

Abstract
Background
Segmental duplications at breakpoints (BP4–BP5) of chromosome 15q13.2q13.3 mediate a recurrent genomic imbalance syndrome associated with mental retardation, epilepsy, and/or EEG abnormalities.
Patients
DNA samples from 1,445 unrelated patients submitted consecutively for clinical array comparative genomic hybridisation (CGH) testing at Children’s Hospital Boston and DNA samples from 1,441 individuals with Autism from 751 families in the Autism Genetic Resource Exchange (AGRE) repository.
Results
We report the clinical features of five patients with a BP4-BP5 deletion, three with a BP4–BP5 duplication, and two with an overlapping but smaller duplication identified by whole genome high resolution oligonucleotide array CGH. These BP4–BP5 deletion cases exhibit minor dysmorphic features, significant expressive language deficits, and a spectrum of neuropsychiatric impairments that include autism spectrum disorder, ADHD, anxiety disorder, and mood disorder. Cognitive impairment varied from moderate mental retardation to normal IQ with learning disability. BP4–BP5 covers ~1.5Mb (chr15:28.719–30.298Mb) and includes 6 reference genes and 1 miRNA gene, while the smaller duplications cover ~500 kb (chr15:28.902–29.404 Mb) and contain 3 reference genes and one miRNA gene. The BP4–BP5 deletion and duplication events span CHRNA7, a candidate gene for seizures. However, none of these individuals reported here have epilepsy, although two have an abnormal EEG.
Conclusions
The phenotype of chromosome 15q13.2q13.3 BP4–BP5 microdeletion/duplication syndrome may include features of autism spectrum disorder, a variety of neuropsychiatric disorders, and cognitive impairment. Recognition of this broader phenotype has implications for clinical diagnostic testing and efforts to understand the underlying etiology of this syndrome.
Keywords: array CGH, autism, language delay, microdeletion/duplication, neuropsychiatric disorders
INTRODUCTION
Recurrent microdeletion or microduplication events are a common cause of developmental delay and mental retardation.1, 2 Most of these events are mediated by recombination between segmentally duplicated sequences through an established mechanism of non-allelic homologous recombination, or NAHR.3 Clinical genetic testing of individuals with developmental delay, mental retardation, and autism spectrum disorders using whole genome high resolution array comparative genomic hybridisation (CGH) has revealed the clinical importance of ever smaller microdeletions and microduplications such as deletions in the range of 500–650 kb at 17q21.3 associated with developmental delay4–6 and at 16p11.2 among individuals with autism.7–9
The proximal portion of chromosome 15q is a well known region of genomic instability that contains many segmental duplications.5, 10–14 Deletions at 15q11–q13 that result in Prader-Willi syndrome and Angelman syndrome (PWS/AS) are typically ~4Mb, and are mediated by repetitive elements with clustered breakpoints (BP) at either of two proximal sites (BP1 and BP2) and one distal site (BP3).15, 16 Genomic imbalance on proximal 15q has been associated with developmental delay and autism, most notably maternally inherited duplications of the PWS/AS region at 15q11q1314, 17–19 and also a large duplication from BP1–BP5 in a simplex case of autism.20
A recurrent microdeletion syndrome mediated by more distal segmental duplication breakpoints on chromosome 15q13.2q13.3, designated BP4–BP514 at chromosome 15q13.2q13.3 was recently described.21 Minor dysmorphic features, mental retardation, epilepsy, and/or EEG abnormalities were common in the reported cases of BP4–BP5 deletion. This deletion is ~1.5Mb and includes six reference genes (MTMR15, MTMR10, TRPM1, KLF13, OTUD7A and CHRNA7) and a miRNA gene (hsa-mir-211). The alpha7-nicotinic receptor subunit gene (CHRNA7) is a candidate gene for seizures based on several lines of evidence: the presence of seizures in 7 out of 9 probands with BP4–BP5 deletions that include CHRNA7,21 genetic linkage to 15q among subjects with epilepsy,22, 23 a Chrna7 knockout mouse model that has an abnormal EEG,24 and genetic linkage studies supporting susceptibility to juvenile myoclonic epilepsy.25
We present eight patients with BP4–BP5 deletions and duplications, and two with a smaller duplication within BP4–BP5, identified by whole genome high resolution array CGH on patients undergoing clinical genetics evaluations from Children’s Hospital Boston, or by genome-wide oligonucleotide genotyping arrays on samples from the Autism Genetic Resource Exchange (AGRE). We report a broader phenotype than originally recognized, including autism spectrum disorder and other neuropsychiatric disorders including anxiety, attention deficits, mood disorder, and cognitive impairment with or without mental retardation and without epilepsy.
MATERIALS AND METHODS
Children’s Hospital Boston Samples
We performed whole genome high resolution oligonucleotide array CGH on 1,445 consecutively submitted clinical samples with referring diagnoses including developmental delay (DD; n=639), mental retardation (MR) or learning disability (LD; n=49 for MR/LD), autism spectrum disorder (ASD; n=177) or pervasive developmental disorder (PDD; n=85; total for ASD/PDD= 262), multiple congenital anomalies (n=118), dysmorphic features (n= 224), seizures (n= 49) or undefined/other phenotypes (n=104). All patients with 15q13 imbalance were examined by a developmental specialist and a clinical geneticist. A team of specialists from Clinical Genetics, Neurology, and Developmental Medicine at Children’s Hospital Boston conducted a medical record review approved by the Children’s Hospital Boston Institutional Review Board (IRB).
Array CGH and Confirmatory Studies
CGH was performed according to previously published methods of analysis using the Agilent 244K human genome oligonucleotide CGH microarray; all coordinates reflect human genome build 18 (G4411B, Agilent Technologies, Palo Alto, CA).26 Independent confirmation of deletion/duplication of the 15q13.2q13.3 region was performed by multiplex ligation-dependent probe amplification (MLPA) and fluorescence in situ hybridisation (FISH) according to previously described methods.26
AGRE Samples
DNA samples from 751 multiplex families were obtained from the Autism Genetic Resource Exchange (AGRE) collection of multiplex families27 using previously described sample selection criteria.8 Our final dataset included 1,441 individuals affected with autism spectrum disorders, 1,420 parents, and 132 unaffected/unknown siblings. This study was approved by the Massachusetts Institute of Technology (MIT) IRB.
Genotyping and Confirmatory Studies
AGRE samples were genotyped on Affymetrix 5.0 arrays at the Genetic Analysis Platform of the Broad Institute, and analyzed for copy number variants with the COPPER and Birdseye algorithms.8, 28 SNP genotype data and raw intensity files have been released to AGRE, and are available to the research community under AGRE guidelines. Independent confirmation of deletion/duplication of the 15q13.2q13.3 region among AGRE samples was performed using Agilent 244k array CGH and MLPA at Children’s Hospital Boston.
RESULTS
We identified ten patients with genomic imbalance at chromosome 15q13.2q13.3, including five with BP4–BP5 microdeletions from the CHB cohort (chr15:28.7Mb to ~30.3Mb; hg18). Patient 5 was identified after the manuscript was originally submitted, and was not included among the original 1,445 DNA samples. We did not find any cases of BP4–BP5 microdeletion among 1,420 parents, and 132 unaffected/unknown siblings in the AGRE samples. We identified three patients with reciprocal BP4–BP5 duplications; and two siblings with a smaller duplication of ~500kb within BP4–BP5 (chr15:28.9–29.4Mb; hg18; Fig. 1). BP4 is more than 1Mb distal to the telomeric breakpoint (BP3) of the 15q11q13 deletion associated with PWS/AS and the reciprocal duplication that has been associated with autism. None of the patients from CHB or AGRE had other clinically significant copy number variants elsewhere in the genome. All CHB patients had normal karyotypes and fragile X testing by Southern blot and PCR. All deletion and duplication events in these samples were confirmed by dye reversal array CGH (Fig. 1) and through a customized MLPA assay or FISH (Supplemental Fig. 1, 2). Genomic coordinates of all deletions and duplications are listed in Tables 1 and 2.
FIGURE 1.

In the top panel, an ideogram of proximal chromosome 15q (15q11q14) shows the PWS/AS region and the more distal 15q13.2q13.3 region between BP4 and BP5. Lower panels show scatter plots of array CGH data for a deletion of ~1.5Mb superimposed with dye-swap scatter plot (note the mirrored distribution of spots). The lower scatter plot represents a duplication of ~500Kb within the BP4–BP5 interval. The relative positions of 7 genes (6 reference genes and one miRNA gene) are shown in the bottom panel (grey bars). The 1.5Mb deletions (chr15:28.719–30.232Mb; hg18) include all 7 genes, while the 500 kb duplications (chr15:28.902–29.404 Mb; hg18) contain 4 genes (MTMR15, MTMR10, TRPM1 and hsa-mir-211) within the BP4–BP5 at chromosome 15q13.2q13.3.
Table 1.
15q13.2q13.3 microdeletions
| Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 | |
|---|---|---|---|---|---|
| Genetics | |||||
| Cohort | CHB | CHB | CHB | CHB | CHB |
| Referring Diagnosis | PDD-NOS | Language Delay | Autistic Spectrum | Mental Retardation | Developmental Language Disorder |
| Deletion size | 1.69 Mb | 1.93 Mb | 1.70 Mb | 1.50 Mb | 1.63 Mb |
| Coordinates (hg18) | 28,719,136–30,405,675 | 28,719,136–30,648,918 | 28,709,202–30,405,675 | 28,719,136–30,298,155 | 28,606,757–30,232,544 |
| Origin | Maternal | Maternal | Unknown (adopted) | Unknown (father not available) | Unknown (father not available) |
| LCR involved | BP4–BP5 | BP4–BP5 | BP4–BP5 | BP4–BP5 | BP4–BP5 |
| Age and Gender | 8y11m Male | 3y10m Female | 10y9m Male | 20y9m Male | 5y11m Male |
| Cognitive/Behavioral Development | |||||
| Developmental Delay | Yes | Yes | Yes | Yes | Yes |
| Mental Retardation | No | No | No | Yes | No |
| ASD | Yes, PDD-NOS | No | No | Yes, PDD-NOS | Autistic features |
| Attention Problems/ADHD | Yes | Yes | Yes, ADHD | Yes, ADHD | Yes |
| Testing | DAS Verbal= 97 (42nd %ile); Nonverbal = 69 (2nd %ile) | WPPSI-III FSIQ=91; | WISC-IV: Verbal 93 (32nd %ile); Working Memory 77 (6th %ile); FSIQ=86 (18th %ile) |
WASI FSIQ below 50 | WPPSI-III: Verbal 72 (3rd %ile); Perceptual Reasoning 75 (5th %ile); |
| Receptive Language | Generally age-appropriate | Severely impaired | Mildly impaired | Impaired (follows simple commands) | Below average |
| Expressive Language | Impaired, but more NVLD | Severely impaired | Mildly impaired | Severely impaired | Below average |
| Written Language | Impaired | N/A | Age appropriate | Impaired | Delayed |
| Repetitive behavior | Yes | No | No | Yes | Yes; hand flapping |
| Poor eye contact | Yes; variable | Yes | Yes | Yes | Yes; variable |
| Social interactions | Impaired | N/A | Impaired | Impaired | Impaired |
| Self-stimulatory behaviors | Yes | No | No | Yes | No |
| Self-injurious behaviors | Yes | Unknown | Unknown | Yes | No |
| Behavioral issues | Yes | Yes | Yes | Yes | Yes |
| Age at walking | 15m | 20m | 18m | 24m | 18m |
| History of Seizures | No | No | No | No | No |
| EEG | Abnormal | Normal | Normal | Mildly abnormal | Not done |
| MRI | Not done | Focal cortical dysplasia | Not done | Normal | Not done |
Bayley = Bayley Scales of Infant Development; BP = breakpoint; CHB = Children’s Hospital Boston; DAS = Differential Ability Scale; EEG = electroencephalogram; FSIQ = Full scale intelligence quotient; GARS = Gilliam Autism Rating Scale; LCR = low-copy repeat; MRI = magnetic resonance imaging; NVLD = non-verbal learning disability; PDD = pervasive developmental disorder (based on DSM-IV criteria); WASI = Wechsler Abbreviated Scale of Intelligence; WISC-IV = Wechsler Intelligence Scale for Children-IV; WPPSI-III = Wechsler Preschool and Primary Scale of Intelligence-III; N/A = not available.
Table 2.
15q13.2q13.3 microduplications
| Patient 6 | Patient 7 | Patient 8 | Patient 9 | Patient 10 | |
|---|---|---|---|---|---|
| Genetics | |||||
| Cohort | CHB | CHB | AGRE | AGRE | AGRE |
| Referring Diagnosis | Autism | Language Delay | Autism | Autism | Autism |
| Duplication size | 1.98 Mb | 1.58Mb | 1.93 Mb | 0.50 Mb | 0.50Mb |
| Coordinates (hg18) | 28,719,136–30,701,432 | 28,719,136–30,298,155 | 28,719,136–30,648,918 | 28,902,339–29,404,603 | 28,902,339–29,404,603 |
| Origin | de novo | Maternal | de novo | Maternal | Maternal |
| LCR involved | BP4–BP5 | BP4–BP5 | BP4–BP5 | Internal to BP4–BP5 | Internal to BP4–BP5 |
| Age and Gender | 20y Male | 2y10m Female | 8y Male | Male | Male |
| Cognitive/Behavioral Development | |||||
| Developmental Delay | Yes | Yes | Yes | N/A | N/A |
| Mental Retardation | Yes | No | Not tested | N/A | N/A |
| ASD | Yes | No | Yes, ADOS, ADI-R | Yes, ADI-R | Yes, ADI-R |
| Cognitive Testing | FSIQ below 50 | Not performed | N/A | N/A | N/A |
| Receptive Language | Severely impaired (follows simple commands | Borderline average | Severely Impaired | N/A | N/A |
| Expressive Language | Severely impaired (nonverbal) | Below average | Severely Impaired | N/A | N/A |
| Repetitive behavior | Yes | Yes | N/A | N/A | N/A |
| Poor eye contact | Yes; variable | Yes | N/A | N/A | N/A |
| Social interactions | Impaired | N/A | N/A | N/A | N/A |
| Self-stimulatory behaviors | Yes | No | N/A | N/A | N/A |
| Self-injurious behaviors | Yes | N/A | N/A | N/A | N/A |
| Behavioral issues | Yes | Yes | N/A | N/A | N/A |
| Age at Walking | 12m | 23m | N/A | N/A | N/A |
| History of Seizures | None | None | N/A | N/A | N/A |
| EEG | Normal | Not done | N/A | N/A | N/A |
| MRI | Normal | Not done | N/A | N/A | N/A |
ADOS = Autism Diagnostic Observational Schedule; ADI-R = Autism Diagnostic Interview-Revised; BP = breakpoint; EEG = electroencephalogram; FSIQ = Full scale intelligence quotient; LCR = low-copy repeat; MRI = magnetic resonance imaging; NVLD = non-verbal learning disability; PDD = pervasive developmental disorder (based on DSM-IV criteria); WISC = Wechsler Intelligence Scale for Children; WPPSI-III = Wechsler Preschool and Primary Scale of Intelligence-III; N/A = not available.
In general, cognitive performance of patients with 15q13.2q13.3 microdeletion/microduplication was variable. Test scores ranged from moderate MR to the normal range. Although some patients had full-scale IQ in the normal range, they all had some degree of language impairment and/or learning disability. Expressive language was consistently more delayed than receptive language. Many of these individuals showed capacity for ongoing improvement in academic and social skills. Neurobehavioral symptoms were very common in microdeletion/microduplication patients. All had difficulties with social interactions. Overall, dysmorphic features were mild (Fig. 2), and the neurobehavioral symptoms were the most significant cause of disability among these patients.
FIGURE 2.
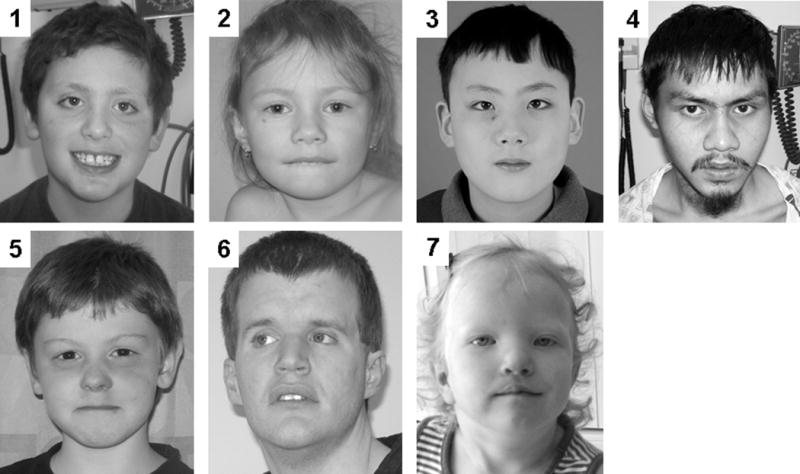
Photographs of five patients with 15q13.2q13.3 BP4–BP5 microdeletion, and two patients with BP4–BP5 microduplication, all from the CHB cohort (numbering corresponds to Patient Number in the text and Supplemental Data). We obtained written consent to publish photographs for each individual included in this figure.
15q13.2q13.3 Microdeletion Patients
Clinical features of the five individuals from the CHB cohort with 15q13.2q13.3 BP4–BP5 deletions are presented in Table 1 and Supplemental Data. All have subtle dysmorphology findings based on examination by a Clinical Geneticist. Cognitive testing was performed on all individuals. Patient 4 has mental retardation, but the other four patients have variable scores in the range of “below average” to “average”. Patients 1 and 3 showed significant nonverbal learning disability. None has a history of developmental regression. All have impaired language skills ranging from mild to severe accounting for a diagnosis of developmental delay in all cases. Those with developed language have significant early expressive language impairment but with better receptive abilities. Profiles commonly included developmental or oro-motor dyspraxia with disarticulation. Older children have good language abilities if they have developed expressive language.
Motor delays were not prominent, especially compared to cognitive and behavioral issues, but were observed among patients with BP4–BP5 deletion. Patients 1–4 had hypotonia that resolved over time, and Patient 5 was delayed in walking until age 18 months, suggesting the possibility of undiagnosed mild hypotonia. Patients 1 and 5 had problems with fine motor coordination. None of the patients with BP4–BP5 deletion had exam findings consistent with cerebral palsy.
The majority of individuals with BP4–BP5 deletion have a diagnosis of an autism spectrum disorder. Others have autistic features such as variably poor eye contact and other difficulties with social interactions. Beyond concerns about autistic features, many of these patients have other behavioral problems. All have some degree of difficulty with attention, hyperactivity, mood regulation, and impulsive behaviors. Patient 3 has ADHD, bipolar disorder, and anxiety disorder. None have problems sleeping at night.
15q13.2q13.3 Microduplication Patients
Clinical features of the five individuals from the CHB and AGRE cohorts with 15q13.2q13.3 BP4–BP5 duplications are presented in Table 2 and Supplemental Data. Four of five patients with the duplications have a diagnosis of autism. Patient 7 does not carry an autism diagnosis, but displays some repetitive behaviors and expressive language delay. Patients 6 and 8 also have severe expressive language delay, but language testing results were not available on Patients 9 and 10. Patient 6 has a history of anxiety spectrum disorder/obsessive-compulsive disorder in addition to autism. Cognitive and behavioral test results are not available for duplication patients from the AGRE cohort, although Patient 8’s Vineland Score suggests he would fall in the range of mental retardation. Clinical examination of Patients 6 and 7 (CHB cohort) did not suggest a consistent pattern of dysmorphology and neither had a history of seizures. Patients 8–10 (AGRE cohort) were not available for exam.
Patient 6 (CHB cohort) and Patient 8 (AGRE cohort) have a de novo duplication and an autism diagnosis. Patient 7 (CHB cohort) has a maternally inherited duplication from an apparently unaffected parent and has developmental delay with a cleft lip and palate. The paternal grandfather also carries the duplication and was apparently unaffected. Patients 9 and 10 (AGRE cohort) are siblings with autism and a smaller duplication nested within BP4–BP5 inherited from their apparently unaffected mother.
DISCUSSION
The recent recognition of genomic imbalance at chromosome 15q13.2q13.3 is certainly due to increasing use of whole genome high resolution array CGH in the evaluation of individuals with developmental delay, mental retardation, and autism spectrum disorder. Our results expand the phenotype of the 15q13.2q13.3 microdeletion/duplication syndrome to include clinically significant developmental delays and features of autism spectrum disorder in a majority of our patients with 15q13.2q13.3 microdeletion/duplication. A larger proportion of patients with duplication had a confirmed diagnosis of autism, while patients with deletion had either PDD-NOS, autistic features, or other neurobehavioral disabilities.
Among Patients 1–7 from the CHB cohort, and patient 8 from AGRE, all had some degree of developmental delay, particularly in expressive language, although not necessarily with mental retardation. These findings are not inconsistent, as verbal cognitive measures have minimal demands for language formulation. For example, they do not consider articulation, pragmatics, and reciprocity. Six of these patients (Patients 1, 4, 6, 8–10) have a clinical diagnosis on the autism spectrum, and another is considered to have autistic features (Patient 5).
Many of these patients also exhibited other neurobehavioral symptoms such as attentional problems, anxiety, mood instability, and impulsivity. Our patients had minor dysmorphic features previously described in this syndrome, but did not show the high prevalence of epilepsy/EEG abnormalities (7 of 9 patients) reported in the earlier study of Sharp et al (2008). In that study, patients were ascertained for mild or moderate mental retardation, but in our study cases were referred for a variety of diagnoses.
We found BP4–BP5 microdeletions in 4/950 (0.4%) and microduplications in 2/950 (0.2%) of CHB cases with developmental delay, mental retardation, or autism spectrum disorder, and 1/751 (0.13%) of AGRE index (proband) cases. Four families had a proband with maternally inherited imbalance; two involved a BP4–BP5 deletion from a mother with learning issues (Patients 1 and 2) and two had duplications also present in apparently unaffected mothers (Patients 7; Patients/siblings 9 and 10). Two BP4–BP5 duplication cases were de novo (Patients 6, 8). BP4–BP5 duplication was also reported in 1/960 (0.1%) European-American controls,21 and an overlapping 2.3Mb duplication (chr15:28,393,128–30,740,356) was reported in 2/776 (0.3%) controls (506 unrelated healthy individuals from Northern Germany, 270 HapMap individuals).29
Based on this sample, the BP4–BP5 microdeletion appears to be fully penetrant with variable expressivity, while the microduplication may not be fully penetrant. All patients with BP4–BP5 deletion showed clinically significant symptoms, including the parents of Patients 1 and 2. Sharp et al. (2008) found no BP4–BP5 deletions among 2,962 unaffected control subjects, and the combined experience from both studies indicates that 14/14 subjects with BP4–BP5 deletion exhibited significant developmental disability. Parent-of-origin effects could account for variable penetrance and expressivity of both deletions and duplications, but we were not able to test this in our cohort. There is no evidence of any parent-of-origin effect based on the prior report.21 The importance of parent-of-origin has been well established for the 15q11 locus.30 By comparison, duplications and other rearrangements in the 15q11–q13 region can result in an autism and mental retardation phenotype, but are also observed in phenotypically normal individuals,31 underscoring the phenotypic variability common in many genomic imbalance disorders such as microduplications at 1q41q42,32 3q29,33 16p11.2,8 and 22q11.34
Further research efforts to understand this variable phenotype should focus on additional imbalance events and the genes located in this region. CHRNA7 is a candidate gene for epilepsy, but is also a candidate for the broader phenotype of neuropsychiatric and neurological disease based on genetic association with schizophrenia,35–39 and bipolar disorder,40 as well as biological studies that support a role for CHRNA7 in neuropsychiatric disease.41 Negative association studies with schizophrenia have also been reported,42, 43 and the literature is likely biased toward more reports of positive association. The 15q13.2q13.3 deletion and duplication events described here show relatively consistent breakpoints at BP4 and BP5, but the smaller duplication suggests that further study may reveal a smaller deletion event that could identify a smallest region of overlap (SRO) that includes CHRNA7.
Awareness of the broad neurobehavioral phenotype will alert clinicians to consider testing for a chromosome 15q13.2q13.3 imbalance on the basis of developmental delay, autistic features, or the other neuropsychiatric issues such as expressive language delay, attention deficit-hyperactivity disorder, anxiety disorders and/or obsessive-compulsive disorder, bipolar or mood disorder, and subclinical EEG or MRI abnormalities. Many of these conditions are known to be common comorbid psychiatric disorders in individuals with autism,44–46 and vice versa.47 The subtlety of physical exam findings underscores the importance of clinical diagnostic tests that include this locus to facilitate an earlier diagnosis and more accurate recurrence risk counseling. In most cases of autism spectrum disorder, some clinical symptoms are apparent before age three, but the current average age at clinical diagnosis is 5 years.48 Clinical genetic testing that leads to early behavioral interventions could significantly improve the developmental outcome for individuals with a chromosome 15q13.2q13.3 imbalance.
Acknowledgments
We would like to thank the families and individuals cited in this work, and also the families from AGRE who agreed to share their information and samples for this research. For helpful discussion: Drs. Orah Platt and Leonard Rappaport from Children’s Hospital Boston. For assistance with clinical evaluations: Drs. Mustafa Sahin and Susan Waisbren from Children’s Hospital Boston and Dr. Eileen Bickford from Salud Family Health Center in Longmont, CO. For technical support of aCGH and MLPA: Va Lip, Xiaoming Sheng, Hong Fang, Hong Shao from Children’s Hospital Boston. For FISH confirmation of clinical cases: Drs. Christa Lese-Martin and David Ledbetter from Emory University; Dr. Arthur R. Brothman and Ms. Emily Aston from University of Utah. For assistance with IRB documentation: Ms. Stephanie Brewster and Dr. Ingrid Holm from Children’s Hospital Boston. We gratefully acknowledge the resources provided by the Autism Genetic Resource Exchange (AGRE) Consortium and the participating AGRE families. The Autism Genetic Resource Exchange is a program of Autism Speaks and is supported, in part, by grant 1U24MH081810 from the National Institute of Mental Health to Clara M. Lajonchere (PI). We also thank the Autism Consortium for their support and enthusiasm.
We acknowledge the following sources of financial support: Young Investigator Award from the Children’s Tumor Foundation (to Y.S.). Ruth L. Kirschstein National Research Service Award (to L.A.W.); National Institutes of Health grant P01-GM061354, Autism Speaks, and NARSAD Distinguished Investigator Award (to J.F.G.), Harvard Scholars in Clinical Science Program K30 grant #RRO22292-07 (to J.T.), grants from the Autism Consortium and the Ellison Foundation (to M.J.D.), and grants from the NIH-CETT Program and the MDA Foundation (to B.L.W.).
Footnotes
DECLARATION
The authors have obtained written consent from all 7 patients and/or his/her legal guardian for publication of their images in print and online with Journal of Medical Genetics.
The Corresponding Author has the right to grant on behalf of all authors and does grant on behalf of all authors, an exclusive licence (or non-exclusive for government employees) on a worldwide basis to the BMJ Publishing Group Ltd and its Licensees to permit this article to be published in Journal of Medical Genetics editions and any other BMJPGL products to exploit all subsidiary rights, as set out in our licence (http://JMG.bmjjournals.com/misc/ifora/licence.pdf).
References
- 1.Shaw-Smith C, Redon R, Rickman L, et al. Microarray based comparative genomic hybridisation (array-CGH) detects submicroscopic chromosomal deletions and duplications in patients with learning disability/mental retardation and dysmorphic features. J Med Genet. 2004;41(4):241–8. doi: 10.1136/jmg.2003.017731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schoumans J, Ruivenkamp C, Holmberg E, et al. Detection of chromosomal imbalances in children with idiopathic mental retardation by array based comparative genomic hybridisation (array-CGH) J Med Genet. 2005;42(9):699–705. doi: 10.1136/jmg.2004.029637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lupski JR. Genomic disorders: structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet. 1998;14(10):417–22. doi: 10.1016/s0168-9525(98)01555-8. [DOI] [PubMed] [Google Scholar]
- 4.Koolen DA, Vissers LE, Pfundt R, et al. A new chromosome 17q21.31 microdeletion syndrome associated with a common inversion polymorphism. Nat Genet. 2006;38(9):999–1001. doi: 10.1038/ng1853. [DOI] [PubMed] [Google Scholar]
- 5.Sharp AJ, Hansen S, Selzer RR, et al. Discovery of previously unidentified genomic disorders from the duplication architecture of the human genome. Nat Genet. 2006;38(9):1038–42. doi: 10.1038/ng1862. [DOI] [PubMed] [Google Scholar]
- 6.Shaw-Smith C, Pittman AM, Willatt L, et al. Microdeletion encompassing MAPT at chromosome 17q21.3 is associated with developmental delay and learning disability. Nat Genet. 2006;38(9):1032–7. doi: 10.1038/ng1858. [DOI] [PubMed] [Google Scholar]
- 7.Kumar RA, KaraMohamed S, Sudi J, et al. Recurrent 16p11.2 microdeletions in autism. Hum Mol Genet. 2008;17(4):628–38. doi: 10.1093/hmg/ddm376. [DOI] [PubMed] [Google Scholar]
- 8.Weiss LA, Shen Y, Korn JM, et al. Association between Microdeletion and Microduplication at 16p11.2 and Autism. N Engl J Med. 2008;358(7):667–75. doi: 10.1056/NEJMoa075974. [DOI] [PubMed] [Google Scholar]
- 9.Marshall CR, Noor A, Vincent JB, et al. Structural variation of chromosomes in autism spectrum disorder. Am J Hum Genet. 2008;82(2):477–88. doi: 10.1016/j.ajhg.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Murdock RL, Wurster-Hill DH. Non-reciprocal translocation (5;15), isodicentric (15) and Prader-Willi syndrome. Am J Med Genet. 1986;25(1):61–9. doi: 10.1002/ajmg.1320250108. [DOI] [PubMed] [Google Scholar]
- 11.Roberts SE, Maggouta F, Thomas NS, et al. Molecular and fluorescence in situ hybridization characterization of the breakpoints in 46 large supernumerary marker 15 chromosomes reveals an unexpected level of complexity. Am J Hum Genet. 2003;73(5):1061–72. doi: 10.1086/379155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sutcliffe JS, Han MK, Amin T, et al. Partial duplication of the APBA2 gene in chromosome 15q13 corresponds to duplicon structures. BMC Genomics. 2003;4(1):15. doi: 10.1186/1471-2164-4-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Klein OD, Cotter PD, Albertson DG, et al. Prader-Willi syndrome resulting from an unbalanced translocation: characterization by array comparative genomic hybridization. Clin Genet. 2004;65(6):477–82. doi: 10.1111/j.0009-9163.2004.00261.x. [DOI] [PubMed] [Google Scholar]
- 14.Sahoo T, Shaw CA, Young AS, et al. Array-based comparative genomic hybridization analysis of recurrent chromosome 15q rearrangements. Am J Med Genet A. 2005;139(2):106–13. doi: 10.1002/ajmg.a.31000. [DOI] [PubMed] [Google Scholar]
- 15.Knoll JH, Nicholls RD, Magenis RE, et al. Angelman syndrome: three molecular classes identified with chromosome 15q11q13-specific DNA markers. Am J Hum Genet. 1990;47(1):149–55. [PMC free article] [PubMed] [Google Scholar]
- 16.Christian SL, Fantes JA, Mewborn SK, et al. Large genomic duplicons map to sites of instability in the Prader-Willi/Angelman syndrome chromosome region (15q11–q13) Hum Mol Genet. 1999;8(6):1025–37. doi: 10.1093/hmg/8.6.1025. [DOI] [PubMed] [Google Scholar]
- 17.Bundey S, Hardy C, Vickers S, et al. Duplication of the 15q11–13 region in a patient with autism, epilepsy and ataxia. Dev Med Child Neurol. 1994;36(8):736–42. doi: 10.1111/j.1469-8749.1994.tb11916.x. [DOI] [PubMed] [Google Scholar]
- 18.Browne CE, Dennis NR, Maher E, et al. Inherited interstitial duplications of proximal 15q: genotype-phenotype correlations. Am J Hum Genet. 1997;61(6):1342–52. doi: 10.1086/301624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Freitag CM. The genetics of autistic disorders and its clinical relevance: a review of the literature. Mol Psychiatry. 2007;12(1):2–22. doi: 10.1038/sj.mp.4001896. [DOI] [PubMed] [Google Scholar]
- 20.Sebat J, Lakshmi B, Malhotra D, et al. Strong association of de novo copy number mutations with autism. Science. 2007;316(5823):445–9. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sharp AJ, Mefford HC, Li K, et al. A recurrent 15q13.3 microdeletion syndrome associated with mental retardation and seizures. Nat Genet. 2008;40(3):322–8. doi: 10.1038/ng.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Elmslie FV, Rees M, Williamson MP, et al. Genetic mapping of a major susceptibility locus for juvenile myoclonic epilepsy on chromosome 15q. Hum Mol Genet. 1997;6(8):1329–34. doi: 10.1093/hmg/6.8.1329. [DOI] [PubMed] [Google Scholar]
- 23.Neubauer BA, Fiedler B, Himmelein B, et al. Centrotemporal spikes in families with rolandic epilepsy: linkage to chromosome 15q14. Neurology. 1998;51(6):1608–12. doi: 10.1212/wnl.51.6.1608. [DOI] [PubMed] [Google Scholar]
- 24.Orr-Urtreger A, Goldner FM, Saeki M, et al. Mice deficient in the alpha7 neuronal nicotinic acetylcholine receptor lack alpha-bungarotoxin binding sites and hippocampal fast nicotinic currents. J Neurosci. 1997;17(23):9165–71. doi: 10.1523/JNEUROSCI.17-23-09165.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Taske NL, Williamson MP, Makoff A, et al. Evaluation of the positional candidate gene CHRNA7 at the juvenile myoclonic epilepsy locus (EJM2) on chromosome 15q13–14. Epilepsy Res. 2002;49(2):157–72. doi: 10.1016/s0920-1211(02)00027-x. [DOI] [PubMed] [Google Scholar]
- 26.Shen Y, Miller DT, Cheung SW, et al. Development of a Focused Oligonucleotide-Array Comparative Genomic Hybridization Chip for Clinical Diagnosis of Genomic Imbalance. Clin Chem. 2007;53(12):2051–9. doi: 10.1373/clinchem.2007.090290. [DOI] [PubMed] [Google Scholar]
- 27.Geschwind DH, Sowinski J, Lord C, et al. The autism genetic resource exchange: a resource for the study of autism and related neuropsychiatric conditions. Am J Hum Genet. 2001;69(2):463–6. doi: 10.1086/321292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Korn JM, Kuruvilla FG, McCarroll SA. Integrated genotype calling and association analysis of SNPs, common copy number polymorphisms, and rare CNVs. 2008 doi: 10.1038/ng.237. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pinto D, Marshall C, Feuk L, et al. Copy-number variation in control population cohorts. Hum Mol Genet. 2007;16(Spec No. 2):R168–73. doi: 10.1093/hmg/ddm241. [DOI] [PubMed] [Google Scholar]
- 30.Maraschio P, Zuffardi O, Bernardi F, et al. Preferential maternal derivation in inv dup(15): analysis of eight new cases. Hum Genet. 1981;57(4):345–50. doi: 10.1007/BF00281681. [DOI] [PubMed] [Google Scholar]
- 31.Goossens E, Decock P, Potgieter S, et al. Mosaic normal/15q11–q13 duplication associated with developmental delay but normal phenotype. Genet Couns. 1999;10(2):133–6. [PubMed] [Google Scholar]
- 32.Shaffer LG, Theisen A, Bejjani BA, et al. The discovery of microdeletion syndromes in the post-genomic era: review of the methodology and characterization of a new 1q41q42 microdeletion syndrome. Genet Med. 2007;9(9):607–16. doi: 10.1097/gim.0b013e3181484b49. [DOI] [PubMed] [Google Scholar]
- 33.Willatt L, Cox J, Barber J, et al. 3q29 microdeletion syndrome: clinical and molecular characterization of a new syndrome. Am J Hum Genet. 2005;77(1):154–60. doi: 10.1086/431653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Derbent M, Bikmaz YE, Yilmaz Z, et al. Variable phenotype and associations in chromosome 22q11.2 microdeletion. Am J Med Genet A. 2006;140(6):659–60. doi: 10.1002/ajmg.a.31120. [DOI] [PubMed] [Google Scholar]
- 35.Freedman R, Leonard S, Gault JM, et al. Linkage disequilibrium for schizophrenia at the chromosome 15q13–14 locus of the alpha7-nicotinic acetylcholine receptor subunit gene (CHRNA7) Am J Med Genet. 2001;105(1):20–2. [PubMed] [Google Scholar]
- 36.Fan JB, Ma J, Li XW, et al. Population-based and family-based association studies of an (AC)n dinucleotide repeat in alpha-7 nicotinic receptor subunit gene and schizophrenia. Schizophr Res. 2006;84(2–3):222–7. doi: 10.1016/j.schres.2006.02.012. [DOI] [PubMed] [Google Scholar]
- 37.Flomen RH, Collier DA, Osborne S, et al. Association study of CHRFAM7A copy number and 2 bp deletion polymorphisms with schizophrenia and bipolar affective disorder. Am J Med Genet B Neuropsychiatr Genet. 2006;141(6):571–5. doi: 10.1002/ajmg.b.30306. [DOI] [PubMed] [Google Scholar]
- 38.Gault J, Hopkins J, Berger R, et al. Comparison of polymorphisms in the alpha7 nicotinic receptor gene and its partial duplication in schizophrenic and control subjects. Am J Med Genet B Neuropsychiatr Genet. 2003;123(1):39–49. doi: 10.1002/ajmg.b.20061. [DOI] [PubMed] [Google Scholar]
- 39.Xu J, Pato MT, Torre CD, et al. Evidence for linkage disequilibrium between the alpha 7-nicotinic receptor gene (CHRNA7) locus and schizophrenia in Azorean families. Am J Med Genet. 2001;105(8):669–74. doi: 10.1002/ajmg.1549. [DOI] [PubMed] [Google Scholar]
- 40.Hong CJ, Lai IC, Liou LL, et al. Association study of the human partially duplicated alpha7 nicotinic acetylcholine receptor genetic variant with bipolar disorder. Neurosci Lett. 2004;355(1–2):69–72. doi: 10.1016/j.neulet.2003.10.043. [DOI] [PubMed] [Google Scholar]
- 41.De Luca V, Likhodi O, Van Tol HH, et al. Regulation of alpha7-nicotinic receptor subunit and alpha7-like gene expression in the prefrontal cortex of patients with bipolar disorder and schizophrenia. Acta Psychiatr Scand. 2006;114(3):211–5. doi: 10.1111/j.1600-0447.2006.00785.x. [DOI] [PubMed] [Google Scholar]
- 42.Ma J, Fan JB, Wu SN, et al. [Association study of an (AC)n dinucleotide repeat and schizophrenia in Asian and European populations] Yi Chuan. 2007;29(10):1207–13. doi: 10.1360/yc-007-1207. [DOI] [PubMed] [Google Scholar]
- 43.Meyer J, Ortega G, Schraut K, et al. Exclusion of the neuronal nicotinic acetylcholine receptor alpha7 subunit gene as a candidate for catatonic schizophrenia in a large family supporting the chromosome 15q13–22 locus. Mol Psychiatry. 2002;7(2):220–3. doi: 10.1038/sj.mp.4000970. [DOI] [PubMed] [Google Scholar]
- 44.Stahlberg O, Soderstrom H, Rastam M, et al. Bipolar disorder, schizophrenia, and other psychotic disorders in adults with childhood onset AD/HD and/or autism spectrum disorders. J Neural Transm. 2004;111(7):891–902. doi: 10.1007/s00702-004-0115-1. [DOI] [PubMed] [Google Scholar]
- 45.Cath DC, Ran N, Smit JH, et al. Symptom overlap between autism spectrum disorder, generalized social anxiety disorder and obsessive-compulsive disorder in adults: a preliminary case-controlled study. Psychopathology. 2008;41(2):101–10. doi: 10.1159/000111555. [DOI] [PubMed] [Google Scholar]
- 46.Levisohn PM. The autism-epilepsy connection. Epilepsia. 2007;48(Suppl 9):33–5. doi: 10.1111/j.1528-1167.2007.01399.x. [DOI] [PubMed] [Google Scholar]
- 47.Ivarsson T, Melin K. Autism spectrum traits in children and adolescents with obsessive-compulsive disorder (OCD) J Anxiety Disord. 2007 doi: 10.1016/j.janxdis.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 48.Wiggins LD, Baio J, Rice C. Examination of the time between first evaluation and first autism spectrum diagnosis in a population-based sample. J Dev Behav Pediatr. 2006;27(2 Suppl):S79–87. doi: 10.1097/00004703-200604002-00005. [DOI] [PubMed] [Google Scholar]