

Abstract
Despite its clinical importance, very little is known about the natural history and molecular underpinnings of lung cancer dissemination and metastasis. Here we employed a genetically-engineered mouse model of metastatic lung adenocarcinoma in which cancer cells are fluorescently marked to determine whether dissemination is an inherent ability or a major acquired phenotype during lung adenocarcinoma metastasis. We find very little evidence for dissemination from oncogenic Kras-driven hyperplasias or most adenocarcinomas. p53 loss is insufficient to drive dissemination but rather enables rare cancer cells in a small fraction of primary adenocarcinomas to gain alterations that drive dissemination. Molecular characterization of disseminated tumors cells indicates that down-regulation of the transcription factor Nkx2-1 precedes dissemination. Finally, we show that metastatic primary tumors possess a highly proliferative sub-population of cells with characteristics matching those of disseminating cells. We propose that dissemination is a major hurdle during the natural course of lung adenocarcinoma metastasis.
Keywords: Metastasis, genetically-engineered mouse model, lung cancer, dissemination, Nkx2-1
INTRODUCTION
Metastasis is a multistep process, and the acquisition of metastatic ability has traditionally been considered a late step in cancer evolution (1). Recent evidence in mouse models of breast and pancreatic cancer suggests that cancer cells within premalignant lesions may already possess the ability to disseminate (1–4). These observations support a model in which cancer cells leave primary tumors early during tumor growth, thereby allowing the primary tumor and disseminated cells to accrue genomic alterations independently (5–7). This paradigm of metastatic progression is consistent with findings of disparate DNA copy number alterations in primary tumors and related disseminated cancer cells and metastases in patients (5–7).
Lung adenocarcinoma is the most common subtype of lung cancer, with over 90,000 patients diagnosed each year with this form of lung cancer in the United States alone (8). Several factors contribute to the poor outcome of lung cancer patients but, as in most solid tumors, the ability of cancer cells to leave the primary tumors and establish inoperable metastases is a major impediment to successful therapy (9). Whether early premalignant lesions in human lungs have an inherent capacity to give rise to disseminated tumor cells (DTCs) is a question of both clinical and biological importance. Despite its significance, systems to address the question of whether lung adenocarcinoma metastatic progression conforms to a traditional late-evolution model or a parallel progression model have not been developed. While these questions would ideally be studied in humans, early lesions are by definition small and exist in undiagnosed patients, making their study difficult if not impossible. Hence, accurate and tractable genetic models are required to investigate this process.
Human lung adenocarcinoma frequently harbors oncogenic mutations in KRAS and inactivation of the p53 tumor suppressor pathway (10, 11) and has been modeled using conditional alleles in mice. Oncogenic KrasG12D has been expressed in lung epithelial cells using transgenic systems, stochastic intrachromosomal recombination, or Cre-mediated deletion of the transcriptional/translational Stop element in loxP-Stop-loxP KrasG12D knock-in mice (KrasLSL-G12D/+). This single event initiates the stepwise development of lung lesions that closely resemble early-stage atypical adenomatous hyperplasias (AAH), adenomas, and early adenocarcinomas (12–14). Concomitant mutation of the p53 tumor suppressor allows the development of overtly invasive and metastatic cancer (15–17). Notably, these genetically-engineered mouse models of human lung cancer recapitulate the genetic events, histological appearance of lesions at both the early and late stages, and metastatic ability of the human disease (12–17).
Here, we use a series of in vivo mouse models to uncover the kinetics of lung tumor cell dissemination, investigate the importance of p53 loss in this process, and characterize the gene expression changes associated with this critical step of the metastatic cascade.
RESULTS
To analyze cancer cell dissemination at defined time points after tumor initiation we used a system to induce fluorescently-labeled lung tumors. To stably mark all cancer cells, we incorporated a Rosa26 knock-in Cre-reporter allele (R26LSL-tdTomato)(18) into the KrasLSL-G12D/+;p53 flox/flox lung adenocarcinoma mouse model to generate KrasLSL-G12D/+;p53flox/flox;R26LSL-tdTomato (KPT) mice (Supplemental Fig. 1). In KPT mice, lung tumors initiated by inhaled viral-Cre express oncogenic KrasG12D, delete p53, and express the red fluorescent protein tdTomato (Fig. 1A, 1B, and Supplemental Fig 1, 2). We have documented excellent specificity and sensitivity to detect Tomatopositive micro- and macro-metastases using fluorescence microscopy and immunohistochemistry and quantifying single Tomatopositive disseminated cancer cells using flow cytometry, confirming stable marker expression in even the most advanced cancer cells (Fig. 1B, 2A and data not shown).
Figure 1. Titratable induction of fluorescently labeled lung tumors enables the analysis of cancer cell dissemination at defined time points after tumor initiation.

A. Infection of KrasLSLG12D/+;p53flox/flox;R26tdTomato/+ (KPT) mice with the indicated viral titer generates KPT-Early and KPT-Late mice. A time course of tumor progression is shown.
B. Representative light and fluorescent images of KPT-Early (~1 month post infection) and KPT-Late lung lobes (~5 month post infection). Scale bar is 5mm.
C. KPT-Early lungs have diffuse hyperplasia throughout the alveolar space. KPT-Late lungs have solid tumors of variable histological presentation among areas of normal lung. Top scale bar in 2mm. Bottom scale bar is 50μm.
D. Expanding Tomatopos cells have histological features of atypical adenomatous hyperplasias (AAHs) and express lung epithelial markers (Nkx2-1 and cytokeratins (pan-CK)). Scale bar is 50μm.
E. KPT-Early and KPT-Late mice have dramatically increased lung weight relative to age matched normal lung. Each dot represents one mouse and the bar indicates the mean.
F. Quantification of the number of Tomatopos tumor cells per lung section in KPT-Early and KPT-Late mice normalized to the average number of Tomatopos cells per lung section in KPT-Early mice. Each dot represents one mouse and the bar indicates the mean. n.s. = not significant.
Figure 2. Lung cancer dissemination is a rare late event.

A. Disseminated tumor cells (DTCs) are detected in the pleural cavity of KPT-Late mice. Representative plots of FSC/SSC gated, viable (DAPIneg), lineageneg cells are shown. Percent of Tomatopos DTCs is indicated.
B. DTCs are present in the pleural fluid of some but not all KPT-Late mice. The number of mice in each group is indicated. Each dot represents a mouse.
C. Quantification of the number of DTCs in KPT-Late mice illustrates that not all late stage tumors have acquired the ability to metastasize, and that DTC number does not correlate with time post-tumor initiation in KPT-Late mice.
D. Representative KPT-Late mice analyzed at the same time point after tumor initiation and with comparable tumor mass have dramatically different numbers of DTCs. H&E staining is shown. Scale bar is 0.5cm.
E. Transplantation of bulk pleural cavity cells from the indicated donor mice into wild-type recipient mice. Pleural cells from KPT-Early (1–1.5 months post initiation), fail to form metastases. Pleural cavity cells from KPT-Late mice have metastasis seeding ability. Data is number of mice with metastases/total number of mice transplanted.
F. Representative light and fluorescent images of a recipient mouse lung lobe with Tomatopos metastases that formed from KPT-Late pleural cavity cells. Scale bar is 2 mm.
G. DTCs are not detected in the pleural cavity of KT-Early mice, or 13/14 KT-Late mice. Number of mice in each group is indicated. Each dot represents a mouse.
H. Transplantation as in 2E. Pleural cells from KT-Early (1.5–2.5 months post initiation), and KT-Late mice (including the mouse with DTCs shown in Figure 2G) fail to form metastases. Data is number of mice with metastases/total number of mice transplanted.
Tumors in KPT mice are initiated synchronously, thus allowing the analysis of cancer cell dissemination at defined time points after tumor initiation (15, 16). To most accurately compare the ability of early-stage and late-stage lung cancer cells to disseminate we generated two separate groups of mice, which we called KPT-Late and KPT-Early mice. Tumor initiation with either 5×106 Adenoviral-Cre or 2×103 Lentiviral-Cre induced 10 to 50 tumors that included small and large adenomas as well as adenocarcinomas with varying degrees of invasion (Fig. 1A–C, Supplemental Fig. 1, and data not shown). These KPT-Late mice survived 4–8 months. To generate KPT-Early mice we infected KPT mice with 3×109 Adeno-Cre (600-fold more than for KPT-Late mice), which induced high numbers of early stage hyperplastic lesions such that the total number of neoplastic cells within the lungs was approximately equal to that in KPT-Late mice. KPT-Early mice became tachypneic approximately one month after tumor initiation and histological analysis indicated that AAHs had replaced almost the entire lung parenchyma (Fig. 1A–D and Supplemental Fig. 1). The Tomatopositive cells that expanded in the lungs of KPT-Early mice displayed histological and cytological features of atypical epithelial hyperplasias and expressed lung epithelial markers (Fig. 1D).
The relative tumor burden in KPT-Early and KPT-Late mice was assessed by measuring total lung weight, semi-quantitative genotyping of total lung DNA for recombination of the KrasLSL-G12D and p53floxed alleles, and direct quantification of Tomatopositive tumor cells by immunohistochemistry (Fig. 1E, F and Supplemental Fig. 2). Collectively, these analyses indicate that similar numbers of neoplastic cells are present in KPT-Early mice with massive numbers of hyperplastic lesions and in KPT-Late mice with distinct solid tumor masses within larger areas of normal lung. We have additionally confirmed a recombination efficiency of >95% for the KrasLSL-G12D and p53floxed alleles in purified Tomatopositive cells from both KPT-Early and KPT-Late mice (Supplemental Fig. 2). By matching both the neoplastic cell number and core genetic alterations in both subsets of mice, this system enables the comparison of DTCs between the two groups.
Dissemination of cancer cells into the blood and lymphatic systems as well as directly into the pleural cavity represents an early stage of metastatic spread. The presence of lung cancer cells in the pleural cavity of human lung adenocarcinoma patients has been assessed by intraoperative pleural lavage cytology (19, 20). Cancer cells in the pleural cavity correlates with lymphatic invasion and is a strong predictor of distant metastatic relapse, even in patients with Stage I disease (19, 20). Malignant pleural effusions as well as the high prevalence of pleural metastases at diagnosis and relapse further suggest that metastatic lung cancer cells frequently invade into the pleural cavity.
The high levels of epithelial hyperplasia in KPT-Early mice represents a large reservoir of “pre-malignant” tumor cells that, if capable of disseminating, should be detectable in the pleural cavity and vascular circulation of these mice. To determine whether lung epithelial tumor cells are inherently endowed with the ability to disseminate we performed pleural lavage and flow cytometry to quantify Tomatopositive DTCs. Despite the very high numbers of neoplastic cells we detected very few if any DTCs in the pleural cavity of KPT-Early mice (Fig. 2A, B). To exclude the possibility that we failed to detect DTCs in the KPT-Early mice for technical reasons related to the flow cytometry preparation we directly transplanted unfractionated pleural cavity cells from these mice into syngeneic recipient animals. None of the recipient mice injected with pleural cavity cells from KPT-Early mice developed tumors (Fig. 2F).
We and others have previously shown that not all KrasLSL-G12D/+;p53 flox/flox late time point tumor bearing mice have macro-metastases (15, 16). However we were surprised to find that not all KPT-Late mice had DTCs, and that the number of DTCs was very diverse across mice within this group (Fig. 2B and Supplemental Fig. 3). The presence and number of DTCs was not related to the total tumor burden or time after tumor initiation but did correlate with the number of metastases (Fig. 2B–D and Supplemental Fig. 4). The presence of metastases seeded through hematogenous spread suggests that circulating tumor cells (CTCs) in the blood should also be detectable using flow cytometry. We identified CTCs in two KPT-Late animals that also exhibited high numbers of pleural cavity DTCs (Supplemental Fig. 4). Despite extremely high total tumor burden, large numbers of tumors, and relatively long times after tumor initiation, approximately half of the KPT-late mice had no DTCs in their pleural cavity (Fig. 2B–D).
To determine whether DTCs from KPT-Late mice have tumor seeding potential we transplanted a fraction of the pleural cells into syngeneic recipient mice. Tomatopositive metastases grew in ~40% of recipient mice (2–19 metastases/mouse; Fig. 2E, F). Histological analyses confirmed that these metastases were epithelial in origin with the characteristic poorly differentiated histology of the metastases that typically form in the KrasLSL-G12D/+;p53 flox/flox autochthonous model (data not shown).
Inactivation of the p53 tumor suppressor is a common event in human lung adenocarcinoma and is associated with more advanced disease, metastatic spread, and poor patient outcome. p53 could function to inhibit dissemination or could strictly limit metastatic seeding and/or growth in distant organs. To clarify how p53 inactivation contributes to cancer progression and metastasis and therefore patient outcome, we generated KrasLSL-G12D/+;R26LSL-tdTomato (KT) mice in which the tumor cells express Tomato but remain p53-proficient. As with KPT mice, we generated cohorts of KT mice infected with high titer Adeno-Cre (3×109; KT-Early mice) or low titer Adeno-Cre (107; KT-Late, Supplemental Fig. 5). Consistent with published studies, KT-Late mice developed large primary tumors (with total tumor burden approaching that of KPT-Late mice) but did not develop macrometastases (12, 16). Flow cytometry and direct fluorescent microscopy indicated that KT-Late mice had neither micro-metastases nor cancer cells in the blood (Supplemental Fig. 4 and data not shown). In addition, pleural DTCs were not detected in KT-Early mice (Fig. 2G). While most KT-Late mice did not have cancer cells in their pleural cavity, we did detect Tomatopositive DTCs in one out of fourteen KT-Late mice (Fig. 2G). This suggests that even in the absence of engineered tumor suppressor loss, very rare tumors can independently evolve mechanisms that allow dissemination within the timeframe of their growth in vivo within this model. Consistent with the general absence of Tomatopositive cancer cells from KT mice, transplantation of pleural cavity cells from these mice into syngeneic recipient did not generate metastases (Fig. 2H). These results indicate that at least one major function of p53 is to directly or indirectly regulate phenotypes associated with cancer cell dissemination. Additionally, our observation that not all KPT-Late mice have DTCs suggests that, contrary to work in other systems (21, 22), p53 loss per se is insufficient to drive dissemination but rather enables rare cancer cells in a small fraction of primary tumors to gain the required alterations that drive dissemination.
Fluorescent marking in KPT mice allows for the accurate detection of DTCs but does not uncover how many individual tumors are the source of disseminating cancer cells at late time points. Systemic effects including the ill health of late stage animals, minor differences in individual’s genetic background, and a single advanced tumor’s ability to alter the host in such a way as to induce malignant progression of otherwise benign primary tumors could induce a large number of tumors to synchronously gain the capacity to disseminate (23). Additionally, local paracrine effects including the inflammatory environment could induce multiple tumors in close proximity to each other to disseminate. Based on these considerations, we used two separate systems to determine whether most or all of the cancer cells in the pleural cavity of each KPT-Late mouse are from a single advanced tumor, or whether large numbers of tumors synchronously gain the capacity to disseminate.
To fluorescently mark distinct tumors we generated a Cre-regulated multicolor Rosa26 knock-in reporter allele based on the recombination of heterotypic lox sites surrounding a stop cassette and three unique fluorescent proteins (R26Motley; Supplemental Fig. 6) (24). Infection of KrasLSL-G12D/+;p53flox/flox;R26Motley/+ (KPM) mice generated red, cyan, and yellow fluorescent tumors (Fig. 3A and Supplemental Fig. 6). Despite the presence of multiple large tumors with typical adenocarcinoma features including high nuclear to cytoplasmic ratio, giant cells, and dedifferentiated solid structure, DTCs in each animal were generally only one or two of the three possible colors (Fig. 3B).
Figure 3. Fluorescent and genetic tumor lineage marking confirms that only rare tumors gain the ability to disseminate.
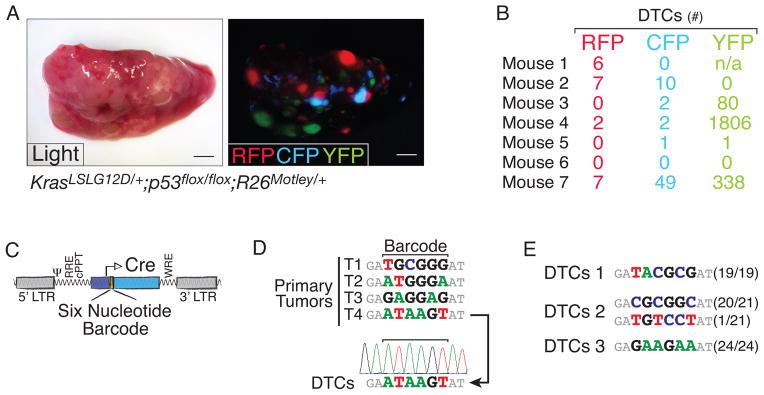
A. Representative light and fluorescent images of a KrasLSL-G12D/+;p53flox/flox;R26Motley/+ lung lobe documents monochromatic tumors. Scale bar is 2mm.
B. Quantification of the number of pleural cavity DTCs of each color in seven KrasLSL-G12D/+;p53flox/flox;R26Motley/+ mice.
C. Schematic of the barcoded lentiviral-Cre vector.
D. Barcode sequencing of purified cancer cells from primary tumors and DTCs. Arrow indicates relationship between Primary Tumor T4 (TMet) and DTCs. Sequencing chromatograph shows barcode homogeneity in the DTCs.
E. Cloning and sequencing of the barcode region confirm that DTCs are largely from a single primary tumor. Barcode sequencing results from three mice are shown as the frequency of that barcode/total number of clones sequenced.
To complement our three-color KPM system, we infected KPT mice with a pool of lentiviral vectors that express Cre and carry a unique nucleotide barcode to generate tumors that each have a stably integrated nucleotide barcode (Fig. 3C; (16)). To determine whether cancer cells in the pleural cavity were from one or more tumors, we isolated these DTCs and sequenced their barcode region. PCR amplification and Sanger sequencing of the barcode region from eight DTC samples indicated that cancer cells in the pleural cavity are seeded from one dominant parental tumor (Fig. 3D). Cloning and sequencing of the barcode amplicon confirmed that DTCs are largely derived from a single primary tumor (Fig. 3E). Collectively, our fluorescent and genetic clonal marking experiments indicate that in lung adenocarcinoma, the ability to disseminate is uniquely gained in a cell-intrinsic manner by rare tumors.
The difficulty in identifying and purifying DTCs from patients and the paucity of methods to isolate these cells from autochthonous mouse models have limited their molecular analysis. We and others have previously identified the transcription factor Nkx2-1 as a critical suppressor of lung adenocarcinoma progression and metastatic ability (16, 25, 26). In our initial study we were unable to determine whether both Nkx2-1positive and Nkx2-1negative cells are equally capable of disseminating but differentially able to seed or grow at distant sites, or whether Nkx2-1negative cells have a unique ability to leave the primary tumor (16) (Supplemental Fig. 7).
To address this question, we purified Tomatopositive cancer cells from primary tumors, disseminated cancer cells, as well as metastases from lymph nodes and pleura. We used the lentiviral-barcode sequences to determine the relationship between the primary tumors, DTCs, and metastases (Fig. 4A). This allowed us to segregate primary tumors into those that had seeded metastases (TMet) and those that had not (TnonMet) and allowed us to choose samples based on the known relationship of the TMet tumors, DTCs, and metastases in individual mice (Fig. 4A). This barcode analysis indicated that these data represent the gene expression state of nine individual metastatic families (Fig. 4A). We determined the expression of Nkx2-1 and its canonical target gene Surfactant protein B (Sftpb) by qRT-PCR on cancer cells from each stage of metastatic progression. In all but one of the families both Nkx2.1 and Sftpb were down-regulated 10-fold or more in DTCs compared to primary tumors (Fig. 4A–C).
Figure 4. Acquisition of an Nkx2.1low state precedes dissemination and is advantageous to the primary tumor.

A. Metastatic primary tumor (TMet), DTC, and metastasis samples are arranged according to their barcode relationship. Samples are named as the mouse number with T = primary tumor, DTC = pleural cavity disseminated tumor cells, LN = lymph node metastasis, Met = pleural metastasis.
B., C., Nkx2-1 (B) and Sftpb (C) expression is high in TMet primary tumors but dramatically down-regulated in DTCs and maintained at low levels in metastases. Gene expression is normalized to the average of all TnonMet tumors = 1. Mean +/− SD of quadruplicate wells is shown.
D. Immunohistochemistry for Nkx2-1 and BrdU incorporation in tumors with both an Nkx2-1neg and Nkx2-1pos area. Left panels show the clear increase in BrdU labeling in the Nkx2-1neg area. Scale bar is 100μm. Middle and right panels are higher magnification images. Scale bar is 50μm.
E., F., Quantification of BrdU+(E) and Ki67+(F) cells in KPT-Late tumors. Each dot represents a tumor area and the bar represents the mean. Data is the number of positive cells per high power (40x) field (#/HPF). *p-value < 0.04. ** p-value < 0.002.
Our ability to identify the metastatic primary tumors (TMet) also allowed us to estimate the fraction of the primary tumor comprised of the metastatic subclone. Nkx2.1 and Sftpb expression was variable in the TMet tumors with three out of four TMet tumors being nearly indistinguishable from TnonMet tumors (Figure 4B, C). Immunohistochemical staining also indicated that the Nkx2-1negative areas comprised diverse proportions in TMet tumors (Supplemental Figure 8). Given the variable size of the metastatic subclones, we investigated whether Nkx2-1negative areas have a selective advantage that drives their expansion within the primary tumor. Immunohistochemistry for BrdU-incorporation, phosphorylated histone H3 (H3P), and Ki67 all uncovered increased proliferation of Nkx2-1negative areas (Figure 4D–F and Supplemental Fig. 8). Importantly, Nkx2-1negative areas were consistently more proliferative than the adjacent Nkx2-1positive areas within the same tumor, and metastases were also more proliferative than Nkx2-1positive primary lung tumors (Figure 4D–F). Conversely, cell death was low and unaltered in Nkx2-1negative primary tumor areas and metastases suggesting that increased proliferation rather than evasion of cell death contributes to the competitive advantage of cancer cells within Nkx2-1negative areas (Supplemental Fig. 8).
DISCUSSION
These studies were initiated to gain insight into whether lung tumors have an inherent ability to disseminate or whether this is a major limiting step of lung adenocarcinoma metastasis. Our results indicate that neither lung epithelial hyperplasias nor most tumors that exist four to eight months after tumor initiation have any ability to shed cancer cells. Our data indicate that the expression of oncogenic Kras and loss of p53 are insufficient to enable lung tumor cells to disseminate, but rather create a permissive genomic context that allows rare tumors to transition to an invasive and metastatic state characterized by down-regulation of the lung differentiation transcription factor Nkx2-1. Importantly, some KPT-Late mice with ~1g of tumor mass have no DTCs, while others have >1000 pleural DTCs derived from a single tumor. We propose that dissemination is a major hurdle during the natural course of lung adenocarcinoma metastasis.
Unlike in pancreatic and breast cancer where tumor cells have been suggested to disseminate from premalignant lesions (2, 3), lung adenocarcinomas begin as benign proliferative lesions with a very limited ability to initiate the metastatic cascade. Our data suggest that even once tumors lose p53 and progress to exhibiting the canonical features of adenocarcinoma, they still require additional alterations to physically escape the primary tumor. While different combinations of oncogenic and tumor suppressor alterations could drive metastasis through unique mechanisms, disruption of the p53 pathways can allow cancer cells to acquire alterations that allow loss of differentiation prior to dissemination.
Our data suggest that the ability to seed metastases early during tumor growth is not a general feature of carcinomas that develop in different organs, but rather that the early dissemination from breast and pancreatic cancer likely represents cell type-specific responses to the initiating oncogenic events or the specific environmental context within those organs or early lesions. We speculate that distinct cancer types and subtypes will have dramatically different thresholds for each step of the metastatic cascade. While pancreatic cancer cells may disseminate from early pancreatic intraepithelial neoplasms, these DTCs appear to have limited to no metastasis seeding ability (3). These primary pancreatic tumors thus require the acquisition of additional traits to allow their DTCs to overcome at least one later step of metastasis. Alternately, for cancers in which dissemination requires a considerable change in cell state, including the fraction of human lung adenocarcinomas driven by the molecular mechanisms described in this autochthonous mouse model, disseminated cancer cells should be viewed as clinically meaningful agents of metastatic spread. These initial disseminated cancer cells may already possess many, if not all, of the attributes required to form metastases. As the molecular characterization of circulating tumor cells in patients become more feasible these types of studies will help in interpreting the clinical significance of these cancer cells.
Most lung adenocarcinoma patients with sub-1cm lung tumors do not present with lymph node metastases or recur with metachronous distant metastatic disease (27–30). Our data suggest that this is likely due to the inability of cells to disseminate, rather than the general inefficiency of each step of metastatic cascade. Unfortunately, lung adenocarcinoma often presents as overtly metastatic disease and even patients that have potentially curative surgery often relapse due to the presence of metastases that were below the detection level at the time of diagnosis. Defining the molecular mediators of lung adenocarcinoma metastasis in this and other genomic context will be an important area of future investigation.
METHODS
Mouse strains
KrasLSL-G12D, p53flox and Rosa26LSL-tdTomato mice have been described (12, 15, 18, 31). We generated mice with the KrasLSL-G12D and the R26LSL-tdTomato in cis on chromosome 6 guaranteeing retention of the R26LSL-tdTomato allele even in genomically unstable tumors. Wildtype male 6–10 week old B6129SF1/J recipients were from The Jackson Laboratory (Stock number: 101043). The Rosa26Moltey knock-in construct was generated using components of the Brainbow-1.1“M” and Brainbow-1.0“L” plasmids (24). The Rosa26Moltey allele was generated from V6.5 ES cells using standard methods. Long-range PCR and Southern blotting identified correctly targeted clones. The MIT and Stanford Institutional Animal Care and Use Committees approved all animal studies and procedures.
Tumor initiation and quantification
Tumors were initiated by intratracheal infection of mice with adenoviral and lentiviral vectors expressing Cre-recombinase as previously described (32). Adenoviral-Cre (Ad5-CMV-Cre) was from the University of Iowa Gene Transfer Core. Lentiviral-Barcode-Cre (Lenti-BC-Cre) was generated as previously described (16). Tumor area, lung area, as well as Nkx2-1positive and Nkx2-1negative areas were determined using ImageJ. Tomatopositive cancer cell number was determined using IHC staining and direct counting. The recombination efficiency of the p53flox and KrasLSL-G12D alleles and the ratio of tumor cells to normal cells at early and late lung adenocarcinoma tumor stages was determined using semi-quantitative PCR.
Cell isolation, flow cytometry, and cell sorting
Cells within the pleural cavity were collected immediately post-euthanasia by making a small incision in the ventral aspect of the diaphragm followed by introduction of 1ml of PBS. One third of the recovered pleural cavity cells were used for flow cytometry. ~0.5mls of blood was collected from the carotid artery. Primary tumors and metastases were dissociated as previously described (16). Cells were stained with antibodies to CD45 (30-F11), CD31 (390), F4/80 (BM8), and Ter119 (all from BioLegend) to exclude hematopoietic and endothelial cells. DAPI was used to exclude dead cells. Cell analysis and sorting were performed on BD LSR II™ analyzers and FACSAria™ sorters (BD Biosciences).
Transplantation
To assess the tumor seeding ability of cancer cells from the pleural cavity one fourth of the total unsorted pleural cells was injected intravenously into the lateral tail vein of syngeneic recipient animals. Recipient mice were analyzed three months after transplantation.
Barcode PCR and RT-qPCR on sorted cancer cells
RNA and genomic DNA were extracted from sorted cancer cells using AllPrep DNA/RNA Micro Kit (Qiagen). Lentiviral barcode sequences were PCR amplified from genomic DNA and directly sequenced or TOPO cloned (Invitrogen) and sequenced. Fluidigm qPCR was performed on 2ng of total RNA from each sample. Site-specific target amplification and real-time PCR was performed on the Fluidigm BioMark HD using Taqman primer-probesets for Nkx2-1(Mm00447558_m1) and Sftpb (Mm00455672_g1)(Applied Biosystems) using standard methods. Samples were run in quadruplicate and normalized to the average of three housekeeping genes (Rpl27(Mm01245874_g1), Rps29(Mm02342448_gH), and Actb(Mm01205647_g1)).
Histologic preparation and immunohistochemistry
Samples were fixed in 4% formalin in PBS overnight and transferred to 70% ethanol until paraffin embedding. Immunohistochemistry was performed on 4μm sections with the ABC Vectastain kit (Vector Laboratories) with antibodies to Nkx2.1 (Epitomics; EP1584Y), Tomato (Rockland Immunochemicals; 600-401-379) Cleaved Caspase 3 (Cell Signaling; 9661), Brdu (BD Pharmingen; 555627), Ki-67 (BD Pharmingen; 550609), SP-B (Santa Cruz Biotechnology; sc-13978), H3P (Serine 10; Millipore; 06-570), and Cytokeratin (Sigma-Aldrich; clone PCK-26). TUNEL staining was performed using the In Situ Cell Death Detection Kit (Roche; 11684817910). Sections were developed with DAB and counterstained with haematoxylin. For in vivo BrdU labeling, mice were injected intraperitoneally with 50 mg/kg BrdU 24 hours before analysis. The number of BrdU, Ki67, H3P, CC3 and TUNEL positive cancer cells was quantified by immunohistochemistry and direct counting, taking into account morphological feature of cancer cells and excluding areas directly adjacent to necrotic areas.
Statistical analysis
The Mann-Whitney test was used except when the outcome of the experiment was a categorical variable in which case the Fisher’s exact test was used.
Supplementary Material
SIGNIFICANCE.
Due to its aggressively metastatic nature, lung cancer is the top cancer killer of both men and women in the United States. We show that, unlike in other cancer types, lung cancer dissemination is a major initial barrier to metastasis. Our findings provide insight into the effect of p53-deficiency and down-regulation of Nkx2-1 during lung adenocarcinoma progression.
Acknowledgments
Grant Support: This work was supported by the National Institutes of Health (R01-CA17533601 to MMW), an American Lung Association Biomedical Research Grant (to MMW), and in part by the Stanford Cancer Institute support grant (P30-CA124435) from the National Cancer Institute. DRC was supported by PHS Grant Number CA09302 from the National Cancer Institute and an NSF Graduate Research Fellowship. C-HC and S-HC were supported by Stanford Dean’s Fellowships, C-HC is additionally funded by an American Lung Association Fellowship. DY is supported by a Stanford Graduate Fellowship. MMW is a Donald E. and Delia B. Baxter Foundation Faculty Fellow and a V Foundation for Cancer Research Martin D. Abeloff, M.D. V Scholar.
We thank Pauline Chu for histology, the Stanford Shared FACS Facility and Protein and Nucleic Acid Facility for expert assistance, Aurora Burds for ES cell targeting, Tyler Jacks for support during the generation of the R26Motley allele, Sopheak Sim at the Stanford Stem Cell Institute Genome Center for Fluidigm qPCR; Jean Livet, Tamily Weissman, David Simpson, Alejandro Sweet-Cordero and Leanne Sayles for reagents; Sean Dolan for administrative support, and David Feldser, Laura Attardi, Steven Artandi and the entire Winslow lab for helpful comments.
Footnotes
Disclosure of Potential Conflicts of Interest: No potential conflicts of interest exist.
References
- 1.Klein CA. Parallel progression of primary tumours and metastases. Nat Rev Cancer. 2009;9:302–12. doi: 10.1038/nrc2627. [DOI] [PubMed] [Google Scholar]
- 2.Husemann Y, Geigl JB, Schubert F, Musiani P, Meyer M, Burghart E, et al. Systemic spread is an early step in breast cancer. Cancer Cell. 2008;13:58–68. doi: 10.1016/j.ccr.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 3.Rhim AD, Mirek ET, Aiello NM, Maitra A, Bailey JM, McAllister F, et al. EMT and dissemination precede pancreatic tumor formation. Cell. 2012;148:349–61. doi: 10.1016/j.cell.2011.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Podsypanina K, Du YC, Jechlinger M, Beverly LJ, Hambardzumyan D, Varmus H. Seeding and propagation of untransformed mouse mammary cells in the lung. Science. 2008;321:1841–4. doi: 10.1126/science.1161621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Klein CA, Seidl S, Petat-Dutter K, Offner S, Geigl JB, Schmidt-Kittler O, et al. Combined transcriptome and genome analysis of single micrometastatic cells. Nat Biotechnol. 2002;20:387–92. doi: 10.1038/nbt0402-387. [DOI] [PubMed] [Google Scholar]
- 6.Schardt JA, Meyer M, Hartmann CH, Schubert F, Schmidt-Kittler O, Fuhrmann C, et al. Genomic analysis of single cytokeratin-positive cells from bone marrow reveals early mutational events in breast cancer. Cancer Cell. 2005;8:227–39. doi: 10.1016/j.ccr.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 7.Stoecklein NH, Hosch SB, Bezler M, Stern F, Hartmann CH, Vay C, et al. Direct genetic analysis of single disseminated cancer cells for prediction of outcome and therapy selection in esophageal cancer. Cancer Cell. 2008;13:441–53. doi: 10.1016/j.ccr.2008.04.005. [DOI] [PubMed] [Google Scholar]
- 8.Siegel R, Ward E, Brawley O, Jemal A. Cancer statistics, 2011: the impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA: a cancer journal for clinicians. 2011;61:212–36. doi: 10.3322/caac.20121. [DOI] [PubMed] [Google Scholar]
- 9.Nguyen DX, Bos PD, Massague J. Metastasis: from dissemination to organ-specific colonization. Nat Rev Cancer. 2009;9:274–84. doi: 10.1038/nrc2622. [DOI] [PubMed] [Google Scholar]
- 10.Takahashi T, Nau MM, Chiba I, Birrer MJ, Rosenberg RK, Vinocour M, et al. p53: a frequent target for genetic abnormalities in lung cancer. Science. 1989;246:491–4. doi: 10.1126/science.2554494. [DOI] [PubMed] [Google Scholar]
- 11.Rodenhuis S, van de Wetering ML, Mooi WJ, Evers SG, van Zandwijk N, Bos JL. Mutational activation of the K-ras oncogene. A possible pathogenetic factor in adenocarcinoma of the lung. N Engl J Med. 1987;317:929–35. doi: 10.1056/NEJM198710083171504. [DOI] [PubMed] [Google Scholar]
- 12.Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, et al. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001;15:3243–8. doi: 10.1101/gad.943001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johnson L, Mercer K, Greenbaum D, Bronson RT, Crowley D, Tuveson DA, et al. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature. 2001;410:1111–6. doi: 10.1038/35074129. [DOI] [PubMed] [Google Scholar]
- 14.Fisher GH, Wellen SL, Klimstra D, Lenczowski JM, Tichelaar JW, Lizak MJ, et al. Induction and apoptotic regression of lung adenocarcinomas by regulation of a K-Ras transgene in the presence and absence of tumor suppressor genes. Genes Dev. 2001;15:3249–62. doi: 10.1101/gad.947701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jackson EL, Olive KP, Tuveson DA, Bronson R, Crowley D, Brown M, et al. The differential effects of mutant p53 alleles on advanced murine lung cancer. Cancer Res. 2005;65:10280–8. doi: 10.1158/0008-5472.CAN-05-2193. [DOI] [PubMed] [Google Scholar]
- 16.Winslow MM, Dayton TL, Verhaak RG, Kim-Kiselak C, Snyder EL, Feldser DM, et al. Suppression of lung adenocarcinoma progression by Nkx2-1. Nature. 2011;473:101–4. doi: 10.1038/nature09881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zheng S, El-Naggar AK, Kim ES, Kurie JM, Lozano G. A genetic mouse model for metastatic lung cancer with gender differences in survival. Oncogene. 2007;26:6896–904. doi: 10.1038/sj.onc.1210493. [DOI] [PubMed] [Google Scholar]
- 18.Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci. 2010;13:133–40. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lim E, Clough R, Goldstraw P, Edmonds L, Aokage K, Yoshida J, et al. Impact of positive pleural lavage cytology on survival in patients having lung resection for non-small-cell lung cancer: An international individual patient data meta-analysis. J Thorac Cardiovasc Surg. 2010;139:1441–6. doi: 10.1016/j.jtcvs.2009.05.048. [DOI] [PubMed] [Google Scholar]
- 20.Buhr J, Berghauser KH, Gonner S, Kelm C, Burkhardt EA, Padberg WM. The prognostic significance of tumor cell detection in intraoperative pleural lavage and lung tissue cultures for patients with lung cancer. J Thorac Cardiovasc Surg. 1997;113:683–90. doi: 10.1016/S0022-5223(97)70225-3. [DOI] [PubMed] [Google Scholar]
- 21.Hwang CI, Matoso A, Corney DC, Flesken-Nikitin A, Korner S, Wang W, et al. Wild-type p53 controls cell motility and invasion by dual regulation of MET expression. Proc Natl Acad Sci U S A. 2011;108:14240–5. doi: 10.1073/pnas.1017536108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Elyada E, Pribluda A, Goldstein RE, Morgenstern Y, Brachya G, Cojocaru G, et al. CKIalpha ablation highlights a critical role for p53 in invasiveness control. Nature. 2011;470:409–13. doi: 10.1038/nature09673. [DOI] [PubMed] [Google Scholar]
- 23.McAllister SS, Gifford AM, Greiner AL, Kelleher SP, Saelzler MP, Ince TA, et al. Systemic endocrine instigation of indolent tumor growth requires osteopontin. Cell. 2008;133:994–1005. doi: 10.1016/j.cell.2008.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Livet J, Weissman TA, Kang H, Draft RW, Lu J, Bennis RA, et al. Transgenic strategies for combinatorial expression of fluorescent proteins in the nervous system. Nature. 2007;450:56–62. doi: 10.1038/nature06293. [DOI] [PubMed] [Google Scholar]
- 25.Hosono Y, Yamaguchi T, Mizutani E, Yanagisawa K, Arima C, Tomida S, et al. MYBPH, a transcriptional target of TTF-1, inhibits ROCK1, and reduces cell motility and metastasis. Embo J. 2012;31:481–93. doi: 10.1038/emboj.2011.416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Berghmans T, Paesmans M, Mascaux C, Martin B, Meert AP, Haller A, et al. Thyroid transcription factor 1--a new prognostic factor in lung cancer: a meta-analysis. Ann Oncol. 2006;17:1673–6. doi: 10.1093/annonc/mdl287. [DOI] [PubMed] [Google Scholar]
- 27.Pashkevich MA, Sigal BM, Plevritis SK. Modeling the transition of lung cancer from early to advanced stage. Cancer Causes Control. 2009;20:1559–69. doi: 10.1007/s10552-009-9401-4. [DOI] [PubMed] [Google Scholar]
- 28.Hamatake D, Yoshida Y, Miyahara S, Yamashita S, Shiraishi T, Iwasaki A. Surgical outcomes of lung cancer measuring less than 1 cm in diameter. Interact Cardiovasc Thorac Surg. 2012;15:854–8. doi: 10.1093/icvts/ivs337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schuchert MJ, Kilic A, Pennathur A, Nason KS, Wilson DO, Luketich JD, et al. Oncologic outcomes after surgical resection of subcentimeter non-small cell lung cancer. Ann Thorac Surg. 2011;91:1681–7. doi: 10.1016/j.athoracsur.2011.01.010. discussion 7–8. [DOI] [PubMed] [Google Scholar]
- 30.Miller DL, Rowland CM, Deschamps C, Allen MS, Trastek VF, Pairolero PC. Surgical treatment of non-small cell lung cancer 1 cm or less in diameter. Ann Thorac Surg. 2002;73:1545–50. doi: 10.1016/s0003-4975(02)03525-7. discussion 50–1. [DOI] [PubMed] [Google Scholar]
- 31.Jonkers J, Meuwissen R, van der Gulden H, Peterse H, van der Valk M, Berns A. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat Genet. 2001;29:418–25. doi: 10.1038/ng747. [DOI] [PubMed] [Google Scholar]
- 32.DuPage M, Dooley AL, Jacks T. Conditional mouse lung cancer models using adenoviral or lentiviral delivery of Cre recombinase. Nature protocols. 2009;4:1064–72. doi: 10.1038/nprot.2009.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.