.1016/s0092-8674(94)90482-0. [DOI] [PubMed] [Google Scholar]
- 171.Aza-Blanc P, Ramirez-Weber FA, Laget MP, Schwartz C, Kornberg TB. Proteolysis that is inhibited by hedgehog targets Cubitus interruptus protein to the nucleus and converts it to a repressor. Cell. 1997;89:1043–1053. doi: 10.1016/s0092-8674(00)80292-5. [DOI] [PubMed] [Google Scholar]
- 172.Gottesman S. Proteases and their targets in Escherichia coli. Annu Rev Genet. 1996;30:465–506. doi: 10.1146/annurev.genet.30.1.465. [DOI] [PubMed] [Google Scholar]
- 173.Gur E, Sauer RT. Recognition of misfolded proteins by Lon, a AAA(+) protease. Genes Dev. 2008;22:2267–2277. doi: 10.1101/gad.1670908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Gur E, Vishkautzan M, Sauer RT. Protein unfolding and degradation by the AAA+ Lon protease. Protein Sci. 2012;21:268–278. doi: 10.1002/pro.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Wohlever ML, Baker TA, Sauer RT. Roles of the N domain of the AAA+ Lon protease in substrate recognition, allosteric regulation and chaperone activity. Mol Microbiol. 2014;91:66–78. doi: 10.1111/mmi.12444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Gur E, Sauer RT. Degrons in protein substrates program the speed and operating efficiency of the AAA+ Lon proteolytic machine. Proc Natl Acad Sci USA. 2009;106:18503–18508. doi: 10.1073/pnas.0910392106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Gottesman S, Halpern E, Trisler P. Role of sulA and sulB in filamentation by lon mutants of Escherichia coli K-12. J Bacteriol. 1981;148:265–273. doi: 10.1128/jb.148.1.265-273.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Mizusawa S, Gottesman S. Protein degradation in Escherichia coli: the lon gene controls the stability of sulA protein. Proc Natl Acad Sci USA. 1983;80:358–362. doi: 10.1073/pnas.80.2.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Van Melderen L, Gottesman S. Substrate sequestration by a proteolytically inactive Lon mutant. Proc Natl Acad Sci USA. 1999;96:6064–6071. doi: 10.1073/pnas.96.11.6064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Vieux EF, Wohlever ML, Chen JZ, Sauer RT, Baker TA. Distinct quaternary structures of the AAA+ Lon protease control substrate degradation. Proc Natl Acad Sci USA. 2013;110:E2002–2008. doi: 10.1073/pnas.1307066110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Wohlever ML, Baker TA, Sauer RT. A Mutation in the N Domain of Escherichia coli Lon Stabilizes Dodecamers and Selectively Alters Degradation of Model Substrates. J Bacteriol. 2013;195:5622–5628. doi: 10.1128/JB.00886-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Leonard AC, Grimwade JE. Regulation of DnaA assembly and activity: taking directions from the genome. Annu Rev Microbiol. 2011;65:19–35. doi: 10.1146/annurev-micro-090110-102934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Collier J. Regulation of chromosomal replication in Caulobacter crescentus. Plasmid. 2012;67:76–87. doi: 10.1016/j.plasmid.2011.12.007. [DOI] [PubMed] [Google Scholar]
- 184.Gorbatyuk B, Marczynski GT. Regulated degradation of chromosome replication proteins DnaA and CtrA in Caulobacter crescentus. Mol Microbiol. 2005;55:1233–1245. doi: 10.1111/j.1365-2958.2004.04459.x. [DOI] [PubMed] [Google Scholar]
- 185.Lesley JA, Shapiro L. SpoT regulates DnaA stability and initiation of DNA replication in carbon-starved Caulobacter crescentus. J Bacteriol. 2008;190:6867–6880. doi: 10.1128/JB.00700-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Jonas K, Chen YE, Laub MT. Modularity of the bacterial cell cycle enables independent spatial and temporal control of DNA replication. Curr Biol. 2011;21:1092–1101. doi: 10.1016/j.cub.2011.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Jonas K, Liu J, Chien P, Laub MT. Proteotoxic stress induces a cell-cycle arrest by stimulating Lon to degrade the replication initiator DnaA. Cell. 2013;154:623–636. doi: 10.1016/j.cell.2013.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Bruel N, Castanie-Cornet MP, Cirinesi AM, Koningstein G, Georgopoulos C, Luirink J, Genevaux P. Hsp33 controls elongation factor-Tu stability and allows Escherichia coli growth in the absence of the major DnaK and trigger factor chaperones. J Biol Chem. 2012;287:44435–44446. doi: 10.1074/jbc.M112.418525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Balaban NQ, Merrin J, Chait R, Kowalik L, Leibler S. Bacterial persistence as a phenotypic switch. Science. 2004;305:1622–1625. doi: 10.1126/science.1099390. [DOI] [PubMed] [Google Scholar]
- 190.Lewis K. Persister Cells. Annu Rev Microbiol. 2010;64:357–372. doi: 10.1146/annurev.micro.112408.134306. [DOI] [PubMed] [Google Scholar]
- 191.Balaban NQ. Persistence: mechanisms for triggering and enhancing phenotypic variability. Curr Opin Genet Dev. 2011;21:768–775. doi: 10.1016/j.gde.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 192.Gerdes K, Maisonneuve E. Bacterial persistence and toxin-antitoxin loci. Annu Rev Microbiol. 2012;66:103–123. doi: 10.1146/annurev-micro-092611-150159. [DOI] [PubMed] [Google Scholar]
- 193.Maisonneuve E, Castro-Camargo M, Gerdes K. (p)ppGpp Controls Bacterial Persistence by Stochastic Induction of Toxin-Antitoxin Activity. Cell. 2013;154:1140–1150. doi: 10.1016/j.cell.2013.07.048. [DOI] [PubMed] [Google Scholar]
- 194.Dubnau D, Losick R. Bistability in bacteria. Mol Microbiol. 2006;61:564–572. doi: 10.1111/j.1365-2958.2006.05249.x. [DOI] [PubMed] [Google Scholar]
- 195.Veening JW, Smits WK, Kuipers OP. Bistability, epigenetics, and bet-hedging in bacteria. Annu Rev Microbiol. 2008;62:193–210. doi: 10.1146/annurev.micro.62.081307.163002. [DOI] [PubMed] [Google Scholar]
- 196.Price MN, Deutschbauer AM, Skerker JM, Wetmore KM, Ruths T, Mar JS, Kuehl JV, Shao W, Arkin AP. Indirect and suboptimal control of gene expression is widespread in bacteria. Mol Syst Biol. 2013;9:660. doi: 10.1038/msb.2013.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Tagkopoulos I, Liu YC, Tavazoie S. Predictive behavior within microbial genetic networks. Science. 2008;320:1313–1317. doi: 10.1126/science.1154456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Mitchell A, Romano GH, Groisman B, Yona A, Dekel E, Kupiec M, Dahan O, Pilpel Y. Adaptive prediction of environmental changes by microorganisms. Nature. 2009;460:220–224. doi: 10.1038/nature08112. [DOI] [PubMed] [Google Scholar]