

Abstract
The EGFR monoclonal antibody cetuximab is the only approved targeted agent for treating head and neck squamous cell carcinoma (HNSCC). Yet resistance to cetuximab has hindered its activity in this disease. Intrinsic or compensatory HER3 signaling may contribute to cetuximab resistance. To investigate the therapeutic benefit of combining MM-121/SAR256212, an anti-HER3 monoclonal antibody, with cetuximab in HNSCC, we initially screened twelve HNSCC cell lines for total and phosphorylated levels of the four HER receptors. We also investigated the combination of MM-121 with cetuximab in preclinical models of HNSCC. Our results revealed that HER3 is widely expressed and activated in HNSCC cell lines. MM-121 strongly inhibited phosphorylation of HER3 and AKT. When combined with cetuximab, MM-121 exerted a more potent anti-tumor activity through simultaneously inhibiting the activation of HER3 and EGFR and consequently the downstream PI3K/AKT and ERK pathways in vitro. Both high and low doses of MM-121 in combination with cetuximab significantly suppressed tumor growth in xenograft models and inhibited activations of HER3, EGFR, AKT and ERK in vivo. Our current work is the first report on this new combination in HNSCC and supports the concept that HER3 inhibition may play an important role in future therapy of HNSCC. Our results open the door for further mechanistic studies to better understand the role of HER3 in resistance to EGFR inhibitors in HNSCC.
Keywords: Cetuximab, MM-121, HNSCC, HER3, EGFR
Introduction
Head and neck cancer (HNC) is the fifth most common form of cancer and causes over 350,000 deaths globally each year (1). Head and neck squamous cell carcinoma (HNSCC) patients account for approximately 90% of all head and neck malignancies (2).
Epidermal growth factor receptor (EGFR) belongs to the human epidermal growth factor receptor (HER) family which includes other three closely related members: HER2, HER3 and HER4. EGFR mRNA (3) and protein (4) are upregulated in 80-90% and 40% of HNSCC cases respectively, and are positively correlated with poor prognosis, advanced disease and reduced survival (5). Cetuximab is a monoclonal anti-EGFR antibody which was approved for the treatment of HNSCC in 2006. Two landmark studies confirmed that treatment with cetuximab plus radiotherapy or chemotherapy resulted in an improvement of survival in comparison with radiotherapy or chemotherapy alone in HNSCC patients (6, 7). Despite the success, the overall response rate to cetuximab as a single agent does not exceed 13% with a response duration less than 70 days (8). In addition, a number of patients frequently display primary resistance to EGFR monoclonal antibodies; acquired resistance may also emerge over time (9, 10). Possible mechanisms for de novo and acquired resistance to cetuximab include mutations in the KRAS, BRAF and NRAS genes (9), a secondary mutation (S492R) in the extracellular domain of EGFR receptor (9, 10), overexpression of the MET proto-oncogene (c-Met) (11), and in HNSCC, the expression of the in-frame deletion mutation of EGFR variant III (12).
Recently, an increasing body of literature has suggested that resistance to anti-EGFR therapy arises frequently through activation of alternative signaling pathways that bypass the original target (13, 14). Compensatory HER3 signaling and sustained PI3K/AKT activation are associated with sensitivity and resistance to anti-EGFR targeted therapies, especially in HNSCC (13-16). Unlike other HER receptors, HER3 has diminished intracellular kinase activity but has known ligands. These characters make HER3 an obligate heterodimerization partner for other HER receptors (16). HER3 contains six PI3K binding sites that are crucial for PI3K/AKT pathway activation (16). A preclinical study reported an association between sensitivity to gefitinib and the overexpression of HER3 in HNSCC cell lines (17). Furthermore, after sustained exposure to gefitinib or erlotinib, cells showed upregulated HER3 and AKT phosphorylation, which correlated with HER3 translocation from the nucleus to the membrane (15). Increased expression of heregulin (HRG), a potent HER3 ligand, also provided a possible mechanism of cetuximab resistance in colorectal cancer (18). There is a recent evidence reported that HER3 signaling plays an important role in acquired resistance to cetuximab, perhaps a more crucial one in comparison with MET in HNSCC and non-small cell lung cancer (13). Direct targeting of HER3 by siRNA in cetuximab-resistant cells has been shown to restore cetuximab sensitivity (13). These data suggest an opportunity to develop combinatorial strategies by using cetuximab and anti-HER3 agent in HNSCC.
MM-121 (SAR256212) is a fully human antibody that directly binds to the extracellular domain of HER3 (19, 20) and induces receptor downregulation resulting in the inhibition of downstream HER3-dependent pathways. As MM-121 has not previously been tested in HNSCC, we were interested in exploring its activity as a single agent and in combination with cetuximab in preclinical models of HNSCC. Overall, we found that HER3 was active in the majority of HNSCC cell lines, a combination of EGFR and HER3 inhibition provided improved antitumor activity relative to either inhibitor alone, and the combination effectively inhibited signaling through both ERK and PI3K/AKT pathways in vitro and in vivo.
Materials and Methods
Cell lines and reagents
Cetuximab was obtained from ImClone (New York, NY) and MM-121/SAR256212 was provided by Merrimack Pharmaceuticals (Cambridge, MA) and Sanofi (Bridgewater, NJ). Human HNSCC Tu212 cell line was provided by Gary L. Clayman (University of Texas M.D. Anderson Cancer Center, Houston, TX) in 2002 (21, 22). Human HNSCC cell lines SCC2, SCC47, JHU-012, 93-VU-147T, PCI-13, PCI-15A, PCI-15B, and UM22B were kindly given by Dr. Robert Ferris in 2012 and SCC090 by Dr. Susanne Gollin from University of Pittsburgh in 2010, respectively. Human HNSCC cell lines SCC1483 and SQCCY1 were obtained from Dr. Shi-Yong Sun at Emory University in 2012. SCC2, SCC47, SCC090, 93-Vu-147T, and SCC1483 cells are positive for human papilloma virus (HPV) (23, 24). Most cell lines were maintained in Dulbecco's modified Eagle's medium (DMEM)/F12 (1:1), SCC090 in Minimum Essential Media (MEM) and JHU-012 in RPMI-1640 medium all supplemented with 10% FBS at 37°C, 5% CO2. All cells were routinely screened for mycoplasma contamination by MycoAlert™ Mycoplasma Detection Kit (Lonza Ltd, Allendale, NJ). The authenticity of cell line Tu212, SCC2, SCC47, SCC090, 93-VU-147T, JHU-012, PCI-13, SCC1483, and SQCCY1 was verified through genomic short tandem repeat (STR) profile by RADIL (Research Animal Dignostic Lab), University of Missouri in September 2009, and by Emory University Integrated Genomics Core (EIGC) in October 2013, respectively. Authenticity of PCI-15A, PCI-15B, and UM22B was not done by the authors, but reported by Zhao et al. in 2011, using the same STR profile (22).
Colony formation assay
Cells were plated in 6-well culture plates at the concentration of 200 per well. After 24h incubation, cells were treated with PBS, 2μg/mL cetuximab, 20μg/mL MM-121 or the cetuximab and MM-121combination (CM combination) for 9 days to form colonies as previously described (25). The dose of cetuximab was chosen from our previous study (25) and the dose of MM-121 was chosen from an escalating serial doses which showed similar trend of synergistic effect in combination with cetuximab (data not shown). Medium was changed every three days. The colonies were then stained with 0.2% crystal violet with buffered formalin (Sigma). Colony numbers were manually counted using Image J software. Cell numbers ≥50 were considered as a colony.
Cell proliferation assay
The inhibition of cell proliferation by cetuximab and MM-121 was analyzed by a cell proliferation assay as previously described (26). Briefly, 2.5 × 105 cells were seeded in 60 mm dishes and incubated overnight. Cells were then treated with PBS, 62μg/mL cetuximab, 125μg/mL MM-121, and the combination for 72 hours. The dose of MM-121 and cetuximab was chosen based on previous studies (19, 25) and our SRB assay (Sulforhodamine B cell proliferation assay) results (Supplementary Fig. S1). Cells were harvested by trypsinization and counted using a cell counter (Beckman Coulter, Fullerton, CA). All the experiments were performed in triplicate.
Flow cytometry analysis of cell cycle and apoptosis
Cells were treated with the two drugs and their combination for 24, 48, 72 and 96 hours, respectively. The doses were chosen based on the previous studies (19, 25) and were consistent with the cell proliferation assay. Cell cycle and apoptosis analysis using a fluorescence-activated cell sorting flow cytometer (Becton Dickinson, Franklin Lakes, NJ) were described previously (25). Briefly, for cell cycle analysis, cells were trypsinized and fixed in pre-cooled 70% ethanol for at least 30 minutes. After spinning down, the cells were resuspended and incubated in 500 μL PI/RNASE staining buffer (BD Pharmingen™) for 15-30 minutes at room temperature (RT) before analysis. For apoptosis assay, cells were collected and prepared by using PE Annexin V Apoptosis Detection Kit (BD Pharmingen™) according to the manufacturer's instructions. FlowJo software was used for data analysis.
Western blot analysis
Cells were treated with the two drugs and their combination with doses consistent with the cell proliferation assay for the indicated time. Cells lysates and xenograft tissue lysates were generated using lysis buffer as previously reported (27). The lysate was centrifuged at 16,000 g at 4°C for 10 min. 50 micrograms of total protein for each sample were separated by 10% SDS-PAGE and transferred onto a Westran S membrane (Whatman Inc. Floham Park, NJ). Desired proteins were probed with corresponding antibodies. Rabbit anti-human AKT, pAKT, pERK, pS6, and pHER3 (1:1000 dilutions) were purchased from Cell Signaling, mouse anti-human β-actin (1:10000 dilution) from Sigma (St. Louis, MO), anti-human EGFR and HER3 antibodies from Santa Cruz (Santa Cruz, CA), and anti-human pEGFR antibody from Millipore (Temcula CA). HRP-conjugated secondary anti-mouse and anti-rabbit IgG (H+L) was obtained from Promega (Madison, WI). Bound antibody was detected using the SuperSignal West Pico Chemoluminescence system (Pierce, Inc., Rockford, IL). Image J software was used for blot quantification.
In vivo xenograft treatment study
The animal experimental protocol was approved by the Institutional Animal Care and Use Committees of Emory University. In brief, 4×106 Tu212 and 2×106 SCC47 cells (1:1 in matrigel) were injected subcutaneously into female nude mice (athymic nu/nu, Taconic, NY) aged 4 to 6 weeks. Mice were randomly divided into 6 groups after tumor formation: PBS control, cetuximab 6.25μg/dose, MM-121 300μg/dose (MM-121.LD), MM-121 600μg/dose (MM-121.HD), combination with low dose MM-121 (comb. LD) and combination with high dose MM-121 (comb. HD) (n=7 for each treatment group). Doses were chosen based on previous studies (19, 25). Drugs were given by intraperitoneal injection (I.P.) twice a week. Tumor volume and bodyweight were measured three times a week. Tumor volume was calculated using the formula: V= π/6×larger diameter ×(smaller diameter)2 as reported previously (25). Major organs were harvested for toxicity evaluation by hematoxylin and eosin (H&E) staining.
Immunohistochemistry (IHC) staining and analysis
Xenograft tissues were harvested, fixed in 10% buffered formalin and embedded in paraffin. Areas of necrosis were quantified under 40× as a percentage on routinely H&E stained slides. Staining of Ki-67 (prediluted; Biomeda, Foster, CA), TUNEL (Promega, Madison, WI) and CD34 (Abcam, Cambridge, MA) were performed as previously described (25). Staining and fluorescent signals from each assay were visualized by Olympus BX41 microscopy. For Ki-67 and TUNEL, the percentage of nuclei labeled cells was counted in 5 randomly and sequentially selected areas from each slide at 100× magnification. Positive CD34 signals were counted in 5 random fields under 100× magnification and microvessels were quantified as described previously (28). Briefly, only vessels containing apparent lumen that were positively stained with CD34 were counted. Necrotic areas were excluded from analysis. These quantifications were determined by at least 2 individuals blindly and independently.
Statistical analyses
Comparison of means from multiple treatment groups was carried out using one-way ANOVA or Kruskal-Wallis test to determine the significance of tumor growth inhibition among treatment groups. A Bonferroni correction was introduced to correct for multiple comparisons. The pairwise comparison was used to compare mean tumor volumes of cell growth inhibition between the different groups over time. Mean values of in vitro colony formation assay, cell proliferation assay and in vivo tumor volumes were used for calculation of the corresponding synergistic indices (SI) using the methods described before (29). A SI of greater than one indicates a synergistic effect. Statistical analyses were conducted using SPSS version 20. All p values were two-sided and p values less than 0.05 were considered significant.
Results
HER3 is widely expressed and activated in HNSCC cell lines
To determine the total and phosphorylated expression levels of HER3 in HNSCC, 12 HNSCC cell lines were screened by immunoblotting in our study. All 12 cell lines expressed total HER3 to variable levels. Phosphorylated HER3 was found in nine out of 12 cell lines (Fig. 1A). Total and phosphorylated levels of the other HER receptors (EGFR, HER2, and HER4) were also detected by immunoblotting (Supplementary Fig. S2A). As our results indicated that HER3 is widely expressed and activated in most HNSCC cell lines, two representative cell lines, SCC47 and Tu212, were chosen for further study as they both exhibited expression of total and phosphorylated HER3.
Fig. 1.
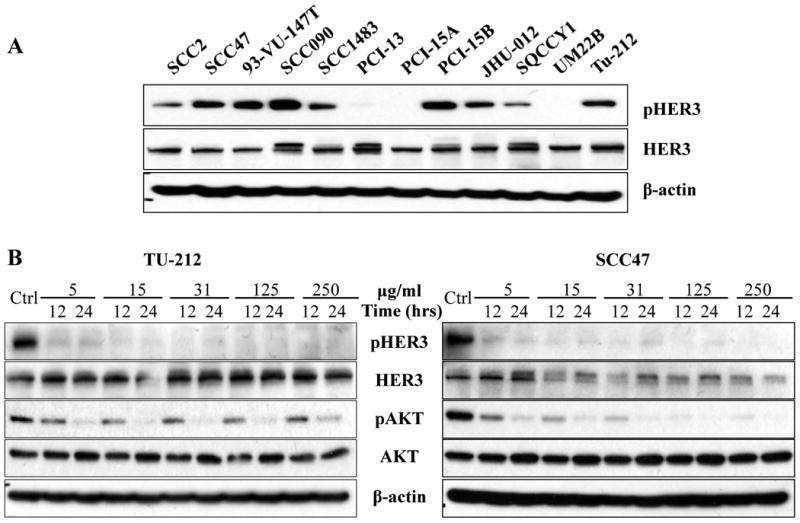
A, total and activated HER3 expression in HNSCC cell lines. Western blot was used to determine the expression of HER3 and pHER3 expression in 12 HNSCC cell lines. B, MM-121 alone inhibits HER3 and AKT activation in vitro. HNSCC cell lines SCC47 and Tu212 were treated with escalating doses of MM-121 as indicated for 12 and 24 hours. Cell lysates were immunoblotted to detect pHER3 (Tyr1289), pAKT (Ser473), total HER3 and AKT. Experiments were repeated three times.
MM-121 alone inhibits activation of HER3 and AKT in HNSCC cancer cells
As the activity of MM-121 has not previously been tested in HNSCC, its inhibitory effect on HER3 activation and downstream signaling through AKT was assessed. Inhibition of HER3 phosphorylation was observed at a dose of 5 μg/mL and higher in both SCC47 and Tu212 cell lines (Fig. 1B). Treatment with MM-121 at a dose of 125 μg/mL for 24 hours completely blocked the activation of HER3 and AKT in both cell lines (Fig. 1B). The results showed that MM-121 has a strong inhibitory activity on phosphorylation of HER3 and AKT as a single drug in HNSCC cancer cell lines. The inhibition of ERK phosphorylation, however, was not observed by MM-121 treatment alone (Supplementary Fig. S2B).
The combination of Cetuximab and MM-121 (CM) provides dual inhibition of PI3K/AKT and ERK signaling pathways
HER family receptors activate a number of important signal transduction pathways including PI3K/AKT and ERK pathways, which are critical for cell growth and survival and are implicated as major factors in many types of cancer. To investigate the combined effect of the HER3 inhibitor MM-121 and the EGFR inhibitor cetuximab on PI3K/AKT and ERK activity, we treated SCC47 and Tu212 cells with MM-121 alone, cetuximab alone and a combination of the two agents. MM-121 potently blocked HER3 phosphorylation at a concentration of 125 μg/mL, whereas cetuximab partially inhibited EGFR and HER3 activation at a concentration of 62 μg/mL in both cell lines (Supplementary Fig. S3). The CM combination substantially inhibited phosphorylation of both HER3 and EGFR after 72 and 96 hours of treatment (Supplementary Fig. S3). Only CM combination simultaneously decreased both AKT and ERK activation after 72 (Fig. 2) and 96 hours (Supplementary Fig. S4) compared to single antibody treatments. Activation of AKT/m-TOR signaling as determined by phosphorylated S6 ribosomal protein (pS6) levels has previously been implicated in progression and poor prognosis of HNSCC (30). Interestingly, in our study, substantial deduction of pS6 level was only observed in the CM combination group (Fig. 2). These results suggest that CM combination substantially inhibits EGFR and HER3 activation and consequently blocks both of the downstream PI3K/AKT and ERK signaling pathways.
Fig. 2. The cetuximab and MM-121 (CM) combination simultaneously inhibits PI3K/AKT and ERK signaling pathways.
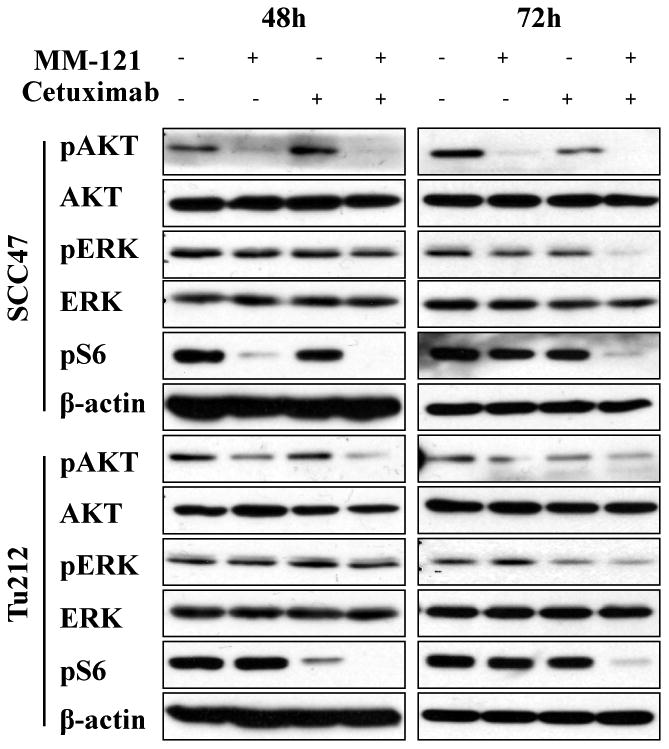
SCC47 and Tu212 cells were treated with 125μg/mL MM-121, 62μg/mL cetuximab and the combination for 48 and 72 hours. Cell lysates were collected and immunoblotted as indicated.
The CM combination effectively inhibits HNSCC cell growth in vitro
The PI3K/AKT and ERK pathways are critical for cell growth and survival. As our study has shown that the combination of cetuximab and MM-121 inhibits PI3K/AKT and ERK signaling pathways, we speculate that the CM combination may provide an improved strategy to inhibit the growth and survival of HNSCC cancer cells. To evaluate this, we initially accessed growth inhibition by either MM-121 or cetuximab alone in eight HNSCC cell lines. Both antibodies alone showed marginal inhibition of cell growth in vitro with concentrations up to 500 μg/mL (Supplementary Fig. S1). These findings are consistent with previous studies which showed that cetuximab alone have limited growth inhibition effect on certain cancer cell lines in vitro with concentrations up to 150 μg/mL (25, 31). A colony formation assay was then carried out to determine the long-term growth-inhibitory activity of the CM combination treatment. In both cell lines, the CM combination showed significantly greater inhibition of colony formation in comparison with each single drug or with control after 9 days of treatment (CM vs. ctrl p<0.001 or vs. C/M single drug p<0.05, Fig. 3A). The synergistic indices (SI) of colony formation assay were 8.25 for Tu212 and 3.92 for SCC47 (Supplementary Table 1); indicating a synergistic inhibitory effect of cetuximab and MM-121 on colony formation ability (29). Cetuximab alone also showed greater growth inhibition in both cell lines compared to control (p<0.001). In addition, similar effects were observed by short-term cell proliferation assay after 72 hours of treatment. The growth of the two cell lines was significantly inhibited by the CM combination compared to single drug treatments (p<0.05). Compared to control, MM-121 and cetuximab as single agents both showed growth inhibition in SCC47 cells (C/M vs. ctrl, p<0.05) and only cetuximab showed growth inhibition in Tu212 cells (p<0.001) (Fig. 3B). Again, a synergistic inhibitory effect was found with SI of 1.46 (Tu212) and 1.64 (SCC47) (Supplementary Table 1).
Fig. 3. The CM combination inhibits HNSCC cell growth in vitro.
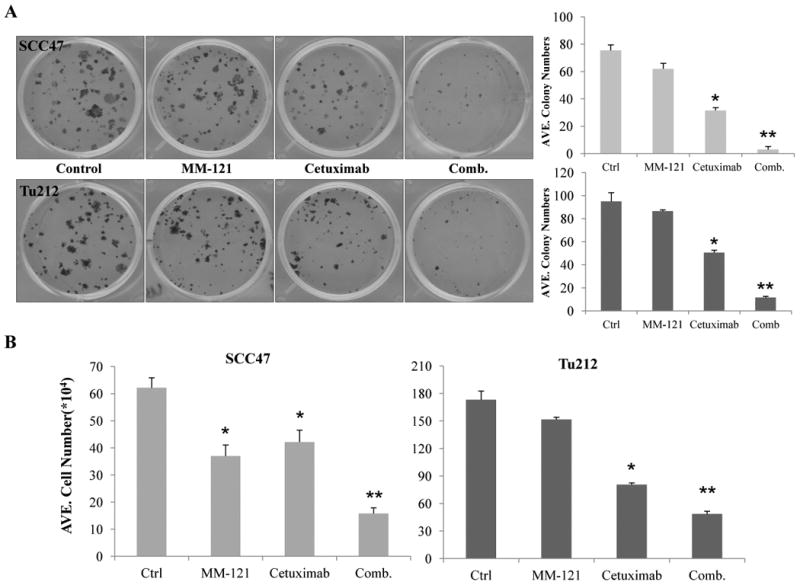
A, for colony formation assay, SCC47 and Tu212 cells at 200/well were treated with 20μg/mL MM-121, 2μg/mL cetuximab or the combination for 9 days. The medium was changed every three days before cells were stained with 0.2% crystal violet. Cell numbers ≥50 were considered as a colony. ImageJ software was used for colony counting. B, cells (2.5 × 105/well) were cultured for overnight and treated with MM-121 125μg/ml, cetuximab 62μg/ml or the combination for 72 hours and then trypsinized and counted. Error bars are mean ± SE from 3 independent experiments. (* indicates p<0.05 vs. ctrl, ** indicates p<0.05 vs. all other groups).
The CM combination induces cell cycle arrest and apoptosis in HNSCC cells
To determine the underlying mechanism of inhibitory effect of CM combination on tumor cell growth, cell cycle analysis and apoptosis assay were carried out. Cell cycle analysis was used to determine if the observed antitumor effect reflected any change in cell cycle distribution. We observed an increased proportion of G1 phase cells and a decreased proportion of S phase cells as early as 24 hours after the CM treatment in both SCC47 and Tu212 cells lines (data not shown). As shown in Fig. 4A, after 72 hours of treatment in the SCC47 cell line, the CM combination induced significantly greater numbers of G0-G1-phase (p<0.01) but reduced S (p<0.01) and G2/M-phase cells (p<0.05) compared to control. Similarly, an increase in G0/G1 cells (p<0.01 vs. ctrl) and a decrease in S-phase cells (p<0.001 vs. ctrl) were observed in Tu212 cells treated with CM combination (Fig. 4A). Overall, the CM treatment most effectively induced cell cycle arrest in G1 relative to control or either agent alone. To investigate the molecular mechanism underlying these changes, we monitored the levels of cyclin D1, and cyclin E by immunoblotting in both cell lines. Consistent with our in vitro observations, treatment with the CM combination resulted in downregulation of these two proteins (Fig. 4B).
Fig. 4. The CM combination induces cell cycle arrest and apoptosis in vitro.
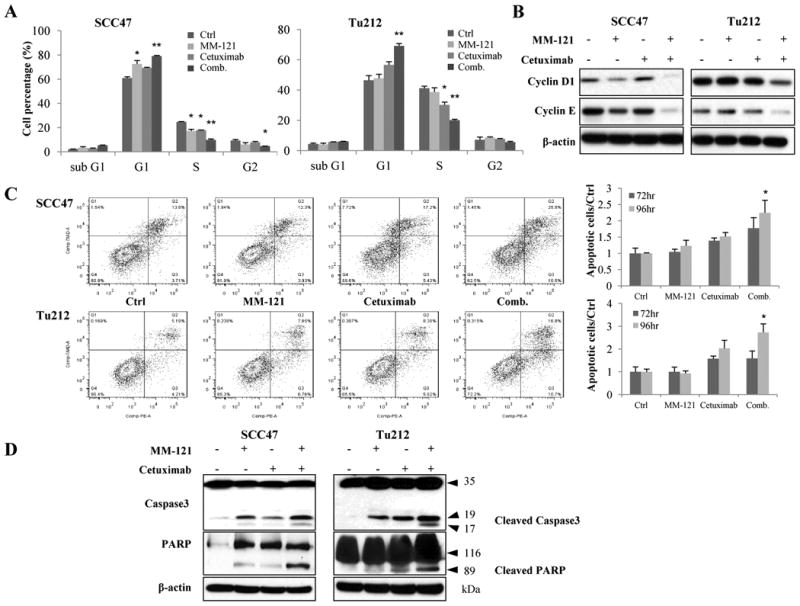
A, for cell cycle analysis, cells (2.5 × 105/well) were treated with 125μg/mL MM-121, 62μg/mL cetuximab and the combination for 72 hours and then analyzed by flow cytometric analysis. The percentage of cells in the G0-G1, S, and G2-M phases of the cell cycle are shown. B, cell lysate in (A) was collected and immunoblotted for cell cycle related cyclinD1 and cyclinE. C, for apoptosis assay, cells were treated for 72 and 96 hours with the same doses of drugs as cell cycle assay and then analyzed by flow cytometry. D, cell lysates from the same treatment as apoptosis assay were collected and immunoblotted for caspase3 and PARP. All data are expressed as mean ± SE from 3 independent experiments. (*indicates p<0.05, ** indicates p<0.01 vs. ctrl).
To assess the effect of the CM combination on apoptosis, PE Annexin V was used as a marker for the early features of apoptosis, whereas 7-AAD staining indicated late-stage apoptosis. Compared to other groups, the CM combination induced significantly more early- and late-stage apoptosis after 96 hours of treatment in the two cell lines (Fig. 4C, p < 0.05). Consistent with these findings, increased levels of cleaved PARP and caspase 3 were also observed in both cell lines (Fig. 4D). These results indicate that the observed anti-proliferative effects of the CM combination likely arise from a combination of cell cycle arrest and apoptosis in HNSCC cell lines.
The CM combination shows potent antitumor effect in HNSCC xenograft models
To expand our findings into the in vivo setting, two xenograft models using HNSCC cell lines (SCC47 and Tu212) were established as previously described (32, 33). Mice were randomly assigned to six treatment groups: PBS control, cetuximab (C), MM-121.LD, MM121.HD, Comb.LD, and Comb.HD and were treated twice a week through intraperitoneal injection.
Consistent with our in vitro observations, the CM combination showed the greatest tumor growth inhibition in Tu212 xenografts. As shown in the tumor volume measurement result, treatment with both high and low doses of CM combination significantly suppressed tumor growth as compared with the PBS control and cetuximab alone (Fig. 5A, p<0.0001 in both cases). Comb. HD showed greater tumor growth inhibition than both doses of MM-121 alone (p<0.05) while Comb. LD showed greater tumor suppression than MM-121.LD (p<0.01). By using the endpoint tumor volumes for calculation (29), a synergistic inhibitory effect was found with the SI of 1.67 (Comb.LD) and 1.56 (Comb.HD) (Supplementary Table 1). After 26 days of treatment, the Comb.LD and Comb.HD inhibited Tu212 tumor growth by 87% and 89% respectively compared to control (defined as tumor weight, Fig. 5B).
Fig. 5. The CM combination inhibits HNSCC xenograft tumor growth in vivo.
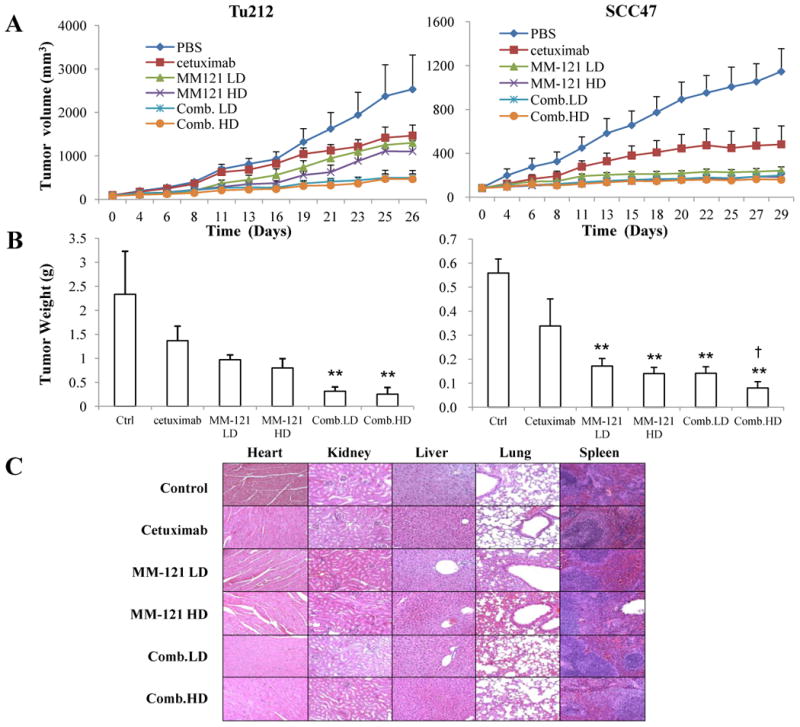
Mice bearing subcutaneous SCC47 and Tu212 tumors of approximately 100 mm3 were treated by intraperitoneal injection (i.p.) twice per week for 4 weeks with: PBS (phosphate-buffered saline) control, cetuximab (6.25μg/dose), MM-121 at low dose (300μg/dose, MM-121.LD), MM-121 at high dose (600μg/dose, MM-121.HD), the combination with LD MM-121 (Comb.LD), and the combination with HD MM-121 (Comb.HD). A, tumor volumes were measured three times a week. B, tumors were harvested and weighed 26 (Tu212) and 29 (SCC47) days after the first treatment. C, major organs were harvested for toxicity evaluation by H&E (Magnification: 100×). Error bars are mean ± SE of 7 mice from each group. (** indicates p<0.01 v.s. Ctrl, † indicates p<0.05 v.s. cetuximab)
The Comb.HD combination was also highly effective in SCC47 xenografts based on tumor volume measurement (Fig. 5A). All of the treatment groups had significantly reduced tumor growth rates compared to the control group (p<0.05). Comb.HD treatment significantly inhibited tumor growth as compared with control (p<0.001), cetuximab (p=0.04) and MM-121.LD (p<0.01). Comb.LD also inhibited tumor growth over time compared to control (p<0.001) and cetuximab (p=0.06). No significant difference was observed between the MM-121 single agent groups and the Comb.LD group (Fig. 5A). After 29 days of treatment, Comb. HD significantly decreased xenograft tumor weight as compared with control (p<0.001) and cetuximab (p=0.04) (Fig. 5B).
The antitumor activity of CM treatment was not accompanied by any side effect or treatment-related weight loss (Supplementary Fig. S5). No cellular abnormalities were observed in the examined organs, including heart, lung, liver, kidney and spleen (n = 3 mice/group), derived from both xenograft mouse models (Fig. 5C).
The CM combination has the greatest effect on HER3/EGFR signaling, tumor proliferation, apoptosis and angiogenesis in vivo
The levels of HER3/pHER3, EGFR/pEGFR and downstream proteins in fresh xenograft tissues were analyzed by Western blot (n=3 per group). Consistent with our in vitro findings, HER3, EGFR, AKT and ERK activation were significantly inhibited by the CM combination (Fig. 6A and Supplementary Fig. S6). pS6 level was also decreased in CM combination treated tissues (Fig. 6A). A similar trend of decreased pHER3 and pAKT expression was observed in xenograft tissues by IHC staining (Supplementary Fig. S7). We then studied Tu212 xenograft tissues and found both high and low dose combination groups showed significantly increased necrotic areas as compared with the untreated control (p < 0.01). Biomarkers of cell proliferation, apoptosis and angiogenesis (Ki67, TUNEL and CD34, respectively) were also examined in the xenograft tissues. Significant reduction in Ki67 signals and increase in TUNEL signals were observed in all treatment groups (p < 0.05 v.s. control), especially in combination groups (p < 0.01 v.s. control). Both high and low dose combinations significantly diminished microvessel density measured by CD34 staining as compared with the control (p < 0.01) (Fig. 6B).
Fig. 6. Effect of the CM combination on HER3 and EGFR signaling and tumor proliferation, apoptosis and angiogenesis in vivo.
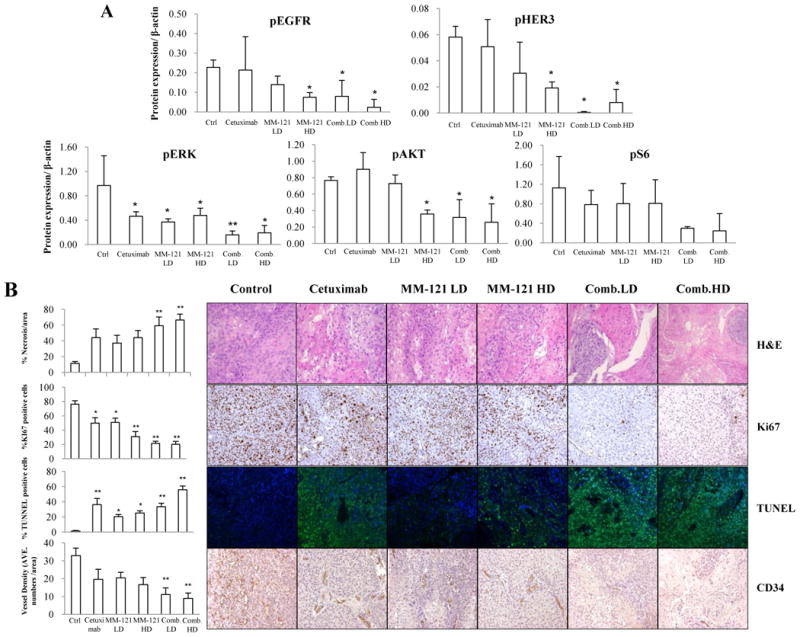
A, the CM combination inhibited HER3 and EGFR signaling in vivo. Fresh tumor tissues were collected and stored at -80°C. Three representative tissue lysates from each group were prepared for Western blot analysis. ImageJ software was used for Western blot quantification. All data are expressed as mean ± SD from 3 tissue samples. B, the CM combination significantly increased necrosis, inhibited proliferation and angiogenesis and induced apoptosis in vivo. Data show the representative tumors with hematoxylin and eosin (H&E) staining, Ki67, TUNEL and CD34 staining (Magnification: 200×). Tissue slides were observed by at least two independent personnel. The percentage of necrotic areas, percentage of positive Ki67 and TUNEL staining and CD34 positive signals were counted from five randomly selected areas in each slide at 100× magnification. For TUNEL assay, green fluorescence indicates positive cells. Error bars are mean ± SE of 7 mice from each group. (* indicates p < 0.05 and ** indicates p < 0.01 v.s. Ctrl).
Discussion
Although better tolerated than cytotoxic chemotherapy, approximately 80% of patients showed de novo resistance and an increasing number of patients become acquired resistant to anti-EGFR therapy in HNSCC (34). Potential strategies to overcome resistance are therefore highly needed. Hyperactivation of HER3 has previously been reported to negatively correlate with response to anti-EGFR therapy (15, 35). Moreover, tumor cells that escape from cetuximab inhibition exhibit EGFR upregulation-dependent HER3 activation (13). Dual inhibition of both EGFR and HER3 is therefore an attractive clinical strategy for treating HNSCC. Anti-HER3 antibody MM-121 alone or in combination with other anti-tumor drugs have been studied previously in several cancers (19, 20, 36-39). In the current study, we explored the activity and the underlying mechanism of MM-121 combined with the anti-EGFR antibody cetuximab in the treatment of preclinical HNSCC models. Our results indicated that the combination of the two antibodies significantly inhibited HNSCC tumor cell growth both in vitro and in vivo. In addition, compared to either antibody alone, the combination inhibited the activation of both EGFR and HER3 and subsequently blocked the activation of AKT, ERK, and S6. Taken together, these findings suggest that the combination of cetuximab with MM-121 may provide potential clinical benefits for patients with HNSCC.
HER3 protein is reportedly over-expressed in HNSCC patients and its membranous expression is correlated with decreased survival (40). In the current study, we assessed the total and phosphorylated levels of all four HER family members in 12 HNSCC cell lines. HER3 was expressed in all 12 cell lines, and pHER3 was observed with EGFR and pEGFR in 9 of the 12 cell lines (Fig. 1A and Supplementary Fig. S2A). Our in vitro activity studies showed that neither HER3 nor EGFR inhibition alone was sufficient to inhibit tumor cell growth and ERK activation in HNSCC (Fig. 2, 3 and Supplementary Fig. S1). As ERK generally promotes cell proliferation, the lack of inhibition on ERK phosphorylation may provide an explanation for the limited growth inhibition. Only dual inhibiting EGFR and HER3 through combined treatment of cetuximab and MM-121 could simultaneously block both PI3K/AKT and ERK pathways in HNSCC (Fig. 2 and Supplementary Fig. S4), which is consistent with a recent study using the dual HER3/EGFR-targeting antibody MEHD7945A (31). Together, these data support the hypothesis that improved tumor inhibition activity can be achieved through simultaneously inhibiting both EGFR and HER3 in HNSCC. Interestingly, several studies reported that IGFR (insulin-like growth factor receptor) activation is involved in resistance to EGFR inhibitors through activating the PI3K/AKT pathway (41-44). It would be of interest to determine the IGFR expression level and target the IGFR and HER pathways together in HNSCC models in future.
Ribosomal protein S6 (S6) is a substrate of ribosomal protein S6 kinase (S6K), which is one of the best-characterized downstream effectors of the PI3K/AKT/mTOR pathway. Phosphorylation of S6 is required for translation initiation and controls cell size, growth, and proliferation in a cell cycle independent fashion (45). In HNSCC, pS6 overactivity persists during tumor progression and cytoplasmic staining of pS6 was detected and positively correlated with poor differentiation (30, 46). Moreover, inhibiting this pathway by rapamycin resulted in rapid tumor regression concomitant with a marked decrease in cell proliferation, enhanced apoptosis both in vitro and in vivo in HNSCC (47). Interestingly, we also observed inhibition of S6 phosphorylation after treatment with CM combination in our study. Moreover, pS6 decreasing was initially observed in MM-121 treated SCC47 and cetuximab treated Tu212 cells at 48 hours. However, this effect was diminished after 72 and 96 hours of treatment regardless of the inhibition status of pAKT at these time points (Fig.2 and Supplementary Fig. S4). Only the CM combination continuously inhibited S6 phosphorylation from 48 to 96 hours. These findings suggest that only simultaneously inhibit EGFR and HER3 could totally and continuously block pathways that phosphorylate S6 and thus contribute to tumor growth inhibition and apoptosis in HNSCC.
HPV is recognized to play a role in the pathogenesis of a subset of HNSCC (48). It is known that this subgroup of patients have a better prognosis compared to their HPV-negative counterparts and could be unnecessarily over-treated by traditional cytotoxic therapy (49, 50). Despite the fact that EGFR expression is inversely correlated with HPV status, current clinical trials are exploring the role of the less toxic cetuximab in treating HPV positive disease (50). The fact that HER3 was expressed and activated in all five HPV positive HNSCC cell lines in our study and the observation of an increased tumor growth inhibition with CM in comparison with C or M alone in the HPV positive SCC47 cell line model, suggests that HER3 inhibition alone or in combination with EGFR inhibition deserves further investigation in HPV positive HNSCC.
Another important issue frequently raised is the toxicity related to single or combined targeted therapy. In our study, the doses of cetuximab and MM-121 were chosen as 312.5 μg/kg per dose and 15 or 30 mg/kg per dose twice a week respectively, based on previous studies (25) (19). We found neither treatment to be associated with body weight loss nor evidence of organ damage in nude mice after treatment for almost a month (Fig. 5C and Supplementary Fig. S5). In order to translate this combination to clinical use, the doses of the two agents need to be further optimized. Furthermore, as combing MM-121 with cetuximab coordinately blocked the downstream signaling pathways of EGFR and HER3, it is not surprising that we observed a significant reduction in microvessel density after the combined treatment (Fig. 6B), which is consistent with our previous observation of angiogenesis inhibition by EGFR blockage in both HNSCC and lung cancer xenograft models (21).
In conclusion, we have demonstrated that simultaneous blockade of the PI3K/AKT and ERK pathways in HNSCC cell line models can be achieved by combining cetuximab and MM-121, which act together to inhibit tumor growth. Although several ongoing clinical trials are investigating MM-121 alone or in combination with other systemic agents in treating cancer, including a Phase I trial currently assessing the safety of combining MM-121 with cetuximab in patients with advanced solid tumors, the mechanism underlies their therapeutic effect remains to be elucidated. Our study provides a molecular mechanism for the antitumor effects of combined EGFR and HER3 inhibition in models of HNSCC and supports the clinical investigation of the combination therapy in HNSCC.
Supplementary Material
Acknowledgments
We thank Dr. Anthea Hammond for her critical reading and editing of the manuscript.
Financial support: This study was supported by funding from Merrimack Pharmaceuticals Inc. to G.Z. Chen and N.F. Saba and partially by grant from the Specialized Program of Excellence (SPORE) in Head and Neck Cancer (P50CA128613) to D.M. Shin.
Footnotes
Disclosure of Potential Conflicts of Interest: This work was funded in part by Merrimack Pharmaceuticals. G. Garcia and G. MacBeath are employees and stockholders of Merrimack Pharmaceuticals.
Note: All of the experiments reported in this manuscript were performed at Emory University.
References
- 1.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 2.Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55:74–108. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- 3.Grandis JR, Tweardy DJ. Elevated levels of transforming growth factor alpha and epidermal growth factor receptor messenger RNA are early markers of carcinogenesis in head and neck cancer. Cancer Res. 1993;53:3579–84. [PubMed] [Google Scholar]
- 4.Ongkeko WM, Altuna X, Weisman RA, Wang-Rodriguez J. Expression of protein tyrosine kinases in head and neck squamous cell carcinomas. Am J Clin Pathol. 2005;124:71–6. doi: 10.1309/BTLN5WTMJ3PCNRRC. [DOI] [PubMed] [Google Scholar]
- 5.Ang KK, Berkey BA, Tu X, Zhang HZ, Katz R, Hammond EH, et al. Impact of epidermal growth factor receptor expression on survival and pattern of relapse in patients with advanced head and neck carcinoma. Cancer Res. 2002;62:7350–6. [PubMed] [Google Scholar]
- 6.Bonner JA, Harari PM, Giralt J, Azarnia N, Shin DM, Cohen RB, et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N Engl J Med. 2006;354:567–78. doi: 10.1056/NEJMoa053422. [DOI] [PubMed] [Google Scholar]
- 7.Vermorken JB, Mesia R, Rivera F, Remenar E, Kawecki A, Rottey S, et al. Platinum-based chemotherapy plus cetuximab in head and neck cancer. N Engl J Med. 2008;359:1116–27. doi: 10.1056/NEJMoa0802656. [DOI] [PubMed] [Google Scholar]
- 8.Vermorken JB, Trigo J, Hitt R, Koralewski P, Diaz-Rubio E, Rolland F, et al. Open-label, uncontrolled, multicenter phase II study to evaluate the efficacy and toxicity of cetuximab as a single agent in patients with recurrent and/or metastatic squamous cell carcinoma of the head and neck who failed to respond to platinum-based therapy. J Clin Oncol. 2007;25:2171–7. doi: 10.1200/JCO.2006.06.7447. [DOI] [PubMed] [Google Scholar]
- 9.Bardelli A, Janne PA. The road to resistance: EGFR mutation and cetuximab. Nat Med. 2012;18:199–200. doi: 10.1038/nm.2646. [DOI] [PubMed] [Google Scholar]
- 10.Montagut C, Dalmases A, Bellosillo B, Crespo M, Pairet S, Iglesias M, et al. Identification of a mutation in the extracellular domain of the Epidermal Growth Factor Receptor conferring cetuximab resistance in colorectal cancer. Nat Med. 2012;18:221–3. doi: 10.1038/nm.2609. [DOI] [PubMed] [Google Scholar]
- 11.Krumbach R, Schuler J, Hofmann M, Giesemann T, Fiebig HH, Beckers T. Primary resistance to cetuximab in a panel of patient-derived tumour xenograft models: activation of MET as one mechanism for drug resistance. Eur J Cancer. 2011;47:1231–43. doi: 10.1016/j.ejca.2010.12.019. [DOI] [PubMed] [Google Scholar]
- 12.Wheeler SE, Suzuki S, Thomas SM, Sen M, Leeman-Neill RJ, Chiosea SI, et al. Epidermal growth factor receptor variant III mediates head and neck cancer cell invasion via STAT3 activation. Oncogene. 2010;29:5135–45. doi: 10.1038/onc.2009.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wheeler DL, Huang S, Kruser TJ, Nechrebecki MM, Armstrong EA, Benavente S, et al. Mechanisms of acquired resistance to cetuximab: role of HER (ErbB) family members. Oncogene. 2008;27:3944–56. doi: 10.1038/onc.2008.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vlacich G, Coffey RJ. Resistance to EGFR-targeted therapy: a family affair. Cancer Cell. 2011;20:423–5. doi: 10.1016/j.ccr.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sergina NV, Rausch M, Wang D, Blair J, Hann B, Shokat KM, et al. Escape from HER-family tyrosine kinase inhibitor therapy by the kinase-inactive HER3. Nature. 2007;445:437–41. doi: 10.1038/nature05474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jiang N, Saba NF, Chen ZG. Advances in Targeting HER3 as an Anticancer Therapy. Chemother Res Pract. 2012;2012:817304. doi: 10.1155/2012/817304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Erjala K, Sundvall M, Junttila TT, Zhang N, Savisalo M, Mali P, et al. Signaling via ErbB2 and ErbB3 associates with resistance and epidermal growth factor receptor (EGFR) amplification with sensitivity to EGFR inhibitor gefitinib in head and neck squamous cell carcinoma cells. Clin Cancer Res. 2006;12:4103–11. doi: 10.1158/1078-0432.CCR-05-2404. [DOI] [PubMed] [Google Scholar]
- 18.Yonesaka K, Zejnullahu K, Okamoto I, Satoh T, Cappuzzo F, Souglakos J, et al. Activation of ERBB2 signaling causes resistance to the EGFR-directed therapeutic antibody cetuximab. Sci Transl Med. 2011;3:99ra–86. doi: 10.1126/scitranslmed.3002442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schoeberl B, Faber AC, Li D, Liang MC, Crosby K, Onsum M, et al. An ErbB3 antibody, MM-121, is active in cancers with ligand-dependent activation. Cancer Res. 2010;70:2485–94. doi: 10.1158/0008-5472.CAN-09-3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sheng Q, Liu X, Fleming E, Yuan K, Piao H, Chen J, et al. An activated ErbB3/NRG1 autocrine loop supports in vivo proliferation in ovarian cancer cells. Cancer Cell. 2010;17:298–310. doi: 10.1016/j.ccr.2009.12.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang X, Zhang H, Tighiouart M, Lee JE, Shin HJ, Khuri FR, et al. Synergistic inhibition of head and neck tumor growth by green tea (-)-epigallocatechin-3-gallate and EGFR tyrosine kinase inhibitor. Int J Cancer. 2008;123:1005–14. doi: 10.1002/ijc.23585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao M, Sano D, Pickering CR, Jasser SA, Henderson YC, Clayman GL, et al. Assembly and initial characterization of a panel of 85 genomically validated cell lines from diverse head and neck tumor sites. Clin Cancer Res. 2011;17:7248–64. doi: 10.1158/1078-0432.CCR-11-0690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wald AI, Hoskins EE, Wells SI, Ferris RL, Khan SA. Alteration of microRNA profiles in squamous cell carcinoma of the head and neck cell lines by human papillomavirus. Head Neck. 2011;33:504–12. doi: 10.1002/hed.21475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kimple RJ, Smith MA, Blitzer GC, Torres AD, Martin JA, Yang RZ, et al. Enhanced radiation sensitivity in HPV-positive head and neck cancer. Cancer Res. 2013;73:4791–800. doi: 10.1158/0008-5472.CAN-13-0587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang H, Yun S, Batuwangala TD, Steward M, Holmes SD, Pan L, et al. A dual-targeting antibody against EGFR-VEGF for lung and head and neck cancer treatment. Int J Cancer. 2012;131:956–69. doi: 10.1002/ijc.26427. [DOI] [PubMed] [Google Scholar]
- 26.Bonner JA, Raisch KP, Trummell HQ, Robert F, Meredith RF, Spencer SA, et al. Enhanced apoptosis with combination C225/radiation treatment serves as the impetus for clinical investigation in head and neck cancers. J Clin Oncol. 2000;18:47S–53S. [PubMed] [Google Scholar]
- 27.Wang D, Muller S, Amin AR, Huang D, Su L, Hu Z, et al. The pivotal role of integrin beta1 in metastasis of head and neck squamous cell carcinoma. Clin Cancer Res. 2012;18:4589–99. doi: 10.1158/1078-0432.CCR-11-3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weidner N, Carroll PR, Flax J, Blumenfeld W, Folkman J. Tumor angiogenesis correlates with metastasis in invasive prostate carcinoma. Am J Pathol. 1993;143:401–9. [PMC free article] [PubMed] [Google Scholar]
- 29.Shin DH, Min HY, El-Naggar AK, Lippman SM, Glisson B, Lee HY. Akt/mTOR counteract the antitumor activities of cixutumumab, an anti-insulin-like growth factor I receptor monoclonal antibody. Mol Cancer Ther. 2011;10:2437–48. doi: 10.1158/1535-7163.MCT-11-0235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Patel V, Marsh CA, Dorsam RT, Mikelis CM, Masedunskas A, Amornphimoltham P, et al. Decreased lymphangiogenesis and lymph node metastasis by mTOR inhibition in head and neck cancer. Cancer Res. 2011;71:7103–12. doi: 10.1158/0008-5472.CAN-10-3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schaefer G, Haber L, Crocker LM, Shia S, Shao L, Dowbenko D, et al. A two-in-one antibody against HER3 and EGFR has superior inhibitory activity compared with monospecific antibodies. Cancer Cell. 2011;20:472–86. doi: 10.1016/j.ccr.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 32.Amin AR, Wang D, Zhang H, Peng S, Shin HJ, Brandes JC, et al. Enhanced anti-tumor activity by the combination of the natural compounds (-)-epigallocatechin-3-gallate and luteolin: potential role of p53. J Biol Chem. 2010;285:34557–65. doi: 10.1074/jbc.M110.141135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Adhim Z, Otsuki N, Kitamoto J, Morishita N, Kawabata M, Shirakawa T, et al. Gene silencing with siRNA targeting E6/E7 as a therapeutic intervention against head and neck cancer-containing HPV16 cell lines. Acta Otolaryngol. 2013;133:761–71. doi: 10.3109/00016489.2013.773405. [DOI] [PubMed] [Google Scholar]
- 34.Harari PM, Wheeler DL, Grandis JR. Molecular target approaches in head and neck cancer: epidermal growth factor receptor and beyond. Semin Radiat Oncol. 2009;19:63–8. doi: 10.1016/j.semradonc.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–43. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 36.Huang J, Wang S, Lyu H, Cai B, Yang X, Wang J, et al. The anti-erbB3 antibody MM-121/SAR256212 in combination with trastuzumab exerts potent antitumor activity against trastuzumab-resistant breast cancer cells. Mol Cancer. 2013;12:134. doi: 10.1186/1476-4598-12-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang S, Huang J, Lyu H, Cai B, Yang X, Li F, et al. Therapeutic targeting of erbB3 with MM-121/SAR256212 enhances antitumor activity of paclitaxel against erbB2-overexpressing breast cancer. Breast Cancer Res. 2013;15:R101. doi: 10.1186/bcr3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liles JS, Arnoletti JP, Kossenkov AV, Mikhaylina A, Frost AR, Kulesza P, et al. Targeting ErbB3-mediated stromal-epithelial interactions in pancreatic ductal adenocarcinoma. Br J Cancer. 2011;105:523–33. doi: 10.1038/bjc.2011.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schoeberl B, Pace EA, Fitzgerald JB, Harms BD, Xu L, Nie L, et al. Therapeutically targeting ErbB3: a key node in ligand-induced activation of the ErbB receptor-PI3K axis. Sci Signal. 2009;2:ra31. doi: 10.1126/scisignal.2000352. [DOI] [PubMed] [Google Scholar]
- 40.Takikita M, Xie R, Chung JY, Cho H, Ylaya K, Hong SM, et al. Membranous expression of Her3 is associated with a decreased survival in head and neck squamous cell carcinoma. J Transl Med. 2011;9:126. doi: 10.1186/1479-5876-9-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chakravarti A, Loeffler JS, Dyson NJ. Insulin-like growth factor receptor I mediates resistance to anti-epidermal growth factor receptor therapy in primary human glioblastoma cells through continued activation of phosphoinositide 3-kinase signaling. Cancer Res. 2002;62:200–7. [PubMed] [Google Scholar]
- 42.Guix M, Faber AC, Wang SE, Olivares MG, Song Y, Qu S, et al. Acquired resistance to EGFR tyrosine kinase inhibitors in cancer cells is mediated by loss of IGF-binding proteins. J Clin Invest. 2008;118:2609–19. doi: 10.1172/JCI34588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jameson MJ, Beckler AD, Taniguchi LE, Allak A, Vanwagner LB, Lee NG, et al. Activation of the insulin-like growth factor-1 receptor induces resistance to epidermal growth factor receptor antagonism in head and neck squamous carcinoma cells. Mol Cancer Ther. 2011;10:2124–34. doi: 10.1158/1535-7163.MCT-11-0294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cortot AB, Repellin CE, Shimamura T, Capelletti M, Zejnullahu K, Ercan D, et al. Resistance to irreversible EGF receptor tyrosine kinase inhibitors through a multistep mechanism involving the IGF1R pathway. Cancer Res. 2013;73:834–43. doi: 10.1158/0008-5472.CAN-12-2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ruvinsky I, Meyuhas O. Ribosomal protein S6 phosphorylation: from protein synthesis to cell size. Trends Biochem Sci. 2006;31:342–8. doi: 10.1016/j.tibs.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 46.Molinolo AA, Hewitt SM, Amornphimoltham P, Keelawat S, Rangdaeng S, Meneses Garcia A, et al. Dissecting the Akt/mammalian target of rapamycin signaling network: emerging results from the head and neck cancer tissue array initiative. Clin Cancer Res. 2007;13:4964–73. doi: 10.1158/1078-0432.CCR-07-1041. [DOI] [PubMed] [Google Scholar]
- 47.Amornphimoltham P, Patel V, Sodhi A, Nikitakis NG, Sauk JJ, Sausville EA, et al. Mammalian target of rapamycin, a molecular target in squamous cell carcinomas of the head and neck. Cancer Res. 2005;65:9953–61. doi: 10.1158/0008-5472.CAN-05-0921. [DOI] [PubMed] [Google Scholar]
- 48.Fakhry C, Gillison ML. Clinical implications of human papillomavirus in head and neck cancers. J Clin Oncol. 2006;24:2606–11. doi: 10.1200/JCO.2006.06.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mirghani H, Amen F, Moreau F, Guigay J, Hartl DM, Lacau St, Guily J. Oropharyngeal cancers: Relationship between epidermal growth factor receptor alterations and human papillomavirus status. Eur J Cancer. 2014 doi: 10.1016/j.ejca.2013.12.018. [DOI] [PubMed] [Google Scholar]
- 50.Psyrri A, Sasaki C, Vassilakopoulou M, Dimitriadis G, Rampias T. Future directions in research, treatment and prevention of HPV-related squamous cell carcinoma of the head and neck. Head Neck Pathol. 2012;6(Suppl 1):S121–8. doi: 10.1007/s12105-012-0361-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.