

Abstract
Complex coacervation is a phenomenon characterized by the association of oppositely charged polyelectrolytes into micron-scale liquid condensates. This process is the purported first step in the formation of underwater adhesives by sessile marine organisms, as well as the process harnessed for the formation of new synthetic and protein-based contemporary materials. Efforts to understand the physical nature of complex coacervates are important for developing robust adhesives, injectable materials, or novel drug delivery vehicles for biomedical applications, however their internal fluidity necessitates the use of in situ characterization strategies of their local dynamic properties—capabilities not offered by conventional techniques such as x-ray scattering, microscopy, or bulk rheological measurements. Herein, we employ the novel magnetic resonance technique Overhauser dynamic nuclear polarization enhanced nuclear magnetic resonance (DNP), together with electron paramagnetic resonance (EPR) lineshape analysis, to concurrently quantify local molecular and hydration dynamics, with species- and site-specificity. We observe striking differences in the structure and dynamics of the protein-based biomimetic complex coacervates from their synthetic analogues, which is an asymmetric collapse of the polyelectrolyte constituents. From this study we suggest charge heterogeneity within a given polyelectrolyte chain to be an important parameter by which the internal structure of complex coacervates may be tuned. Acquiring molecular-level insight to the internal structure and dynamics of dynamic polymer complexes in water through the in situ characterization of site- and species-specific local polymer and hydration dynamics should be a promising general approach that has not been widely employed for materials characterization.
Keywords: Complex coacervates, DNP, EPR, Mytilus Foot Protein, Mussel adhesive protein
INTRODUCTION
The development of aqueous adhesive materials is of tremendous importance due to the biocompatibility, biodegradability, and versatility of these systems.1,2 Such features provide aqueous adhesives with abundant possibilities for biomedical applications.3 Recent efforts in the evolution of such materials have been motivated by nature, where the sessile marine organisms, mussels (Mytilus species) and sandcastle worms (Phragmatopoma californica), demonstrate strong underwater adhesion.4–6 In such organisms, the process of complex coacervation is suggested to be a critical step in the formation of protein-based adhesives that efficiently coat surfaces underwater and harden into plaques that are robust enough to uphold strong currents in oceanic tidal zones.
The adhesion mechanism of the mussel and sandcastle worm involves the expulsion of a multi-component solution of adhesive proteins from their secretory glands at a solid/liquid interface.4 The environment around the protein solution after expulsion induces the formation of complex coacervates. This mechanism allows for high concentrations of protein to be sequestered, precluding their dissolution into surrounding water, and enabling the internal proteins to then spread at the interface.
Complex coacervates form spontaneously upon association of oppositely charged polyelectrolytes (including polypeptides), and exist as micron-scale, dense, liquid spheres suspended in solution. Biological complex coacervates are believed to exhibit interesting physical properties that are key to their function as intermediates in adhesion, such as high internal fluidity, low interfacial tension, and high protein density,7–9 but the origin and the chemical features that influence these properties are still debated.
Efforts to analyze the physical nature of complex coacervates at the molecular level, and to ultimately gain control of their adhesion processes, have proven challenging due to their high fluidity, which negates the utility of typical characterization techniques such as x-ray scattering and microscopy. Most suitable for these dynamic and hydrated systems would be the characterization of their internal motion on sub-nanometer length scales, a traditionally impossible endeavor that can now be accomplished by exploiting recent developments in magnetic resonance technologies.10–13
Besides the limitation of existing techniques in analyzing local dynamic properties of dilute and heterogeneous solution systems in situ, investigations into protein-based complex coacervates have also been limited by the typically low yield obtained in native protein purification (less than 1mg for each purification). Instead, much of our understanding of the physics of complex coacervates comes from experimental investigations of polyelectrolytes that are not derived from adhesive proteins.14,15
The identification of several protein constituents has been achieved in the case of mussel adhesion (mussel foot proteins, mfps). Among the cataloged mfp proteins are two that have high propensity for adhesive and cohesive behavior, these are mfp1 and mfp5, respectively.16,17 Due to difficulties in purification of the adhesive proteins from these organisms, recombinant mussel foot proteins have been used to make the bio-mimetic complex coacervates.18,19 Among the recombinant mussel foot protein-based coacervates, those formed with mfp151 (a high yield recombinant hybrid protein of mfp1 and mfp5 with a terminal RGD peptide sequence)20 show intriguing properties, including five times higher preosteoblast cell growth on coacervate-coated titanium surfaces than bare titanium,21 and facile switching of the rheological properties between shear thinning and shear thickening, an effect that is modulated by tuning the mixing ratio of positively versus negatively charged constituents.22
The internal polymer and hydration dynamics within complex coacervates formed between synthetic polyelectrolytes has been demonstrated,15 but not in biologically relevant complex coacervates between protein- and carbohydrate-based polyelectrolytes. Synthetic complex coacervates offer the potential to scale up aqueous adhesive systems for technological applications, but it is unknown whether their internal dynamics compare to those of protein-based complex coacervates, and what the molecular basis is for their unique physical properties.
In a synthetic complex coacervate system made of the linear polyelectrolytes, poly(aspartic acid) and poly(vinylimidazole), that present homogenous charge distribution, both polyelectrolyte constituents were found to collapse symmetrically into condensed, dynamically restricted, states. Specifically, the internal water dynamics of the surface hydration layer around both polyelectrolyte components is retarded by ~ 5 fold when coacervated. This symmetric behavior likely reflects the homogenous and linear charge densities of each polyelectrolyte constituent, where pseudo-pairwise association may be expected. However, such linear charge homogeneities are not observed in nature, where protein-polycations are rather composed of amino acid residues with varying charges and hydropathies, and are thus prone to folding, driven by a wide range of electrostatic, hydrogen bonding, and hydrophobic intermolecular interactions.16,17,23
Herein, we examine the effects of the charge density distribution of the polycations in biomimetic versus synthetic complex coacervates to illustrate the differences in their dynamic structure. We use the cationic hybrid protein, mfp151, coacervated with the anionic polyelectrolyte, hyaluronic acid (HA), which is a biological polyelectrolyte ubiquitous in the extracellular matrices of most living organisms. We access polyelectrolyte segment dynamics and hydration water dynamics within 5–15 Å of the polyelectrolyte surface of HA and mfp151, by carrying out nitroxide radical spin labeling, and employing the magnetic resonance techniques electron paramagnetic resonance spectroscopy (EPR) and Overhauser dynamic nuclear polarization (DNP) enhanced NMR spectroscopy.
In addition to examining the behavior of each, mfp151 and HA, before versus after complex coacervation, we probe the dynamics at specific sites of another related decapeptide, recombinant mfp1, by examining complex coacervates formed with HA. To achieve site-specific probing along the mfp1 sequence, this mfp1 decapeptide was selectively spin labeled at three specific single-mutation cysteine residues, at the N terminus, the C terminus, and in the middle of the protein chain. Finally, poly(aspartic acid) (PAsp) was used as an alternative polyanion to HA, to test the generality of the structural and dynamic properties observed from within the mfp151/HA complex coacervates.
EXPERIMENTAL SECTION
Protein and polyelectrolyte preparation
Recombinant mfp151 was kindly donated from the group of Prof. Cha (POSTECH). Mfp1 and their mutants were prepared as previous described.24 Hyaluronic acid (35 kDa) was purchased from Lifecore Biomedical. For magnetic resonance studies, nitroxide radical spin labels were incorporated into each polyelectrolyte. Spin labeling of mfp151 was carried out by addition of MTSL (S-(2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-3-yl)methyl methanesulfonothioate) in 2-fold excess, resulting in the quantitative functionalization of mfp151 at its single cysteine residue. Recombinant mfp1s with a single cysteine at either of three sites, at its N terminus, C terminus, or in the middle of the protein were prepared with a site-directed mutagenesis kit (Strategene, CA, USA). Each mfp1 was functionalized with MTSL in an analogous way to the spin labeling of mfp151. HA was labeled by the EDC (1-ethyl-3-(3-dimethylaminopropyl) carbodiimide) -mediated coupling of amino-TEMPO to HA’s carboxylate functionalities. HA was spin labeled at < 10% by repeat unit, and was controlled stoichiometrically. Poly(aspartic acid) (PAsp) was synthesized according to a previously reported procedure,15 and spin labeled at < 10% per repeat unit.
Preparation of complex coacervates
Complex coacervates of proteins and HA, and of the spin labeled counterparts of each, were prepared by addition of HA into protein solution at varying molar ratios. Each solution was prepared by dissolving the polyelectrolyte in sodium acetate buffer (10 mM and pH 5), and the protein concentration was confirmed spectroscopically using Beer’s law, with the mfp151 molar absorption coefficient of ɛ = 9.95 × 103 M−1 cm−1 at 290 nm. The overall protein concentration was kept constant, and the extent of coacervation was determined by examining the amplitude of turbidity at 600 nm upon addition of HA, around the stoichiometric charge ratio. The coacervation optimum of mfp151/HA occurs at 60 mass % mfp151. The coacervation optimum of mfp1/HA, in contrast, was found to be at 75 mass % mfp1.
EPR
Cw EPR spectra were measured on a Bruker EMX X-band spectrometer equipped with a cylindrical (TE011) cavity. Samples were irradiated at 9.8 GHz with the center field set at 3400 G and sweep width of 200 G. The field modulation amplitude was kept below 0.2 times the center peak linewidth to acquire the intrinsic EPR lineshapes and amplitudes. N2 gas was streamed through the cavity at 14 L min−1, and all spectra were acquired at 298 K. Sample volumes of 4 μL were used, and the total spin label concentrations were kept at ~ 500 μM. Samples were analyzed in PTFE capillaries, rather than quartz, to minimize spontaneous spreading of the coacervates at the sample-wall interface. The PTFE capillaries were sealed at each end with wax.
EPR lineshape analysis
Cw EPR spectra were fit using the microscopic order macroscopic disorder (MOMD) model implemented by Freed et al. in their non-linear least squares stochastic Liouville (NLSL) program.25 With the rbar values obtained from each fit, the rotational diffusion rates, kr, were calculated as kr = 10rbar. Fits were performed with initial fitting parameters both above and below the final values to ensure convergence to global minima. Final NLSL fits all exhibited > 99.0 % correlation to the experimental EPR spectral lineshape.
DNP-enhanced NMR
The local water diffusion coefficient within 5–15 Å of nitroxide radical-based spin labels were measured by Overhauser DNP-enhanced NMR spectroscopy. The same samples and instrument were used as described for X-band EPR experiments. The magnetic field and frequency for irradiation were set to the center resonance of the nitroxide EPR spectra. Samples were positioned in a home-built U-shaped NMR coil (Cu wire, 28 AWG) tuned to 14.8 MHz and connected to a broadband channel of a Bruker Avance NMR spectrometer, as described in detail elsewhere.10,26 T1 relaxation of the 1H of water, in the absence and presence of the nitroxide spin labels functionalized to the polymers, was carefully measured using an inversion recovery pulse sequence in order to determine the leakage factor. The 1H NMR signal enhancement of water was recorded as a function of increasing microwave power that was varied using a home-built X-band microwave amplifier with a power output between 0.1 mW and 1.5 W. The 1H NMR spectra obtained were integrated over their absorption peak and the absolute values were plotted versus input microwave power. From the 1H NMR signal enhancement extrapolated to infinitely high microwave power, together with the determined leakage factor, the key DNP parameter termed the coupling factor was computed, which corresponds to the dipolar electron-1H cross relaxation efficiency with respect to all 1H relaxation processes. From the DNP coupling factor, using the appropriate molecular dynamics model and spectral densities, the dipolar correlation time for translational diffusion dynamics of the local water in the dipolar coupling vicinity of the spin label, was determined. From the dipolar correlation time, τ, the water diffusion rate, D was determined from the relation, τ = (d2/D), where d = 3.0 Å represents the distance of closest approach of the water to the radical electron spin label.13,26,27 Error bars on the DNP measurements are approximated to fall between 3–5%, as estimated from the quality of Emax and T1 fitting curves.
RESULTS
The spin-labeling procedures used for mfp151 and HA are shown in Figures 1a and 1b, respectively. Positively charged amino acids at pH 5.0 (R, H, and K) and negatively charged amino acids (D and E) in mfp1 are heterogeneously distributed on its primary sequence, and yield an overall net positive charge, whereas the negative charges of the glucuronic acid groups are homogeneously distributed across the HA backbone. When mfp151 and HA are mixed at a specific ratio, the turbidity sharply increases where the coacervation conditions are met (Figure 1c), indicating the formation of complex coacervates. The validity of the formation of liquid complex coacervates, as opposed to solid precipitates, is confirmed by light microscopy that displays perfectly spherical droplets suspended in solution (Figure 1d). The turbidity measurements show that the mass fraction of mfp151 with respect to HA, at which the maximal extent of complex coacervation occurs, is 0.6. When spin labels are incorporated into each component, the optimal coacervation ratio is unchanged, indicating that the spin labels do not measurably affect the coacervation process or condition.
Figure 1.
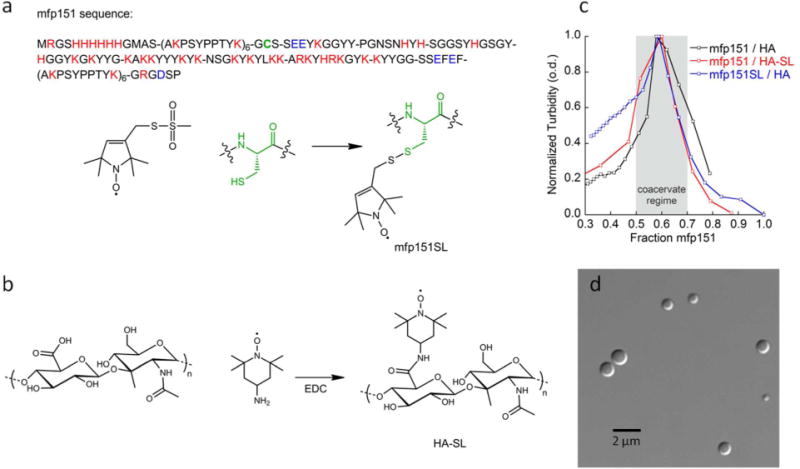
(a) mfp151 sequence, where mfp1 segments (AKPSYPPTYK)6 flank the mfp5 mid-segment. Cationic residues are indicated by red and anionic residues are indicated by blue. The single cysteine is indicated in green. Mfp151 is spin labeled (mfp151SL) by covalent functionalization of the single cysteine by the spin label MTSL; (b) Hyaluronic acid (35 kDa) is spin labeled (HA-SL) by addition of 4-amino-TEMPO in the presence of EDC; (c) Turbidity of mfp151 / HA shows maximum coacervation occurs at 60 % mfp151 by mass; (d) Differential interference contrast (DIC) micrograph of mfp151 / HA complex coacervate suspension in water.
Continuous wave (cw) EPR line shape analysis was used to monitor the dynamics of each spin-labeled polyelectrolyte in aqueous solution, and also of the complex coacervate suspension formed by mixing oppositely charged polyelectrolytes at specific ratios. Representative cw EPR spectra are shown in Figure 2. The three peaks in the EPR spectra are characteristic of nitroxide radicals where the nuclear magnetic moment of the nitroxide 14N nucleus (ml = 1) induces hyperfine splitting of each electron spin state of the nitroxide probe (ms = ½) in an external magnetic field, resulting in three allowed transitions and three EPR absorption peaks. Due to the large anisotropy of the g (electron spin Zeeman) and A (hyperfine) tensors of the nitroxide probe in an external magnetic field, spectral broadening is observed when the molecular motion of the nitroxide is restricted.25 In Figure 2a, mfp151SL exhibits a narrow EPR spectrum, consistent with the fast motion characteristic of a solubilized protein, and especially of a protein like mfp151 that is expected to be largely unstructured and extended in water. Upon complex coacervation by addition of HA, the EPR spectrum of spin labeled mfp151 (mfp151SL) broadens, congruous with a less dynamic state. In contrast to the behavior of mfp151SL, Figure 2b shows that the spectrum of spin labeled HA (HA-SL) is narrow, and indicates undistinguishable fast motion, both in solution and after coacervation with mfp151. This difference in dynamics between mfp151 and HA in complex coacervation suggests asymmetric behavior between the constituent polyelectrolytes in complex coacervates, which is in stark contrast to what was observed in fully synthetic complex coacervates with homogenous charge density.15
Figure 2.
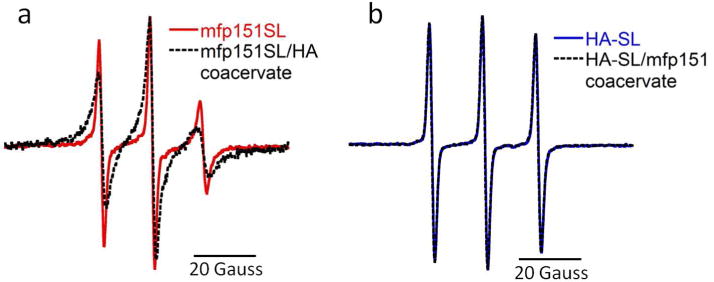
(a) EPR of mfp151SL alone (red) and after coacervation with HA (black); (b) EPR of HA-SL alone (blue) and after coacervation with mfp151 (black).
Rotational diffusion rates, describing the averaged motion of the spin label, were determined from the cw EPR spectra via the NLSL program25 of each polyelectrolyte alone and after complex coacervation (Figure 2). The spin label’s rotational diffusion rates reflect the time-averaged orientation of the spin label, and are correlated to polyelectrolyte flexibility, inter-chain interactions, and packing. Thus, at the surface of a freely solvated polyelectrolyte, the spin label and the polyelectrolyte dynamics contribute to one average rate constant.
These average rotational diffusion rates (kr), extracted from the fitting parameters to describe the EPR line shape, are presented in Table 1. Here, kr of mfp151SL alone is 383 MHz, a value on the order of that of freely dissolved polyelectrolytes, while after complex coacervation with HA, a slow component of 69 MHz emerges and constitutes 79 % of the spin label’s motion on mfp151SL. The observation of this slow component confirms that, upon complex coacervation, the dominant population of mfp151 is in a motionally more restricted state. Similar to dissolved mfp151SL alone, HA-SL by itself in solution exhibits high mobility (kr = 409 MHz). However, in contrast to mfp151, HA-SL maintains this high rotational diffusion rate of 409 MHz upon complex coacervation, indicating that neither intramolecular collapse nor dynamic restriction due to stronger inter-polyelectrolyte packing of the HA polymer chains occurs.
Table 1.
Rotational diffusion rates determined by EPR spectral simulations of mfp151 and HA with each component spin labeled, alone and after complex coacervation.
| kr (MHz) | kr (MHz) | ||
|---|---|---|---|
|
|
|
||
| mfp151SL | 383 | HA-SL | 409 |
|
|
|
||
| mfp151SL/HA Coacervated[a] |
69 (79%) 383 (21%) |
mfp151/HA-SL Coacervated |
409 |
|
|
|
||
EPR spectral simulation returned a two component fit
While the polymer chain mobility determined by EPR reveals the extent of collapse of the polyelectrolyte chains before and after complex coacervation, Overhauser dynamic nuclear polarization (DNP) spectroscopy determines the rate at which local hydration water in the vicinity of the spin labels diffuses around each polyelectrolyte. The hydration landscape can provide a more sensitive measure on the dynamic state of the polyelectrolyte, given that longer-range interactions (up to 15 Å from the spin label) than with spin label rotational dynamics can be detected. These DNP measurements reveal the local surface hydration layer fluidity of each polymer chain. Figure 3 shows the measured water diffusion coefficient, on the surface of mfp151 or HA polyelectrolytes, respectively, as a function of the fraction of mfp151 over HA. Here, as the optimal complex coacervation condition (fraction mfp151 = 0.6) is approached, water surrounding mfp151SL (red) is shown to diffuse more slowly with a 5.5-fold retardation with respect to mfp151SL alone, which is an expected feature if mfp151 resumes a more compact state when complex coacervated. Interestingly, upon complex coacervation, the local hydration water moves measurably more quickly around HA-SL (blue). As an important reference measurement, HA-SL is probed alone as a function of its concentration in the absence of mfp151. The surface water diffusion coefficient of HA-SL alone reaches a value close to D = 1.14 × 10−9 m2 sec−1. This value is higher than the water diffusion coefficient measured at the HA-SL surface when interacting with mfp151 at any measured ratio, in solution or coacervated state. This implies that HA-SL experiences an overall less fluidic hydration shell in the presence of mfp151 (at fractions between 0.2–0.8) regardless of their complexation state. Given this feature, the observed increase in water diffusion coefficient around HA-SL under optimal coacervation conditions suggests that HA becomes liberated from the electrostatic interactions with mfp151, allowing for HA’s local hydration shell to exhibit higher dynamics.
Figure 3.
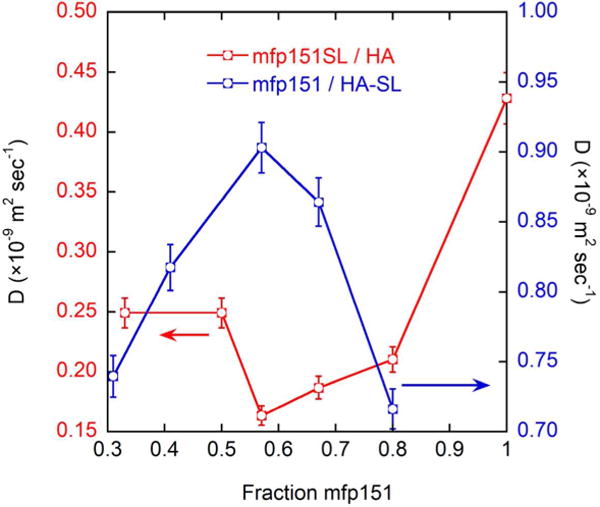
Water diffusion coefficients, determined by dynamic nuclear polarization enhanced NMR spectroscopy (DNP), show that under optimal coacervation conditions (0.6 fraction mfp151) water diffusion is minimized around mfp151SL and maximized around HA-SL.
The generality of this trend is explored by examining three different sites on a recombinant construct with 12 tandem repeats of the mfp1 consensus decapeptide, i.e. (AKPSYPPTYK)12 that mimics the flanking domains of mfp151. Figure 4a shows the schematic of mfp1SL with a cysteine mutation near the N terminus (mfp1SL-N), in the middle of the sequence (mfp1SL-m), and near the C terminus (mfp1SL-C). Upon spin labeling of the different mfp1 proteins by covalent functionalization of the cysteine residues with the spin label MTSL (Figure 4a), their EPR spectra shown in Figure 4b reveal similarly high rotational diffusion rates as seen with mfp151SL, at each spin labeled position of mfp1SL in solution with kr ~ 730 MHz.
Figure 4.
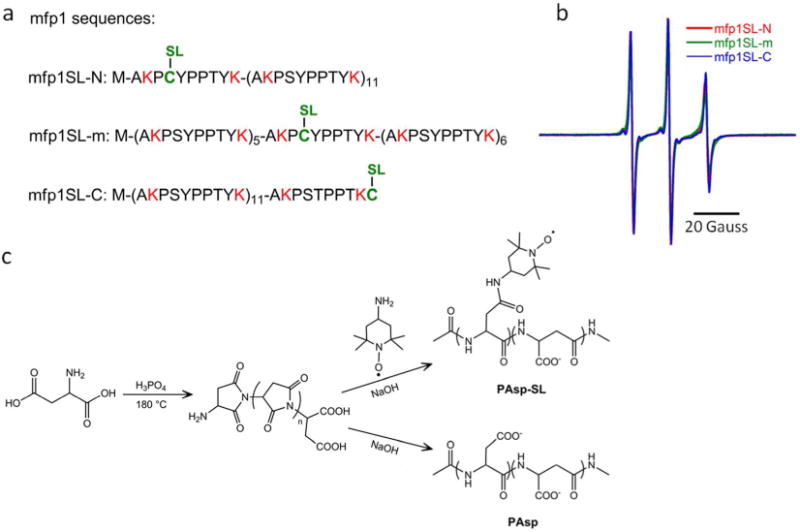
(a) The mfp1 protein mutated at three select sites is then spin labeled (SL) by addition of MTSL, yielding mfp1SL-N, mfp1SL-m, and mfp1SL-C. Positively charged residues are designated in red and the single cysteine site at which the spin label is installed is indicated in green. No negatively charged residues occur in the mfp1 sequence. (b) EPR of each mfp1SL species indicates similar local environments when the polymer chains are freely dissolved in water; (c) Synthetic routes to poly(aspartic acid) (PAsp) and spin labeled poly(aspartic acid) (PAsp-SL).
Water dynamics measurements by DNP of the mfp1 protein spin labeled at the three different positions also confirm the earlier obtained results with mfp151. Here, under optimized coacervation conditions with HA, the water diffusion rate reaches a minimum with an approximately 3-fold retardation with respect to non-coacervated mfp1, as the mfp1 protein collapses in the complex condensate. Similar to the case of mfp151/HA complex coacervation, the water diffusion rate on the HA surface in mfp1/HA reaches a maximum value under optimal coacervation conditions, substantiating our earlier hypothesis of the liberation of HA from electrostatic inter-actions with mfp1 when coacervated (Figure 5). While these general trends are the same as those observed in the mfp151/HA system, the absolute values for the surface water dynamics of the polyelectrolyte constituents in the mfp1/HA systems are greater than those observed in the mfp151/HA system. This difference may be due to the absence of the highly charged mfp5 (~ 30 % charged residues) mid-segment in mfp1. The absence of this highly charged mid-segment could be weakening the intermolecular interactions between mfp1 and HA as compared to mfp151 and HA, resulting in an overall more fluidic hydration shell of the polyelectrolyte constituents.
Figure 5.
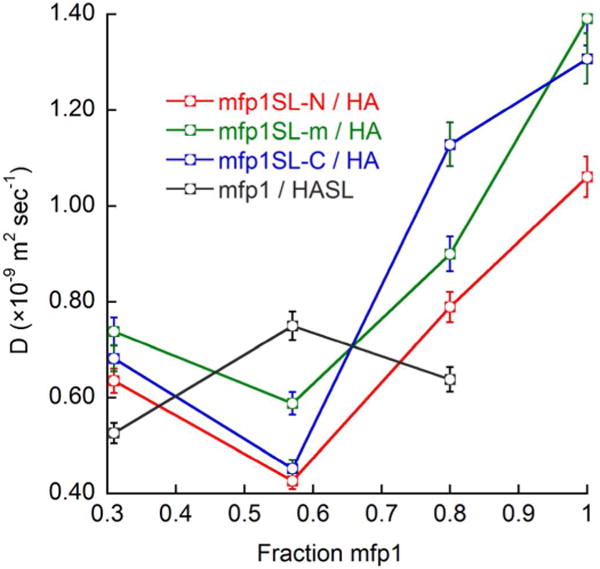
Water diffusion coefficients determined by DNP show the collapse of mfp1 upon complex coacervation with HA at each of three selected sites, near the N-terminus, the middle of the protein chain, and the C-terminus. In contrast, the water diffusion coefficient of HA-SL is shown to increase upon complex coacervation with mfp1.
Figure 5 also shows small, but measurable, differences in the surface water diffusion coefficient between the three mfp1SL species. Under optimal coacervation conditions, the spin label near the N and C termini of mfp1 measures a water diffusion coefficient down to D = 0.4 × 10−9 m2 sec−1, whereas the spin label positioned in the middle of mfp1SL measures a higher water diffusion coefficient of D = 0.6 × 10−9 m2 sec−1 (± ~ 0.01 × 10−9 m2 sec−1). This dispersion in water diffusion indicates that the collapse of mfp1 protein chains upon complex coacervation occurs with some degree of intramolecular structural organization. This non-random folding pattern may be due to the uneven charge distribution across mfp1, where uncharged and hydrophobic residues are dispersed through its sequence (Figure 4a).
The measurement of the local hydration water dynamics on polyelectrolyte surfaces confirms there is an asymmetric behavior for the polycation versus polyanion upon complex coacervation, with a collapse of the polycationic mfp151SL or mfp1SL and the liberation of the HA-SL chains from their interaction with mfp151 or mfp1. The final investigation into the generality of this asymmetric behavior that we attribute to the charge heterogeneity of mfp proteins (Figure 2a) versus the charge homogeneity of HA (Figure 2b) was performed by testing another polyelectrolyte, poly(aspartic acid) (PAsp), as the counter polyanion to undergo complex coacervation with mfp151. The scheme shown in Figure 4c was employed to synthesize PAsp, and complex coacervates were formed between PAsp-SL and mfp151 at the optimum coacervation condition (75 mass % mfp1) determined by turbidity measurements. The rotational diffusion rates (kr) as determined by EPR spectral simulations, and water diffusion coefficients (D) determined by DNP, are reported in Table 2.
Table 2.
Rotational diffusion rates (kr) determined by EPR spectral simulations, and water diffusion rates (D) determined by DNP, of spin labeled poly(aspartic acid) (PAsp-SL) and PAsp-SL/mfp151 complex coacervates.
| kr (MHz) | D (× 10−9 m2 sec−1) | |
|---|---|---|
|
| ||
| PAsp-SL | 380 (100 %) | 0.63 |
|
| ||
| PAsp-SL / mfp151 Initial[a] |
30.0 (72%) 403 (28%) |
0.69 |
|
| ||
| PAsp-SL / mfp151 Equilibrium (2 d) |
540 (100%) | 1.02 |
|
| ||
EPR spectral simulation returned a two component fit
Interestingly, the rotational diffusion rates observed by EPR in the PAsp/mfp151 coacervate system reveal a previously unobserved time dependence. While the local water dynamics and spin label’s rotational diffusion rates on PAsp-SL alone in solution (Table 2) are similar to those observed in other pure polyelectrolyte solutions (including HA), upon addition of mfp151, the initial rotational diffusion rate drops by > 10 fold to 30 MHz. This initial restriction of the PAsp chain mobility then gradually increases over the course of 2 days to a value greater than the initial rotational diffusion rate. DNP again reveals an initially fast water diffusion coefficient around PAsp-SL alone of 0.63 × 10−9 m2 sec−1, as expected. Upon complex coacervation, the water diffusion rate first increases only slightly to 0.69 × 10−9 m2 sec−1. After equilibration the water diffusion rate increases further, as the PAsp-SL chains are liberated analogously to the behavior found with HA-SL chains under optimal coacervation, reaching high values of 1.02 × 10−9 m2 sec−1. This observation indicates that coacervated PAsp-SL after equilibration is exposed to more dynamic water, similar to observations made with HA-SL. These results, once again, depict asymmetric behavior when a polyelectrolyte with heterogeneous charge distribution is incorporated into a complex coacervate, as is observed in protein-based systems. Figure 6 presents a cartoon description of how an asymmetric collapse of the mfp151 or mfp1 proteins in their complex coacervate phase with HA or PAsp is envisioned.
Figure 6.
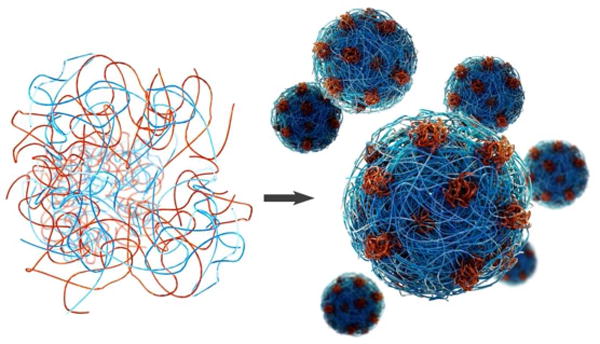
Illustration of the collapse of cationic protein (red) and the liberation of anionic biopolymer (blue) that occur upon complex coacervation of biomimetic polyelectrolytes.
CONCLUSIONS
Recombinant hybrid proteins based on adhesive protein sequences from Mytilus edulis (blue mussel) were expressed in high yields and coacervated with the biological anionic polyelectrolyte, hyaluronic acid, and also with poly(aspartic acid). Unique insight into the internal molecular structure and dynamics of polyelectrolytes, before and upon complex coacervation, was obtained through the in situ determination of local rotational and hydration water dynamics around small nitroxide-based spin labels tethered to the polyelectrolyte chains. Unlike the study of folded proteins with well-defined structures, the molecular-level study of polymer assemblies is much more challenging, given the high degree of dynamics, variability, and heterogeneity. Presented here is the elucidation of protein and water dynamics within these biological complex coacervates. To accomplish this, spin labeling was employed with site- and species-specificity along with the novel magnetic resonance technique, Overhauser dynamic nuclear polarization enhanced NMR spectroscopy. Concurrently, EPR spectra were acquired to quantify polymer chain rigidity through EPR spectral analyses, yielding rotational diffusion rates of the tethered spin label. This approach should be generally applicable to the molecular-level study of the internal structure and dynamics of other macromolecular assemblies.
Biomimetic complex coacervates based on recombinant hybrid proteins, mfp151 and mfp1, exhibit asymmetric collapse of the proteinaceous polycation and liberation of the anionic biopolymer. This behavior can be attributed to the mismatch in linear charge density distributions between the homogeneously charged polyanion and heterogeneously charged protein. The liberation of polyelectrolytes with linear charge density upon complex coacervation is generalizable, to the extent that both HA and PAsp exhibit the same behavior upon equilibration. However, PAsp complex coacervates un-ergo a time-dependent equilibration that takes over ~ 2 d. The origin of this time-dependence is still unclear, however one possible explanation could be due to the dramatically different linear charge densities of HA versus PAsp. Here, the negatively charged pendent groups of PAsp reside at ~ every 3 to 4 carbons along the backbone while the carboxylate pendant groups in HA occur at approximately every tenth carbon atom along the backbone. With such a great charge density of poly(aspartic acid), the initial interaction between poly(aspartic acid) and mfp151 is likely fundamentally different from the initial interaction between mfp151 and HA.
We have identified the heterogeneity of polyelectrolyte charge density as a key parameter in the internal structure of complex coacervates, where complex coacervates composed of polyelectrolytes with both homogeneous and heterogeneous charge densities undergo asymmetric collapse that results in heterogeneous internal structure. Given that natural complex coacervates are composed of proteins with intrinsically complicated charge density profiles, it is likely that biological complex coacervates exhibit heterogeneities in their internal structure as a general rule. The next key question is how such heterogeneous structure imparts particular physical properties on biomimetic and biological complex coacervates, and what adaptive function is served in the particular case of aqueous adhesion systems.
Supplementary Material
Acknowledgments
This work was financially supported by the National Science Foundation (NSF) through the MRSEC Program DMR-1121053 (MRL-UCSB) for all authors. This work utilized the MRL Central Facilities supported by the MRSEC Program of the NSF under DMR-1121053; a member of the NSF-funded Materials Research Facilities Network (www.mrfn.org). J.M.F. acknowledges the Elings Fellowship through the California NanoSystems Institute at UCSB. S.H. was also supported by the Packard Fellowship for Science and Engineering and the 2012 NIH innovator award. J.H.W acknowledges support from the US National Institutes of Health (R01 DE018468). J.H.O. acknowledges support from the NSF Division for Materials Research Graduate Student Fellowship (MRSEC Award No. DMR05-20415). D.S.H. acknowledges Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (2011-0007605).
The authors thank Scott D. Auerbach (UCSB) for assisting in recombinant mfp1 preparations and H.J. Cha (POSTECH) for supplying mfp151. This work was completed at the UCSB Materials Research Laboratory (MRL) facilities. The authors acknowledge Jerry Hu for contributions to the instrumentation.
ABBREVIATIONS
- NMR
Nuclear magnetic resonance
- DNP
Dynamic nuclear polarization enhanced NMR
- EPR
Electron paramagnetic resonance
- Mfp
mytilus foot protein
- SL
spin label
- PTFE
poly(1,1,2,2-tetrafluoroethylene)
Footnotes
SUPPORTING INFORMATION AVAILABLE
Supporting Information describes the experimental details of dynamic nuclear polarization (DNP) experiments used to measure water diffusion rates around polymers or proteins in solution and within complex coacervates. A representative data set corresponding to an experiment discussed in the manuscript is shown, and the calculations required to convert experimental parameters to the final measure of water diffusion are explained. This information is available free of charge via the Internet at http://pubs.acs.org/.
References
- 1.Mooney DJ, Silva EA. Tissue Engineering: A glue for biomaterials. Nature materials. 2007;6(5):327–328. doi: 10.1038/nmat1896. [DOI] [PubMed] [Google Scholar]
- 2.Hammer DA, Tirrell M. Biological adhesion at interfaces. Annual Review of Materials Science. 1996;26(1):651–691. [Google Scholar]
- 3.Stewart RJ. Protein-based underwater adhesives and the prospects for their biotechnological production. Applied microbiology and biotechnology. 2011;89(1):27–33. doi: 10.1007/s00253-010-2913-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Waite J. Nature’s underwater adhesive specialist. International Journal of Adhesion and Adhesives. 1987;7(1):9–14. [Google Scholar]
- 5.Winslow BD, Shao H, Stewart RJ, Tresco PA. Biocompatibility of adhesive complex coacervates modeled after the sandcastle glue of Phragmatopoma californica for craniofacial reconstruction. Biomaterials. 2010;31(36):9373–9381. doi: 10.1016/j.biomaterials.2010.07.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Waite JH, Andersen NH, Jewhurst S, Sun C. Mussel adhesion: finding the tricks worth mimicking. The journal of adhesion. 2005;81(3–4):297–317. [Google Scholar]
- 7.de Kruif CG, Weinbreck F, de Vries R. Complex coacervation of proteins and anionic polysaccharides. Current opinion in colloid & interface science. 2004;9(5):340–349. [Google Scholar]
- 8.Weinbreck F, De Vries R, Schrooyen P, De Kruif C. Complex coacervation of whey proteins and gum arabic. Biomacromolecules. 2003;4(2):293–303. doi: 10.1021/bm025667n. [DOI] [PubMed] [Google Scholar]
- 9.Overbeek JTG, Voorn M. Phase separation in polyelectrolyte solutions. Theory of complex coacervation. Journal of Cellular and Comparative Physiology. 1957;49(S1):7–26. [PubMed] [Google Scholar]
- 10.Ortony JH, Cheng CY, Franck JM, Kausik R, Pavlova A, Hunt J, Han S. Probing the hydration water diffusion of macromolecular surfaces and interfaces. New Journal of Physics. 2011 13, 015006. [Google Scholar]
- 11.Armstrong BD, Choi J, López C, Wesener DA, Hubbell W, Cavagnero S, Han S. Site-specific hydration dynamics in the nonpolar core of a molten globule by dynamic nuclear polarization of water. Journal of the American Chemical Society. 2011;133(15):5987–5995. doi: 10.1021/ja111515s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kausik R, Han S. Dynamics and state of lipid bilayer-internal water unraveled with solution state 1H dynamic nuclear polarization. Phys Chem Chem Phys. 2011;13(17):7732– 7746. doi: 10.1039/c0cp02512g. [DOI] [PubMed] [Google Scholar]
- 13.Armstrong BD, Han S. Overhauser dynamic nuclear polarization to study local water dynamics. Journal of the American Chemical Society. 2009;131(13):4641–4647. doi: 10.1021/ja809259q. [DOI] [PubMed] [Google Scholar]
- 14.Srivastava A, Waite JH, Stucky GD, Mikhailovsky A. Fluorescence Investigations into Complex Coacervation between Polyvinylimidazole and Sodium Alginate. Macromolecules. 2009;42(6):2168–2176. doi: 10.1021/ma802174t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kausik R, Han S. Ultrasensitive detection of interfacial water diffusion on lipid vesicle surfaces at molecular length scales. J Am Chem Soc. 2009;131(51):18254. doi: 10.1021/ja9060849. [DOI] [PubMed] [Google Scholar]
- 16.Rzepecki LM, Hansen KM, Waite JH. Characterization of a cystine-rich polyphenolic protein family from the blue mussel Mytilus edulis L. The Biological Bulletin. 1992;183(1):123–137. doi: 10.2307/1542413. [DOI] [PubMed] [Google Scholar]
- 17.Waite JH, Qin X. Polyphosphoprotein from the adhesive pads of Mytilus edulis. Biochemistry. 2001;40(9):2887–2893. doi: 10.1021/bi002718x. [DOI] [PubMed] [Google Scholar]
- 18.Hwang DS, Gim Y, Yoo HJ, Cha HJ. Practical recombinant hybrid mussel bioadhesive fp-151. Biomaterials. 2007;28(24):3560–3568. doi: 10.1016/j.biomaterials.2007.04.039. [DOI] [PubMed] [Google Scholar]
- 19.Lim S, Choi YS, Kang DG, Song YH, Cha HJ. The adhesive properties of coacervated recombinant hybrid mussel adhesive proteins. Biomaterials. 2010;31(13):3715–3722. doi: 10.1016/j.biomaterials.2010.01.063. [DOI] [PubMed] [Google Scholar]
- 20.Hwang DS, Sim SB, Cha HJ. Cell adhesion biomaterial based on mussel adhesive protein fused with RGD peptide. Biomaterials. 2007;28(28):4039–4046. doi: 10.1016/j.biomaterials.2007.05.028. [DOI] [PubMed] [Google Scholar]
- 21.Hwang DS, Waite JH, Tirrell M. Promotion of osteoblast proliferation on complex coacervation-based hyaluronic acid-recombinant mussel adhesive protein coatings on titanium. Biomaterials. 2010;31(6):1080–1084. doi: 10.1016/j.biomaterials.2009.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hwang DS, Zeng H, Srivastava A, Krogstad DV, Tirrell M, Israelachvili JN, Waite JH. Viscosity and interfacial properties in a mussel-inspired adhesive coacervate. Soft matter. 2010;6(14):3232–3236. doi: 10.1039/C002632H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Floriolli RY, von Langen J, Waite JH. Marine surfaces and the expression of specific byssal adhesive protein variants in Mytilus. Marine Biotechnology. 2000;2(4):352–363. doi: 10.1007/s101269900032. [DOI] [PubMed] [Google Scholar]
- 24.Zeng H, Hwang DS, Israelachvili JN, Waite JH. Strong reversible Fe3+-mediated bridging between dopa-containing protein films in water. Proceedings of the Nationa l Academy of Sciences. 2010;107(29):12850–12853. doi: 10.1073/pnas.1007416107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Budil DE, Lee S, Saxena S, Freed JH. Nonlinear Least-Squares Analysis of Slow-Motional EPR Spectra in One and Two Dimensions Using a Modified Levenberg-Marquardt Algorithm. J Magn Res A. 1996;120:155. [Google Scholar]
- 26.Armstrong BD, Han S. A new model for Overhauser enhanced nuclear magnetic resonance using nitroxide radicals. The Journal of Chemical Physics. 2007;127(10):104508. doi: 10.1063/1.2770465. [DOI] [PubMed] [Google Scholar]
- 27.McCarney ER, Armstrong BD, Kausik R, Han S. Dynamic nuclear polarization enhanced nuclear magnetic resonance and electron spin resonance studies of hydration and local water dynamics in micelle and vesicle assemblies. Langmuir. 2008;24(18):10062–10072. doi: 10.1021/la800334k. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.