

Abstract
Pancreatic ductal adenocarcinoma (PDAC) is associated with pronounced fibrosis that contributes to chemoresistance, in part, through increased histone acetylation. Since bromodomain (BRD) and extra terminal domain (BET) proteins are ‘readers’ of histone acetylation marks, we targeted BET proteins in PDAC cells grown in three-dimensional collagen. We show that treatment with BET inhibitors decreases growth of PDAC cells (AsPC1, CD18 and Panc1) in collagen. Transfection with siRNA against BRD4, which is increased in human PDAC tumors, also decreases growth of PDAC cells. BET inhibitors additionally decrease growth in collagen of PDAC cells that have undergone epithelial-to-mesenchymal transition or have become resistant to chemotherapy. Although BET inhibitors and BRD4 siRNA repress c-MYC only in AsPC1 and CD18 cells, downregulating c-MYC decreases growth of all three PDAC cell lines in collagen. FOSL1, which is also targeted by BET inhibitors and BRD4 siRNA in AsPC1, CD18 and Panc1 cells, additionally regulates growth of all three PDAC cell lines in collagen. BET inhibitors and BRD4 siRNA repress HMGA2, an architectural protein that modulates chromatin state and also contributes to chemoresistance, in PDAC cells grown in collagen. Importantly, we show that there is a statistically significant correlation between BRD4 and HMGA2 in human PDAC tumors. Significantly, overexpression of HMGA2 partially mitigates the effect of BET inhibitors on growth and c-MYC and/or FOSL1 expression in collagen. Overall, these results demonstrate that BET inhibitors block growth of PDAC cells in collagen and that BET proteins may be potential targets for the treatment of pancreatic cancer.
Keywords: JQ1, I-BET151, BRD4, collagen, c-MYC, FOSL1, HMGA2, pancreatic cancer
INTRODUCTION
The prognosis of patients diagnosed with pancreatic ductal adenocarcinoma (PDAC) continues to remain abysmal. Patients presenting with metastases have a median survival of ~6 months, while patients who undergo resection of their localized disease have a median survival of ~17–22 months (1, 2). These dismal outcomes are attributed to the fact that PDAC is a highly chemoresistant cancer, in part, due to concurrent activation of multiple signaling pathways that limit the efficacy of current therapies (3, 4). In addition, both primary tumors and distant metastases exhibit pronounced fibrosis (5, 6), which limits the delivery of chemotherapy to cancer cells and also activates signaling pathways that mitigate the effects of chemotherapy (7–9). Previously, we reported that collagen induces PDAC expression of high mobility group A2 (HMGA2), an architectural protein that regulates chromatin structure (10, 11), to attenuate the effects of chemotherapy (7). Recently, we have shown that PDAC cells in three-dimensional collagen through HMGA2 upregulate histone acetyltransferase (HAT) expression to attenuate the effect of chemotherapy (12). Moreover, we have shown that areas of fibrosis in human PDAC tumors are associated with increased acetylation (12), suggesting that epigenetic changes contribute to chemotherapy resistance in human PDAC tumors as well.
Although there has been increasing interest in targeting epigenetic changes to overcome chemotherapy resistance of PDAC tumors, inhibitors of histone deacetylases (HDACs; ‘erasers’ of chromatin marks) have demonstrated limited success against solid tumors in clinical trials (13, 14). PDAC patients treated with the HDAC inhibitor CI-994 with chemotherapy did not demonstrate increased response compared to chemotherapy alone (15). In contrast to HDAC inhibitors (13, 14), only a limited number of HAT (‘writers’ of chromatin marks) inhibitors have been developed (16). The clinical utility of HAT inhibitors has been limited due to low metabolic stability or poor cellular permeability (16, 17). Recently, there has been increasing interest in targeting ‘readers’ of histone acetylation, in particular members of bromodomain (BRD) and extra terminal domain (BET) family of proteins, in a number of different cancer types (18–22).
BET proteins, which include BRD2, BRD3, BRD4 and the testis specific BRDT, are important ‘reader’ molecules that bind to acetylated histones and regulate transcription of genes involved in growth, fibrosis, inflammation and cancer (18–23). A number of selective inhibitors of BET proteins [e.g., JQ1 and I-BET151 (22, 24)] have been developed that compete with the acetyl-binding pockets of BRD proteins (22, 25). These compounds potently inhibit growth of leukemia and lymphoma cell lines (20, 21), mainly through repression of c-MYC and downstream transcriptional targets (20, 21). They have also been shown to inhibit growth of glioblastoma and neuroblastoma cell lines through MYC suppression (18, 26). However, in lung cancer cells, the effects of JQ1 on cell growth occurred through repression of the oncogenic transcription factor FOS-like antigen 1 (FOSL1) rather than through c-MYC repression (19).
In this report, we examined the role of BET proteins in pancreatic cancer. We show that BET inhibitors decrease growth in three-dimensional collagen of AsPC1, CD18 and Panc1 cells, including PDAC cells that have undergone epithelial-to-mesenchymal transition or have become resistant to chemotherapy. Although BET inhibitors decrease c-MYC only in AsPC1 and CD18 cells, downregulating c-MYC decreases growth of all three PDAC cell lines in three-dimensional collagen. FOSL1, which is also targeted by BET inhibitors in AsPC1, CD18 and Panc1 cells, also regulates growth of all three PDAC cell lines in three-dimensional collagen. Significantly, BET inhibitors repress HMGA2 levels in all three PDAC cell lines grown in three-dimensional collagen. Importantly, overexpression of HMGA2 partially mitigates the effect of BET inhibitors on growth and c-MYC and/or FOSL1 expression. Overall, these results demonstrate that BET inhibitors block growth of PDAC cells in three-dimensional collagen and that BET inhibitors may be potential therapeutic agents for the treatment of pancreatic cancer.
MATERIALS AND METHODS
Chemicals/Reagents
General tissue culture materials were obtained from VWR International. Antibodies against Snail and BRD4 were obtained from Abcam. Antibodies against c-MYC, p21 and FOSL1 were purchased from Cell Signaling, HMGA2 antibody was from Biocheck Inc, while vimentin antibody was from Abcam. Alpha-tubulin antibody was obtained from Santa Cruz, while E-cadherin antibody was from BD Bioscience. Secondary antibodies were purchased from Sigma. The EZ-Chip and EZ-Zyme Chromatin Prep kits were from Millipore. The anti-BRD4 antibody for ChIP assay was purchased from Bethyl Laboratories, while the anti-RNA polymerase II antibody and control IgG antibody were from Millipore. BET inhibitor JQ1 was obtained from BPS Bioscience, while I-BET151 was acquired from Tocris Bioscience. BRD4, c-MYC, and FOSL1 siRNAs were purchased from Life Technologies.
Cell culture
AsPC1, CD18/HPAF-II and Panc1 cells were obtained from American Type Culture Collection (ATCC; Manassas, VA) in 2008. AsPC1 and Panc1 cells were last authenticated by STR profiling at the Johns Hopkins Genetic Resources Core Facility in 2010, while CD18 cells were authenticated by STR profiling in 2013. Cells were maintained in DMEM containing 10% FBS and antibiotics (100 U/ml Penicillin and 100 μg/ml Streptomycin).
AsPC1 and CD18 cells expressing Snail were generated by the Munshi Lab as detailed previously (27). Similarly, CD18 and Panc1 cells expressing HMGA2 were created by the Munshi Lab as previously described (7). AsPC1-vector, AsPC1-Snail, CD18-vector, and CD18-Snail cells have not been previously authenticated.
Chemoresistant CD18 (CD18-CR) cells were generated by treating parental CD18 (CD18-P) cells with increasing concentration of 5-fluorouracil (5-FU) over a period of 3 months. The surviving cells were maintained in 10 μM concentration of 5-FU. CD18-P and CD18-CR cells were authenticated by STR profiling at the Johns Hopkins Genetic Resources Core Facility in 2013.
Embedding and examination of cells in three-dimensional type I collagen gels
Collagen mixture (2 mg/mL) was made by adding the appropriate volumes of sterile water, 10X DMEM and NaOH and kept on ice until needed (27). Cells were then suspended in the collagen solution and allowed to gel at 37°C. For RNA extraction, the gel containing cells was processed using RNeasy extraction kit (Qiagen) and then processed for qRT-PCR analysis. For morphological examination of cells, cell colonies in three-dimensional collagen were examined using a Zeiss Axiovert 40 CFL microscope and pictures taken with a Nikon Coolpix 4500 camera (27). The relative size of individual colonies was measured using Photoshop.
Transfection
Cells were transfected with siRNA against BRD4, c-MYC, FOSL1 or control siRNA using RNAimax (Invitrogen) according to manufacturer’s instructions before plating into collagen.
Quantitative Real Time-PCR analysis
Quantitative gene expression was performed with gene specific Taqman probes, TaqMan Universal PCR Master Mix and the 7500 Fast Real-time PCR System from Applied Biosystems. The data were then quantified with the comparative CT method for relative gene expression.
Oncomine Analysis
The relative expression of BRD2, BRD3, BRD4 and BRDT was determined by searching the publicly accessible Oncomine database version 4.4.3.
Human PDAC tissue analysis
Pancreatic tissue was obtained from patients with pancreatic adenocarcinoma on an IRB-approved protocol. Using de-identified human pancreatic tissue specimens, RNA was isolated from human PDAC tumors previously stored in RNAlater and analyzed by qRT-PCR (7, 27).
Immunoblotting
Immunoblotting for Snail, E-cadherin, vimentin, BRD4, c-MYC, FOSL1, HMGA2 and α-tubulin was done as previously described (27).
Chromatin Immunoprecipitation
Panc1 cells treated with DMSO or JQ1 for 24 hours were treated with formaldehyde to create DNA-protein cross-links. Chromatin fragments were prepared using EZ-Zyme Chromatin Prep kit and then ChIP performed using the EZ ChIP kit and anti-BRD4 antibody or anti-RNA polymerase II antibody. Purified DNA was then analyzed by PCR using primers specific for c-MYC or FOSL1 locus (28, 29), and the PCR products visualized on a 2%-agarose gel.
Statistical analysis
All statistical analyses were done using Microsoft Excel or GraphPad Instat.
RESULTS
Decreased BET bromodomain activity inhibits growth of PDAC cells in three-dimensional collagen
Recently, small molecule inhibitors have been developed to specifically block BET bromodomain (22, 25). As one of the hallmarks of PDAC is a pronounced fibrotic tumor microenvironment (5, 6), we determined the effect of the highly specific BET bromodomain inhibitor JQ1 on PDAC cells grown in three-dimensional collagen. AsPC1, CD18 and Panc1 cells grown in three-dimensional collagen were treated with JQ1 every other day for 7 days and the effect on colony size was measured. As shown in Fig. 1A, treatment with JQ1 decreased the growth of all 3 cell lines, with AsPC1 and CD18 cells being more sensitive to growth inhibition than Panc1 (Fig. 1B). I-BET151, another highly specific BET bromodomain inhibitor (22, 25), also decreased the growth of all 3 PDAC cell lines in three-dimensional collagen (Supplementary Fig. S1). Since expression of BRD4, and not BRD2, BRD3 or BRDT, is increased in human pancreatic tumors relative to normal tissue in the Oncomine database (30–33), we also evaluated the role of BRD4 in regulating growth in three-dimensional collagen using siRNA against BRD4. The siRNA specific for BRD4 successfully knocked down BRD4 expression (Fig. 1C), and decreased the growth of AsPC1, CD18 and Panc1 cells in three-dimensional collagen (Figs. 1D,E).
Figure 1. Decreased BET bromodomain activity inhibits growth of PDAC cells in three-dimensional collagen.

A, B. AsPC1, CD18 and Panc1 cells were grown in three-dimensional collagen gels and fresh serum-containing medium supplemented with DMSO or JQ1 was added every other day for 7 days. The effect on colony size was examined by phase contrast microscopy (A), and size of the individual colonies measured (B). C–E. PDAC cells were transfected with control siRNA (siCtrl) or BRD4-specific siRNAs (siBRD4), allowed to recover for 48 hours, and then plated in collagen gels. The specific knockdown of BRD4 was determined by qRT-PCR using GAPDH as normalization control and by Western blotting using α-tubulin as loading control (C). The effect on colony size in three-dimensional collagen was examined by phase contrast microscopy (D), and size of the individual colonies measured (E). The results are representative of at least three independent experiments. *, p < 0.05.
JQ1 decreases growth of PDAC cells that have undergone epithelial-to-mesenchymal transition (EMT)
It is being increasingly recognized that cells that have undergone EMT are resistant to standard chemotherapy (34, 35). Snail, one of the key regulators of EMT (27), is associated with metastasis in human PDAC tumors (36, 37). Thus, we evaluated the effect of JQ1 on CD18 cells that were made to undergo EMT following Snail overexpression. Consistent with previous characterization of cells that have undergone EMT (34, 38), expression of Snail in CD18 cells decreased E-cadherin protein and mRNA expression and increased vimentin mRNA levels (Fig. 2A). Compared to CD18-vector cells that have an epithelial-like cobblestone appearance CD18-Snail cells demonstrate a scattered mesenchymal-like phenotype (Fig. 2A). Importantly, treatment with JQ1 (Figs. 2B,C) and I-BET151 (Supplementary Fig. S2) decreased the growth of both CD18-vector and CD18-Snail cells grown in three-dimensional collagen equally. JQ1 and I-BET151 also equally decreased growth of AsPC1-Snail cells in three-dimensional collagen compared to AsPC1-vector cells (Supplementary Fig. S3). These results indicate that BET bromodomain inhibitors are effective against epithelial cells as well as against cells that have undergone EMT.
Figure 2. JQ1 decreases growth of PDAC cells that have undergone epithelial-to-mesenchymal transition (EMT).

A–C. Lysates from CD18-Vector and CD18-Snail cells were analyzed for Snail, E-cadherin and vimentin by Western blotting, and the effect on E-cadherin and vimentin mRNA by qRT-PCR (A). CD18-Vector and CD18-Snail cells growing on tissue culture plastic were examined by phase microscopy. CD18-Vector and CD18-Snail cells were grown in three-dimensional collagen gels and fresh serum-containing medium supplemented with DMSO or JQ1 was added every other day for 7 days. The effect of JQ1 on colony size in three-dimensional collagen was examined by phase contrast microscopy (B), and size of the individual colonies measured (C). JQ1 equally decreases growth of chemoresistant PDAC cells in three-dimensional collagen. D–F. The mRNA samples from parental CD18 (CD18-P) cells and chemo-resistant CD18 (CD18-CR) cells growing on tissue culture plastic were analyzed for E-cadherin, vimentin and ZEB1 by qRT-PCR (D). CD18-P and CD18-CR cells growing on tissue culture plastic were examined by phase microscopy. CD18-P and CD18-CR cells were grown in three-dimensional collagen gels and fresh serum-containing medium supplemented with DMSO or JQ1 was added every other day for 7 days. The effect of JQ1 on colony size in three-dimensional collagen was examined by phase contrast microscopy (E), and size of the individual colonies measured (F). The results are representative of at least three independent experiments. *, p < 0.05.
JQ1 equally decreases growth of chemoresistant PDAC cells in three-dimensional collagen
Previous studies have found that cells that have become resistant to standard chemotherapy demonstrate evidence of EMT (39, 40). In order to investigate whether JQ1 was able to limit the growth of chemoresistant PDAC cells, CD18 cells were treated with increasing concentration of 5-FU to create CD18-CR cells. Consistent with previous findings (39, 40), CD18-CR cells show a pronounced mesenchymal phenotype, with reduced E-cadherin and increased vimentin expression (Fig. 2D). Although CD18-CR cells do not show a significant increase in Snail expression (data not shown), CD18-CR cells have increased levels of ZEB1 (Fig. 2D), another well-known regulator of EMT (34). Similar to its effect on CD18-Snail cells, treatment with JQ1 (Figs. 2E,F) and I-BET151 (Supplementary Fig. S4) decreased the growth of CD18-CR cells in three-dimensional collagen. These results indicate that the BET bromodomain inhibitors are also effective against cells that have become resistant to standard chemotherapy.
BET proteins regulate expression of c-MYC in AsPC1 and CD18 cells, but not in Panc1 cells
As BET bromodomain inhibitors can repress c-MYC expression (20, 21), we evaluated the effect of BET protein inhibition on c-MYC levels in PDAC cells grown in three-dimensional collagen. Treatment with JQ1 (Fig. 3A) and I-BET151 (Supplementary Fig. S5A) for 48 hours decreased c-MYC mRNA levels in AsPC1 and CD18 cells, but not in Panc1 cells. Significantly, the c-MYC mRNA levels in AsPC1 and CD18 cells were repressed within 8 hours of treatment with JQ1 (Supplementary Fig. S5B). Importantly, BET inhibitors also decreased c-MYC levels in cells that have undergone EMT and in PDAC cells that have become resistant to chemotherapy (Supplementary Fig. S5C,D). Treatment with JQ1 also decreased c-MYC protein levels in AsPC1 and CD18 cells, but had minimal effect in Panc1 cells (Fig. 3B). Although treatment with JQ1 did not affect p21 protein levels in AsPC1 cells, JQ1 increased p21 protein levels in CD18 and Panc1 cells (Fig. 3B). We also evaluated the effect of siRNA against BRD4 on c-MYC levels in PDAC cells. Similar to the findings with JQ1 and I-BET151 treatment, transfection with siRNA against BRD4 decreased c-MYC levels in AsPC1 and CD18 cells, but not in Panc1 cells (Fig. 3C). To determine to what extent BRD4 may regulate c-MYC in human PDAC tumors, we evaluated the relationship between BRD4 and c-MYC expression in human PDAC tumor samples. As shown in Fig. 3D, c-MYC levels significantly correlate with BRD4 levels in human PDAC tumor samples (n=15, r=0.78, p=0.0006).
Figure 3. BET proteins regulate expression of c-MYC in AsPC1 and CD18 cells, but not in Panc1 cells.

A,B. PDAC cells were grown in three-dimensional type I collagen gels in the presence of DMSO (vehicle control), or JQ1 (0.5 μM) for 48 hours. The effect on c-MYC mRNA expression was analyzed by qRT-PCR using GAPDH as normalization control (A). *, p < 0.05. The effect on c-MYC and p21 protein expression was analyzed by Western blotting using α-tubulin as loading control (B). C. Effect of siBRD4 on c-MYC mRNA expression was analyzed by qRT-PCR using GAPDH as normalization control. The results are representative of three independent experiments. *, p < 0.05. D. De-identified PDAC samples (n=15) were collected on an IRB-approved protocol. The samples were processed for BRD4 and c-MYC mRNA by qRT-PCR using GAPDH and the relationship between BRD4 and c-MYC was analyzed using Pearson correlation coefficient. c-MYC regulates growth of AsPC1, CD18 and Panc1 cells in three-dimensional collagen. E,F. PDAC cells were transfected with control siRNA (siCtrl) or c-MYC-specific siRNAs (si c-MYC), allowed to recover for 48 hours, and then plated in collagen gels. The specific knockdown of c-MYC was determined by qRT-PCR using GAPDH as normalization control and by Western blotting using α-tubulin as loading control (E). The effect on colony size was examined by phase contrast microscopy, and size of the individual colonies measured (F). The results are representative of at least three independent experiments. *, p < 0.05.
c-MYC regulates growth of AsPC1, CD18 and Panc1 cells in three-dimensional collagen
Since JQ1 did not affect c-MYC levels in Panc1 cells (Fig. 3A) but decreased growth of Panc1 cells in three-dimensional collagen (Fig. 1), we evaluated to what extent suppression of c-MYC was sufficient to inhibit growth of PDAC cells. Knockdown of c-MYC decreased growth of all three PDAC cell lines in three-dimensional collagen (Fig. 3E,F). Even though c-MYC is able to regulate growth of Panc1 cells in three-dimensional collagen, the lack of effect of JQ1 on c-MYC levels indicates that the inhibitory effect of JQ1 on Panc1 cells must involve additional targets of BET inhibitors.
FOSL1 regulates growth of AsPC1, CD18 and Panc1 cells in three-dimensional collagen
It has been shown that the oncogenic transcription factor FOSL1 is a target of JQ1 in lung cancer cells (19). Thus, we determined to what extent BET protein inhibition in PDAC cells affected FOSL1 levels. Treatment with JQ1 (Fig. 4A), I-BET151 (Supplementary Fig. S6A) or transfection with BRD4 siRNA (Supplementary Fig. S6B) decreased FOSL1 in PDAC cells, including in Panc1 cells. Significantly, the FOSL1 mRNA levels in PDAC cells were repressed within 8 hours of treatment with JQ1 (Supplementary Fig. S6C). We next evaluated to what extent siRNA against FOSL1 (Fig. 4B) attenuated growth of PDAC cells in three-dimensional collagen. As shown in Figs. 4C and 4D, FOSL1 siRNA decreased the growth of AsPC1, CD18 and Panc1 cells. These results suggest that BET protein inhibition may also decrease growth of PDAC cells through modulation of FOSL1 expression.
Figure 4. FOSL1, a target of BET bromodomain inhibitor, regulates growth of PDAC cells in three-dimensional collagen.
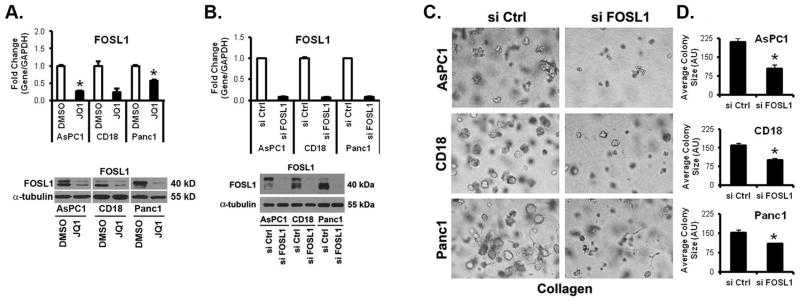
A. PDAC cells were grown in three-dimensional type I collagen gels in the presence of DMSO (vehicle control) or JQ1 (0.5 μM) for 48 hours. The effect on FOSL1 expression was analyzed by qRT-PCR using GAPDH as normalization control and by Western blotting using α-tubulin as loading control. B–D. PDAC cells were transfected with control siRNA (si Ctrl) or FOSL1-specific siRNAs (si FOSL1), allowed to recover for 48 hours, and then plated in collagen gels. The specific knockdown of FOSL1 was determined by qRT-PCR using GAPDH as normalization control and by Western blotting using α-tubulin as loading control (B). The effect on colony size was examined by phase contrast microscopy (C), and size of the individual colonies measured (D).
Targeting BET bromodomain decreases high mobility group A2 (HMGA2) levels in PDAC cells
We have previously shown that expression of HMGA2, an architectural protein that modulates chromatin state, is induced when PDAC cells are grown in three-dimensional collagen (7, 12). Significantly, we demonstrated that HMGA2 could overcome the effects of gemcitabine in three-dimensional collagen (7, 12). Thus, we determined whether BET protein inhibition affected HMGA2 levels in PDAC cells. AsPC1, CD18 and Panc1 cells in three-dimensional collagen were treated with JQ1 and the effect on HMGA2 levels was determined. Treatment with JQ1 decreased HMGA2 levels in PDAC cells at both protein and mRNA levels (Figs. 5A and 5B). Similar to the findings with JQ1 treatment, transfection with BRD4 siRNA decreased HMGA2 levels in AsPC1, CD18 and Panc1 cells (Fig. 5C). To determine to what extent BRD4 may regulate HMGA2 in human PDAC tumors, we evaluated the relationship between BRD4 and HMGA2 expression in human PDAC tumor samples. As shown in Fig. 5D, HMGA2 levels significantly correlate with BRD4 levels in human PDAC tumor samples (n=21, r=0.52, p=0.02). These results suggest that the in vivo effects of BET bromodomain proteins may, in part, also be mediated through regulation of HMGA2 expression.
Figure 5. Targeting BET bromodomain decreases high mobility group A2 (HMGA2) levels in PDAC cells.
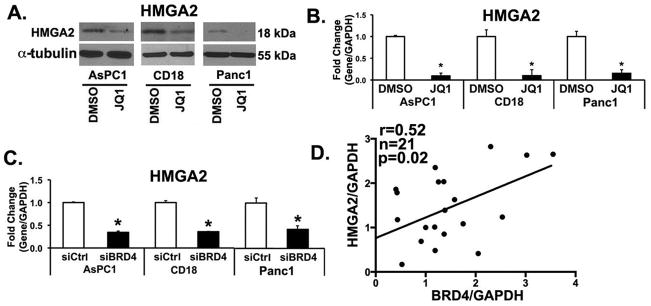
A,B. PDAC cells were grown in three-dimensional type I collagen gels in the presence of DMSO (vehicle control) or JQ1 (0.5 μM) for 48 hours and the effect on HMGA2 expression analyzed by Western blotting and by qRT-PCR using GAPDH as normalization control. *, p < 0.05. C. Effect of siBRD4 on HMGA2 mRNA expression was analyzed by qRT-PCR using GAPDH as normalization control. The results are representative of three independent experiments. *, p < 0.05. D. De-identified PDAC samples (n=21) were obtained on an IRB-approved protocol. The samples were processed for BRD4 and HMGA2 mRNA by qRT-PCR using GAPDH and the relationship between BRD4 and HMGA2 was analyzed using Pearson correlation coefficient.
Overexpression of HMGA2 partly mitigates the effects of JQ1 on growth and c-MYC and FOSL1 expression
To determine the extent to which HMGA2 modulates the effect of JQ1 in collagen, we generated CD18 and Panc1 cells overexpressing HMGA2 (Fig. 6A). Initially, we examined the effect of HMGA2 overexpression on JQ1-mediated repression of growth in three-dimensional collagen. CD18-Vector, CD18-HMGA2, Panc1-Vector and Panc1-HMGA2 cells in three-dimensional collagen were treated with low levels of JQ1 for 7 day. Overexpression of HMGA2 in CD18 cells partly mitigated the effect of BET bromodomain inhibition at 0.125 μM concentration of JQ1; however, JQ1 at the 0.25 μM concentration overwhelmed the protective effects of HMGA2 (Fig. 6B,C). HMGA2 overexpression in Panc1 cells also partly mitigated the effects of BET bromodomain inhibition at 0.25 μM concentration of JQ1 (Fig. 6B,C); however, JQ1 at the 0.5 μM concentration overwhelmed the protective effects of HMGA2 in Panc1 cells (data not shown). These results indicate that HMGA2 overexpression can partly protect PDAC cells from the effects of JQ1 in three-dimensional collagen.
Figure 6. Overexpression of HMGA2 partly mitigates the effect of JQ1 on growth and c-MYC and FOSL1 expression.

A. Lysates from CD18-Vector, CD18-HMGA2, Panc1-Vector and Panc1-HMGA2 cells grown on tissue culture plastic were analyzed for HMGA2 by Western blotting. B,C. CD18-Vector, CD18-HMGA2, Panc1-Vector and Panc1-HMGA2 cells were grown in three-dimensional type I collagen gels in the presence of DMSO (vehicle control) or JQ1 for 7 days. The effect of JQ1 on colony size was examined by phase contrast microscopy (B), and size of the individual colonies measured (C). D. CD18-Vector and CD18-HMGA2 cells were grown in three-dimensional type I collagen gels in the presence of DMSO (vehicle control) or JQ1 (0.125 μM) for 48 hours and the effect on c-MYC and FOSL1 analyzed by qRT-PCR using GAPDH as normalization control. E. Panc1-Vector and Panc1-HMGA2 cells were grown in three-dimensional type I collagen gels in the presence of DMSO (vehicle control) or JQ1 (0.25 μM) for 48 hours and the effect on c-MYC and FOSL1 analyzed by qRT-PCR using GAPDH as normalization control. The results are representative of three independent experiments. *, p < 0.05. ns, not significant.
Since we have found that c-MYC and FOSL1 regulate growth of these cells in three-dimensional collagen (Figs. 3 and 4), we next evaluated the effect of HMGA2 overexpression on JQ1-mediated changes in c-MYC and FOSL1. As shown previously in Fig. 3A, treatment of CD18-Vector cells with JQ1 decreased c-MYC mRNA levels (Fig. 6D, left). Importantly, the suppressive effect of JQ1 on c-MYC levels was mitigated in CD18-HMGA2 cells compared to CD18-Vector cells. Consistently with our findings in Fig. 4, treatment of CD18-Vector cells with JQ1 decreased FOSL1 levels (Fig. 6D, right). Significantly, HMGA2 overexpression not only increased FOSL1 levels, but co-treatment with JQ1 resulted in even further increase in FOSL1 levels in CD18 cells. Similarly, we examined the effect of HMGA2 overexpression on JQ1-mediated changes in c-MYC and FOSL1 levels in Panc1 cells. Although JQ1 did not affect c-MYC mRNA in Panc1-Vector cells (Fig. 6E left and Fig. 3A), treatment of Panc1-HMGA2 cells with JQ1 increased c-MYC levels by ~1.7-fold compared to Panc1-Vector cells treated with JQ1. As shown previously in Fig. 4, treatment of Panc1-Vector cells with JQ1 decreased FOSL1 levels (Fig. 6E, right). Importantly, HMGA2 overexpression increased FOSL1 levels and partially reversed the effects of JQ1 on FOSL1 levels in Panc1 cells.
DISCUSSION
Recently, there has been an increasing interest in understanding the contribution of epigenetic changes to cancer development and how best to target these epigenetic changes (18–22). It is now well-established that pancreatic cancer is associated with epigenetic changes resulting, in part, from alteration in histone methylation and acetylation (41, 42). Previously, we have shown that the collagen microenvironment, which is particularly pronounced in human PDAC tumors (5), contributes to pancreatic cancer progression through epigenetic changes (7, 12). We have reported that there is increased histone acetylation in areas of fibrosis in human PDAC tumors and that PDAC cells grown in three-dimensional collagen demonstrate increased histone acetylation (12). Significantly, we demonstrated that targeting HATs (‘writers’ of chromatin marks) with siRNA sensitizes PDAC cells grown in three-dimensional collagen to chemotherapy (12); however, HAT inhibitors have had limited clinical success because of low metabolic stability and/or poor cellular permeability (16, 17). In this report, we show that targeting ‘readers’ of histone acetylation marks instead could be an effective strategy in the treatment of pancreatic cancer. We show that BET bromodomain inhibitors and siRNA against BRD4, which is increased in human tumors compared to adjacent normal tissue (30–33), block growth of PDAC cells in three-dimensional collagen.
Our data suggest that BET proteins mediate some of the effects of epigenetic changes observed in PDAC cells, including in PDAC cells that have undergone EMT. Significantly, it has been demonstrated that there are profound epigenetic changes associated with EMT (43, 44). For example, there is increased H3K9 acetylation in breast cancer cells following Snail-driven EMT (44). Furthermore, cells that have undergone EMT can demonstrate stem-cell like properties (34, 35), and it was recently shown that BET inhibitors could eliminate stem- and progenitor-cells in acute leukemia (45). Here, we show that the BET inhibitors are equally effective against cells that have undergone EMT induced either by expression of Snail transcription factor or by selecting cells that have become resistant to chemotherapy. Additionally, we show that the BET inhibitors block growth of chemoresistant PDAC cells equally as well as parental cells, suggesting that the BET inhibitors may have efficacy against pancreatic cancer stem cells.
Mechanistically, it has been shown that a significant effect of BET bromodomain inhibitors is through repression of MYC expression. BET bromodomain inhibitors repress MYC in leukemia, lymphoma, neuroblastoma and glioblastoma cell lines (18, 20, 21, 26). However, overexpression of c-MYC in glioblastoma and leukemia cells failed to overcome the effects of JQ1 treatment, suggesting that BET bromodomain inhibitors can have both c-MYC-dependent and c-MYC-independent effects (20, 26). We have found that neither JQ1 nor BRD4 siRNA represses c-MYC levels in Panc1 cells, even though both JQ1 and BRD4 siRNA block growth of Panc1 cells in three-dimensional collagen, suggesting that the effects of JQ1 in Panc1 cells are independent of c-MYC. This was also found to be the case with a number of lung cancer cell lines that were sensitive to the effects of JQ1, but showed no repression of c-MYC following JQ1 treatment (19). Instead, the effect of JQ1 in lung cancer cells was mediated by FOSL1 repression (19). We also show that JQ1 and BRD4 siRNA can repress FOSL1 in PDAC cells and that FOSL1 siRNA blocks growth of these cells in three-dimensional collagen. However, as with c-MYC overexpression in glioblastoma and leukemia cells (20, 26), FOSL1 overexpression in lung cancer cells was also not sufficient to overcome the effects of JQ1 (19).
Our findings suggest that the differential effect of JQ1 on FOSL1 and c-MYC mRNAs is not due to failure of JQ1 to block BRD4 binding to the c-MYC locus in Panc1 cells. We have found that JQ1 in fact decreases the recruitment of BRD4 at both the c-MYC and the FOSL1 loci (Supplementary Fig. S7A). However, in contrast to the effect of JQ1 blocking recruitment of RNA polymerase II to the FOSL1 locus, JQ1 does not block recruitment of RNA polymerase II to the c-MYC locus in Panc1 cells (Supplementary Fig. S7B). These findings are consistent with a recent report showing that JQ1 decreases BRD4 occupancy on the c-MYC locus not only in the JQ1-sensitive cells – cells that show c-MYC mRNA repression following JQ1 treatment – but also in the JQ1-insensitive cells (46). JQ1 blocked BRD4 recruitment to the c-MYC locus even in cells that did not demonstrate any changes in c-MYC mRNA following JQ1 treatment (46). However, in contrast to JQ1-sensitive cells, JQ1 did not decrease occupancy of polymerase II on the c-MYC locus in JQ1-insensitive cells (46), indicating that in the JQ1-insensitive cells a BRD4-independent mechanism functions to regulate c-MYC transcription.
We also show in this report that JQ1 and BRD4 siRNA can additionally repress HMGA2, an architectural protein that we have identified previously to mediate chemoresistance in pancreatic cancer cells (7, 12). Significantly, we demonstrate that HMGA2 overexpression can partially reverse the inhibitory effects of JQ1 on growth of PDAC cells in three-dimensional collagen. Moreover, HMGA2 overexpression can partially reverse the repressive effects of JQ1 on c-MYC and/or FOSL1. Given that HMGA2 can act as a global chromatin switch (47), it is also possible that the ability of HMGA2 to counteract JQ1 may be through its effect on global changes in gene expression rather than modulation of c-MYC and/or FOSL1. Indeed, it has been shown that HMGA2 can regulate > 1,000 genes, either directly by binding to promoter sequences or indirectly through activation of signaling pathways (47, 48). Although it is beyond the scope of this study, we plan in future studies to examine the effect of HMGA2 overexpression on changes in global gene expression following JQ1 treatment.
Finally, it was recently shown that MYC inhibition in transgenic mouse models was sufficient to eradicate K-Ras driven lung cancers (49). Since >90–95% of human pancreatic cancers have mutant K-Ras (3, 50), which has been challenging to directly target (51, 52), one way to mitigate the effects of mutant K-Ras in pancreatic cancer could be by blocking MYC expression using BET inhibitors. Importantly, these inhibitors cause minimal toxicity to normal mouse tissue (20, 22), likely due to disruption by BET inhibitors of oncogene-driven super-enhancers that are present in malignant cells and absent in normal cells (53, 54). Since there is increasing interest in developing BET inhibitors for human studies (22, 25), it will be important to examine the efficacy of JQ1 and related compounds in transgenic mouse models of pancreatic cancer, and ultimately in patients with pancreatic cancer.
Supplementary Material
Acknowledgments
Financial Support: This work was supported by grant R01CA126888 (to H.G. Munshi) from the NCI, a Merit award I01BX001363 (to H.G. Munshi) from the Department of Veterans Affairs, and the Rosenberg Family Fund (to H.G. Munshi). This research was also supported by the training grants T32CA070085 (to C.R. Chow) and T32CA079447 (to S.S. Raza) from the NCI.
Footnotes
Conflict of Interest: The authors disclose no potential conflicts of interest
References
- 1.Hidalgo M. Pancreatic cancer. N Engl J Med. 2010;362:1605–17. doi: 10.1056/NEJMra0901557. [DOI] [PubMed] [Google Scholar]
- 2.Vincent A, Herman J, Schulick R, Hruban RH, Goggins M. Pancreatic cancer. Lancet. 2011;378:607–20. doi: 10.1016/S0140-6736(10)62307-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maitra A, Hruban RH. Pancreatic cancer. Annu Rev Pathol. 2008;3:157–88. doi: 10.1146/annurev.pathmechdis.3.121806.154305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Simeone DM. Pancreatic cancer stem cells: implications for the treatment of pancreatic cancer. Clin Cancer Res. 2008;14:5646–8. doi: 10.1158/1078-0432.CCR-08-0584. [DOI] [PubMed] [Google Scholar]
- 5.Shields MA, Dangi-Garimella S, Redig AJ, Munshi HG. Biochemical role of the collagen-rich tumor microenvironment in pancreatic cancer progression. Biochem J. 2012;441:541–52. doi: 10.1042/BJ20111240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bardeesy N, DePinho RA. Pancreatic cancer biology and genetics. Nature reviews. 2002;2:897–909. doi: 10.1038/nrc949. [DOI] [PubMed] [Google Scholar]
- 7.Dangi-Garimella S, Krantz SB, Barron MR, Shields MA, Heiferman MJ, Grippo PJ, et al. Three-Dimensional Collagen I Promotes Gemcitabine Resistance in Pancreatic Cancer through MT1-MMP-Mediated Expression of HMGA2. Cancer Res. 2011;71:1019–28. doi: 10.1158/0008-5472.CAN-10-1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324:1457–61. doi: 10.1126/science.1171362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR. Enzymatic Targeting of the Stroma Ablates Physical Barriers to Treatment of Pancreatic Ductal Adenocarcinoma. Cancer Cell. 2012;21:418–29. doi: 10.1016/j.ccr.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pfannkuche K, Summer H, Li O, Hescheler J, Droge P. The high mobility group protein HMGA2: a co-regulator of chromatin structure and pluripotency in stem cells? Stem Cell Rev. 2009;5:224–30. doi: 10.1007/s12015-009-9078-9. [DOI] [PubMed] [Google Scholar]
- 11.Fusco A, Fedele M. Roles of HMGA proteins in cancer. Nat Rev Cancer. 2007;7:899–910. doi: 10.1038/nrc2271. [DOI] [PubMed] [Google Scholar]
- 12.Dangi-Garimella S, Sahai V, Ebine K, Kumar K, Munshi HG. Three-Dimensional Collagen I Promotes Gemcitabine Resistance In Vitro in Pancreatic Cancer Cells through HMGA2-Dependent Histone Acetyltransferase Expression. PLoS One. 2013;8:e64566. doi: 10.1371/journal.pone.0064566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ouaissi M, Giger U, Sielezneff I, Pirro N, Sastre B, Ouaissi A. Rationale for possible targeting of histone deacetylase signaling in cancer diseases with a special reference to pancreatic cancer. J Biomed Biotechnol. 2011;2011:315939. doi: 10.1155/2011/315939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baylin SB, Jones PA. A decade of exploring the cancer epigenome - biological and translational implications. Nat Rev Cancer. 2011;11:726–34. doi: 10.1038/nrc3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Richards DA, Boehm KA, Waterhouse DM, Wagener DJ, Krishnamurthi SS, Rosemurgy A, et al. Gemcitabine plus CI-994 offers no advantage over gemcitabine alone in the treatment of patients with advanced pancreatic cancer: results of a phase II randomized, double-blind, placebo-controlled, multicenter study. Ann Oncol. 2006;17:1096–102. doi: 10.1093/annonc/mdl081. [DOI] [PubMed] [Google Scholar]
- 16.Dekker FJ, Haisma HJ. Histone acetyl transferases as emerging drug targets. Drug Discov Today. 2009;14:942–8. doi: 10.1016/j.drudis.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 17.Eliseeva ED, Valkov V, Jung M, Jung MO. Characterization of novel inhibitors of histone acetyltransferases. Mol Cancer Ther. 2007;6:2391–8. doi: 10.1158/1535-7163.MCT-07-0159. [DOI] [PubMed] [Google Scholar]
- 18.Puissant A, Frumm SM, Alexe G, Bassil CF, Qi J, Chanthery YH, et al. Targeting MYCN in Neuroblastoma by BET Bromodomain Inhibition. Cancer Discov. 2013;3:308–23. doi: 10.1158/2159-8290.CD-12-0418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lockwood WW, Zejnullahu K, Bradner JE, Varmus H. Sensitivity of human lung adenocarcinoma cell lines to targeted inhibition of BET epigenetic signaling proteins. Proc Natl Acad Sci U S A. 2012;109:19408–13. doi: 10.1073/pnas.1216363109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146:904–17. doi: 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. 2011;478:524–8. doi: 10.1038/nature10334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, et al. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–73. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tang X, Peng R, Ren Y, Apparsundaram S, Deguzman J, Bauer CM, et al. BET bromodomain proteins mediate downstream signaling events following growth factor stimulation in human lung fibroblasts and are involved in bleomycin-induced pulmonary fibrosis. Mol Pharmacol. 2013;83:283–93. doi: 10.1124/mol.112.081661. [DOI] [PubMed] [Google Scholar]
- 24.Dawson MA, Prinjha RK, Dittmann A, Giotopoulos G, Bantscheff M, Chan WI, et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature. 2011;478:529–33. doi: 10.1038/nature10509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Matzuk MM, McKeown MR, Filippakopoulos P, Li Q, Ma L, Agno JE, et al. Small-molecule inhibition of BRDT for male contraception. Cell. 2012;150:673–84. doi: 10.1016/j.cell.2012.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cheng Z, Gong Y, Ma Y, Lu K, Lu X, Pierce LA, et al. Inhibition of BET Bromodomain Targets Genetically Diverse Glioblastoma. Clin Cancer Res. 2013 doi: 10.1158/1078-0432.CCR-12-3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shields MA, Dangi-Garimella S, Krantz SB, Bentrem DJ, Munshi HG. Pancreatic Cancer Cells Respond to Type I Collagen by Inducing Snail Expression to Promote Membrane Type 1 Matrix Metalloproteinase-dependent Collagen Invasion. J Biol Chem. 2011;286:10495–504. doi: 10.1074/jbc.M110.195628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin L, Zhang JH, Panicker LM, Simonds WF. The parafibromin tumor suppressor protein inhibits cell proliferation by repression of the c-myc proto-oncogene. Proc Natl Acad Sci U S A. 2008;105:17420–5. doi: 10.1073/pnas.0710725105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zippo A, Serafini R, Rocchigiani M, Pennacchini S, Krepelova A, Oliviero S. Histone crosstalk between H3S10ph and H4K16ac generates a histone code that mediates transcription elongation. Cell. 2009;138:1122–36. doi: 10.1016/j.cell.2009.07.031. [DOI] [PubMed] [Google Scholar]
- 30.Badea L. Extracting gene expression profiles common to colon and pancreatic adenocarcinoma using simultaneous nonnegative matrix factorization. Pac Symp Biocomput. 2008:267–78. [PubMed] [Google Scholar]
- 31.Grutzmann R, Pilarsky C, Ammerpohl O, Luttges J, Bohme A, Sipos B, et al. Gene expression profiling of microdissected pancreatic ductal carcinomas using high-density DNA microarrays. Neoplasia. 2004;6:611–22. doi: 10.1593/neo.04295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ishikawa M, Yoshida K, Yamashita Y, Ota J, Takada S, Kisanuki H, et al. Experimental trial for diagnosis of pancreatic ductal carcinoma based on gene expression profiles of pancreatic ductal cells. Cancer Sci. 2005;96:387–93. doi: 10.1111/j.1349-7006.2005.00064.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pei H, Li L, Fridley BL, Jenkins GD, Kalari KR, Lingle W, et al. FKBP51 affects cancer cell response to chemotherapy by negatively regulating Akt. Cancer Cell. 2009;16:259–66. doi: 10.1016/j.ccr.2009.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Krantz SB, Shields MA, Dangi-Garimella S, Munshi HG, Bentrem DJ. Contribution of epithelial-to-mesenchymal transition and cancer stem cells to pancreatic cancer progression. J Surg Res. 2012;173:105–12. doi: 10.1016/j.jss.2011.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–15. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hotz B, Arndt M, Dullat S, Bhargava S, Buhr HJ, Hotz HG. Epithelial to mesenchymal transition: expression of the regulators snail, slug, and twist in pancreatic cancer. Clin Cancer Res. 2007;13:4769–76. doi: 10.1158/1078-0432.CCR-06-2926. [DOI] [PubMed] [Google Scholar]
- 37.Yin T, Wang C, Liu T, Zhao G, Zha Y, Yang M. Expression of snail in pancreatic cancer promotes metastasis and chemoresistance. J Surg Res. 2007;141:196–203. doi: 10.1016/j.jss.2006.09.027. [DOI] [PubMed] [Google Scholar]
- 38.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–90. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 39.Yang AD, Fan F, Camp ER, van Buren G, Liu W, Somcio R, et al. Chronic oxaliplatin resistance induces epithelial-to-mesenchymal transition in colorectal cancer cell lines. Clin Cancer Res. 2006;12:4147–53. doi: 10.1158/1078-0432.CCR-06-0038. [DOI] [PubMed] [Google Scholar]
- 40.Arumugam T, Ramachandran V, Fournier KF, Wang H, Marquis L, Abbruzzese JL, et al. Epithelial to mesenchymal transition contributes to drug resistance in pancreatic cancer. Cancer Res. 2009;69:5820–8. doi: 10.1158/0008-5472.CAN-08-2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wei Y, Xia W, Zhang Z, Liu J, Wang H, Adsay NV, et al. Loss of trimethylation at lysine 27 of histone H3 is a predictor of poor outcome in breast, ovarian, and pancreatic cancers. Mol Carcinog. 2008;47:701–6. doi: 10.1002/mc.20413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Manuyakorn A, Paulus R, Farrell J, Dawson NA, Tze S, Cheung-Lau G, et al. Cellular histone modification patterns predict prognosis and treatment response in resectable pancreatic adenocarcinoma: results from RTOG 9704. J Clin Oncol. 2010;28:1358–65. doi: 10.1200/JCO.2009.24.5639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McDonald OG, Wu H, Timp W, Doi A, Feinberg AP. Genome-scale epigenetic reprogramming during epithelial-to-mesenchymal transition. Nat Struct Mol Biol. 2011;18:867–74. doi: 10.1038/nsmb.2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dhasarathy A, Phadke D, Mav D, Shah RR, Wade PA. The transcription factors Snail and Slug activate the transforming growth factor-beta signaling pathway in breast cancer. PLoS One. 2011;6:e26514. doi: 10.1371/journal.pone.0026514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Herrmann H, Blatt K, Shi J, Gleixner KV, Cerny-Reiterer S, Mullauer L, et al. Small-molecule inhibition of BRD4 as a new potent approach to eliminate leukemic stem- and progenitor cells in acute myeloid leukemia AML. Oncotarget. 2012;3:1588–99. doi: 10.18632/oncotarget.733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fowler T, Ghatak P, Price DH, Conaway R, Conaway J, Chiang CM, et al. Regulation of MYC expression and differential JQ1 sensitivity in cancer cells. PLoS One. 2014;9:e87003. doi: 10.1371/journal.pone.0087003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zha L, Wang Z, Tang W, Zhang N, Liao G, Huang Z. Genome-wide analysis of HMGA2 transcription factor binding sites by ChIP on chip in gastric carcinoma cells. Mol Cell Biochem. 2012;364:243–51. doi: 10.1007/s11010-012-1224-z. [DOI] [PubMed] [Google Scholar]
- 48.Yu KR, Park SB, Jung JW, Seo MS, Hong IS, Kim HS, et al. HMGA2 regulates the in vitro aging and proliferation of human umbilical cord blood-derived stromal cells through the mTOR/p70S6K signaling pathway. Stem Cell Res. 2013;10:156–65. doi: 10.1016/j.scr.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 49.Soucek L, Whitfield JR, Sodir NM, Masso-Valles D, Serrano E, Karnezis AN, et al. Inhibition of Myc family proteins eradicates KRas-driven lung cancer in mice. Genes Dev. 2013;27:504–13. doi: 10.1101/gad.205542.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4:437–50. doi: 10.1016/s1535-6108(03)00309-x. [DOI] [PubMed] [Google Scholar]
- 51.Gysin S, Salt M, Young A, McCormick F. Therapeutic strategies for targeting ras proteins. Genes Cancer. 2011;2:359–72. doi: 10.1177/1947601911412376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Baines AT, Xu D, Der CJ. Inhibition of Ras for cancer treatment: the search continues. Future Med Chem. 2011;3:1787–808. doi: 10.4155/fmc.11.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Loven J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153:320–34. doi: 10.1016/j.cell.2013.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153:307–19. doi: 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.