

Abstract
We developed a three-dimensional (3D) cellular microarray platform for the high-throughput (HT) analysis of human neural stem cell (hNSC) growth and differentiation. The growth of an immortalized hNSC line, ReNcell VM, was evaluated on a miniaturized cell culture chip consisting of 60 nl spots of cells encapsulated in alginate, and compared to standard 2D well plate culture conditions. Using a live/dead cell viability assay, we demonstrated that the hNSCs are able to expand on-chip, albeit with lower proliferation rates and viabilities than in conventional 2D culture platforms. Using an in-cell, on-chip immunofluorescence assay, which provides quantitative information on cellular levels of proteins involved in neural fate, we demonstrated that ReNcell VM can preserve its multipotent state during on-chip expansion. Moreover, differentiation of the hNSCs into glial progeny was achieved both off- and on-chip six days after growth factor removal, accompanied by a decrease in the neural progenitor markers. The versatility of the platform was further demonstrated by complementing the cell culture chip with a chamber system that allowed us to screen for differential toxicity of small molecules to hNSCs. Using this approach, we showed differential toxicity when evaluating three neurotoxic compounds and one antiproliferative compound, and the null effect of a non-toxic compound at relevant concentrations. Thus, our 3D high-throughput microarray platform may help predict, in vitro, which compounds pose an increased threat to neural development and should therefore be prioritized for further screening and evaluation.
Keywords: human neural stem cells, microarray cell culture, high throughput screening, differentiation, toxicology
1. Introduction
Neurotoxicity detection is a major challenge due to the complexity of the central and peripheral nervous systems. The majority of commercial chemicals have not been evaluated for developmental neurotoxicity, at least partly due to the high cost of animal testing (National Research Council 2007). Regulatory agencies have traditionally used in vivo methods for adult and developmental neurotoxicity testing, including neurobehavioral evaluation of cognitive, sensory and motor functions accompanied by neuropathological studies, with no specific studies of the underlying cell biology (Bal-Price et al. 2010). There is also a need to test large sets of compounds to comply with specific regulatory requirements (Breier et al. 2010; Andersen & Krewski 2009). To this end, there is pressure to develop alternative test strategies, which are rapid, economical, and, most critically, highly predictive (Breier et al. 2010).
An often overlooked aspect of neurotoxicity is the impact of chemicals, as well as drugs and drug candidates, on neural stem cells and their terminally differentiated lineages. Stem cells have been shown to exhibit differential sensitivities to both non-toxic (e.g., serum) and toxic compounds, as compared to terminally differentiated cells (Trosko & Chang 2010; Dietrich et al. 2006). Broad knowledge of the toxicity of such compounds to stem cells in comparison to other cell types in a given tissue can provide fundamental information critical for assessing the safety of new drug candidates and the health effects of environmental agents. Thus, the development of new high-throughput screening tools that enable the study of these differential effects on stem cells and their differentiated progeny, should encompass not only endpoints that assess chemical toxicity, but also allow us to determine stem cell fate. This is generally achieved by following protein markers of multipotency and differentiation.
With this in mind, we have developed a three-dimensional (3D) cellular microarray platform for the high throughput analysis of hNSC differentiation and toxicity screening (Fig. S1). Our system has the ability to expand our knowledge of neurotoxicity by discriminating between toxic and nontoxic compounds. It can also detect differentiation stage-specific toxicities. Knowledge of differences in molecular toxicity to stem cells in comparison to other cell types is critical for assessing safety of new drug candidates and health effects of environmental agents (Laustriat et al. 2010). We demonstrated herein the differentiation of the ReNcell VM hNSC line into glial progeny on a 3D cellular microarray platform. This platform was then used to screen dose-dependent toxicity of a number of neurotoxic compounds, leading to identification of compounds with differential toxicity to hNSCs in relation to the differentiated glial progeny.
2. Materials and Methods
2.1 Cell culture
ReNcell VM (Millipore) is an immortalized neural progenitor cell line derived from the ventral mesencephalon region of a 10-week human fetal brain. All cells used in this investigation were from passage 31 or lower; previous work (Donato et al. 2007) has shown that these cells maintain a stable karyotype past 45 passages. Cells were cultured according to the manufacturer’s instructions. Briefly, the ReNcell VM cells were expanded in expansion medium (ReNcell NSC Maintenance Medium (Millipore) supplemented with 20 ng/ml of epidermal growth factor (EGF, Millipore) and 20 ng/ml of basic fibroblast growth factor (bFGF, Millipore)) on laminin-coated (1.7 μg/cm2) TC-treated culture flasks at 37°C in a 5% CO2 humidifier incubator. The medium was renewed every two days during proliferation, and the cells subcultured approximately every five days (90% confluence) by detaching them with Accutase® (Millipore). After each passage, cell concentration and viability was determined by counting with a hemocytometer (Hauser Scientific) using the trypan blue dye (Invitrogen) exclusion test, and the cells were once again seeded at 5 × 104 cells/ml in freshly coated flasks. Differentiation of the cells was accomplished by adding fresh differentiation medium (ReNcell NSC Maintenance Medium without growth factors) to confluent monolayers of cells. Unless otherwise indicated, cells were incubated for 6 days in differentiation medium, replenishing the medium every 2 days.
2.2 Preparation of 3D microarray cultures
ReNcell VM cells were cultured in three-dimensional (3D) microarrays by embedding them in alginate, and printing the gel mixture in 60 nl spots on hydrophobic glass slides. Briefly, a poly-L-lysine (PLL)–BaCl2 mixture was made by mixing a sterile 0.1 M BaCl2 solution in deionized water with a 0.01% (w/v) sterile PLL solution (Sigma) in a 1:2 volume ratio. The PLL-Ba2+ mixture was spotted onto poly(styrene-co-maleic anhydride) (PSMA) coated glass slides using a MicrosSys 5100-4SQ microcontact microarray spotter (Genomics Solutions), making a 6 × 8 × 8 patterned array of 60 nl spots on the chip and allowing them to dry. Each spot had a diameter of ca. 600 μm and a height of ca. 70 μm. A ReNcell VM cell suspension in media was then mixed with a low viscosity alginic acid (Sigma) solution in deionized sterile water such that the final alginate concentration was 1% (w/v), and the concentration of cells in suspension was 5 × 106 cells/ml, unless otherwise indicated. Subsequently, the cell-alginate mixture was printed (60 nl/spot, 300 cells/spot) on top of the dried PLL-Ba2+ spots, which allowed the nearly instantaneous gelation of the alginate matrix. During the printing process, the humidity in the microarray chamber was maintained above 90% to prevent water evaporation of the spots. Following printing, the slide was fitted with a commercially available 8-well polystyrene medium chamber coated with a biocompatible adhesive (Lab-Tek II, Nunc) that allowed firm attachment of the chip to the chamber, and physically separated groups of spots within the slide. Each of the wells, containing 48 cell culture microarray spots, was filled with 250 μl of ReNcell expansion medium. Following expansion, differentiation, exposure to drugs and/or assaying the chambers could be easily removed with a slide separator.
2.3 Cell viability assay
Cell viability on the 3D microarrays was assessed via a Live/Dead® viability/cytotoxicity kit for mammalian cells (Invitrogen). The slides were separated from the chamber, and subsequently rinsed twice with a wash solution (140 mM NaCl (Sigma-Aldrich) and 20 mM CaCl2 (Sigma) at pH 7). Then, they were immersed in a 0.5 μM calcein AM/1 μM ethidium homodimer 1 solution to detect viable and dead cells via green and red fluorescence, respectively. After 45 min incubation at room temperature, the dyes were removed and the chips rinsed thoroughly with the wash solution and then dried. The slides were scanned with a GenePix 4200A scanner (Molecular Devices) using a 488 nm laser for excitation, with a standard blue filter and a 645AF75/594 filter for detection of the green and red dye, respectively. The fluorescence intensity from the resulting image was quantified with the GenePix Pro 6.0 software (Molecular Devices).
To quantify viability in 2D cultures within 96-well microtiter plates we used the methylthiazolyldiphenyl-tetrazolium bromide (MTT) assay. Cells seeded at a density of 5,000 cells/well were allowed to attach for at least 5 h. To assay the cells, 50 μl of a 2.5 mg/ml MTT (Sigma) reagent solution prepared in sterile conditions were added to each well, and incubated for 3 h under culture conditions. The culture supernatant was subsequently removed and the formazan crystals generated in metabolically active cells were dissolved with 150 μl of dimethylsulfoxide (DMSO, Sigma-Aldrich). The absorbance was read using a SpectraMax M5 Multi-Mode Microplate Reader (Molecular Devices) at 590 nm with a reference filter at 690 nm.
2.4 Western blotting analysis of marker proteins
ReNcell VM cells were seeded and expanded or differentiated on a T-75 flask as described above. After detachment of nearly confluent monolayers of differentiated or non-differentiated cells, whole-cell extracts were prepared by lysing the cells with NP40 lysis buffer (Boston BioProducts) supplemented with a protease/phosphatase inhibitor cocktail (Cell Signaling Technology). The lysates were collected and the protein content was estimated using a BCA assay (Thermo Scientific Pierce), allowing equal amounts of protein to be loaded in each lane. The samples were resolved in 4–20% precast SDS-PAGE gradient gels (Precise™ Protein Gels, Thermo Scientific) for 30 min at 150 V and electrotransferred to a nitrocellulose membrane (Bio-Rad). For the transfer, the gel was first equilibrated for 1 h in the transfer buffer (Bio-Rad), and then transferred to the membrane for 60 min at 100 V and 4 °C. The membrane was subsequently immersed in blocking buffer (5% (w/v) skim milk and 0.05% (v/v) Tween-20 in phosphate-buffered saline (PBS)) for 1 h at room temperature.
The primary antibody solutions were then prepared in the blocking buffer and incubated with the membrane for 1 h at room temperature. The antibodies used were mouse monoclonal anti-GalC (1:300 dilution, EMD Millipore), mouse monoclonal anti-GAPDH (1:3000 dilution, Abcam), mouse monoclonal anti-GFAP (1:1500 dilution, Abcam), mouse monoclonal anti-Nestin (1:500 dilution, BD BioScience), mouse monoclonal anti-βIII-Tubulin (1:1000 dilution, Covance), and rabbit polyclonal anti-γ-Tubulin (1:1000 dilution, Sigma-Aldrich). Following primary antibody incubation, the membrane was washed thoroughly with 0.05% (v/v) Tween-20 in PBS (PBS-T) and incubated with the peroxidase-conjugated goat anti-mouse or goat anti-rabbit IgG secondary antibody (1:1000 dilution, Invitrogen) in blocking buffer for 1h at room temperature. The membrane was once again washed thoroughly with PBS-T and immunoreactivity was visualized using a chemiluminescence reagent (SuperSignal, Pierce).
2.5 On-Chip, in-cell immunofluorescence assay
Cellular immunofluorescence assays were performed on both non-differentiated ReNcell VM cells (+EGF, +FGF-2) and differentiated cells (−EGF, −FGF-2) cultured both in the 3D microarrays (on-chip) and in conventional 2D culture flasks (off-chip), for which the cells were printed immediately before assaying. Non-differentiated cells (on-chip) were printed and incubated for 6 days in expansion medium to permit their expansion in microarray cultures, while the cells in the flask (off-chip) were expanded to 90% confluence, detached, and subsequently printed. Differentiated cells (on-chip) were printed, allowed to expand for 3 days in expansion medium and subsequently incubated in differentiation medium for 6 days. For off-chip differentiation, cells seeded in flasks were expanded to 90% confluence and were then switched to differentiation medium for 6 more days, after which they were printed and assayed.
Following expansion or differentiation, the chips were rinsed three times with a wash buffer (Tris-buffered saline (TBS) containing 10 mM of CaCl2) and subsequently fixed with a 3.7% (v/v) formaldehyde (Sigma) solution in the washing buffer for 20 min. The cells were then permeabilized for 10 min with 0.15% (v/v) Triton X-100 (Sigma), rinsed three times with the wash buffer, and incubated overnight at 4°C with a blocking solution (TBS Superblock from Pierce). After blocking, the chips were rinsed three times with wash buffer containing 0.1% (v/v) Tween-20 (MP Biomedicals) and incubated for 2 h at room temperature with the various primary antibodies diluted in 1% (w/v) bovine serum albumin (BSA, Santa Cruz Biotechnology) in the wash buffer. The antibodies used were rabbit polyclonal anti-GalC (1:250 dilution, Santa Cruz Biotechnology), mouse monoclonal anti-GAPDH (1:100 dilution, Abcam), mouse monoclonal anti-GFAP (1:100 dilution, Abcam), mouse monoclonal anti-Nestin (1:100 dilution, BD BioScience), mouse monoclonal anti-βIII-Tubulin (1:400 dilution, Covance), and rabbit polyclonal anti-γ-Tubulin (1:1000 dilution, Sigma-Aldrich). After washing the cells thoroughly, the peroxidase-conjugated goat anti-mouse or goat anti-rabbit IgG secondary antibody (1:1000 dilution, Invitrogen) was added, and the cells were incubated at room temperature for 1.5 h. A tyramide signal amplification kit (Invitrogen) was subsequently used following the manufacturer’s instructions to detect the presence of the target proteins through fluorescence analysis. The microarrays were scanned with a slide scanner as described above (at 488 nm with a standard blue laser). The experiment was repeated at least three times, with 48 replicates per marker probed in each chip. The fluorescence signal from GAPDH was used as internal control, while the signal from cells exposed to the secondary antibody and tyramide dye alone was used as a negative control. Similar results were obtained when using anti-γ-Tubulin to normalize the fluorescence signals.
2.6 Immunocytochemistry
Following growth and/or differentiation on laminin-coated 35-mm glass-bottom cell culture dishes (Greiner Bio-One), the medium was removed and the cells were washed twice with PBS. The cells were fixed with a cold 3.7% (v/v) formaldehyde (Sigma) solution in PBS for 10 min, and then washed three times with PBS. Cells were then permeabilized with PBS containing 0.1% Triton-X100 at room temperature and washed three times with PBS. The cells were blocked in PBS containing 1% BSA for 20 min at room temperature. Alexa Fluor 488 phalloidin (1:40 dilution, Life Technologies) in blocking solution was used to detect filamentous actin (F-actin), cells were incubated for 20 min in the dark at room temperature, then washed three times with PBS, counterstained with 10 μg/ml Hoechst 33342 (Sigma) for 1 min, in the dark, and washed once with PBS. To preserve fluorescence, all samples were coated with Prolong Gold antifade reagent (Life Technologies).
2.7 Cytotoxicity of test compounds in ReNcell VM cell line and derived differentiated cells
To probe differential cytotoxicity in ReNcell VM cells and their differentiated progeny, we examined the effect on cell viability of three known developmental neurotoxicants, retinoic acid (Adams & Lammer 1991), cadmium chloride hydrate (Costa et al. 2007), and dexamethasone (Baud 2004). In addition to these compounds, we also tested the effect of 5-fluorouracil and acetaminophen on cytotoxicity. The former is a known antiproliferative compound (Bogdahn et al. 1987; Longley et al. 2003), while the latter is an analgesic approved for use during pregnancy and presumed to have no effect on the developing central nervous system (Buzanska et al. 2009; Saunders et al. 1980). All compounds were purchased from Sigma, and were dissolved in DMSO (Sigma) to prepare stock solutions, which were stored in frozen aliquots. Immediately before use, the stocks were diluted in media to a concentration corresponding to the highest dose tested for each compound. The remaining doses were prepared by 4-fold serial dilutions of the highest dose. A vehicle concentration of 0.5% DMSO was used in all doses for the five compounds.
For determination of non-differentiated ReNcell VM cytotoxicity, cells were printed in the 3D microarrays and incubated for 3 days in expansion medium, after which the media was removed and the cells incubated in expansion medium containing varying doses of each drug. In the case of the differentiated cells, the printed 3D microarrays were similarly incubated for 3 days in expansion medium, subsequently cultured in differentiation medium for 6 more days, and finally incubated in differentiation medium containing the drug solutions. Following a 24 h incubation period with the compounds, the drug solutions were removed and the cells were cultured with either expansion or differentiation media for an additional 48 h. Cytotoxicity was determined via a live/dead cell viability assay, as described previously. Cells incubated in media with 0.5% DMSO served as our positive “live cell” control, and cells treated with 5% saponin for 30 min served as the negative “dead cell” control. To produce the conventional dose-response curve, green fluorescence intensity values (calcein AM signal) were normalized with these controls and plotted against the logarithm of the tested concentrations. The IC50 values were obtained by fitting the data to a sigmoidal dose-response curve (variable slope) using Prism 4 (GraphPad Software). Each experiment was repeated at least three times, having 48 replicates per concentration of each chemical.
2.8 Statistical Analysis
Changes in marker expression and chemosensitivity upon differentiation were determined using a one-way analysis of variance (ANOVA) followed by planned comparisons using Student’s t test. For all comparisons, results were considered significant if p < 0.05. Data are represented graphically as the mean ± SEM.
3. Results and Discussion
3.1 Proliferation of ReNcell VM cell line in 2D monolayers and 3D microarrays
The influence of the 3D alginate environment on cell proliferation was first investigated by comparing cell growth on-chip to that in 2D cultures. The on-chip growth was performed as described by Meli et al. (2012) and shown in Fig. S1. The expansion of the hNSC ReNcell VM cell line was assessed in conventional 2D cultures using an MTT assay to detect metabolically active cells in a 96-well microtiter plate. The cells expanded readily in monolayer culture, with doubling times of 22 h in the exponential phase of growth (Fig. 1A). The growth of ReNcell VM cells embedded in the 3D microarrays, and monitored using a live/dead cell viability assay, however, shows a striking 4-day lag period. This is followed by growth at much slower rate than in 2D culture, with doubling times of 84 h (Fig. 1A).
Figure 1.
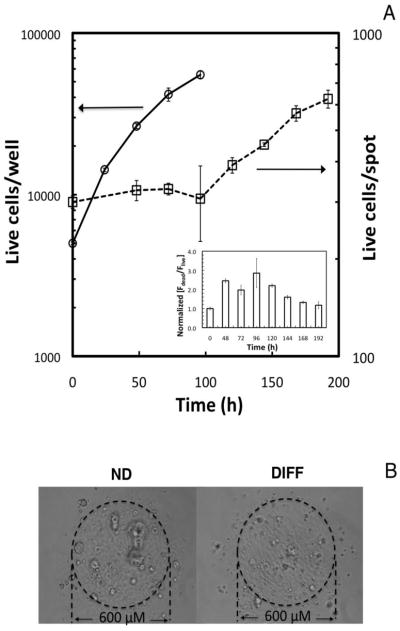
Comparison of ReNcell VM cell expansion in the 3D cellular microarray platform with expansion in conventional 2D culture platforms. (A) Cells seeded at 160 cells/mm2 in a 96-well microtiter plate (5000 cells/well; 2.5 × 104 cells/ml medium) grow exponentially with doubling times of 22 h, and no lag phase observed (open circles, right axis). Cells seeded in microcultures in alginate spots at 5000 cells/mm3 (5.8 × 104 cells/ml medium) demonstrate a 4-day lag, followed by a growth phase with slower proliferation (tD = 84 h) (open squares, left axis). The inset shows the changes in ReNcell VM viability during 3D culture, measured indirectly by following the ratio of average red-to-green fluorescence intensity with time, normalized to the first measurement taken after printing (typically 4 h after cell detachment). The data indicate a decrease in viability during the lag phase. Data represent the mean ± SEM. (B) Bright field images of the alginate microcultures (300 cells/spot; 5 × 106 cells/ml) after cell expansion on-chip using expansion medium left panel), and cells similarly expanded and subsequently differentiated for 6 days in differentiation medium (right panel). ND refers to non-differentiated cells, DIFF refers to differentiated cells.
Changes in cell viability in the 3D microarrays were gauged indirectly by following the ratio of average red-to-green fluorescence intensity with time, normalized to time zero, as seen in the inset of Fig. 1A (proportional to dead-to-live cell ratio). These data indicate that cell viability immediately after printing is ca. 50%. The viability then decreases during the lag phase of the growth curve, which is followed by a recovery to initial viability once cell death ceases.
Previous studies have shown similar sharp decreases in the viability of human embryonic stem cells (hESC) embedded in alginate microcapsules of roughly 500 μm in diameter, i.e., 3-orders of magnitude larger than our microarray spots (Chayosumrit et al. 2010). Specifically, the ability of the hESCs to maintain viability, proliferate and form clusters depended on the concentration and type of divalent cation used to form the alginate matrix (e.g., calcium or barium). These parameters are thought to affect viability by altering the permeability of the matrix to nutrients and growth factors. Additionally, the proliferation rates of the hNSCs in microarrays could be influenced by the chemistry of the 3D milieu and culture dimensionality. For example, Kisaalita and co-workers (Cheng et al. 2008; Wang & Kisaalita 2010) showed that neural progenitor cells (NPCs) cultured in 3D polystyrene scaffolds in 96-well plate assays grew as clusters with lower neurite density and shorter length than observed in 2D cultures, and also grew more slowly (approx. 50% of that in 2D). Additionally, Ashton et al. (2007) suggested that the growth rate of NPCs in an alginate hydrogel was slowed due to the small pore size. Regardless of the cause of decreased initial hNSC viability in the alginate-based microarrays, the cells undergoing expansion in the 3D microarrays do show formation of cell clusters. These clusters are seen in the bright field image of ReNcell VM cells encapsulated in an alginate microarray spot and cultured in expansion medium for 6 days (left-hand side of Fig. 1B). The figure also contains an image of a ReNcell microculture where the cells have been expanded and subsequently differentiated for 6 days (right-hand side of Fig 1B). In this case, cell clusters are seldom observed.
3.2 Validation of in-cell, on-chip microarray immunofluorescence assay for studying ReNcell VM fate
To quantify the levels of specific marker proteins that could help us determine the state of differentiation of the cells, we employed an in-cell, on-chip immunofluorescence methodology, similar to that used previously for mouse ESCs and several human carcinoma cell lines (Fernandes et al. 2009; Fernandes et al. 2008; Meli et al. 2012). As a validation of our cell-based assay, we first studied assay sensitivity and range for several marker proteins after printing the non-differentiated ReNcell VM hNSCs. We observed an increase in the fluorescence intensity of the SOX2 neural stem cell marker and the GAPDH internal control marker with increasing number of non-differentiated cells printed per spot (Fig. S2A). The number of cells per spot correlated linearly with the green fluorescence intensity for both markers over a range of cell densities (0–400 cells/spot) (Fig. S2B).
Similar results were obtained for the hNSCs subjected to a standard monolayer differentiation protocol for 6 days prior to microarray printing using Nestin (hNSC) and GFAP (astrocyte) markers, as well as GAPDH (Fig. S2C). It is important to note that when the marker signals are normalized to the internal control, GAPDH, the response is essentially constant regardless of the cell number (Fig. S2D). It is also important to note that the extent of nonspecific binding of the secondary antibody was evaluated by reproducing the immunofluorescence protocol, but omitting addition of a primary antibody. The weak response obtained was used to define background levels that were subtracted from the marker signal of interest prior to normalization with the internal control.
We then proceeded to use the in-cell, on-chip immunofluorescence assay to compare the levels of several neurodevelopmental stage-specific markers in hNSCs cultured on laminin-coated flasks in their proliferative non-differentiated state, to those in cells differentiated by removal of growth factors for 6 days. This relatively short induction process yielded striking differences in the morphological characteristics of the cells, which transition from a cobblestone morphology when confluent, to more rounded bodies with extensive outgrowths (Fig. 2A). These morphological changes were highlighted by staining the cells with Alexa Fluor 488 conjugated phalloidin (Fig 2E).
Figure 2.

Cell-based microarray immunofluorescence assay for quantification of marker proteins in ReNcell VM cells cultured on 2D platforms. (A) Bright field microscopy images of ReNcell VM cells expanded in monolayer culture for 3 days (upper panel) using expansion medium, and cells similarly expanded and subsequently differentiated for 6 days in differentiation medium (lower panel). ND indicates non-differentiated cells; DIFF indicates differentiated cells. (B) Western blot analysis showing astrocytic lineage commitment after 6 days of differentiation, as evidenced by the decrease in Nestin expression, and concomitant increase in GFAP signal (γ-Tubulin and GAPDH were used as loading control). (C) On-chip immunofluorescence assay of marker proteins in ReNcell VM cells expanded or differentiated in 2D culture platforms (off-chip) and subsequently printed for analysis with 300 cells/spot (5 × 106 cells/ml). Each spot has a diameter of 600 μm, as indicated. (D) Quantification of the normalized fluorescence intensity signal obtained from microarrays such as those illustrated in (C), using GAPDH as internal control (similar results obtained with γ-Tubulin normalization). Statistically significant differences between the normalized intensity of differentiated and non-differentiated cells are represented with (*) for p < 0.05. Data represent the mean ± SEM, with n ≥ 3 experiments, each with 48 replicate microcultures. (E) Expression of F-actin (green) by ReNcell VM cells expanded in monolayer culture for 3 days (upper panel) using expansion medium, and cells similarly expanded and subsequently differentiated for 6 days in differentiation medium (lower panel). Cell nuclei were counterstained with Hoechst 33342 (blue). ND indicates non-differentiated cells; DIFF indicates differentiated cells. Scale bars: 20 μm.
The ReNcell VM cell line has been shown to stain positive for the neural stem cell marker, Nestin, in its non-differentiated state (Donato et al. 2007). This was indeed the case for our cells, which show a relatively weak but distinct Nestin signal (normalized to GAPDH) in their proliferative state immediately after printing the cells onto the microarray (Fig. 2C and 2D). This signal is lost (p < 0.05) in cells that are not supplemented with growth factors, suggesting that the decrease in this multipotency marker is a sign of cell differentiation. As a reference, we compared our method with standard Western blotting (Fig. 2B). The cells show a strong band corresponding to Nestin when expanded in the presence of growth factors, whereas in the absence of growth factors the band disappears.
Concomitant with the decrease in Nestin, we observe a sharp increase in the levels of the GFAP glial marker, both in Western analysis and via in-cell, on-chip immunofluorescence. This increase signals astrocytic commitment of the ReNcell VM progeny after applying the monolayer differentiation protocol. Interestingly, ReNcell VM is GFAP+ even prior to mitogen withdrawal, while still exhibiting neural progenitor cell characteristics and expressing Nestin. Similar marker profiles have been reported during the early stages of neural commitment of stem cells (Rieske et al. 2007; Buzanska et al. 2009). For example, Rieske et al. (Rieske et al. 2007) demonstrated that a population of GFAP+ cells derived from fetal human brain parenchyma express both Nestin and βIII-Tubulin in their initial proliferative phase, and can later generate two distinct progenies, one of which is of glial lineage with up-regulated GFAP expression and down-regulated Nestin and βIII-Tubulin expression. Indeed, the expression of the neuronal marker βIII-Tubulin is evident in ReNcell VM prior to differentiation (Fig. 2), when the cells are still supplemented with growth factors. The signal decreases substantially as the cells commit to a glial lineage. In this regard, it should be noted that in the case of hESC-derived neural progenitors, it has been suggested by Krencik et al. (2011) that progenitors first give rise to neurons and then to astrocytes. While this may seem to indicate that this procedure is limited to glial differentiation, Donato et al. (2007), working with the same cell line, showed that it is possible to generate mainly neurons by altering the differentiation process to include a pre-aggregation step in the differentiation process.
The hNSCs cultured under both expansion and differentiation conditions appear to express low levels of the oligodendrocyte marker GalC. Western blot results suggest very little change of GalC upon differentiation, while in-cell, on-chip immunofluorescence indicates a small, but statistically significant, decrease in GalC expression. However, as shown in Fig. 2C, the anti-GalC primary antibody used causes significantly more nonspecific binding than the other antibodies, particularly in the film surrounding the alginate spots, and actually leads to negative values of the signal after differentiation when subtracting the background (secondary-only control) from the primary signal (Fig. 2D). It should be noted that the choice of antibodies for the in-cell, on-chip immunofluorescence assays is critical, especially with regard to non-specific binding, which can result in significant levels of background fluorescence, as can be seen with GalC (Figs. 2C and 3A). This increased background may impact in-cell data, affecting its ability to correlate quantitatively with Western data.
Figure 3.

On-chip, in-cell immunofluorescence assay for quantification of marker proteins in ReNcell VM cells cultured in the 3D microarray platform. (A) Scanning images of portions of the microcultures containing ReNcell VM cells (seeded at 300 cells/spot) after expansion or differentiation on-chip, followed by immunoassay of marker proteins. (B) Quantification of marker protein expression in differentiated and non-differentiated cells through analysis of the normalized fluorescence intensity signal obtained from the scanning microarray images using GAPDH as the internal control. Statistically significant differences between the normalized intensity of differentiated and non-differentiated cells is represented with (*) for p < 0.05. Data represent the mean ± SEM, with n ≥ 3 experiments, each with 48 replicate microcultures.
Once we had demonstrated the concordance between the in-cell, on-chip immunofluorescence assay and Western blotting, we endeavored to assess stem cell fate on-chip. To that end, we sought to evaluate how established marker profiles might change when the cells were cultured on-chip with or without growth factor supplementation. Cells were allowed to overcome the lag phase of growth observed in the microarrays by expanding them for 6 days on-chip before assaying. In the case of the differentiated cells, they were cultured on-chip for 3 days in expansion medium before inducing differentiation for an additional 6 days, and subsequently assayed. Qualitatively similar marker profiles are observed in the proliferative and differentiated cell populations cultured on-chip (Fig. 3) as those observed when the cells are cultured off-chip in laminin-coated flasks and then printed immediately before assaying (Fig. 2C and 2D). Specifically, we observe a decrease in Nestin and βIII-Tubulin expression and a large increase in GFAP expression upon differentiation, suggesting once again the generation of a cell population primarily composed of cells differentiated towards astrocytic phenotype after induction. Interestingly, on-chip expansion appears to lead to higher βIII-Tubulin and GalC signals than cells proliferating off-chip. The decrease in expression of the neuronal marker upon differentiation is less abrupt in the 3D microarrays than in conventional cultures, whereas the GalC signal drops off to zero, in both cases.
The marker profile results depicted in Fig. 2D and 3B do not change substantially when using γ-Tubulin to normalize the marker signals (results not shown), indicating that GAPDH serves as an adequate control for the ReNcell VM cells and their progeny. This is further illustrated in Fig. S3, where we show the evolution of Nestin and GFAP expression during expansion and differentiation, using three different internal controls to normalize the signals, GAPDH, γ-Tubulin, and COXIV, all showing similar trends.
It is also important to note that the viability of cells cultured on-chip tends to be lower than that of their off-chip counterparts, particularly in the case of differentiated cells, where a 3-fold increase in red-to-green fluorescence intensity is observed (results not shown). Besides the lag phase in growth and the slower proliferation rates observed on-chip, this is also likely a result of the entrapment of dead cells within the alginate matrix. For the off-chip cultures, detached dead cells are regularly removed as the medium is replenished, whereas on-chip these cells are entrapped in the 3D network until they degrade. This is of particular importance during differentiation, where we observed significant cell death and no proliferation. In addition to modifying the matrix chemistry and permeability, the incorporation of survival factors, like GDNF and BDNF, in the differentiation medium could help address the low viability observed (Miljan 1992).
3.3 Differential effect of test compounds on cytotoxicity of ReNcell VM and differentiated progeny
There is a rapidly emerging need to develop in vitro, high-throughput screens to assess neurotoxicity of drugs and chemicals at different stages of neural development. Cell-based models that mimic the in vivo environment are ideal to predict the differential response of various populations of cells in the brain to a given compound, at different stages of development.
In this context, we wanted to assess whether our cell-based platform could be used to probe in vitro cytotoxicity of a population of hNSCs and its differentiated progeny in a high throughput manner, when challenged with five model compounds. We chose three developmental neurotoxicants (cadmium chloride, retinoic acid and dexamethasone), an antiproliferative anti-cancer agent (5-fluorouracil), and a compound that should have no effect on the developing central nervous system (acetaminophen).
As before, the printed cells were allowed to overcome the lag phase of growth and, in the case of the cells differentiated towards an astrocytic phenotype, differentiated for 6 days before being challenged with the compounds for a period of 24 h. This was followed by an additional incubation with either expansion or differentiation medium for 2 days, before determining cytotoxicity via a live/dead cell viability assay. MTT analysis indicated that the DMSO at the concentration used in these analyses (0.5%) was not toxic, yielding cell viability of 100.8 ± 4.6%, compared with cells grown in the absence of DMSO, which had a cell viability of 100.0 ± 11.3%.
The dose-response curves of the five compounds for both populations of cells are depicted in Fig. 4 and a summary of IC50 values are given in Table 1. No statistically significant difference was observed between the log IC50 values of the two cell populations for retinoic acid, cadmium chloride, and 5-fluorouracil, with the latter two being the most toxic chemicals overall. The effect of retinoic acid and cadmium chloride on hNSCs, as measured by the IC50 values, is consistent with the results of Breier et al. (Breier et al. 2008a) who studied the closely related ReNcell CX neural progenitor cell line (non-differentiated) in 2D well plate experiments. On the other hand, these authors reported that 5-fluorouracil inhibited proliferation without affecting viability after 24 h of incubation with the drug. The difference may be related to the fact that in our experimental protocol the cells are incubated in medium for 2 days after being challenged with the compounds for 24 h, and the dose responses are quantified in terms of the percentage of live cells (relative to the growth of the cells in the absence of the chemicals). Thus, our cytotoxicity assays involve both inhibition of cell proliferation and decrease in cell viability. Importantly, the IC50 values of 5-fluorouracil, extracted from the dose response curves of the ReNcell VM and its astrocytic derivatives (8.4 and 5.2 μM, respectively), coincide well with literature values for various cancer cell lines (IC50 = 1 – 85 μM) (Chauffert & Dimanche-Boitrel 1998; Lee et al. 2008; Meli et al. 2012). Finally, we compared dexamethasone and acetaminophen in 2D ReNcell VM cultures in a 96-well plate format. The IC50 values were 215 and 1760 μM, respectively. These numbers are lower than that obtained in 3D (Table 1), which is largely consistent with our previous studies comparing 2D and 3D for cytotoxicity (Lee et al. 2008).
Figure 4.

Comparison of the dose response of ReNcell VM cells and their differentiated progeny (following 6 days of growth factor removal) to 3 developmental neurotoxicants (retinoic acid-(A), cadmium chloride-(B) and dexamethasone-(C)), and a non-neurotoxicant (acetaminophen-(E)). Cytotoxicty was determined with a live/dead cell viability assay and the data were fitted to a sigmoidal dose response to obtain the IC50 values. The figures show representative curves where each data point is the mean of 48 replicates ± SEM.
Table 1.
Response of ReNcell VM cells and their differentiated progeny to various compounds, as measured by their IC50 values.
| Compound | Non-differentiated | Differentiated | ||
|---|---|---|---|---|
| log (IC50 μM) | IC50 (μM) | log (IC50 μM) | IC50 (μM) | |
| 5-Fluorouracil | 0.92 ± 0.23 | 8.40 | 0.70 ± 0.06 | 5.20 |
| CdCl | 0.87 ± 0.49 | 7.36 | 0.98 ± 0.04 | 10.2 |
| Dexamethasone * | 3.11 ± .09 | 1920 | 1.60 ± 0.09 | 58.5 |
| Retinoic acid | 1.18 ± 0.38 | 15.6 | 1.00 ± 0.81 | 10.4 |
| Acetaminophen* | 3.38 ± 0.02 | 2450 | 4.11 ± 0.08 | 14100 |
Statistically significant changes in log IC50 values after differentiation were observed for dexamethasone and acetaminophen (p < 0.05, represented with (*)). The data represents the mean ± SEM, with n=3 experiments, where each concentration had 48 replicate microcultures.
Proliferating ReNcell VM cells challenged with high concentrations of acetaminophen (0.02–20 mM) show greater susceptibility to this analgesic than the cells differentiated towards an astrocytic phenotype (IC50 values are 2.4 and 14.1 mM, respectively). Both of these concentrations, however, are higher than the toxic blood concentrations required to cause hepatotoxic effects (~ 0.2 – 1 mM) (Clemedson et al. 2007). As expected, the viability of the cells below the hepatotoxic range of concentrations remains unaffected for both the hNSC line and its progeny, and is consistent with reports in the literature (Breier et al. 2008b; Buzanska et al. 2009). Additionally, acetaminophen has previously been shown to be non-toxic to a hepatoma cell line (Hep3B), when P450 metabolism was absent (Lee et al. 2008).
Interestingly, the differentiated cells were more sensitive to dexamethasone than the hNSC population. Specifically, there is a marked differentiation-specific toxicity to dexamethasone, where the IC50 value of the differentiated cells is two orders of magnitude lower than that of the hNSCs (0.06 mM to 2 mM, respectively). The relatively high chemoresistance of the hNSCs observed is consistent with the negligible effect on viability and limited impact on proliferation seen by Brier and coworkers for the ReNCell CX cell line challenged with concentrations of dexamethasone of up to 100 μM (Breier et al. 2008b).
The panel of compounds chosen for testing helps illustrates the capabilities of our ReNcell VM-based microarray platform for neurotoxicity screening. Our results demonstrate the ability of the 3D cellular microarray system to discriminate between toxic and non-toxic compounds. This is evidenced by the low toxicity shown by the microarray cultures to acetaminophen (extremely high IC50 values). In contrast, the response to the antiproliferative and neurotoxic compounds tested seems to be consistent overall with published data on hNSC lines, thus demonstrating adequate sensitivity to detect adverse effects of target compounds (Breier et al. 2008b; Buzanska et al. 2009; Longley et al. 2003). Moreover, the statistically significant difference between the IC50 values of dexamethasone and acetaminophen in non-differentiated hNSCs and differentiated glial cells suggests our platform is capable of detecting cell type-dependent toxicities.
It is important, however, to point out that our expectation was to find higher sensitivity of the proliferating hNSC population to these compounds (Paquette et al. 2008; Buzanska et al. 2009), since all of them have been reported to exhibit antiproliferative activity (Breier et al. 2008b; Buzanska et al. 2009), and as such might be expected to compromise cell division and perhaps induce differentiation (Dietrich et al. 2006). This is particularly the case with retinoic acid, as this compound is known to induce differentiation in a wide variety of neural model systems and embryonic stem cells (Martin-Ibanez et al. 2007; Rajasingh & Bright 2006). Nonetheless, precedence exists for studies that show toxicity is not necessarily limited to rapidly dividing cells. For example, Noble and coworkers demonstrated that non-dividing oligodendrocytes were as sensitive as neural progenitors to chemotherapeutic agents carmustine and cisplatin (Nutt et al. 2000). Moreover, the authors showed that cell division by itself was insufficient to confer vulnerability, as rapidly dividing hNSCs were more resistant than progenitor cells.
4. Conclusions
The characterization and safety assessment of drugs, drug candidates, environmental chemicals, etc., requires extensive resources, particularly when animal studies are employed. Neurotoxicity detection is particularly tedious, since regulatory authorities solely use in vivo methods for adult and developmental neurotoxicity testing (Bal-Price et al. 2010). The majority of commercial chemicals have not been evaluated for developmental neurotoxicity. Thus, there is a need to develop new in vitro techniques to predict which compounds pose an increased threat to human health and should therefore be prioritized for further screening and evaluation.
In this work, we used a 3D cellular microarray platform for high throughput analysis of hNSC differentiation and toxicity screening. The hNSC microarray-based system presents several advantages over more conventional approaches. First of all, it uses an immortalized hNSC line, ReNcell VM, that proliferates readily and allows us to generate a large number of cells of human origin. This cell line has also been shown to differentiate into all three neural subtypes (Donato et al. 2007), yielding mainly GFAP+ cells, followed by βIII-tubulin+ cells and a small minority of O1+ cells upon growth factor removal. In the same paper, Donato et al. used a pre-aggregation step to yield mainly dopaminergic neurons. Given this ability to produce both cell types in abundance and their immortalized nature, the ReNcell VM cell line offers a good source of neural cells and as such could represent an appropriate model for the developing brain, as well as for neuronal and glial lineages. Moreover, the technology uses sensitive fluorescence-based immunoassays and live/dead cell viability assays specifically modified for the microarray format to screen relevant endpoints, such as cytotoxicity/viability, self renewal/differentiation, and changes in protein expression levels that indicate shifts in important metabolic and signaling pathways. All of this is done in a miniaturized 3D culture platform that is expected to more closely mimic in vivo environments and allows the screening of compounds in a high throughput, highly parallel fashion.
Using this approach we showed that the ReNcell VM cell line can be cultivated in 3D microarray alginate spots, and preserve its proliferative progenitor state during on-chip expansion, albeit with lower proliferation rates and viabilities than those observed in conventional 2D culture platforms. Additionally, through the use of an in-cell, on-chip immunofluorescence assay to monitor levels of neurodevelopmental markers, we demonstrated that the ReNcell VM cells initiate differentiation into an astrocytic lineage following a standard 6 day differentiation protocol that consists of growth factor removal. Moreover, the population of cells generated on-chip has a similar marker prolife to cells that are cultured and differentiated in 2D monolayers. Finally, we showed that our platform can serve for high throughput predictive toxicology screening of drugs and environmental chemicals by demonstrating the cell type-dependent detection of adverse effects of neurotoxic and antiproliferative compounds on cell proliferation and viability, as well as the null effect of a non-toxic compound at relevant concentrations. A better understanding of the differential responses of human stem cells compared with terminally differentiated cell types against chemicals is vital to identify the toxic effects of such chemicals on the human body and in specific organs, and to ensure retention of healthy adult stem cell populations, which play a critical role in tissue function and homeostasis. Future endeavors will focus on the effects of potential neurotoxic compounds on key signaling pathways involved in hNSC proliferation (self-renewal) and differentiation; and efforts to obtain pure populations of differentiated cells.
Supplementary Material
Highlights.
We employed a new 3D platform for neural stem cell growth and differentiation.
We showed that this microplatform can discern differential small molecule toxicity.
We followed stem cell differentiation using an on-chip immunofluorescence assay.
We studied differential responses of stem cells vs. differentiated cells.
Acknowledgments
The authors would like to thank Dr. Sandrine Lavenus and Dr. Seok Joon Kwon for important discussions. This research was supported by the National Institutes of Health (ES-020903). HSCB acknowledges support from the Portuguese Government (Fundação para a Ciência e a Tecnologia).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams J, Lammer EJ. Relationship between dysmorphology and neuropsychological function in children exposed to isotretinoin “in utero”. In: Fujii T, Boer CJ, editors. Functional Neuroteratology of Short-Term Exposure to Drugs. Tokyo: Teikyo University Press; 1991. pp. 59–70. [Google Scholar]
- Andersen ME, Krewski D. Toxicity testing in the 21st century: bringing the vision to life. Toxicological sciences : an official journal of the Society of Toxicology. 2009;107(2):324–30. doi: 10.1093/toxsci/kfn255. [DOI] [PubMed] [Google Scholar]
- Ashton RS, et al. Scaffolds based on degradable alginate hydrogels and poly(lactide-co-glycolide) microspheres for stem cell culture. Biomaterials. 2007;28(36):5518–5525. doi: 10.1016/j.biomaterials.2007.08.038. [DOI] [PubMed] [Google Scholar]
- Bal-Price AK, et al. Relevance of in vitro neurotoxicity testing for regulatory requirements: challenges to be considered. Neurotoxicology and teratology. 2010;32(1):36–41. doi: 10.1016/j.ntt.2008.12.003. [DOI] [PubMed] [Google Scholar]
- Baud O. Postnatal steroid treatment and brain development. Archives of Disease in Childhood: Fetal And Neonatal Edition. 2004;89:F96–100. doi: 10.1136/adc.2003.028696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdahn U, et al. Therapy of malignant brain tumors: Comparison of the in vitro activities of vidarabin-monophosphate, BCNU and 5-fluorouracil. Acta Neurologica Scandinavica. 1987;75:28–36. doi: 10.1111/j.1600-0404.1987.tb07885.x. [DOI] [PubMed] [Google Scholar]
- Breier JM, et al. Development of a high-throughput screening assay for chemical effects on proliferation and viability of immortalized human neural progenitor cells. Toxicological sciences : an official journal of the Society of Toxicology. 2008;105(1):119–33. doi: 10.1093/toxsci/kfn115. [DOI] [PubMed] [Google Scholar]
- Breier JM, et al. Neural progenitor cells as models for high-throughput screens of developmental neurotoxicity: state of the science. Neurotoxicology and teratology. 2010;32(1):4–15. doi: 10.1016/j.ntt.2009.06.005. [DOI] [PubMed] [Google Scholar]
- Buzanska L, et al. A human stem cell-based model for identifying adverse effects of organic and inorganic chemicals on the developing nervous system. Stem cells (Dayton, Ohio) 2009;27(10):2591–601. doi: 10.1002/stem.179. [DOI] [PubMed] [Google Scholar]
- Chauffert B, Dimanche-Boitrel M. New insights into the kinetic resistance to anticancer agents. Cytotoxicology. 1998;27:225–235. doi: 10.1023/A:1008025124242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chayosumrit M, Tuch B, Sidhu K. Alginate microcapsule for propagation and directed differentiation of hESCs to definitive endoderm. Biomatierials. 2010;31:505–514. doi: 10.1016/j.biomaterials.2009.09.071. [DOI] [PubMed] [Google Scholar]
- Cheng K, Lai Y, Kisaalita WS. Three-dimensional polymer scaffolds for high throughput cell-based assay systems. Biomaterials. 2008;29:2802–2812. doi: 10.1016/j.biomaterials.2008.03.015. [DOI] [PubMed] [Google Scholar]
- Clemedson C, Kolman A, Forsby A. The integrated acute systemic toxicity project (ACuteTox) for the optimisation and validation of alternative in vitro tests. Alternatives to Lab Animals. 2007;35:33–38. doi: 10.1177/026119290703500102. [DOI] [PubMed] [Google Scholar]
- Costa LG, Fattori V, Giordano G. An in vitro approach to assess the toxicity of certain food contaminants: Methylmercury and poly-chlorinated biphenyls. Toxicology. 2007;237:65–76. doi: 10.1016/j.tox.2007.05.003. [DOI] [PubMed] [Google Scholar]
- Dietrich J, et al. CNS progenitor cells and oligodendrocytes are targets of chemotherapeutic agents in vitro and in vivo. Journal of Biology. 2006;5(22):1–23. doi: 10.1186/jbiol50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donato R, et al. Differential development of neuronal physiological responsiveness in two human neural stem cell lines. BMC neuroscience. 2007;8:36. doi: 10.1186/1471-2202-8-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes TG, et al. High-throughput cellular microarray platforms: applications in drug discovery, toxicology and stem cell research. Trends in biotechnology. 2009;27(6):342–9. doi: 10.1016/j.tibtech.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes TG, et al. On-chip cell-based microarray immunofluorescence assay for high-throughput analysis of target proteins. Analytical Chemistry. 2008;80(17):6633–6639. doi: 10.1021/ac800848j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krencik R, et al. Specification of transplantable astroglial subtypes from human pluripotent stem cells. Nature biotechnology. 2011;29(6):528–34. doi: 10.1038/nbt.1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laustriat D, Gide J, Peschanski M. Human pluripotent stem cells in drug discovery and predictive toxicology. Biochemical Society transactions. 2010;38(4):1051–7. doi: 10.1042/BST0381051. [DOI] [PubMed] [Google Scholar]
- Lee M-Y, et al. Three-dimensional cellular microarray for high-throughput toxicology assays. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(1):59–63. doi: 10.1073/pnas.0708756105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longley DB, Harkin DP, Johnston PG. 5-Fluorouracil: Mechanisms of Action and Clinical Strategies. Nature reviews. Cancer. 2003;3(5):330–8. doi: 10.1038/nrc1074. [DOI] [PubMed] [Google Scholar]
- Martin-Ibanez R, et al. Interplay of leukemia inhibitory factor and retinoic acid on neural differentiation of mouse embryonic stem cells. Journal of Neuroscience Research. 2007;85:286–2701. doi: 10.1002/jnr.21228. [DOI] [PubMed] [Google Scholar]
- Meli L, et al. Influence of a three-dimensional, microarray environment on human cell culture in drug screening systems. Biomaterials. 2012;33(35):9087–96. doi: 10.1016/j.biomaterials.2012.08.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miljan EA. ReNcell human neural progenitors : Renewable and Consistent Supply of Human Functional Neurons. Millipore Technical Publications; 1992. [Google Scholar]
- National Research Council. Toxicity testing in the 21st century: a vision and a strategy. Washington, DC: National Academies Press; 2007. [Google Scholar]
- Nutt CL, et al. Differential expression of drug resistance genes and chemosensitivity in glial cell lineages correlate with differential response of oligonderogliomas and astrocytomas to chemotherapy. Cancer Research. 2000;60:4812–4818. [PubMed] [Google Scholar]
- Paquette JA, et al. Assessment of the embryonic stem cell test and application and use in the pharmaceutical industry. Birth Defects Research Part B. 2008;83:104–111. doi: 10.1002/bdrb.20148. [DOI] [PubMed] [Google Scholar]
- Rajasingh J, Bright JJ. 15-Deoxy-delta12, 14-prostaglanding J2 regulates leukemia inhibitory factor signaling through JAK-STAT pathway in mouse embryonic stem cells. Experimental Cell Research. 2006;312:2538–2546. doi: 10.1016/j.yexcr.2006.04.010. [DOI] [PubMed] [Google Scholar]
- Rieske P, et al. A population of human brain parenchymal cells express markers of glial, neuronal and early neural cells and differentiate into cells of neuronal and glial lineages. The European journal of neuroscience. 2007;25(1):31–7. doi: 10.1111/j.1460-9568.2006.05254.x. [DOI] [PubMed] [Google Scholar]
- Saunders JB, Wright N, Lewis KO. Predicting outcome of paracetmol poisoning by using 14C-aminopyrine breath test. British Medical Journal. 1980;280:279–280. doi: 10.1136/bmj.280.6210.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trosko JE, Chang C. Factors to consider in the use of stem cells for pharmaceutic drug development and for chemical safety assessment. Toxicology. 2010;270:18–34. doi: 10.1016/j.tox.2009.11.019. [DOI] [PubMed] [Google Scholar]
- Wang L, Kisaalita WS. Characterization of micropatterned nanofibrous scaffolds for neural network. Journal of Biomedical Materials Research Part B: Applied Biomaterials. 2010;94B:238–249. doi: 10.1002/jbm.b.31646. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.