

SUMMARY
Tumor suppressor PTEN controls genomic stability and inhibits tumorigenesis. The N-terminal phosphatase domain of PTEN antagonizes the PI3K/AKT pathway, but its C-terminal function is less defined. Here we describe a knock-in mouse model of a nonsense mutation that results in deletion of the entire Pten C-terminal region, referred to as PtenΔC. Mice heterozygous for PtenΔC develop multiple spontaneous tumors, including cancers and B cell lymphoma. Heterozygous deletion of the Pten C-terminal domain also causes genomic instability and common fragile site rearrangement. We found that Pten C terminal disruption induces p53 and its downstream targets. Simultaneous depletion of p53 promotes metastasis without influencing initiation of tumors, suggesting that p53 mainly suppresses tumor progression. Our data highlight the essential role of the PTEN C-terminus in the maintenance of genomic stability and suppression of tumorigenesis.
Keywords: PTEN, Tumor, Chromosome Instability
INTRODUCTION
PTEN is one of the most frequently mutated genes in human cancer (Li et al., 1997; Steck et al., 1997) and cancer-associated PTEN mutations are found scattered over the entire PTEN gene (Song et al., 2012). The phosphatase domain in the N-terminal region of PTEN is responsible for antagonizing the PI3 kinase/AKT pathway, the primary signaling pathway for cell survival and proliferation (Maehama and Dixon, 1998). A large number of PTEN mutations occur in the C-terminus, suggesting that this region may also play a critical role in tumor suppression. We previously demonstrated that the C-terminal region is required for PTEN function in maintaining chromosomal integrity (Shen et al., 2007). The C-terminal sequence of PTEN is also critical for its nuclear localization (Terrien et al., 2012) and regulation of anchorage-independent growth (Georgescu et al., 1999) and cell migration (Leslie et al., 2007). These findings suggest that C-terminal PTEN plays a significant role in suppressing neoplasm, but until now there has been no direct proof of this in vivo. Animal models are therefore necessary for elucidation of whether the C-terminus of PTEN possesses a tumor suppressor function.
In addition to somatic PTEN alterations in a variety of sporadic tumor types, germline PTEN mutations are associated with a range of hereditary tumor predisposition syndromes such as Cowden syndrome (Marsh et al., 1998a; Marsh et al., 1998b). Cowden syndrome is an autosomal dominant disorder characterized by multiple hamartomas and high risk of breast and thyroid cancers (Farooq et al., 2010; Liaw et al., 1997). Genetic and clinical data have been combined to interrogate how PTEN mutations in Cowden syndrome increase cancer risk (Bubien et al., 2013; Marsh et al., 1998a; Tan et al., 2012). Knockout of the Pten gene in mice increases incidence of tumors that partially resemble the spectrum of neoplasia in Cowden syndrome (Di Cristofano et al., 1998; Podsypanina et al., 1999; Stambolic et al., 2000; Suzuki et al., 1998). Moreover, knock-in of mutant alleles that affect PTEN phosphatase activity can cause distinct yet Cowden syndrome-like tumor phenotypes in a mutation-specific manner (Wang et al., 2010).
The majority of germline PTEN mutations are nonsense or frame-shift mutations that result in premature stop codons and C-terminus truncations (Bubien et al., 2013; Marsh et al., 1998a). According to a recent report, germline PTEN truncation mutations preferentially target the C-terminal region and comprise over 80% of the total mutations in this region (Bubien et al., 2013). These data suggest that C-terminal PTEN mutation or deletion may contribute to the cancer susceptibility phenotype of Cowden syndrome. Among the variety of germline truncated PTEN mutations identified in Cowden syndrome, c.565A>T, a nonsense point mutation, is of particular interest because it causes truncation at Arg189, resulting in loss of the entire PTEN C-terminal region (ΔC).
In this study, we modeled this nonsense point mutation in mice to examine the role of the PTEN C-terminus in tumor suppression. Pten+/ΔC mice exhibit chromosomal instability and develop multiple tumors. We also found that the p53 signaling pathway is activated in Pten+/ΔC mice and depletion of p53 in Pten+/ΔC mice facilitates malignancy and promotes metastasis. These data suggest that C-terminal PTEN functions to prevent tumorigenesis whereas p53 acts mainly at later stages to suppress tumor progression. Our study demonstrates that the PTEN C-terminal region is essential for the maintenance of genomic stability and suppression of tumor development.
RESULTS
Heterozygous Pten C-terminal Deletion Causes Early Mortality and Spontaneous Tumor Development in Multiple Organs
To determine whether the C-terminal truncated PTEN mutation gives rise to Cowden syndrome-like phenotypes, we generated a mouse model by knocking in this point mutation (c.565A>T) preceded by a FLAG tag (Figure 1A). Pten C-terminal truncation, referred to as PtenΔC, was confirmed in targeted ES cells by Southern blot and DNA sequencing, and subsequently in heterozygous mice by genotyping (Figures 1B, S1A and S1B). The C-terminal truncated Pten protein, PtenΔC, is expressed in various tissues in Pten+/ΔC mice (Figures 1C) but not in Pten+/+ or Pten+/- mouse tissues (Figure S1C). We observed reduced levels of Pten expression in tissues from both Pten+/ΔC and Pten+/- mice, which results in elevated pAkt levels only in some Pten+/- tissues. The majority of Pten+/ΔC tissues express comparable levels of pAkt as counterpart Pten+/+ tissues (Figure S1D), indicating that there is a similar level of phosphatase active Pten in Pten+/ΔC mice as in wild-type cells. This suggests that Pten+/ΔC mice contain a phosphatase active PtenΔC protein that is capable of suppressing Akt phosphorylation. To determine whether PtenΔC retains the phosphatase activity, we overexpressed this truncated protein in Pten-/- MEFs. We found that PtenΔC can reduce Akt phosphorylation whereas PTEN mutants lacking the N-terminus (ΔN) or phosphatase activity (C124S) fail to do so (Figure S1E). An in vitro dephosphorylation assay further confirmed these results (Figure S1F) and suggests that PtenΔC retains the canonical phosphatase activity of Pten.
Figure 1. Knock-in of C-terminal truncated Pten reduces mouse lifespan due to spontaneous tumor development.
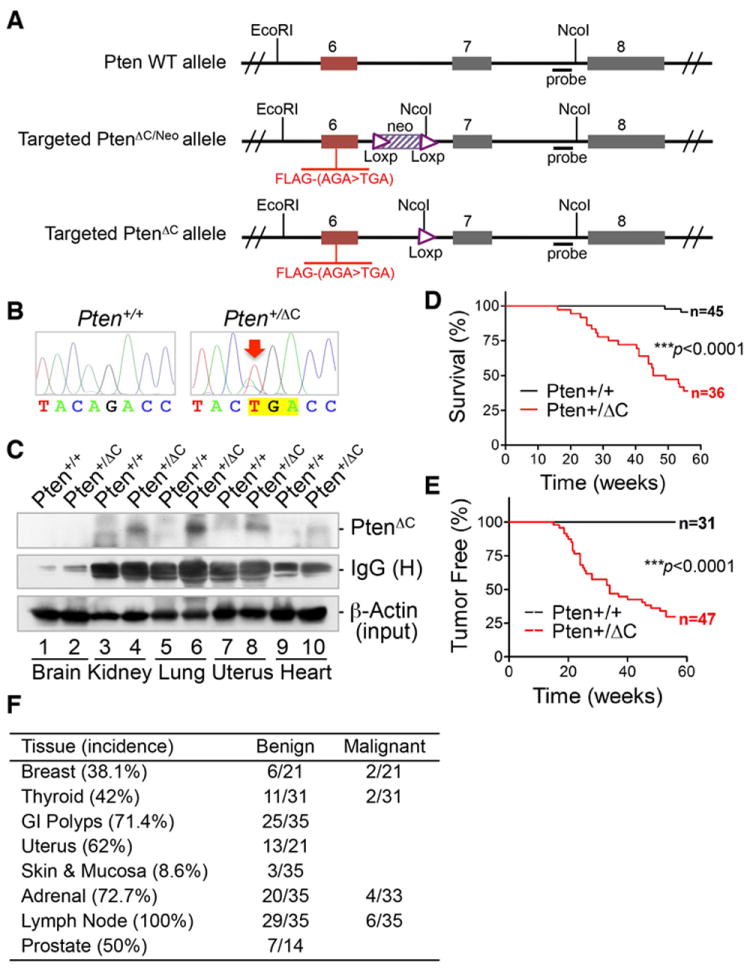
(A) Schematic diagram of strategy for knock-in of the Pten point mutation, c.565A>T, leading to deletion of the Pten C-terminal domain (aa189-403). (B) Confirmation of the point mutation of c.565A>T by direct sequencing of Pten exon 6 in Pten+/+ and Pten+/ΔC ES clones. (C) The expression of the C-terminal truncated PtenΔC in various tissues of Pten+/ΔC mice examined by immunoprecipitation with an N-terminus-specific PTEN antibody prior to FLAG immunoblotting. Corresponding tissues from wild-type mice were included as controls. (D) Kaplan-Meier plot for overall survival of Pten+/+ and Pten+/ΔC mice. (E) Kaplan-Meier plot showing significantly shorter latency of tumor development in Pten+/ΔC mice as compared with wild-type control mice. (F) Incidence of tumor in different tissues. Pten+/ΔC mice aged from 12 to 60 weeks (n=35, 14 males and 21 females) were subjected to extensive histological evaluation. Enlarged lymph nodes were found in every mouse analyzed. Pheochromocytoma occurred in all Pten+/ΔC mice older than six months with a 12% frequency of pulmonary metastasis. See also Figure S1.
In more than 50 litters, there were no viable homozygous PtenΔC mutant mice, suggesting that homologous deletion of the Pten C-terminus is lethal. In a cohort of 36 heterozygous PtenΔC mutation knock-in mice (Pten+/ΔC) and 45 wild-type control mice followed over 60 weeks for survival analysis, we found that knock-in of the PtenΔC mutation leads to earlier death (Figure 1D) mainly due to tumor formation in multiple organs (Figures 1E and 1F).
Pten+/ΔC mice develop spontaneous tumors or demonstrate premalignant lesions in multiple organs, including breast, thyroid, and endometrium, all of which show predisposition to cancer in human Cowden syndrome (Pilarski, 2009). The primary clinical concern in Cowden syndrome is elevated overall cancer incidence, and the most affected organs are breast and thyroid (Pilarski, 2009). Individuals with PTEN germline mutations have an 85.2% lifetime risk for breast cancer and a 35.2% risk for thyroid cancer (Tan et al., 2012). Consistent with human Cowden syndrome, breast and thyroid cancers are the most prominent malignancies in Pten+/ΔC mice (Figures 1F, 2A and 2B). Mammary adenoma and carcinoma occur in female Pten+/ΔC mice with frequencies of 28.6% and 9.5%, respectively (37.5% and 12.5% in female mice older than 9 months). The frequency of follicular thyroid adenoma in Pten+/ΔC mice is 35.5%, and thyroid malignancy is found in 6.5% of PtenΔC mutation knock-in mice. Five out of twenty one Pten+/ΔC female mice (23.8%) develop both mammary and thyroid tumors, and malignancy usually emerges in 9-13.5 months. Diagnostic criteria of Cowden syndrome also include macrocephaly and mental retardation, thyroid goiter, gastrointestinal polyps and mucocutaneous lesions (Farooq et al., 2010). In addition to breast and thyroid cancers, intestinal polyps occur in almost all Pten+/ΔC mice older than 7 months (Figures 1F and 2C). Pten+/ΔC mice also exhibit skin lesions such as orthokeratotic hyperkeratosis or sebaceous adenoma (Figure 2D), as well as endometrial hyperplasia (Figure 2E) and macrocephaly (Figure 2F). Knock-in of PtenΔC, a mutation derived from Cowden syndrome patients, endows Pten+/ΔC mice with all the characteristic phenotypes precisely imitating those in human Cowden syndrome. Our PtenΔC mutation knock-in mice therefore provide an exemplary animal model for Cowden syndrome.
Figure 2. Pten+/ΔC mice exhibit Cowden syndrome-like tumors in multiple tissues.
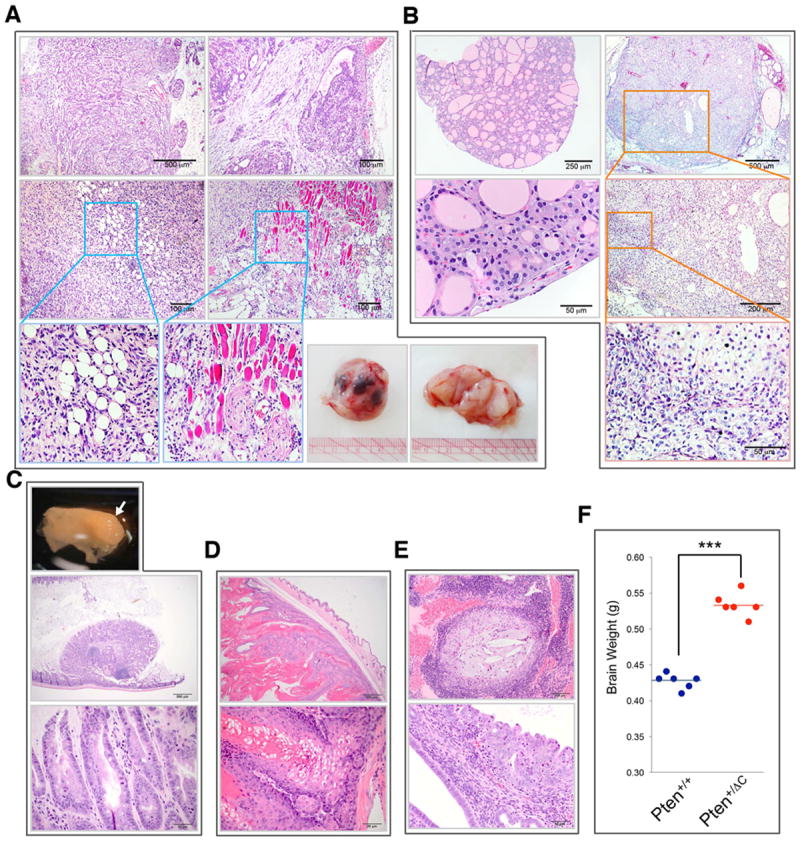
(A) Benign and malignant mammary tumors. Top panels, complex mammary adenoma; Middle and bottom left panels, carcinoma with areas magnified to show infiltrative growth of adipose tissue and skeletal muscle; Bottom right panel, gross view of a breast tumor with multiple foci of hemorrhagic necrosis. (B) Follicular adenoma of the thyroid (left panels) and thyroid carcinoma (right panels). The carcinoma consists of a large expansile nodule. Selected areas of the tumor are magnified to show follicular cells arrayed in disorderly, multilobulated nodules with atypia and very little follicle formation reflecting lack of differentiation in this carcinoma. (C) A polyp of the large intestine showing features of a hamartomatous polyp shown in gross view (top, white arrow) and histological views (middle and bottom). (D) Sebaceous adenoma of the skin consisting of a pseudoencapsulated expansile neoplasm showing features of skin appendage architecture. (E) Endometrial epithelial hyperplasia with dysplasia showing endometrium expanded by moderate cystic dilation of endometrial glands and disorderly growth of epithelial cells. (F) Macrocephaly manifested by significantly increased brain weight in Pten+/ΔC mice as compared with age- and gender-matched wild-type control mice. ***, p<0.0001. See also Figure S2.
In addition to the typical phenotypes associated with Cowden syndrome, Pten+/ΔC mice exhibit other abnormalities and lesions, such as thyroid goiter, adrenal pheochromocytoma with a 12% metastatic rate, polypoid adenoma of gallbladder mucosa, prostatic intraepithelial neoplasia, and epididymitis as shown in Figures S2A-S2E. The location and malignant status of these lesions are summarized in Figure 1F. The high incidence of neoplasm particularly in the premalignant stage in Pten+/ΔC mice offers opportunity for investigation of genetic and molecular events during tumor development and progression.
Epithelial to mesenchymal transition (EMT) is associated with acquisition of malignancy in epithelial tumor development (Nieto and Cano, 2012; Scheel and Weinberg, 2012). We evaluated for EMT in the development of breast cancer in Pten+/ΔC mice and found that a typical transition from epithelium to mesenchyme is indeed evident in mammary tissues at the adenoma stage. As compared with normal breast tissue from wild-type mice, mammary adenomas from Pten+/ΔC mice exhibit a significant decrease in an epithelial marker, E-cadherin, with a simultaneous increase in expression of a marker for fibroblast-like cells, FSP-1 (Figure S3A). These results suggest that heterozygous deletion of the Pten C-terminal region may elicit malignancy by promoting epithelial plasticity. A variant isoform of CD44, CD44v6, has been implicated in tumor progression and may induce mesenchymal and metastatic phenotypes (Zoller, 2011). Breast tissues from Pten+/ΔC mice, particularly at the adenoma stage, express significantly higher levels of CD44v6, as compared with normal tissues from wild-type mice (Figure S3B). These mesenchymal phenotypes in Pten+/ΔC mice, together with the widespread occurrence of pathological changes in multiple tissues, suggest that the C-terminal truncated Pten mutation may result in fundamental genomic alterations.
Mice with the Pten C-terminal Truncated Allele Develop B cell Lymphoma
Heterozygous deletion of the whole Pten gene can result in development of lymphoma, but mice with heterozygous Pten deletion develop only T cell lymphomas (Podsypanina et al., 1999; Stambolic et al., 2000; Suzuki et al., 1998). In contrast, knock-in of the Pten C-terminal truncated allele gives rise to lymphomas of predominant B cell lineage in mice. Pten+/ΔC mice also exhibit lymphoproliferative change in various sites. Splenomegaly is often found associated with abnormal splenic architecture, with expansion of white pulp and infiltration, and compression of the red pulp (Figure 3A). This splenic expansion in Pten+/ΔC mice involves B220+ B cells, resulting in displacement of CD3+ T cells, and many regions with B cell expansion have a high index of proliferation as measured by Ki67 staining (Figure 3B). Lymphocytic infiltration is also often found in non-lymphoid tissues such as lung and kidney (Figure 3A). In lymph nodes and non-lymphoid tissues with lymphocytic infiltration, strong Ki67 signals are found in regions of B cell expansion representing neoplasm or preneoplastic change (Figures 3C and S4A). Bcl-6 is frequently activated in B cell lymphomagenesis (Basso and Dalla-Favera, 2010) and plays a causal role in development of diffuse large B cell lymphoma (Cattoretti et al., 2005). In Pten+/ΔC mice, Bcl-6 staining is found consistently in regions of B cell proliferation (Figures 3B, 3C and S5A), and these features together are diagnostic of B cell lymphoma. Further analysis of clonality in rearranged IgH genes from 10 biopsies confirms these regions are clonal B cell expansions (Figure 3D). These same biopsies also display a clonal rearrangement of the TCR genes (Figure S4B), suggesting that lymphomas in Pten+/ΔC mice are of mixed lineage.
Figure 3. Pten+/ΔC mice develop lymphoma predominantly of B cell lineage.
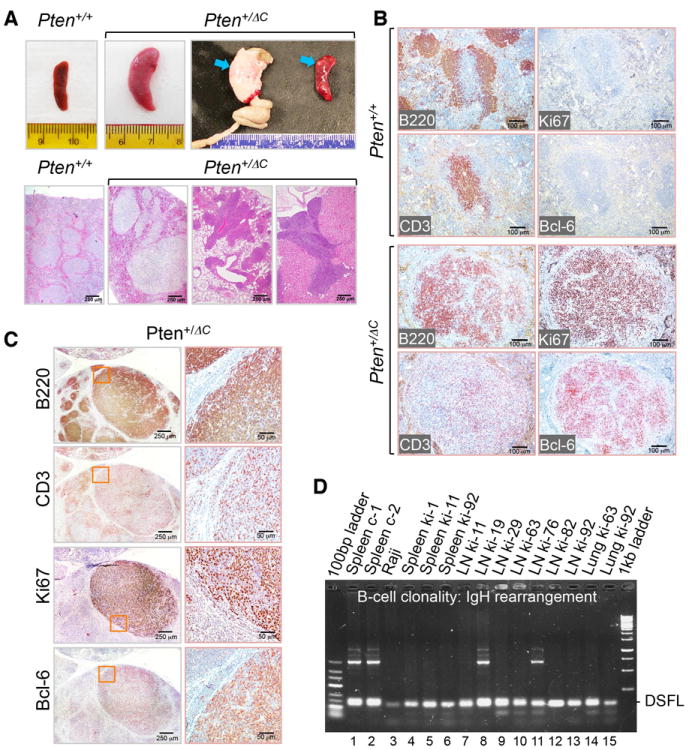
(A) Splenomegaly and lymphoma. The upper panel shows gross views of enlarged spleen (arrows point to a mesenteric lymph node and nodular aggregates in the spleen) in Pten+/ΔC mice. The lower panel displays histological images of abnormal splenic architecture (lower mid-left) and lymphoma in the lung (lower mid-right) and kidney (lower right). Normal spleen from a wild-type mouse is shown (lower left) as a control. (B) Lymphoma in the spleen from a Pten+/ΔC mouse. Immunohistochemical analysis of B (B220), T (CD3), proliferation (Ki67) cell markers and Bcl-6 in spleens from Pten+/+ and Pten+/ΔC mice. (C) B cell lymphoma with strong expression of the Bcl-6 marker. Lymph nodes from Pten+/ΔC mice were subjected to immunohistochemical staining for indicated cell markers. (D) Validation of B cell lymphoma lineage derivation by clonality analysis. Clonal B cell expansion is identified by monotonous D-J rearrangement of IgH in 10 out of 12 biopsies (lanes 3-7, 9, 10, 12-15). See also Figure S4.
Pten C-terminal Haploinsufficiency Leads to Chromosome Instability and Genomic Alterations
PTEN is critical for chromosome integrity and loss of Pten results in centromere instability (Shen et al., 2007). In order to determine whether C-terminal truncation of Pten also leads to centromere instability, we performed cytogenetic analysis for potential numerical and structural chromosome aberrations in our mouse model. As expected, there is a significantly higher frequency of tetraploidy in primary mouse embryonic fibroblasts (MEFs) with heterozygous Pten C-terminal deletion, as compared to wild-type MEFs (Figures 4A and 4B). Aneuploidy is found in Pten+/ΔC MEFs but not in Pten+/+ cells. In addition to aberrant chromosome numbers, Pten+/ΔC cells exhibit prominent structural chromosome abnormalities. This is manifested by centromeric aberrations such as centromeric fragments (CF), acentric chromosomes (AC), premature centromeric separation (PCS) and Robertsonian centromeric fusion (Rob), as shown in Figure 4C and summarized in Figure 4D. Accordingly, the ability of PTEN to physically interact with the integral centromere protein CENP-C is impaired in Pten+/ΔC cells and tissues (Figure 4E). We also compared the DNA damage status using γH2AX as a DNA double strand break (DSB) marker and found that Pten+/ΔC MEFs carry a significantly increased frequency of DSBs as compared with Pten+/+ or Pten-/- cells (Figure S5A and S5B). These data collectively suggest that the Pten C-terminal region is important for maintaining numerical and structural stability of the chromosome.
Figure 4. Knock-in of PtenΔC causes chromosomal instability.
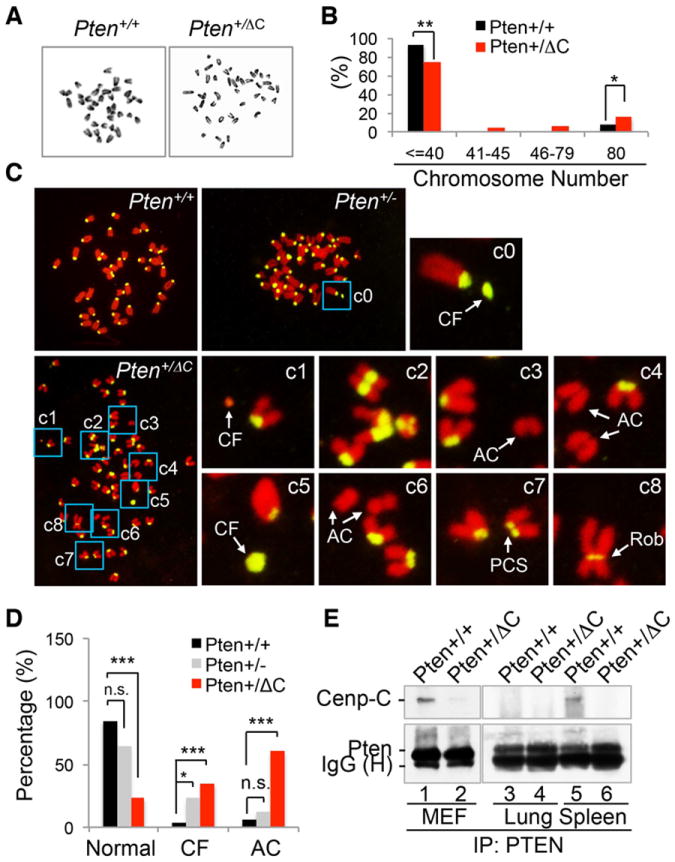
(A and B) Aneuploidy and polyploidy. Chromosome number was examined in metaphase spreads prepared from Pten+/+ (n=101) and Pten+/ΔC (n=126) MEFs (A) and data are summarized in the histogram (B). Images shown above the histogram are representative metaphase spreads. *, p<0.05; **, p<0.01. (C) Chromosomal aberrations featured by centromeric abnormalities. Metaphase spreads from Pten+/+ (n=32), Pten+/- (n=48) and Pten+/ΔC (n=43) MEFs were hybridized with a pan-centromeric probe (yellow) and DNA was stained with propidium iodide. Areas with aberrant chromosomes are magnified for visual enhancement. CF, centromeric fragment; AC, acentric chromosome fragment; PCS, premature centromeric separation; Rob, Robertsonian chromosomal fusion. (D) A summary of frequencies of centromeric fragments (CF, >2 per cell) and acentric chromosomes (AC). (E) Loss of physical association with CENP-C. Protein lysates from Pten+/+ and Pten+/ΔC cells or tissues were immunoprecipitated with a PTEN monoclonal antibody followed by immunoblotting with a rabbit anti-CENP-C antibody. See also Figure S5.
To further investigate genetic alterations caused by Pten C-terminal truncation, we examined genome-wide copy number changes using a comparative genomic hybridization array (aCGH). The CGH array identified over 50 genes with copy number variations, a subset of which is listed in Table S1. Two of these genes, Fhit and Wwox, are located in chromosomal regions prone to deletion and translocation known as common fragile sites (Iliopoulos et al., 2006). Common fragile sites constitute preferential targets for oncogenic chromosome damage especially in pre-neoplastic lesions, and instability of these sites represents a driving event in tumor development and progression (Gorgoulis et al., 2005; Tsantoulis et al., 2008). Altered splicing and translocation of both Fhit and Wwox result in abnormal transcripts, which occur frequently in multiple tumor types and precancerous lesions (Huebner and Croce, 2001; Matsuyama et al., 2004). In order to assess genetic alterations in these fragile genes, we examined abnormal transcripts of Fhit and Wwox in different tissues from Pten+/ΔC mice. As shown in Figures 5A and S6A, multiple aberrant Fhit and Wwox transcripts are found in different Pten+/ΔC tissues. Direct sequencing of these variant transcripts reveals multiple patterns of rearrangement, including alternative splicing and segmental deletion/insertion (Figures 5B, 5C, 5D and S6B). For example, in some adrenal and prostate tissues harboring the PtenΔC allele, a large segmental deletion removes the exons 3-7 of Fhit and short sequence segments from upstream of the Fhit start codon insert between exons 2 and 8 instead (Figures 5C and 5D). In order to evaluate whether these altered transcripts influence protein expression, we examined the levels of Fhit and Wwox in various tissues from Pten+/+ and Pten+/ΔC mice. Both Fhit and Wwox are down regulated in most tissues with PtenΔC knock-in, as compared with counterpart wild-type tissues (Figures 5E and S6C). An additional faster migrating protein band accompanies reduced expression of Fhit in almost every tissue from Pten+/ΔC mice (Figure 5E), which is likely the product of aberrant transcripts. Both FHIT and WWOX are tumor suppressor genes and biomarkers of genomic instability during early malignant transformation (Aqeilan et al., 2007; Iliopoulos et al., 2006; Pichiorri et al., 2008). Aberrant transcripts and deregulation of these genes suggest that PtenΔC knock-in causes extensive genetic alterations indicative of pre-neoplastic or neoplastic change. These results suggest that genomic instability caused by this truncated Pten mutant may be a driving force for tumorigenesis.
Figure 5. Knock-in of PtenΔC results in common fragile site instability.
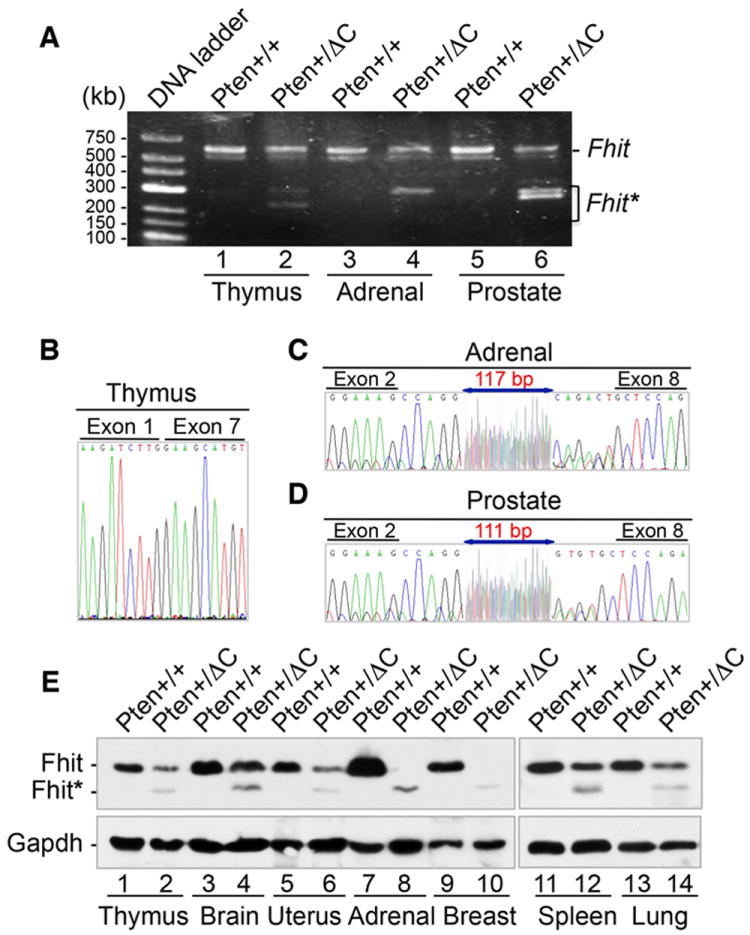
(A-D) Aberrant transcripts of Fhit in different tissues from Pten+/ΔC mice due to alternative splicing and translocation. Transcripts of Fhit were amplified with nested PCR from cDNA of indicated tissues (A) and abnormal transcripts were purified for sequencing (B-D). (E) Reduction of normal Fhit expression (~17 kDa) and production of a smaller Fhit protein variant (~15 kDa). Various pairs of tissues from Pten+/+ and Pten+/ΔC mice as indicated were analyzed for Fhit expression by Western blot. See also Figure S6 and Table S1.
The p53 Pathway is Upregulated in Pten+/ΔC Mice
p53 is the most frequently mutated genes in human cancer and plays important roles in cell cycle control, apoptosis and genomic stability. It has been reported that PTEN deficiency can activate p53 (Chen et al., 2005; Kim et al., 2007). In order to determine whether Pten C-terminal deletion may activate p53, we examined the levels of p53 in cells and tissues lacking the Pten C-terminal region. Pten+/ΔC MEFs express a higher level of p53 and accordingly, two well-known p53 transcriptional targets, the apoptosis mediator Apaf1 and the cell cycle inhibitor p21, are both similarly induced (Figure 6A). Further analysis in various Pten+/ΔC tissues also confirmed the increased levels of p53 as compared to counterpart control tissues from wild type mice (Figure 6B). Immunohistochemistry analysis of p53 signaling illustrates that thyroid tissues from Pten+/ΔC mice express elevated levels of p53, p21 and Apaf1, as compared with wild type mice (Figure 6C). These data suggest that the p53 pathway is activated by heterozygous Pten C-terminal loss, which may represent a defensive response to genetic stress resulting from Pten C-terminal deficiency.
Figure 6. Pten C-terminal deletion induces the p53 signaling pathway.
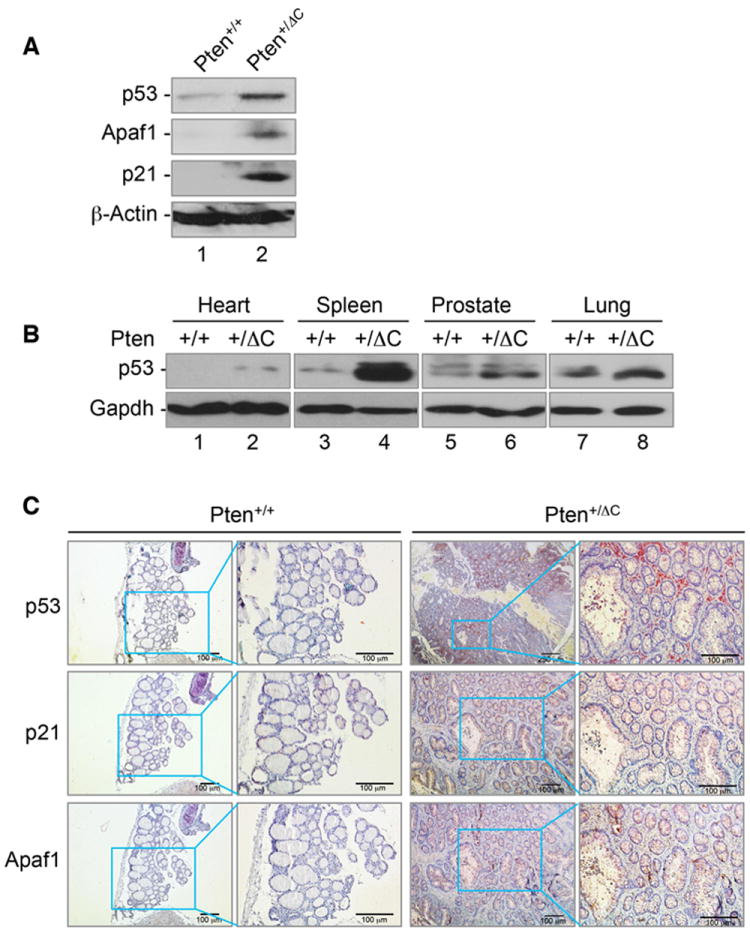
(A) Upregulation of p53 and its downstream targets in Pten+/ΔC MEFs as compared with wild type cells. Western blot analysis of p53 and its target genes in Pten+/+ and Pten+/ΔC MEFs using specific antibodies as indicated. (B) p53 induction in various tissues lacking the Pten C-terminal region. Various tissues from mice with and without Pten C-terminal deletion were subjected to immunoblotting analysis of p53 expression. (C) Thyroid tissues from Pten+/+ and Pten+/ΔC mice were stained by immunohistochemistry for p53, p21 and Apaf1, showing increased signals in PtenΔC knock-in tissues.
Simultaneous Disruption of p53 and Pten C-terminus Promotes Thyroid Tumor Progression
In order to determine whether depletion of p53 may enhance tumor development in Pten+/ΔC mice, we generated p53 and Pten C-terminus dual deficient (Pten+/ΔCp53+/-) mice by crossing Pten+/ΔC mice with p53+/- mice (Figure 7A). We found that as compared with Pten+/ΔC mice, dual deficient mice develop similar types of tumors in comparable frequencies in most tumor-prone organs except for thyroid gland (Figure 7B). Malignant thyroid tumors occur at earlier ages (5 months versus 13 months) and higher frequencies (27.8% versus 6.5%) in dual deficient mice than in single Pten C-terminal deficient mice. Moreover, thyroid tumor lung metastasis has been observed only in Pten+/ΔCp53+/- mice (Figure 7C), but not in Pten+/ΔC mice. These observations suggest that p53 activation in Pten+/ΔC mice (Figure 6C) plays an important role in preventing thyroid tumor progression and metastasis. Other than the difference in thyroid tumor, Pten+/ΔCp53+/- mice develop sarcoma and T cell lymphoma, whereas no sarcoma was found in Pten+/ΔC mice. Single p53 deficient (p53+/-) mice also develop sarcoma and T cell lymphoma, but at older ages (Donehower, 1996) as compared with Pten+/ΔCp53+/- mice, indicating that Pten C-terminal deficiency facilitates tumor development in p53+/- mice. All these results demonstrate that PTEN and p53 can collaborate with each other in tumor suppression. Our data suggest that p53 deficiency promotes tumor progression at later stages including metastasis in certain tumor types without affecting the initiation of tumor development.
Figure 7. Inactivation of p53 in Pten+/ΔC mice facilitates malignancy and promotes metastasis of thyroid tumors.
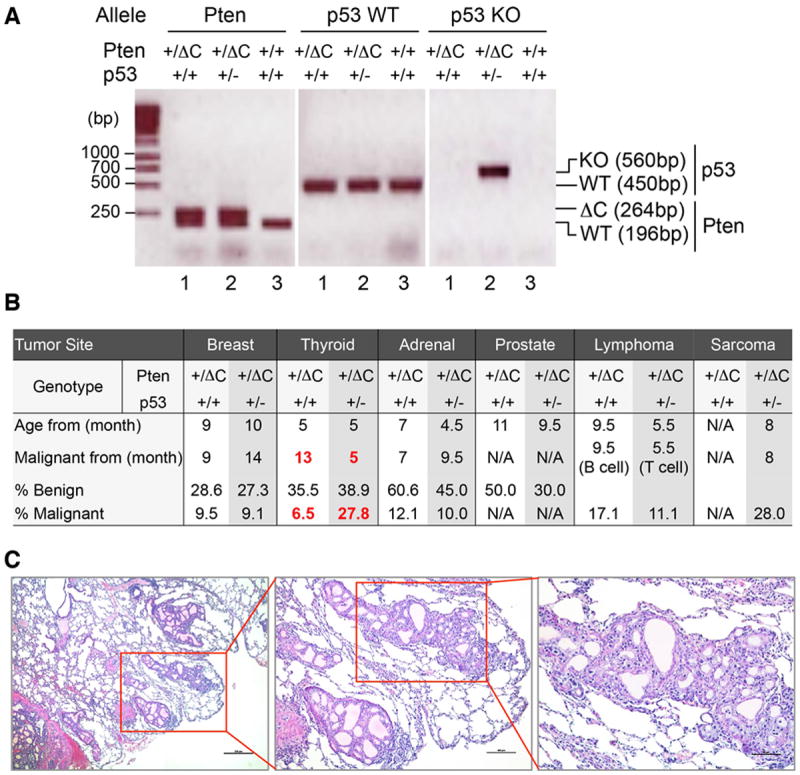
(A) Generation of double knockout mice with heterozygous p53 and PtenΔC alleles. Genotyping PCR analysis of tail DNA from Pten+/+ p53+/+, Pten+/ΔC p53+/+ and Pten+/ΔC p53+/- mice. (B) A summary of tumor incidence based on extensive histological evaluation. (C) Representative histological images showing thyroid tumor metastasis in lung.
DISCUSSION
PTEN function in tumor suppression has been attributed primarily to its phosphatase activity in antagonizing the PI3K/AKT survival pathway (Simpson and Parsons, 2001). In this study, we generated a mouse model in which the entire C-terminal region of Pten is deleted. Data generated from our Pten+/ΔC mice demonstrate that even in the presence of the intact N-terminal phosphatase domain, heterozygous segmental deletion of the Pten C-terminus leads to a wide spectrum of tumors. In addition to the identification of tumor phenotypes in PtenΔC knock-in mice, our study reveals genetic and molecular alterations that may drive tumor development. For example, PtenΔC knock-in mice exhibit extensive genomic instability in multiple organs manifested by common fragile site rearrangement, representing an overall fragile genome susceptible to genotoxic stress or DNA damage and subsequent neoplastic transformation. Therefore, our data highlight the important role of the PTEN C-terminal region in genomic stability and tumor suppression, and suggest that genomic instability is a driving force of tumorigenesis.
One of the important features of Pten+/ΔC mice is that neoplastic phenotypes emerge at different stages, ranging from hyperplasia, dysplasia, and premalignancy to malignant neoplasia. This mimics cancer susceptibility in Cowden syndrome and offers the opportunity for tracking genetic and molecular alterations occurring at different stages of tumor development and progression. At the adenoma stage, PtenΔC knock-in mammary tissues exhibit EMT, suggesting that epithelial alteration may be an early manifestation of cellular deterioration, leading to initiation of malignancy. Common fragile site rearrangement is often found in precancerous lesions and Fhit is the most frequent target for allelic deletion and translocation (Saldivar et al., 2012; Saldivar et al., 2010). Indeed, the expression of Fhit is reduced in almost all tissues harboring the PtenΔC allele, even those without obvious features for neoplasm. Therefore, common fragile site instability may represent an early genetic trait or genomic signature for cancer susceptibility.
Our study demonstrates that a PTEN germline mutation associated with Cowden disease results in genetic instability and cancer susceptibility in a mouse model. We found that haploinsufficiency of the Pten C-terminal region results in a series of phenotypes that encompass almost all the characteristic pathological features of Cowden syndrome. Thus the PtenΔC knock-in mouse is a bona fide Cowden syndrome model. Our PtenΔC knock-in mouse not only successfully models human disease-associated truncation mutations that predominantly disrupt the PTEN C-terminus, but also illustrates that PTEN C-terminal haplosufficiency may shift the genomic and tissue programming towards cancer susceptibility.
Knock-in of PtenΔC not only disrupts one Pten allele, but also creates a truncated phosphatase active PtenΔC protein, which is distinct from existing Pten heterozygous deletion animal models (Di Cristofano et al., 1998; Podsypanina et al., 1999; Stambolic et al., 2000; Suzuki et al., 1998). Gain of oncogenic function is an important feature of mutation in a typical tumor suppressor. A comparison between our Pten+/ΔC mouse model and different Pten+/- mouse strains (Table S3) suggests that Pten+/ΔC mice develop tumors of various types not only similar to those reported in Pten+/- strains but we also found some heretofore unidentified phenotypes such as B cell lymphoma. Lymphoma development has been described in mice with heterozygous Pten deletion (Podsypanina et al., 1999; Stambolic et al., 2000; Suzuki et al., 1998). However, these previously reported lymphomas are T cell type (Table S3). Although a reduction of PTEN expression has recently been correlated with diffuse large B-cell lymphoma in human samples (Pfeifer et al., 2013), there is no evidence in any available model systems to suggest a direct link between PTEN deficiency and B cell lymphomagenesis. Even conditional deletion of Pten specifically in B cells does not cause B cell lymphoma (Anzelon et al., 2003). Most likely, loss of Pten is insufficient to induce lymphoma of B cell lineage. Therefore, lymphomagenesis with clonal B cell expansion in Pten+/ΔC mice represents a newly acquired phenotypic feature originated from Pten C-terminal truncation and subsequent genomic instability. We propose that PtenΔC is a gain-of-function oncogenic mutation leading to a dominant negative mutator phenotype, of which B cell lymphoma is only one presentation.
PTEN and p53 are both powerful tumor suppressors. However, they may function at distinct stages to prevent tumor development and progression. In our mouse models, dual deficiency of both p53 and the Pten C-terminus only promotes thyroid tumor progression without affecting tumor initiation or progression of other tumor types in mice with single Pten C-terminal deficiency. These observations suggest that PTEN deficiency contributes to the initial stage of tumor development whereas p53 mainly acts at later stages of tumor progression. Our data also suggest that p53 activation in Pten+/ΔC mice may represent a defensive response to lack of Pten function. As p53 mutation is very common in human cancer, maintaining functional integrity of the p53 pathway ensures a favorable response to cancer treatment. Therefore, activation of the p53 pathway in response to Pten C-terminal truncation may benefit future therapeutic studies using our Pten+/ΔC mice.
This study establishes the important role of C-terminal PTEN in genomic stability and tumor suppression. Our work also reveals early genetic and molecular alterations, such as common fragile site instability and EMT, which contribute to tumorigenesis and malignancy. Data obtained from PtenΔC knock-in mice provide insight into the process of cellular transformation in which a driver mutation leads to systemic genomic instability and tumorigenesis. Moreover, the p53 pathway is activated in our Pten C-terminal truncated mice, which suggests that this mouse model would be ideal for preclinical trials of chemotherapeutic compounds to treat human cancers or cancer predisposition diseases.
EXPERIMENTAL PROCEDURES
Mice
To generate Pten C-terminal truncation (PtenΔC) knock-in mice, C57BL6/129S6 mouse ES cells were electroporated with targeting vectors containing a 5’ homology region with Pten exons 4-5, a neo cassette flanked by two loxP sites (LNL), and a 3’ homology region containing exon 6 with the desired mutation. ES cells were selected by G418 and screened for homologous recombination by PCR. Southern blot was used to further identify PtenΔC knock-in ES clones. Positive targeted ES clones containing the targeted allele with desired FLAG and stop codon replacement were injected into C57BL/6 blastocysts, and the blastocysts were implanted into pseudopregnant females to generate chimeras. The chimeras were bred with EIIA-Cre transgenic female mice to delete the loxP-flanked neomycin resistance gene (neo) from the targeted allele. The resultant neo-deleted mosaic Pten+/ΔC mice were genotyped by PCR. Positive mosaic mice were crossed with the C57BL/6 strain to generate heterozygous mice. To further validate FLAG and stop codon replacement in Pten+/ΔC mice, we sequenced Pten mRNA from the original ES cells to confirm the knock-in. The offspring were PCR genotyped using a primer set (forward, 5’-GCCTATCAGGGAGTCACAATTC-3’; reverse, 5’-AAAGTTTTCCGACACACAGACA-3’). p53+/- mice (B6.129S2-Trp53tm1Tyj/J) were purchased from the Jackson Laboratory. Double heterozygous Pten+/ΔCp53+/- mice were generated by crossing Pten+/ΔC mice with p53+/- mice.
Primary MEF Cells
Primary MEFs were isolated from embryos harvested at 13.5 days post coitus. Individual embryos were minced and digested by trypsin, and embryonic cells were collected by centrifugation before plating in MEM supplemented with 10% FBS. MEFs from each embryo were genotyped as described above and early passage cells were used for chromosomal analysis.
Histopathology and immunohistochemistry (IHC)
Animals were sacrificed or autopsied and all tissues were examined regardless of their pathologic status. Tissue samples were fixed in 10% buffered formalin and embedded in paraffin for preparation of 5-μm sections. Sections were stained with hematoxylin and eosin (H&E) by standard methods. For IHC, tissue sections were processed for immunohistochemical evaluation of B220, CD3, Ki67, Bcl6, E-cadherin, FSP1, or CD44v6 following standard procedures.
Antibodies
Polyclonal antibodies against mouse Fhit and Wwox were produced by immunizing rabbits with specific peptides DELQKHDREEEDSPAFWC (aa115-131 of Fhit) and CTVDDNPTKPTTRQRYDG(Amd) (aa93-109 of Wwox). Antisera from the immunized rabbits were affinity purified using their respective peptides.
Centromeric fluorescent in situ hybridization (C-FISH)
Metaphase spreads were prepared from early passage (<3) MEFs and C-FISH was performed using a FITC-labeled pan-centromeric probe (Cambio) following the procedures described previously(Shen et al., 2007). DNA was counter-stained with propidium iodide.
Comparative genomic hybridization array (aCGH)
Genomic DNA extracted from Pten+/+ and Pten+/ΔC MEFs was subjected to oligonucleotide array-CGH using Agilent SurePrint G3 mouse 1×1M CGH microarrays, following the manufacturer’s instructions. These arrays include 963261 probes with a 1.8-kb overall median probe space. Hybridized slides were scanned using a microarray scanner (G2505B, Agilent), and the image data were extracted and converted to text files using Agilent Feature Extraction software. Following data normalization, segmentation and analysis, segments with log2 ratios higher than 0.2 or lower than -0.2 were selected and analyzed for copy number variation.
Common fragile site aberrant transcripts analysis
RNA was prepared from different tissues of Pten+/+ and Pten+/ΔC mice and cDNA was synthesized by reverse transcription. Nested PCR was performed to amplify transcripts of Fhit or Wwox using the following primers: mFhit-1F, CTTGCCTTTCATTCCAGC; mFhit-9R, GGGGTCCATTTTCTTAT; mFhit-2F, TGAAGCCCAGCAAAGAAG; mFhit-8R, AAAGTAGACCCGCAGAGC; mWwox-1F, TTGAAGCCAGAGTGAGTTGCTAG; mWwox-9R, GATGGGGCTGACTGTGATGAGG; mWwox-2F, AAAACCGGCAAGAGGAAACG; and mWwox-8R, GTTATAAGCCAGCATGGCCC. Abnormal transcripts were purified and sequenced.
Clonality analysis
Genomic DNA was prepared from different tissues of Pten+/+ and Pten+/ΔC mice. Clonal rearrangement of the IgH segment (DSFL, B-cell lineage) or TCR 8 chain segment (VgH10, T-cell lineage) was amplified by PCR using the following primers: DSFL forward, 5’-AG GGATCCTTGTGAAGGGATCTACTACTGTG-3’; DSFL reverse, 5’-AAAGACCTGCAGAGGCCATTCTTACC-3’; VgH10 forward: 5’-AAGCAACAAAGTGGAGGCAAGAAAG-3’; VgH10 reverse: 5’-GAAGTTACTATGAGCTTAGTCCCTT-3’.
Statistics
Overall and tumor-free survival curves were calculated with the Kaplan-Meier method and analyzed with Gehan-Breslow-Wilcoxon test for statistical significance. Brain weight was measured in different age and gender groups, and the paired t-test was used to analyze differences between Pten+/ΔC and Pten+/+ mice in each littermate group. Chromosome numbers and mitotic indices were compared using Pearson’s chi-square test (χ2). All statistical tests were two-sided and p values less than 0.05 (*), 0.01 (**) or 0.0001 (***) were considered statistical significant.
Supplementary Material
HIGHLIGHTS.
Loss of Pten C-terminus leads to multiple tumors including B cell lymphoma
Pten C-terminus is required for genomic stability and tumor suppression
Disruption of Pten activates the p53 pathway
PTEN suppresses tumorigenesis whereas p53 mainly inhibits tumor progression
Acknowledgments
We thank Sebastien Monette at the Center of Comparative Medicine and Pathology for his assistance in mouse phenotype analysis. We also thank the Genomics Core Laboratory at Memorial Sloan-Kettering Cancer Center for CGH analysis. We are grateful to Ms. Pan Wang for pAkt analysis and Dr. Michael McNutt for critical reading of the manuscript. This work was supported by NIH grants CA133008 (Y.Y.) and GM100478 (W.H.S.), and China National Major Scientific Program (973 Project-2010CB912202), National Natural Science Foundation of China (Key Project-30930021), Beijing Natural Science Foundation (Major Project-5100003), and Shu Fan Education and Research Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anzelon AN, Wu H, Rickert RC. Pten inactivation alters peripheral B lymphocyte fate and reconstitutes CD19 function. Nat Immunol. 2003;4:287–294. doi: 10.1038/ni892. [DOI] [PubMed] [Google Scholar]
- Aqeilan RI, Trapasso F, Hussain S, Costinean S, Marshall D, Pekarsky Y, Hagan JP, Zanesi N, Kaou M, Stein GS, et al. Targeted deletion of Wwox reveals a tumor suppressor function. Proc Natl Acad Sci U S A. 2007;104:3949–3954. doi: 10.1073/pnas.0609783104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basso K, Dalla-Favera R. BCL6: master regulator of the germinal center reaction and key oncogene in B cell lymphomagenesis. Adv Immunol. 2010;105:193–210. doi: 10.1016/S0065-2776(10)05007-8. [DOI] [PubMed] [Google Scholar]
- Bubien V, Bonnet F, Brouste V, Hoppe S, Barouk-Simonet E, David A, Edery P, Bottani A, Layet V, Caron O, et al. High cumulative risks of cancer in patients with PTEN hamartoma tumour syndrome. J Med Genet. 2013;50:255–263. doi: 10.1136/jmedgenet-2012-101339. [DOI] [PubMed] [Google Scholar]
- Cattoretti G, Pasqualucci L, Ballon G, Tam W, Nandula SV, Shen Q, Mo T, Murty VV, Dalla-Favera R. Deregulated BCL6 expression recapitulates the pathogenesis of human diffuse large B cell lymphomas in mice. Cancer Cell. 2005;7:445–455. doi: 10.1016/j.ccr.2005.03.037. [DOI] [PubMed] [Google Scholar]
- Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, Koutcher JA, Scher HI, Ludwig T, Gerald W, et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. 2005;436:725–730. doi: 10.1038/nature03918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Cristofano A, Pesce B, Cordon-Cardo C, Pandolfi PP. Pten is essential for embryonic development and tumour suppression. Nat Genet. 1998;19:348–355. doi: 10.1038/1235. [DOI] [PubMed] [Google Scholar]
- Donehower LA. The p53-deficient mouse: a model for basic and applied cancer studies. Semin Cancer Biol. 1996;7:269–278. doi: 10.1006/scbi.1996.0035. [DOI] [PubMed] [Google Scholar]
- Farooq A, Walker LJ, Bowling J, Audisio RA. Cowden syndrome. Cancer Treat Rev. 2010;36:577–583. doi: 10.1016/j.ctrv.2010.04.002. [DOI] [PubMed] [Google Scholar]
- Georgescu MM, Kirsch KH, Akagi T, Shishido T, Hanafusa H. The tumor-suppressor activity of PTEN is regulated by its carboxyl-terminal region. Proc Natl Acad Sci U S A. 1999;96:10182–10187. doi: 10.1073/pnas.96.18.10182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorgoulis VG, Vassiliou LV, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, Venere M, Ditullio RA, Jr, Kastrinakis NG, Levy B, et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434:907–913. doi: 10.1038/nature03485. [DOI] [PubMed] [Google Scholar]
- Huebner K, Croce CM. FRA3B and other common fragile sites: the weakest links. Nat Rev Cancer. 2001;1:214–221. doi: 10.1038/35106058. [DOI] [PubMed] [Google Scholar]
- Iliopoulos D, Guler G, Han SY, Druck T, Ottey M, McCorkell KA, Huebner K. Roles of FHIT and WWOX fragile genes in cancer. Cancer Lett. 2006;232:27–36. doi: 10.1016/j.canlet.2005.06.048. [DOI] [PubMed] [Google Scholar]
- Kim JS, Lee C, Bonifant CL, Ressom H, Waldman T. Activation of p53-dependent growth suppression in human cells by mutations in PTEN or PIK3CA. Mol Cell Biol. 2007;27:662–677. doi: 10.1128/MCB.00537-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie NR, Yang X, Downes CP, Weijer CJ. PtdIns(3,4,5)P(3)-dependent and -independent roles for PTEN in the control of cell migration. Curr Biol. 2007;17:115–125. doi: 10.1016/j.cub.2006.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, Puc J, Miliaresis C, Rodgers L, McCombie R, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943–1947. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- Liaw D, Marsh DJ, Li J, Dahia PL, Wang SI, Zheng Z, Bose S, Call KM, Tsou HC, Peacocke M, et al. Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat Genet. 1997;16:64–67. doi: 10.1038/ng0597-64. [DOI] [PubMed] [Google Scholar]
- Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998;273:13375–13378. doi: 10.1074/jbc.273.22.13375. [DOI] [PubMed] [Google Scholar]
- Marsh DJ, Coulon V, Lunetta KL, Rocca-Serra P, Dahia PL, Zheng Z, Liaw D, Caron S, Duboue B, Lin AY, et al. Mutation spectrum and genotype-phenotype analyses in Cowden disease and Bannayan-Zonana syndrome, two hamartoma syndromes with germline PTEN mutation. Hum Mol Genet. 1998a;7:507–515. doi: 10.1093/hmg/7.3.507. [DOI] [PubMed] [Google Scholar]
- Marsh DJ, Dahia PL, Caron S, Kum JB, Frayling IM, Tomlinson IP, Hughes KS, Eeles RA, Hodgson SV, Murday VA, et al. Germline PTEN mutations in Cowden syndrome-like families. J Med Genet. 1998b;35:881–885. doi: 10.1136/jmg.35.11.881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuyama A, Croce CM, Huebner K. Common fragile genes. Eur J Histochem. 2004;48:29–36. [PubMed] [Google Scholar]
- Nieto MA, Cano A. The epithelial-mesenchymal transition under control: global programs to regulate epithelial plasticity. Semin Cancer Biol. 2012;22:361–368. doi: 10.1016/j.semcancer.2012.05.003. [DOI] [PubMed] [Google Scholar]
- Pfeifer M, Grau M, Lenze D, Wenzel SS, Wolf A, Wollert-Wulf B, Dietze K, Nogai H, Storek B, Madle H, et al. PTEN loss defines a PI3K/AKT pathway-dependent germinal center subtype of diffuse large B-cell lymphoma. Proc Natl Acad Sci U S A. 2013;110:12420–12425. doi: 10.1073/pnas.1305656110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichiorri F, Palumbo T, Suh SS, Okamura H, Trapasso F, Ishii H, Huebner K, Croce CM. Fhit tumor suppressor: guardian of the preneoplastic genome. Future Oncol. 2008;4:815–824. doi: 10.2217/14796694.4.6.815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilarski R. Cowden syndrome: a critical review of the clinical literature. J Genet Couns. 2009;18:13–27. doi: 10.1007/s10897-008-9187-7. [DOI] [PubMed] [Google Scholar]
- Podsypanina K, Ellenson LH, Nemes A, Gu J, Tamura M, Yamada KM, Cordon-Cardo C, Catoretti G, Fisher PE, Parsons R. Mutation of Pten/Mmac1 in mice causes neoplasia in multiple organ systems. Proc Natl Acad Sci U S A. 1999;96:1563–1568. doi: 10.1073/pnas.96.4.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldivar JC, Miuma S, Bene J, Hosseini SA, Shibata H, Sun J, Wheeler LJ, Mathews CK, Huebner K. Initiation of genome instability and preneoplastic processes through loss of Fhit expression. PLoS Genet. 2012;8:e1003077. doi: 10.1371/journal.pgen.1003077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldivar JC, Shibata H, Huebner K. Pathology and biology associated with the fragile FHIT gene and gene product. J Cell Biochem. 2010;109:858–865. doi: 10.1002/jcb.22481. [DOI] [PubMed] [Google Scholar]
- Scheel C, Weinberg RA. Cancer stem cells and epithelial-mesenchymal transition: concepts and molecular links. Semin Cancer Biol. 2012;22:396–403. doi: 10.1016/j.semcancer.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen WH, Balajee AS, Wang J, Wu H, Eng C, Pandolfi PP, Yin Y. Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell. 2007;128:157–170. doi: 10.1016/j.cell.2006.11.042. [DOI] [PubMed] [Google Scholar]
- Simpson L, Parsons R. PTEN: life as a tumor suppressor. Exp Cell Res. 2001;264:29–41. doi: 10.1006/excr.2000.5130. [DOI] [PubMed] [Google Scholar]
- Song MS, Salmena L, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol. 2012;13:283–296. doi: 10.1038/nrm3330. [DOI] [PubMed] [Google Scholar]
- Stambolic V, Tsao MS, Macpherson D, Suzuki A, Chapman WB, Mak TW. High incidence of breast and endometrial neoplasia resembling human Cowden syndrome in pten+/- mice. Cancer Res. 2000;60:3605–3611. [PubMed] [Google Scholar]
- Steck PA, Pershouse MA, Jasser SA, Yung WK, Lin H, Ligon AH, Langford LA, Baumgard ML, Hattier T, Davis T, et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet. 1997;15:356–362. doi: 10.1038/ng0497-356. [DOI] [PubMed] [Google Scholar]
- Suzuki A, de la Pompa JL, Stambolic V, Elia AJ, Sasaki T, del Barco Barrantes I, Ho A, Wakeham A, Itie A, Khoo W, et al. High cancer susceptibility and embryonic lethality associated with mutation of the PTEN tumor suppressor gene in mice. Curr Biol. 1998;8:1169–1178. doi: 10.1016/s0960-9822(07)00488-5. [DOI] [PubMed] [Google Scholar]
- Tan MH, Mester JL, Ngeow J, Rybicki LA, Orloff MS, Eng C. Lifetime cancer risks in individuals with germline PTEN mutations. Clin Cancer Res. 2012;18:400–407. doi: 10.1158/1078-0432.CCR-11-2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terrien E, Chaffotte A, Lafage M, Khan Z, Prehaud C, Cordier F, Simenel C, Delepierre M, Buc H, Lafon M, et al. Interference with the PTEN-MAST2 interaction by a viral protein leads to cellular relocalization of PTEN. Sci Signal. 2012;5:ra58. doi: 10.1126/scisignal.2002941. [DOI] [PubMed] [Google Scholar]
- Tsantoulis PK, Kotsinas A, Sfikakis PP, Evangelou K, Sideridou M, Levy B, Mo L, Kittas C, Wu XR, Papavassiliou AG, et al. Oncogene-induced replication stress preferentially targets common fragile sites in preneoplastic lesions. A genome-wide study. Oncogene. 2008;27:3256–3264. doi: 10.1038/sj.onc.1210989. [DOI] [PubMed] [Google Scholar]
- Wang H, Karikomi M, Naidu S, Rajmohan R, Caserta E, Chen HZ, Rawahneh M, Moffitt J, Stephens JA, Fernandez SA, et al. Allele-specific tumor spectrum in pten knockin mice. Proc Natl Acad Sci U S A. 2010;107:5142–5147. doi: 10.1073/pnas.0912524107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoller M. CD44: can a cancer-initiating cell profit from an abundantly expressed molecule? Nat Rev Cancer. 2011;11:254–267. doi: 10.1038/nrc3023. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.