

Abstract
Background
Previous genome-wide association studies (GWAS) have identified a large number of genetic variants for obesity and its related traits, representing a group of potential key genes in the etiology of obesity. Emerging evidence suggests that epigenetics may play an important role in obesity. It has not been explored whether the GWAS-identified loci contribute to obesity through epigenetics (e.g., DNA (deoxyribonucleic acid) methylation) in addition to genetics.
Method
A multi-stage cross-sectional study was designed. We did a literature search and identified 117 genes discovered by GWAS for obesity and its related traits. Then we analyzed whether the methylation levels of these genes were also associated with obesity in two genome-wide methylation panels. We examined an initial panel of seven adolescent obese cases and seven age-matched lean controls, followed by a second panel of 48 adolescent obese cases and 48 age- and gender-matched lean controls. The validated CpG sites were further replicated in two independent replication panels of youth (46 vs. 46 and 230 cases vs. 413 controls, respectively) and a general population of youth, including 703 healthy subjects.
Results
One CpG site in the lymphocyte antigen 86 (LY86) gene, which showed higher methylation in the obese in both the initial (p = .009) and second genome-wide DNA methylation panel (p = .008), was further validated in both replication panels (meta p = .00016). Moreover, in the general population of youth, the methylation levels of this region were significantly correlated with adiposity indices (p ≤.02), insulin resistance (p = .001), and inflammatory markers (p < .001).
Conclusion
By focusing on recent GWAS findings in genome-wide methylation profiles, we identified a solid association between LY86 gene DNA methylation and obesity.
Keywords: DNA methylation, obesity, GWAS, insulin resistance, inflammation
Obesity is an important risk factor for various diseases, including cardiovascular diseases (CVD) (Poirier et al., 2006), type 2 diabetes (T2D; Wild et al., 2004), and certain types of cancer (Anderson & Caswell, 2009). Its epidemic has imposed a huge burden on public health worldwide (Danaei et al., 2009; Misra & Khurana, 2008). As a typical, common complex disease, obesity is the result of the interplay between external (environmental) and internal (genetic) factors (Catenacci et al., 2009). Recent fruitful genome-wide association studies (GWAS) have identified a large number of genetic variants contributing to obesity (Loos, 2012). However, a majority of these loci have only a small effect on obesity susceptibility and explain just a fraction of total variance (Loos, 2012). There is growing evidence suggesting that epigenetics, a plastic and heritable (during cell division) mechanism to record cues from external and internal environments (Petronis, 2010) may play an important role in obesity (Drong et al., 2012; Feinberg et al., 2010; Wang et al., 2010). It has not been explored if the GWAS-identified loci contribute to obesity through epigenetics (e.g., DNA (deoxyribonucleic acid) methylation) in addition to genetics.
Taking advantage of our previous genome-wide methylation profilings (Wang et al., 2010; Xu et al., 2013), the current study focused on a group of candidate genes identified by previous GWAS for obesity and its related traits, and examined whether these genes showed differential DNA methylation between the obese and lean controls. Using this approach,we successfully identified a repeatable association of the DNA methylation level of one obesity GWAS gene (LY86) with obesity. This is the first study to report the association between LY86 gene methylation and obesity.
Materials and Methods
Study Design
The design of our multistage experiment is shown in Figure 1. We first conducted a literature search for obesity genes previously identified on DNA sequence variants. We then mapped these genes on our initial genome-wide DNA methylation panel (Illumina 27k chip) of seven obese cases versus seven lean controls (i.e., the initial epigenome-wide association studies [EWAS] panel). The CpG sites within these genes showing differential methylation between cases and controls with a raw p < .01 in this initial EWAS panel were further checked for validation in our second genome-wide DNA methylation panel (Illumina 450k chip) of 48 obese cases versus 48 lean controls (i.e., the second EWAS panel). The CpG sites that survived the second EWAS panel with a raw p < .01 were taken forward to the replication stage. The replicated CpG site was further carried over to a third stage, with the goal of examining the relationships of the CpG sites with more accurate adiposity indices, as well as obesity-related metabolic traits and inflammation in a general population sample of youth.
Figure 1.
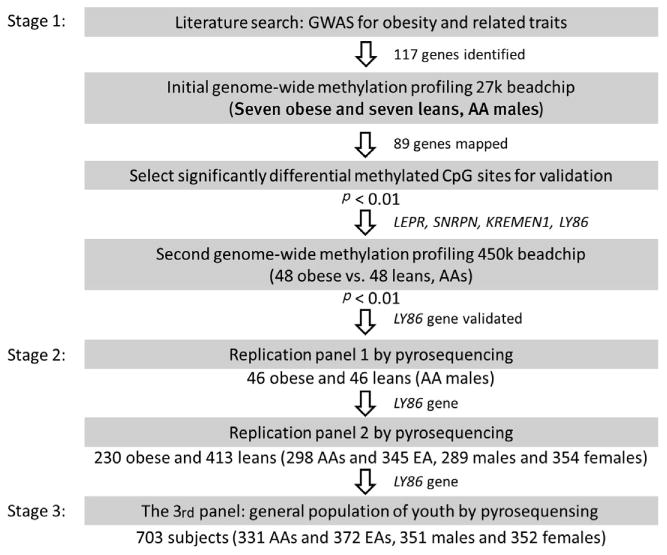
Flow chart describing the design of the multi-stage experiment. Note: AA = African-American; EA = European-American.
Literature Search
We focused on three categories: (1) monogenic obesity, typically caused by a single gene mutation with severe early-onset obesity as the main symptom; (2) syndromic obesity, in which patients are clinically obese, yet are additionally distinguished by mental retardation and other developmental abnormalities; and (3) polygenic or common obesity, which is likely caused by the interaction between numerous genes and environmental factors (McCarthy, 2010). The genetic bases for some of the extreme Mendelian human obesity (monogenic and syndromic) have been partially or completely elucidated and well reviewed previously (Ramachandrappa & Farooqi, 2011). For polygenic obesity, we included those genes reported in previous GWAS in terms of obesity and obesity-related traits, including body mass index (BMI), waist-hip ratio (WHR), waist circumference (WC), and weight. The full gene list (N = 117) and references are shown in Supplementary Table 1.
Study Subjects
Both the initial EWAS panel and the second EWAS panel have been described previously (Wang et al., 2010; Xu et al, 2013). Briefly, the initial EWAS panel included seven obese and seven age-matched lean controls, all being African-American (AA) males aged 14–18 years, living in the Augusta, Georgia area. The obese cases had a BMI ≥ 99th percentile and lean controls had a BMI ≤ 10th percentile for age and gender. The second EWAS panel included 48 obese and 48 age- and gender-matched lean controls, all being AAs aged 14–20 years, and with 50% males. Obese cases had a BMI ≥ 95th percentile and lean controls had a BMI < 50th percentile for age and gender. There were two replication panels. The first one included 46 obese (BMI ≥ 30 kg/m2 or BMI ≥ 95th percentile for age and gender if age ≤ 18 years) and 46 lean (BMI ≤ 22 kg/m2 or BMI ≤ 40th percentile for age and gender if age ≤ 18 years) youths; all were AA males aged 14–30 years recruited from the same area. The second replication panel included 230 obese (BMI ≥ 30 kg/m2 or BMI ≥ 95th percentile for age and gender if age ≤ 18 years) and 413 lean (BMI ≤ 25 kg/m2 or BMI ≤ 50th percentile for age and gender if age ≤ 18 years) subjects (aged 13–34 years) recruited from the same area, including both AAs (N = 298) and European-Americans (EAs, N = 345), as well as males (N = 289) and females (N = 354). There were no subjects overlapped among the initial EWAS panel, the second EWAS panel, and two replications panels. The third stage involved 703 subjects (aged 13–19 years, 372 EAs, 331 AAs, 351 males, and 352 females) with a BMI ranging from 14.6 to 45.9 kg/m2, of which 12 subjects (six obese vs. six lean) were included in the initial EWAS panel (two subjects excluded here because no DNA was available for this panel) and 221 subjects (53 obese vs. 168 lean) were included in the replication stage. More detailed information of the initial EWAS panel, the second EWAS panel, the two replication panels, and the third panel is given in the supplementary material. All subjects from these panels were overtly healthy, free of any acute or chronic illness on the basis of self-report and parental report (if subjects were younger than 18 years), and were not on anti-hypertensive, lipid lowering, anti-diabetic, or anti-inflammatory medications. The general characteristics are shown in Table 1. The Institutional Review Board at the Medical College of Georgia approved the studies. Written informed consent was obtained from all subjects and by parents if subjects were less than 18 years old.
Table 1. General Characteristics of the Subjects.
| Initial EWAS panel | Second EWAS panel | Replication panel 1 | Replication panel 2 | General population panel | |||||
|---|---|---|---|---|---|---|---|---|---|
|
|
|
|
|
||||||
| Cases | Controls | Cases | Controls | Cases | Controls | Cases | Controls | ||
| N | 7 | 7 | 48 | 48 | 46 | 46 | 230 | 413 | 703 |
| Males (%) | 100 | 100 | 50 | 50 | 100 | 100 | 37.8 | 48.9 | 49.9 |
| AAs (%) | 100 | 100 | 100 | 100 | 100 | 100 | 58.3 | 39.7 | 47.1 |
| Age (years) | 15.8 ± 1.0 | 15.9 ± 1.4 | 17.5 ± 1.8 | 17.7 ± 1.8 | 20.3 ± 5.0 | 17.6 ± 3.1 | 20.6 ± 4.6 | 19.1 ± 3.9 | 16.2 ± 1.2 |
| BMI(kg/m2) | 39.0 ± 1.7 | 17.0 ± 0.7 | 42.0 ± 7.5 | 18.9 ± 1.3 | 37.4 ± 6.1 | 18.9 ± 1.2 | 35.8 ± 6.6 | 20.3 ± 2.2 | 22.9 ± 4.9 |
| BMI percentile (%)* | 99.6 ± 0.1 | 4.8 ± 2.2 | 99.2 ± 0.6 | 20.3 ± 11.1 | 98.6 ± 1.0 | 19.4 ± 9.4 | 97.5 ± 1.6 | 29.2 ± 14.2 | 60.4 ± 28.3 |
Note: EWAS = Epigenome-wide association studies; AAs = African-Americans; Means ± SD (range);
BMI percentile for age and gender if age ≤ 18.
Measurement of DNA Methylation
For all subjects, peripheral blood samples were collected at the visit time. The buffy coat and plasma samples were separated within two hours and stored at -80°C immediately. DNA was extracted from the buffy coat using Qiagen® QiAamp® Blood Kit. The HumanMethylation27 BeadChip and the HumanMethylation450 BeadChip from Illumina (San Diego, CA, USA) were used for the genome wide DNA methylation profiling in the initial and the second EWAS panels respectively, as described previously (Wang et al., 2010; Xu et al, 2013). The methylation levels of the selected CpG sites for replications were determined by pyrosequencing technology (Wang et al, 2010), which is based on the principle of sequencing by synthesis. For methylation analysis, pyrosequencing not only offers individual methylation high-resolution results for all CpG sites, but also has built-in control for bisulfite treatment to improve accuracy and reproducibility. For each CpG site, a 100–350-bp region was amplified by polymerase chain reaction (PCR) using a pair of primers complementary to the bisulfite-treated DNA sequence. Therefore, in addition to the targeted CpG site, the pyrosequencing assay may involve several surrounding CpG sites.
Additional Measurements
A wide range of metabolic traits were measured in the third panel (N = 703), including fasting glucose and insulin levels, systolic and diastolic blood pressure (SBP and DBP), plasma levels of total cholesterol (TC), high-density lipoprotein cholesterol (HDL-C), low-density lipoprotein cholesterol (LDL-C), and triglyceride (TG), as well as fibrinogen and C-reactive protein (CRP). Insulin sensitivity was assessed by quantitative insulin sensitivity check index (QUICKI), calculated as: QUICKI = 1/(Log (fasting insulin, μU/ml) + Log (fasting glucose, mg/dl)). In addition, more accurate indices for adiposity as derived from dual-energy X-ray absorptiometry (DEXA) scans, including visceral adipose tissue (VAT), subcutaneous abdominal adipose tissue (SAAT), and percentage of body fat (%BF), were also available in the same population (Gutin et al, 2007). Detailed information is provided in the supplementary material.
Statistical Analyses
The genome wide methylation analysis has been described previously (Wang et al, 2010; Xu et al., 2013). The Limma package (Smyth, 2004) was used to analyze each CpG site for differential methylation between the obese and lean subjects. Each CpG site was assigned a raw p value based on a moderated t statistic. The successfully validated CpG sites were further replicated in the second replication panel. In this panel, linear regression was used to adjust for the potential effects of age, gender, and race on methylation levels. The gender × group (obese vs. lean) and race × group interaction terms were also built into the analyses to test whether the observed methylation differences between the obese and lean groups were race and gender dependent. The weighted z score-based meta-analysis approach implemented in the package METAL (Willer et al, 2010) was used to combine the results from the two replication panels, as well as the initial and the second EWAS panels. The relationships of the validated CpG sites with more accurate adiposity indices, as well as obesity-related metabolic traits in the third panel, were examined by using partial correlation analyses with adjustment for age, gender, and race. As this panel included samples with pyrosequencing conducted at different stages (221 samples at the second stage and 482 samples at the third stage), the potential batch effect on the methylation levels of each CpG site was removed by using linear regression and the residuals were used in the partial correlation analyses. All statistical analyses were performed using STATA 12 (StataCorp, College Station, TX, USA) and p ≤ .05 is considered as significance. We also used the program TFSEARCH to search for putative transcription factor binding sites in this promoter region, which is a simple routine searching highly correlated sequence fragments versus TFMATRIX transcription factor binding site profile database (Heinemeyer et al, 1998).
Results
A total of 117 genes were identified from the literature, of which 89 genes were mapped onto the Illumina 27K Beadchip. Four CpG sites showed significant differential methylation between the obese and lean subjects (p < .01) in the initial panel, located at the LEPR, SNRPN, KREMEN1 and LY86 genes (Table 2). In the second EWAS panel, only the CpG site in LY86 gene showed significant differential methylation between cases and controls with p < .01. In both EWAS panels, the obese cases had higher methylation levels of the CpG site in LY86 gene than the lean controls. Pyrosequencing assays were designed to validate the association of this CpG site with obesity in the first replication panel (46 obese vs. 46 lean controls). In addition to the targeted CpG site, the pyrosequencing assays involved five surrounding CpG sites. As shown in Ta b l e 3, the methylation level of the targeted CpG site was significantly higher in the obese than that in the lean controls (56.7% vs. 52.2%, p = .009). The methylation levels of the surrounding five CpG sites varied from 20% to 50%, but always showed higher methylation levels in the obese group in comparison with the lean group. All six CpG sites of the LY86 gene were further replicated in the second replication panel (230 obese vs. 413 lean controls). After adjusting for age, gender, and race, all six CpG sites showed significant higher methylation levels in the obese compared to the controls (Table 3). No significant interaction of obesity status with race or gender was found on the methylation levels of these six CpG sites. As expected, the meta-analysis with the EWAS panels resulted in stronger associations of the methylation levels with obesity (p = .00016 for LY86_5, the targeted CpG site).
Table 2. CpG Sites Selected From the Initial and the Second EWAS Panel.
| Gene | CpG site | Methylation level (%) | p | ||
|---|---|---|---|---|---|
|
| |||||
| Cases | Controls | Difference | |||
| Initial panela | N = 7 | N = 7 | |||
| LEPR | cg21655790 | 18.6 | 12.6 | 6.0 | .002 |
| SNRPN | cg26033681 | 44.3 | 49.4 | -5.1 | .009 |
| KREMEN1 | cg01791232 | 23.7 | 18.1 | 5.6 | .009 |
| LY86 | cg02212836 | 51.5 | 43.0 | 8.5 | .009 |
| Second panela | N = 48 | N = 48 | |||
| LEPR* | cg21655790 | — | — | — | — |
| SNRPN | cg26033681 | 53.9 | 52.9 | 1.0 | .169 |
| KREMEN1 | cg01791232 | 25.0 | 23.9 | 1.1 | .028 |
| LY86 | cg02212836 | 58.4 | 54.7 | 3.7 | .008 |
Note:
Adjusted for age in the initial panel and adjusted for age and gender in the second panel;
Not included in the 450K chip.
Table 3. DNA Methylation of LY86 and Obesity in the Replication Panels.
| Gene | CpG site | Methylation level (%) | p | p (adjusted)a | p (meta)b | ||
|---|---|---|---|---|---|---|---|
|
| |||||||
| Cases | Controls | Difference | |||||
| Replication panel 1 | N = 46 | N = 46 | |||||
| LY86 | LY86_1 | 41.8 | 38.1 | 3.7 | .03 | .08 | — |
| LY86_2 | 44.2 | 39.0 | 5.2 | <.001 | .005 | — | |
| LY86_3 | 24.6 | 20.8 | 3.8 | .001 | .009 | — | |
| LY86_4 | 56.1 | 51.5 | 4.6 | .008 | .05 | — | |
| LY86_5* | 56.7 | 52.2 | 4.5 | .009 | .05 | — | |
| LY86_6 | 51.4 | 46.8 | 4.6 | .003 | .03 | — | |
| Replication panel 2 | N = 230 | N = 413 | |||||
| LY86 | LY86_1 | 44.3 | 42.4 | 1.9 | .002 | .01 | .003 |
| LY86_2 | 46.3 | 44.6 | 1.7 | .002 | .01 | <.001 | |
| LY86_3 | 24.8 | 23.5 | 1.3 | .003 | .02 | .001 | |
| LY86_4 | 49.3 | 47.0 | 2.3 | .001 | .01 | .002 | |
| LY86_5* | 49.7 | 47.7 | 2.0 | .006 | .03 | .007 | |
| LY86_6 | 51.9 | 49.7 | 2.2 | .001 | .02 | .004 | |
Note:
LY86_5 is the targeted CpG site cg02212836;
Adjusted for age in the replication panel 1 and adjusted for age, gender, and race in the replication panel 2;
Meta analysis on the replication panels 1 (age-adjusted) and 2 (age-, gender- and race-adjusted).
In the general population panel, we found significant positive correlations of the methylation levels of the LY86 gene with BMI, and additional indices of adiposity, including %BF, VAT, and SAAT (Table 4). As the methylation levels of these six CpG sites are highly correlated (Pearson r > 0.8, Supplementary Table 2), we also performed a principal component (PC) analysis to combine the six CpG sites into one PC score (LY86_pc), which explained 90% of the variance for all six CpG sites. Similarly, this component was significantly associated with all four indices of adiposity, with the partial correlation coefficients being 0.096 (p = .01), 0.099 (p = .009), 0.138 (p = .004), and 0.109 (p = .02) for BMI, %BF, VAT, and SAAT respectively, after adjusting for age, gender, race, and batch. These results indicate that the association of LY86 gene methylation is consistent across different measures of adiposity.
Table 4. Associations of Methylation Levels of the LY86 Gene With Body Mass Index and Additional Indices for Adiposity in the Third Panel (N = 703) Representing the General Population of Youth.
| CpG sitesa | BMI | % body fat | VATb | SAATb | ||||
|---|---|---|---|---|---|---|---|---|
|
|
|
|
|
|||||
| Partial correlationc | p | Partial correlationc | p | Partial correlationc | p | Partial correlationc | p | |
| LY86_1 | .131 | <.001 | .138 | <.001 | .147 | .002 | .130 | .006 |
| LY86_2 | .100 | .008 | .097 | .01 | .151 | .001 | .137 | .004 |
| LY86_3 | .089 | .02 | .090 | .02 | .120 | .01 | .105 | .03 |
| LY86_4 | .094 | .01 | .087 | .02 | .126 | .008 | .094 | .05 |
| LY86_5 | .088 | .02 | .093 | .01 | .137 | .004 | .097 | .04 |
| LY86_6 | .067 | .08 | .077 | .04 | .127 | .008 | .085 | .08 |
| LY86_pc1d | .096 | .01 | .099 | .009 | .138 | .004 | .109 | .02 |
Note: BMI = body mass index; VAT = visceral adipose tissue; SAAT = subcutaneous abdominal adipose tissue;
Adjusted for batch;
Log-transformed;
Adjusted for age, gender, and race;
The first principal component score calculated based on the methylation levels of six CpG sites, adjusted for batch.
We further examined the associations of the methylation levels of the CpG sites with several obesity-related metabolic traits and two inflammatory markers. As shown in Table 5, the PC score of the six CpG sites was significantly associated with fasting glucose (partial r = 0.107, p = .007), fasting insulin (partial r = 0.123, p = .002), and insulin sensitivity (partial r = -0.129, p = .001), as well as fibrinogen (partial r = 0.167, p < .001) and CRP (partial r = 0.147, p < .001), after adjusting for age, gender, race, and batch. These associations attenuated somewhat, but remained significant after further adjustment for BMI, indicating that part of the effects of LY86 gene methylation on insulin resistance and inflammation are independent of body weight. The associations of the metabolic traits with the six individual CpG sites were similar to the results obtained from the PC score (Supplementary Table 3).
Table 5. Associations of Methylation Levels of the LY86 gene (LY86_pc) with the Obesity-Related Metabolic Traits and Inflammatory Markers in the Third Panel (N = 703) Representing the General Population of Youth.
| Metabolic traitsa | LY86_pcb | ||||
|---|---|---|---|---|---|
|
| |||||
| Model 1c | Model 2d | ||||
|
|
|
||||
| Partial correlation | p | Partial correlation | p | ||
| Fasting glucose | .094 | .02 | .082 | .04 | |
| Fasting insulin | .121 | .002 | .086 | .03 | |
| QUICKI | -.124 | .002 | -.091 | .02 | |
| SBP | .092 | .02 | .067 | .08 | |
| DBP | .072 | .06 | .068 | .08 | |
| TC | .030 | .44 | .024 | .55 | |
| HDL-C | -.058 | .14 | -.032 | .41 | |
| LDL-C | .064 | .10 | .048 | .22 | |
| TG | .050 | .21 | .025 | .52 | |
| Fibrinogen | .170 | <.001 | .145 | <.001 | |
| CRP | .143 | <.001 | .114 | .005 | |
Note:
All traits are log-transformed except QUICKI and DBP;
The first principal component score calculated based on the methylation levels of six CpG sites, adjusted for batch;
Adjusted for age, gender and race;
Adjusted for age, gender, race and BMI.
To further elucidate the role of methylation in this region on potential function of the LY86 gene, we performed an in silico analysis of the region, including these six CpG sites (122 bases), using TFSEARCH software (Heinemeyer et al., 1998; Figure 2). There were 21 transcription factor-binding sites that passed the threshold (score > 85.0), of which 12 could be found in humans. Interestingly, the third CpG site is located in a region potentially binding with many transcription factors, including USF, c-Myc, and Max. The sixth CpG site is within a binding site for AML-1a. These results indicate that methylation of these CpG sites may affect gene expression by blocking these transcription factor binding sites.
Figure 2.

TFSEARCH search result of the six CpG sites (underlined) in the promoter region of the LY86 gene (122 bases). There were 21 transcription factor binding sites that passed the threshold (score > 85.0), of which 12 could be found in human. Interestingly, the third CpG site is located in a region potentially binding with many transcription factors, including USF, c-Myc, and Max. The sixth CpG site is within a binding site for AML-1a.
Discussion
Taking advantage of our previous genome-wide methylation profiling on obesity (Wang et al., 2010; Xu et al., 2013), in this study we aimed to integrate this data with a hypothesis-based list of obesity genes based on evidence from monogenic, syndromic, and common obesity. Among the 89 GWAS genes mapped on the Illumina 27K methylation platform, the higher methylation level of the LY86 gene in obese cases was successfully confirmed in another genome-wide methylation panel and two replication panels. In a well-phenotyped general population cohort of youth, we further showed that LY86 gene methylation was not only associated with all adiposity indices but also with insulin resistance and inflammation markers, indicating that LY86 gene methylation may also play a role in obesity-related metabolic disorders.
The protein encoded by the LY86 gene is the ‘lymphocyte antigen 86’, also known as protein MD-1, which is a secreted glycoprotein physically associated with RP105 (a toll-like receptor (TLR) family protein) and playing a crucial role in B cell surface expression of RP105 (Nagai et al., 2002). The RP105/MD-1 complex is expressed on immune cells, including B cells, macrophages, and dendritic cells. Mice deficient in either RP105 or MD-1 exhibit reduced lipopolysaccharide (LPS) responsiveness in B cells (Nagai et al., 2002; Ogata et al., 2000). On the other hand, the expression of RP105/MD-1 in dendritic cells and macrophages has been shown to serve as a negative regulator of TLR4/MD-2 in the LPS response (Divanovic et al., 2005). Growing evidence suggests that LY86 may be involved in the (patho)physiological regulation of the innate immune system and inflammation (Sasaki et al., 2012). Most recently, in a mice model, Watanabe et al. (2012) reported that MD-1 may contribute to high-fat, diet-induced obesity, adipose tissue inflammation, and insulin resistance.
Whether the expression of the LY86 gene is regulated by DNA methylation remains unknown. According to the most recent release of Genome Browser, the CpG sites showing higher methylation levels in the obese subjects from the current study are located in the first exon with a distance of 61–133 bp (first and sixth CpG sites) to the transcription start site. Further in silico analysis of this region revealed that the third and sixth CpG sites are located at known binding sites for several transcription factors. For example, USF expression has been associated with metabolic traits in mice, including obesity, plasma lipid, and glucose/insulin ratio (Wu et al., 2010). In addition, the same study showed that binding of this factor to its DNA target is controlled by the methylation level of a single CpG dinucleotide included in its targeted sequence. Similarly, c-Myc binds its target sequence in a methylation-dependent manner, and over-expression of c-Myc in the liver has been reported to prevent obesity and insulin resistance in mice (Riu et al., 2003). The enrichment of transcription binding sites in this region and the methylation-dependent binding manner provide a possibility that methylation of these CpG sites in the LY86 gene may alter gene expression by blocking transcription factor binding. Transcriptional profiling studies in peripheral blood leukocytes may provide some evidence on the relationship between LY86 gene methylation and gene expression. We searched the Gene Expression Omnibus (GEO) database and identified a dataset (Ghosh et al., 2010; GSE18897) that included genome-wide gene expression data in peripheral blood samples from 20 obese and 20 lean adults. In line with our findings that obese cases had higher methylation levels of the LY86 gene, in this gene expression dataset, the expression level of LY86 gene was lower in the obese cases than in the lean controls, although the difference did not reach significance (309 ± 78 vs. 340 ± 75, p = .18).
The fact that LY86 is one of the GWAS-identified genes for obesity traits raised a possibility that the observed methylation difference between obese cases and lean controls may result from the obesity-associated single nucleotide polymorphisms(SNPs). TheGWAS-identifiedSNP is rs1294421, which is associated with WHR (adjusted for BMI) and is located 87kb from the LY86 gene. Data on both the genotype of this SNP and LY86 gene methylation are available in 345 of the current study samples. We did not observe the genotype of this SNP was associated with the methylation levels of these CpG sites (data not shown), indicating that the observed methylation difference between cases and controls is not caused by this SNP. However, we cannot exclude the possibility that other genetic variants in or near LY86 gene may influence the methylation levels of these CpG sites.
There are several strengths to this study. First, by focusing on youth and young adults at a pre-disease stage, we were optimizing chances to detect disease-specific epigenetic alterations, as presumably they are not yet masked by the background of age-related and medication-arising epigenetic ‘drift’ (Groom et al., 2011). Second, the two replication panels, with large samples recruited from the same area, enabled us to replicate the initial findings not only in AA males, but also in EAs and females. As shown in the result section, we did not observe significant interactions between obesity status and race or gender on the methylation levels of LY86 gene. Further stratified analyses by race and gender revealed similar significant findings in EAs (p = .001) and females (p = .003). In addition, involving the third panel enables us to extend our findings to a general population of youth. Third, the approach of integrating previous genomic and epigenomic data may help in prioritizing the signals for further validation. Our approach of replicating candidate genes is hypothesis driven, thus not requiring the same multiple testing penalty as the genome-wide approach.
This study also has some limitations. First, epigenetic regulation is tissue specific. In this regard, the target tissue to identify epigenetic variations responsible for obesity should preferably be adipose tissue or the hypothalamus. However, there is increasing evidence that biomarkers derived from blood may provide an alternative to tissue biopsy for the diagnosis and prognosis of disease (Dzau & Liew, 2007). In addition, obesity is multifactorial in origin and several organs have been implicated in metabolic regulation. In recent years, compelling evidence has emerged pointing to the involvement of low-grade inflammation in the pathophysiology of obesity and suggests that the immune system might be the missing mechanism that links these various organs with peripheral blood leukocytes as an important mediator (Pradhan, 2007). Furthermore, methylation profiles are typically established during early developmental stages prior to major cell differentiation, and subsequently maintained through cell divisions (Petronis, 2010). Thus, individual-specific, disease-related methylations may appear not only in the affected tissue(s), but across the human body, including accessible tissues such as blood. In this case, to interpret the function of the current finding on LY86 gene methylation, it will be necessary to conduct gene expression analysis not only in peripheral blood cells but also in adipose and hypothalamic or other organs implicated in the metabolic regulation of obesity. Second, we used the DNA from leukocytes, which represent different cell populations with distinct epigenetic profiles. A potential concern is that the results might be biased if the case group and control group display different cell compositions. Recently, a series of studies (Houseman et al., 2012; Koestler et al, 2012; Liu et al, 2013; Reinius et al, 2012) were conducted to address this concern by identifying differentially methylated CpG sites among leukocyte subtypes (including CD4+ T cells, CD8+ T cells, CD56+ NK cells, B cells, monocytes and granulocytes) as well as using these CpG sites to predict the cell compositions and adjusting them in the data analysis. The LY86 gene CpG site is within the top 500 CpG sites showing differential methylation among leukocyte subtypes (Supplementary Figure 1; Koestler et al, 2012). We predicted the cell compositions for both the initial and the second EWAS panels using Houseman's algorithm (Houseman et al, 2012) and observed that the obese group had a higher percentage of granulocytes in the initial panel and a lower percentage of natural killer (NK) cells in both the initial and the second genome-wide panels (Supplementary Tables 4 and 5). We conducted further analysis in the second genome-wide panel adjusting for the predicted cell compositions using linear regression models as suggested by Liu et al. (2013) and observed that this CpG site showed an even stronger association with obesity status (p = 3.49 × 10−7, Supplementary Table 6), indicating that our significant finding on the LY86 gene was not caused by shifts in these leukocyte subpopulations. Due to the limited sample size of the first panel (seven cases vs. seven controls) and the request of five covariates for the adjustment of cell compositions, we did not conduct this analysis in the first panel because we did not have enough sample size to provide reliable correlation estimates. Future studies on DNA methylation profiling of various types of cell populations of leukocytes are warranted to gain a greater understanding of the epigenetic dysregulation in obesity.
In conclusion, by focusing on previous genomic findings on monogenic, syndromic, and common obesity in our genome-wide methylation profiling, for the first time we identified a solid association between LY86 gene methylation and obesity in three stages with over 1,000 samples. Further studies are warranted to determine the functional meaning of such methylation changes. Such studies will have the ability to identify new insight into disease etiology and provide new targets for prevention of obesity related diseases such as CVD and T2D.
Supplementary Material
Acknowledgments
We are grateful to all participants and staffs who contributed to all cohorts used by our work. The participants in this study were recruited by several NIH-funded projects including HL041781, HL069999, HL056622, HL064225, HL077230, and HL64157. This publication was supported by grants from the National Heart, Lung and Blood Institute (HL078216) and the South Carolina Clinical and Translational Research Institute, with an academic home at the Medical University of South Carolina, Clinical and Translational Science Award, NIH/National Center for Research Resources (grant UL1RR029882). The current study is funded by NIH HL105689. S.S. is also funded by the American Heart Association (09SDG2140117) and the NIH (HL106333-01A1). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Supplementary Material: To view supplementary material for this article, please visit http://dx.doi.org/10.1017/thg.2014.22.
References
- Anderson AS, Caswell S. Obesity management — An opportunity for cancer prevention. Surgeon. 2009;7:282–285. doi: 10.1016/s1479-666x(09)80005-x. [DOI] [PubMed] [Google Scholar]
- Catenacci VA, Hill JO, Wyatt HR. The obesity epidemic. Clinics in Chest Medicine. 2009;30:415–444. doi: 10.1016/j.ccm.2009.05.001. [DOI] [PubMed] [Google Scholar]
- Danaei G, Ding EL, Mozaffarian D, Taylor B, Rehm J, Murray CJ, Ezzati M. The preventable causes of death in the United States: Comparative risk assessment of dietary, lifestyle, and metabolic risk factors. PLoS Medicine. 2009;6:e1000058. doi: 10.1371/journal.pmed.1000058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Divanovic S, Trompette A, Atabani SF, Madan R, Golenbock DT, Visintin A, Karp CL. Negative regulation of Toll-like receptor 4 signaling by the Tolllike receptor homolog RP105. Nature Immunology. 2005;6:571–578. doi: 10.1038/ni1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drong AW, Lindgren CM, McCarthy MI. The genetic and epigenetic basis of type 2 diabetes and obesity. Clinical Pharmacology & Therapeutics. 2012;92:707–715. doi: 10.1038/clpt.2012.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzau VJ, Liew C. Cardiovascular genetics and genomics for the cardiologist. Oxford: Blackwell Publishing; 2007. [Google Scholar]
- Feinberg AP, Irizarry RA, Fradin D, Aryee MJ, Murakami P, Aspelund T, Fallin MD. Personalized epigenomic signatures that are stable over time and covary with body mass index. Science Translational Medicine. 2010;2:49ra–67. doi: 10.1126/scitranslmed.3001262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, Dent R, Harper ME, Gorman SA, Stuart JS, McPherson R. Gene expression profiling in whole blood identifies distinct biological pathways associated with obesity. BMC Medical Genomics. 2010;3:56. doi: 10.1186/1755-8794-3-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groom A, Elliott HR, Embleton ND, Relton CL. Epigenetics and child health: Basic principles. Archives of Disease in Childhood. 2011;96:863–869. doi: 10.1136/adc.2009.165712. [DOI] [PubMed] [Google Scholar]
- Gutin B, Johnson MH, Humphries MC, Hatfield-Laube JL, Kapuku GK, Allison JD, Barbeau P. Relationship of visceral adiposity to cardiovascular disease risk factors in black and white teens. Obesity (Silver Spring) 2007;15:1029–1035. doi: 10.1038/oby.2007.602. [DOI] [PubMed] [Google Scholar]
- Heinemeyer T, Wingender E, Reuter I, Hermjakob H, Kel AE, Kel OV, Kolchanov NA. Databases on transcriptional regulation: TRANSFAC, TRRD and COMPEL. Nucleic Acids Research. 1998;26:362–367. doi: 10.1093/nar/26.1.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, Kelsey KT. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics. 2012;13:86. doi: 10.1186/1471-2105-13-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koestler DC, Marsit CJ, Christensen BC, Accomando W, Langevin SM, Houseman EA, Kelsey KT. Peripheral blood immune cell methylation profiles are associated with nonhematopoietic cancers. Cancer Epidemiology, Biomarkers & Prevention. 2012;21:1293–1302. doi: 10.1158/1055-9965.EPI-12-0361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Aryee MJ, Padyukov L, Fallin MD, Hesselberg E, Runarsson A, Feinberg AP. Epigenome-wide association data implicate DNA methylation as an intermediary of genetic risk in rheumatoid arthritis. Nature Biotechnology. 2013;31:142–147. doi: 10.1038/nbt.2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loos RJ. Genetic determinants of common obesity and their value in prediction. Best Practice & Research Clinical Endocrinology & Metabolism. 2012;26:211–226. doi: 10.1016/j.beem.2011.11.003. [DOI] [PubMed] [Google Scholar]
- McCarthy MI. Genomics, type 2 diabetes, and obesity. New England Journal of Medicine. 2010;363:2339–2350. doi: 10.1056/NEJMra0906948. [DOI] [PubMed] [Google Scholar]
- Misra A, Khurana L. Obesity and the metabolic syndrome in developing countries. Journal of Clinical Endocrinology & Metabolism. 2008;93:S9–30. doi: 10.1210/jc.2008-1595. [DOI] [PubMed] [Google Scholar]
- Nagai Y, Shimazu R, Ogata H, Akashi S, Sudo K, Yamasaki H, Miyake K. Requirement for MD-1 in cell surface expression of RP105/CD180 and B-cell responsiveness to lipopolysaccharide. Blood. 2002;99:1699–1705. doi: 10.1182/blood.v99.5.1699. [DOI] [PubMed] [Google Scholar]
- Ogata H, Su I, Miyake K, Nagai Y, Akashi S, Mecklenbrauker I, Tarakhovsky A. The tolllike receptor protein RP105 regulates lipopolysaccharide signaling in B cells. Journal of Experimental Medicine. 2000;192:23–29. doi: 10.1084/jem.192.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petronis A. Epigenetics as a unifying principle in the aetiology of complex traits and diseases. Nature. 2010;465:721–727. doi: 10.1038/nature09230. [DOI] [PubMed] [Google Scholar]
- Poirier P, Giles TD, Bray GA, Hong Y, Stern JS, Pi-Sunyer FX, Eckel RH. Obesity and cardiovascular disease: Pathophysiology, evaluation, and effect of weight loss: An update of the 1997 American Heart Association Scientific Statement on Obesity and Heart Disease from the Obesity Committee of the Council on Nutrition, Physical Activity, and Metabolism. Circulation. 2006;113:898–918. doi: 10.1161/CIRCULATIONAHA.106.171016. [DOI] [PubMed] [Google Scholar]
- Pradhan A. Obesity, metabolic syndrome, and type 2 diabetes: Inflammatory basis of glucose metabolic disorders. Nutrition Reviews. 2007;65:S152–156. doi: 10.1111/j.1753-4887.2007.tb00354.x. [DOI] [PubMed] [Google Scholar]
- Ramachandrappa S, Farooqi IS. Genetic approaches to understanding human obesity. Journal of Clinical Investigation. 2011;121:2080–2086. doi: 10.1172/JCI46044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinius LE, Acevedo N, Joerink M, Pershagen G, Dahlen SE, Greco D, Kere J. Differential DNA methylation in purified human blood cells: Implications for cell lineage and studies on disease susceptibility. PLoS One. 2012;7:e41361. doi: 10.1371/journal.pone.0041361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riu E, Ferre T, Hidalgo A, Mas A, Franckhauser S, Otaegui P, Bosch F. Overexpression of c-myc in the liver prevents obesity and insulin resistance. FASEB Journal. 2003;17:1715–1717. doi: 10.1096/fj.02-1163fje. [DOI] [PubMed] [Google Scholar]
- Sasaki S, Nagai Y, Yanagibashi T, Watanabe Y, Ikutani M, Kariyone A, Takatsu K. Serum soluble MD-1 levels increase with disease progression in autoimmune prone MRL (lpr/lpr) mice. Molecular Immunology. 2012;49:611–620. doi: 10.1016/j.molimm.2011.10.008. [DOI] [PubMed] [Google Scholar]
- Smyth GK. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Statistical Applications in Genetics and Molecular Biology. 2004;3 doi: 10.2202/1544-6115.1027. Article3. [DOI] [PubMed] [Google Scholar]
- Wang X, Zhu H, Snieder H, Su S, Munn D, Harshfield G, Shi H. Obesity related methylation changes in DNA of peripheral blood leukocytes. BMC Medicine. 2010;8:87. doi: 10.1186/1741-7015-8-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y, Nakamura T, Ishikawa S, Fujisaka S, Usui I, Tsuneyama K, Nagai S. The radioprotective 105/MD-1 complex contributes to diet-induced obesity and adipose tissue inflammation. Diabetes. 2012;6:1199–1209. doi: 10.2337/db11-1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wild S, Roglic G, Green A, Sicree R, King H. Global prevalence of diabetes: Estimates for the year 2000 and projections for 2030. Diabetes Care. 2004;27:1047–1053. doi: 10.2337/diacare.27.5.1047. [DOI] [PubMed] [Google Scholar]
- Willer CJ, Li Y, Abecasis GR. METAL: Fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26:2190–2191. doi: 10.1093/bioinformatics/btq340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Mar-Heyming R, Dugum EZ, Kolaitis NA, Qi H, Pajukanta P, Drake TA. Upstream transcription factor 1 influences plasma lipid and metabolic traits in mice. Human Molecular Genetics. 2010;19:597–608. doi: 10.1093/hmg/ddp526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Su S, Barnes VA, De Miguel C, Pollock J, Ownby D, Wang X. A genome-wide methylation study on obesity: Differential variability and differential methylation. Epigenetics. 2013;8:522–533. doi: 10.4161/epi.24506. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.