

Abstract
microRNAs (miR) can act as oncogenes and tumor suppressors and several miRs are associated with cancer development and progression through the modulation of multiple cellular processes. miR-26b is down regulated in several cancers and tumors and miR-26b directly targets the Lef-1 3'UTR and inhibits endogenous Lef-1 expression. We report that miR-26b expression is associated with human colon cancer through the regulation of LEF-1 expression in colon cancer cells. Analyses of multiple colon cancer cell lines revealed an inverse correlation between miR-26b and LEF-1 expression. Normal human colon cells express low levels of LEF-1 and high levels of miR-26b, however human colon cancer cells have decreased miR-26b expression and increased LEF-1 expression. We demonstrate that miR-26b expression is a potent inhibitor of colon cancer cell proliferation and significantly decreases LEF-1 expression. The LEF-1 regualted genes Cyclin D1 and c-Myc were indirectly repressed by miR-26b and this was consistent with decreased proliferation. miR-26b overexpression in SW480 colon cancer cells also inhibited tumor growth in nude mice and this was due to decreased tumor growth and not apoptosis. Analyses of human colon cancer databases also demonstrated a link between miR-26b and LEF-1 expression. c-Myc expression is associated with multiple cancers and we propose that miR-26b may act as a potential therapeutic agent in reducing cancer cell proliferation through repressing LEF-1 activation of c-Myc and Cyclin D1 expression.
Introduction
MicroRNAs (miRs) are a group of endogenous non-coding RNAs that post-transcriptionally regulate expression of protein-coding genes by recognizing specific mRNAs with complementary sequence (1, 2). miRs imperfectly match the 3'UTR of target mRNAs and inhibit translation and/or promote degradation. miR's can also target regions other than 3'UTR to trigger degradation of target mRNAs or repress protein synthesis. miRs are estimated to represent about 1% of all genes in human genome and the importance of their biological functions are being intensively investigated (3–5). miRs are associated with cancer as either oncogenes or tumor-suppressor genes and in cancer cells they are either up or down regulated (6–8). Many miRs up regulated in colon adenomas or adenocarcinomas have been reported (6). Recent reports have shown that miR-26b is under expressed in human breast cancer, parathyroid tumor, oral lichen planus disease, glioma cells, hepatocellular carcinomas, nasopharyngeal carcinomas, primary squamous cell lung carcinomas and squamous cell carcinoma of the tongue (9–16). Recently, two new target genes of miR-26b have been reported in glioma cells (EphA2) and human breast cancer cells (SLC7A11) (9, 12). EphA2 is an erythropoietin-producing hepatocellular (EPH) A receptor and activation of this receptor tyrosine kinase is associated with cancer cell growth (12). SLC7A11 is a solute carrier family seven member eleven that may play a role in providing resistance to apoptosis in cells (9). Lastly, human embryonic stem cells and metastatic colorectal cancer cells also express miR-26b, which may regulate TAF12, PTP4A1, CHFR and ALS2CR2 gene expression (17).
Wnt signaling is one of the most important and best characterized signaling pathways involved in embryonic development, cell proliferation and cancer progression. The canonical Wnt signaling pathway is activated when Wnt ligand binds to cell surface receptor Frizzled and LRP, which activate Dishevelled and releases β-catenin from its destroying complex formed by Axin, APC and GSK3β. The released β-catenin enters nucleus and binds to Lef/Tcf transcription factors to activate downstream gene expression (18). Among those components, many of them are potential targets of miR regulation.
In the case of colorectal cancer, mutation of Wnt signal components are estimated to cause approximately 90% of the cancer (19)(19). Most are associated with truncated APC and point mutations of phosphorylation sites in β-catenin involved in its degradation. Targeting the Wnt/β-catenin pathway, especially the downstream transcriptional regulation, would be effective in therapeutic treatment. We previously demonstrated that miR-26b was a potent inhibitor of LEF-1 expression (20). Given the critical role of LEF-1 in canonical Wnt signaling, colon cancer progression (21) and human sebaceous tumors (22), we asked if miR-26b repression of LEF-1 would inhibit colon cancer cell proliferation.
Materials and Methods
Expression and reporter constructs
hsa-miR-26b precursor (addition of six thymine deoxyriboside at 3') was cloned from the genomic DNA of HEK 293 cell and was linked to U6 promoter in pLL3.7 backbone. Human LEF-1 cDNA was cloned from total mRNA of HEK 293 cell and was inserted downstream of EF1α promoter in pWPI backbone. The 7xTopFlash reporter plasmid was constructed into luciferase vector by inserting seven LEF/TCF binding sites upstream of the minimal TK promoter (20). The 7xFopFlash negative control contains mutations within each LEF/TCF binding site. All constructs were confirmed by DNA sequencing.
Lentiviral vector generation and cell proliferation assays
A second generation Lentiviral vector system was used. The packaging vectors are psPAX2 and pMDG. The gene expression vectors were transfected with packaging vectors into HEK 293 FT cells to generate lentiviral vectors as previously described (23). The virus containing cell medium was harvested 48 hours after transfection. The viral vectors were concentrated by ultracentrifuge at 26,000 rpm for 2 hours at 4°C and added to the SW480 cell medium with 8 μg/ml polybrene. Infected cells were subject to the cell proliferation assay. Cells (150,000) were seeded in 60 mm plates on day 0 and then trypsinized and counted after 24, 48, 72 and 96 hours by a Coulter Z1 cell counter as previously described (24). Experiments were run in triplicate.
Cell culture, transient transfections, luciferase and β-galactosidase assays
CCD 841 CoN, SW480, DLD-1, Colo 320 and HEK 293 cells were obtained from ATCC (cultured and frozen immediately for storage and used within 3 months; cell cultures used in revision experiments were subsequently thawed and cultured and genotypes were confirmed to previous cell cultures) and cultured in DMEM supplemented with 10% FBS and penicillin/streptomycin. SW480 cells were seeded in 12 well plates and transfected by Fugene HD (Roche) with 500 ng of reporter plasmid, 500 ng of expression plasmid and 50 ng SV-40 driven β-galactosidase plasmid. Transfected cells were incubated for 48 hours and then lysed for reporter activities and protein content by Bradford assay (Bio-Rad) as previously described (25). Luciferase activity was measured with reagents from Promega and β-galactosidase activity was assessed by Galacto-Light Plus reagents (Tropix). Experiments were run in triplicate and all luciferase activities were normalized to β-galactosidase activity.
Real-time PCR analyses
Cells were cultured in T-75 flasks and 2 flasks of 80% confluent cells were harvested by scraping and miR isolation. Total RNA including miR from cells was prepared using the miRNeasy Mini Kit (Qiagen). Mulitple isolations were performed and separate qPCR assays were run on each sample (N=3). The amount and integrity of the RNA samples were assessed by measurement at 260 and 280 nm and gel analyses. Quantitative real-time PCR for miR-26b mature expression was done with TaqMan MicroRNA assay probes (Applied Biosystems), including U6B as a reference gene. Total RNA was reverse transcribed into cDNA by iScript Select cDNA Synthesis kit (BioRad). Real time PCR was carried out in a total reaction of 25 μl containing 12.5 μl iQ SYBR Green Supermix, 0.1 μM forward primer, 0.1 μM reverse primer, 0.25 μl cDNA template in the MyiQ Single color Real-Time Detection System and analyzed by the MyiQ Optical System Software 2.0 (BioRad). β-actin served as a reference gene for normalization of LEF-1, c-MYC and cyclin D1 mRNA levels and delta-delta Ct values are reported. The thermal cycling profile consisted of 95°C for 4 min followed by 40 cycles of denaturation at 95°C for 30 sec, annealing at 60°C for 30 sec and elongation at 72°C for 18 sec. Samples were run in triplicate. No-template control was run in each experiment. Melting curve analyses were performed to confirm amplification specificity of the PCR products. PCR primers are LEF-1 F 5'-TTCCTTGGTGAACGAGTCTG-3', R 5'-CTCTGGCCTTGTCGTGGTAG-3'; cyclin D1 F 5'-ACACGCGCAGACCTTCGTTG-3', R 5'-GTAGGACAGGAAGTTGTTGG-3'; c-MYC F 5'-GATTCTCTGCTCTCCTCGAC-3', R 5'- GTGATCCAGACTCTGACCTT-3'. β-actin primers were described previously (26).
Western blot assays
Endogenous LEF-1 isoforms were identified in colon cells and HEK 293 cells using the LEF-1 antibody (Cell Signaling, C12A5). c-MYC and cyclin D1 expressions in colon cells were detected using c-MYC antibody (Cell Signaling, D84C12) and cyclin D1 antibody (Cell Signaling, 92G2). GAPDH antibody was from Santa Cruz (sc-32233). Approximately 20–30 μg of cell lysates were analyzed in Western blots. Following SDS gel electrophoresis, the protein were transferred to PVDF filters (Millipore), immunoblotted and detected by specific antibodies and ECL plus reagents from GE HealthCare.
Xenograft model
Male nude mice (Foxn1nu, aged 6–7weeks) were purchased from Harlan (Indianapolis, IN) were maintained in laminar flow cabinets under pathogen free conditions. SW480 cells (1×107 cells) stably expressing scramble shRNA (control) or miR-26b in serum-free DMEM were injected into either side of flank area of nude mice. The mice were weighed, and tumor sizes were measured every third day with calipers for calculation of tumor volumes, V=LW2/2 where L and W were length and width, respectively. After 33 days, mice were sacrificed and the tumors were collected for weighting and analysis of the expression levels of miR-26b and other related genes. Tumors were flash frozen in LN2, tissue was crushed and lysed and RNA extracted using the Qiagen miRNeasy extraction kit (Qiagen).
Tunel Assay
SW480 cells stably expressing scramble shRNA or miR-26b were grown to 75% confluence on glass slides and incubated for 2 hours with or without 0.5 mM H2O2. The TUNEL assay was performed with the DeadEnd™ Fluorometric TUNEL System (Promega) according to the manufacturer's instruction.
Statistics
Statistics of growth curves were performed by the repeated measures analysis of variance (ANOVA). Other statistics were performed by two-sample t-test. p-values less than 0.05 were considered to be significant.
Results
miR-26b expression is inversely correlated with LEF-1 expression in several colon cancer cell lines
The non-tumor colon epithelial cell line (CCD 841 CoN, from ATCC) was set as a control to compare the relative expression of LEF-1 and miR-26b in non-cancer colon vs. colon cancer cells. miR-26b was significantly down regulated in colon cancer cell lines compared to the normal cells. miR-26b expression in normal cells was approximately 7.4, 12.3 and 37 times higher than observed in DLD-1, SW480, Colo 320 colon cancer cells, respectively (Fig. 1A). In contrast, LEF-1 isoform expression was up regulated in the cancer cells at both the mRNA level and protein level (Fig. 1B, C). A gradient of LEF-1 expression was detected where normal cells <DLD-1 <SW480 <Colo 320 (Fig. 1B, C) and the data indicated an inverse relationship between miR-26b and LEF-1 isoform expression in these cell lines. c-Myc, a LEF-1 target gene was also correlated with increased LEF-1 expression (Fig. 1B). This inverse correlation suggests an inhibitory role of miR-26b on LEF-1 expression and indicated that miR-26b may have a potent regulatory role during cancer progression. miR-26b targets all LEF-1 isoforms as they contain identical 3'UTRs and miR-26b binding sites. Furthermore, we analyzed miR-26b expression in 54 human colon cancer specimens compared to 20 normal colon specimens from the NCBI's Gene Expression Omnibus public database (GSE30454) (27). These data show decreased miR-26b expression (p<0.005) in human colon cancer tissue (Fig. 1D). Because SW480 cells exhibited moderate expression of both LEF-1 and miR-26b we used these cells for over expression and knockdown treatments of both genes in cell proliferation assays.
Fig. 1. Endogenous LEF-1 and miR-26b expression among colon cancer cell lines.

A, B) Relative expression of miR-26b and LEF-1, c-Myc mRNA reveals endogenous miR-26b and LEF-1,c-Myc transcripts in a non-cancer colon epithelial cell line (CCD 841 CoN) and colon cancer cell lines DLD-1, SW480 and Colo 320. miR-26b expression is inversely correlated with LEF-1 and c-Myc expression. C) Western blot of LEF-1 expression in HEK 293 and colon cell lysates. The colon cancer cells express multiple LEF-1 isoforms. The relative protein levels were consistent with mRNA levels. D) miR-26b expression in human colon cancer tissue and normal tissue obtained from NCBI gene expression Omnibus database. * p-value < 0.05; ** p-value < 0.01; *** p-value < 0.005.
miR-26b represses colon cancer cell proliferation
To test whether miR-26b effected colon cancer proliferation, we established a variety of overexpression, knockdown and rescue cell lines and analyzed their proliferation (Fig. 2). Human miR-26b and control scramble shRNA were inserted downstream of the U6 promoter in pLL3.7 plasmid. Human LEF-1 FL mRNA (full length isoform) was inserted downstream of the EF1α promoter in pWPI plasmid (Fig. 2A). The gene expression vectors were transfected with packaging vectors into HEK 293 FT cells to generate lentiviral vectors. The virus containing cell medium was harvested 48 hours after transfection. The viral vectors were concentrated and added to the SW480 cell medium with 8 μg/ml polybrene. Approximately 100% infection efficiency was achieved indicated by expression of selection marker EGFP in virus treated cells (Fig. 2B). Three stable cell lines were established: control scramble cell line, miR-26b over expression cell line, and LEF-1 FL over expression cell line. These cell lines, together with SW480 cells transfected with anti-miR™ miRNA inhibitor (anti-miR-26b, Ambion) and empty vector (mock) cells, were subjected to cell proliferation assays and the growth rates over 96 hours are summarized in Fig. 2C. LEF-1 FL over expression in SW480 cells significantly increased growth compared to the control cells. In contrast, miR-26b overexpression decreased growth. The anti-miR-26b treated cells also exhibited increased growth albeit not as high as cells in which LEF-1 FL was overexpressed. These data suggest that miR-26b is a potent inhibitor of colon cancer cell growth through regulation of LEF-1 expression.
Fig. 2. miR-26b represses colon cancer cell proliferation.

A) Schematic of miR-26b and LEF-1 overexpression lentiviral vectors. Human miR-26b and scrambled miR expression are driven by the U6 promoter of pLL3.7 backbone. Human LEF-1 FL mRNA (full length isoform) is driven by EF1α promoter of pWPI backbone. All vectors have GFP selection marker. B) GFP fluorescence pictures showing that infected SW480 cells have GFP expression. C) Growth curves of SW480 cells with indicated genes expression. 150,000 cells were seeded in 60 mm plates on day 0 and counted after 24, 48, 72 and 96 hours. Experiments were run in triplicate. Error bars indicate S.E.M. Photos of cells in the plates on day 4 are shown on the right side. LEF-1 FL cell growth was compared to mock transfected cells and miR-26b and anti-miR-26b cells were compared to scrambled control, **: p-value < 0.01
miR-26b represses endogenous LEF-1 expression in colon cancer cells
To demonstrate that LEF-1 isoform expression was targeted by miR-26b in the cells used for cell proliferation assays, LEF-1 expression was analyzed by real time PCR and Western blots. LEF-1 mRNA levels were significantly decreased in the miR-26b overexpressing cells compared to control cells, whereas LEF-1 mRNA was significantly increased in cells transfected with anti-miR-26b or overexpressing LEF-1 FL (Fig. 3A). Western blots showed that LEF-1 protein and mRNA levels were similar in these cell lines (Fig. 3B). A Topflash reporter assay was used to examine the overall Wnt/β-catenin signal activity. SW480 cells were transfected with 7xTopflash reporter plasmid and indicated expression plasmids, together with SV-40 β-galactosidase as a control. Transfection with miR-26b decreased Topflash activity compared with control cells, while anti-miR-26b and LEF-1 transfection increased reporter activity. The Fopflash plasmid with a mutation in each LEF/TCF binding site was used as a negative control (Fig. 3C). These data indicated that miR-26b had a potent inhibitory effect on LEF-1 expression as well as Wnt/β-catenin signal activity in colon cancer cells.
Fig. 3. miR-26b represses endogenous LEF-1 expression in SW480 cells.

A) LEF-1 mRNA levels were significantly decreased in miR-26b over expression cells, while it was increased in anti-miR-26b treated cells. LEF-1 mRNA levels were significantly increased in LEF-1 over expression cells as expected, while co-expression of LEF-1 and miR-26b reduced LEF-1 expression to normal levels. Real time PCR normalized to β-actin, N=3. B) Western blot showing that LEF-1 protein levels were reduced in miR-26b over expression cells compared with control cells, while it was increased in anti-miR-26b treated cells and LEF-1 over expression cells. LEF-1 and miR-26b combined over expression cells revealed no change in LEF-1 expression. C) Topflash reporter assays show that transfection of miR-26b vector reduced Topflash activity, whereas transfection of anti-miR-26b and LEF-1 vector increased Topflash activity. Mutated reporter (Fopflash) served as a negative control. SV-40 promoter driven β-galactosidase activity served as a control for transfection efficiency, N=3. Error bars indicate S.E.M.; *: p-value < 0.05; **: p-value < 0.01; NS: not significant.
miR-26b represses expression of the LEF-1 target genes cyclin D1 and c-MYC
Wnt/β-catenin signaling regulates cell proliferation through several different mechanisms (18, 19). One of the most important mechanisms is through regulation of Cyclin D1 and c-Myc, which are essential regulators of cell proliferation and direct targets of the LEF-1/β-catenin transcription complex (28–30). Therefore, we wanted to determine whether expression of these two genes was altered in our lentiviral-infected cells, which exhibit different rates of cell proliferation (Fig. 2C). Real-time PCR showed that Cyclin D1 and c-Myc mRNA levels were decreased in miR-26b overexpressing cells compared to control cells, while they were increased in anti-miR-26b transfected and LEF-1 FL overexpressing cells (Fig. 4A, B). Western blots showed that there were parallel changes in c-Myc and Cyclin D1 protein and mRNA levels (Fig. 4C), suggesting that miR-26b repressed c-Myc and Cyclin D1 expression through inhibition of LEF-1 in colon cancer cells. As a control miR-26b expressing cells were transfected with the LEF-1 FL cDNA, which is not targeted by miR-26b to demonstrate Lef-1 FL expression by the Western blot (Fig. 4C).
Fig. 4. miR-26b repressed expression of LEF-1 target genes Cyclin D1 and c-Myc.

A, B) Real-time PCR results showing that Cyclin D1 and c-Myc mRNA levels were reduced in miR-26b over expression cells, while they were increased in anti-miR-26b treated cells. LEF-1 over expression cells had increased expression of Cyclin D1 and c-Myc as expected, while co-expression of LEF-1 and miR-26b reduced their expression to normal levels. C) A Western blot with Cyclin D1 and c-Myc antibodies showing that the protein levels were changed similar to mRNA levels. miR-26b cells transfected with LEF-1 FL cDNA was used as a positive control for LEF-1 FL expression, which is not targeted by miR-26b.
miR-26b inhibits tumor growth in a xenograft nude mouse model
To test the effect of miR-26b inhibition on growth of colon cancer cells in vivo, we injected male nude mice (Foxn1nu, aged 6–7weeks) with SW480 cells (1×107 cells) stably expressing scramble shRNA (control) or miR-26b in serum-free DMEM at either side of the flank area. Tumor sizes were measured every third day with calipers to calculate tumor volumes (V=LW2/2, L:length, W:width) (Fig. 5A). After 33 days, mice were sacrificed and the tumors were collected for weighing and expression of miR-26b and other related genes. The average weight of miR-26b tumors were about 50% of the control tumors (Fig. 5B, C) and real-time PCR analyses showed that miR-26b expression was significantly increased in miR-26b tumors, while LEF-1, c-Myc and Cyclin D1 were significantly decreased in tumors derived from miR-26b overexpressing SW480 cells (Fig. 5D, E). These data were consistent with the in vitro cell proliferation assay and real-time PCR results.
Fig. 5. Effect of miR-26b inhibition on growth of SW480 cells in xenograft model.
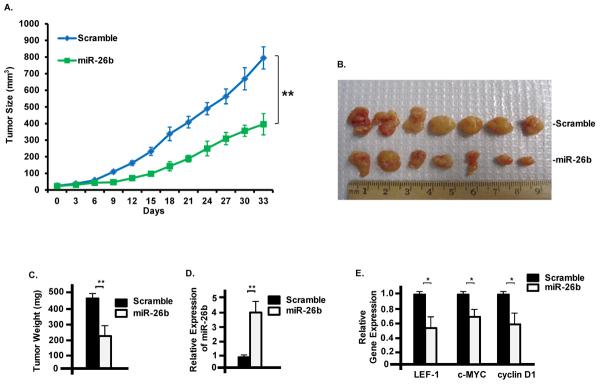
A) Growth curves of control and miR-26b tumors. Male nude mice were injected with SW480 cells (1×107 cells) stably expressing scramble shRNA (control) or miR-26b in DMEM at either side of flank area. Tumor sizes were measured every third day to calculate tumor volumes (V=LW2/2, L:length, W:width). The two curves were analyzed by ANOVA and the p-value<0.01. B) Tumors collected for weighting and analyses of gene expression. C) The average weight of miR-26b tumors were about half of the control tumors. D) miR-26b expression was significantly increased in miR-26b tumors measured by real-time PCR. N=3. E) LEF-1, c-Myc and cyclin D1 were significantly decreased in miR-26b tumors measured by real-time PCR. N=3, experiments were run in triplicate. Error bars indicate S.E.M.; n=7; * p-value < 0.05; ** p-value < 0.01.
Tumor growth inhibition due to decreased proliferation not apoptosis
To test whether miR-26b inhibited colon cancer cells through apoptosis, we performed TUNEL assays. SW480 cells stably expressing scramble miR (control) or miR-26b were stained with Promega TUNEL assay kit and almost no apoptosis was detected in either control or miR-26b cell lines (Fig. 6A,B). The same cell lines were treated with 0.5 mM H2O2 for 2 hours as positive controls. After H2O2 treatment, apoptosis was induced in both cell lines (Fig. 6A,B). These data suggest that miR-26b expression does not affect apoptosis of colon cancer cells in the presence or absence of oxidative stress indicating that miR-26b primarily regulates colon cancer cell/tumor proliferation and not survival.
Fig. 6. Apoptosis is not affected by miR-26b.

A) SW480 cells stably expressing scramble shRNA (control) or miR-26b were treated with or without 0.5 mM H2O2 for 2 hours and stained with Promega TUNEL assay kit. B) almost no apoptosis was detected in either control or miR-26b cell lines without H2O2. After H2O2 treatment, apoptosis was detected in both cell lines. N=6; NS: not significant. C) A schematic model illustrates the underlying mechanism of miR-26b repression of cancer cell proliferation through regulation of LEF-1, cyclin D1 and c-Myc.
Discussion
The role of LEF-1 in colon cancer
We previously demonstrated that miR-26b not only targets the β-catenin responsive isoform of LEF-1 FL but also the dominant negative isoform (LEF-1ΔN) and intermediate isoform because they share the same 3'UTR. Thus, miR-26b also reduces expression of the dominant negative isoform. However, many colon cancer cells lack expression of the dominant negative isoform of LEF-1 (31). The full-length LEF-1 isoform is responsive to Wnt/β-catenin signaling in colon cancer cells (19). LEF-1 and TCF belong to a family of DNA binding transcription factors that interact with β-catenin to activate genes involved in cell proliferation, morphogenesis, epithelial-mesenchymal transition and stemness, which can lead to cancer progression (32–36). The LEF-1 full-length and intermediate isoforms are produced in colon cancers through Wnt signaling and the LEF-1ΔN isoform is produced by an alternative promoter, which is inactive in some cancers (31, 37). Furthermore, β-catenin also activates Cyclin D1 expression in colon carcinoma cells through TCF/LEF-1 binding elements in the Cyclin D1 promoter (29, 30). The c-Myc promoter contains multiple Tcf-4 binding sites and is activated by β-catenin and presumably by LEF-1 (28). These data demonstrate the role for LEF-1/TCF and β-catenin in activating Cyclin D1 and c-Myc expression in cancer cells and our studies demonstrate that miR-26b targets LEF-1 expression, which in turn regulates Cyclin D1 and c-Myc expression.
c-Myc regulation of miRs and cancer
Myc is a well-known oncogenic transcription factor that controls many cellular processes including cell growth, differentiation, cell adhesion and motility. Aberrant Myc activity is associated with many human cancers and Myc-mediated transactivation of target genes results in deregulated gene expression. Interestingly, some of these Myc target genes are miRs. Myc directly activates transcription of the polycistronic miR-17-92 cluster and miR-9 (38–40). The miR-17-92 cluster is classified as an oncogene as its expression is associated with several solid tumors (for reviews see (41, 42)). Conversely, Myc represses several miRNAs that are tumor suppressors including let-7 family members, miR-34a, miR-29 members, miR-26a and miR-15a/16-1 (43). Myc-regulated miRs control cell cycle programs, apoptosis, angiogenesis, metabolism and therefore tumorigenesis through a variety of molecular mechanisms and gene targets (41, 42). miR-26a over expression has been used in a MYC-driven mouse hepatocellular carcinoma model to reverse disease progression (44). The molecular mechanism of miR-26a involved decreased expression of Cyclin D1 and D2 and induced cell cycle arrest (44). Our data suggests that miR-26b overexpression could potentially be used in patients as a therapeutic, targeting hepatocellular carcinoma in these patients or a combination of miR-26a and b. Similar to miR-26a, which is expressed at low levels in human liver cancer samples, miR-26b is also expressed at low levels in colon cancer cells (Fig. 1A) (17). Interestingly, there is a direct correlation to the severity of colon cancer with a decrease miR-26b expression and a concomitant increase in LEF-1 expression suggesting that miR-26b could be an effective therapeutic agent in treating this disease. miR-26b was also decreased in LoVo colon cancer cells and miR-26b over expression in these cells suppressed cell growth and tumor growth in vivo (17). However, LEF-1 was not included as a miR-26b target in the LoVo cells and tumors.
In summary, miR-26b is decreased and LEF-1 expression is increased in colon cancer cells. We show an inverse correlation between miR-26b and LEF-1 expression in multiple colon cell lines (Fig. 1A and 1B) and that miR-26b has a potent inhibitory effect on colon cancer cell proliferation (Fig. 2). Furthermore, in patient colon cancer samples miR-26b expression is decreased (Fig. 1D). Endogenous LEF-1 expression is significantly decreased by miR-26b over expression and the Wnt/β-catenin target genes Cyclin D1 and c-Myc are also repressed, and this represents the underlying mechanism of miR-26b-dependent inhibition of colon cancer cell proliferation. Thus, miR-26b is a tumor repressor that is a potent inhibitor of colon cancer cell proliferation that blocks Wnt/β-catenin dependent activation of LEF-1 and a model for tumor suppressor-like activity of miR-26b is illustrated in figure 6C. These findings also have important therapeutic implications.
Given the prevalence of increased Wnt/β-catenin signaling in colon cancer patients, miR-26b can be a potential candidate for gene therapy of multiple cancer types that express LEF-1. The efficient delivery of miRs and other nucleic acids are being extensively investigated with some clinical trials already in progress (4, 5).
ACKNOWLEDGMENTS
Grant Support, this work was supported by the National Institutes of Health grant DE13941 to B.A.A.
Financial Support: NIH grant DE13941 to B.A. Amendt
Footnotes
Conflict of Interest: The authors have no conflicts to disclose
REFERENCES
- 1.He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet. 2004;5:522–31. doi: 10.1038/nrg1379. [DOI] [PubMed] [Google Scholar]
- 2.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–97. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 3.Nelson P, Kiriakidou M, Sharma A, Maniatak E, Mourelatos Z. The microRNA world: small is mighty. Trends Biochem Sci. 2003;28:534–40. doi: 10.1016/j.tibs.2003.08.005. [DOI] [PubMed] [Google Scholar]
- 4.Nana-Sinkam SP, Croce CM. Clinical applications for microRNAs in cancer. Clin Pharmacol Ther. 2013;93:98–104. doi: 10.1038/clpt.2012.192. [DOI] [PubMed] [Google Scholar]
- 5.Mendell JT, Olson EN. MicroRNAs in Stress Signaling and Human Disease. Cell. 2012;148:1172–87. doi: 10.1016/j.cell.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Visone R, Croce CM. MiRNAs and Cancer. AJP. 2009;174:1131–8. doi: 10.2353/ajpath.2009.080794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang B, Pan X, Cobb GP, Anderson TA. microRNAs as oncogenes and tumor suppressors. Dev Biol. 2007;302:1–12. doi: 10.1016/j.ydbio.2006.08.028. [DOI] [PubMed] [Google Scholar]
- 8.Esquela-Kerscher A, Slack FJ. Oncomirs- microRNAs with a role in cancer. Na Rev. 2006;6:259–69. doi: 10.1038/nrc1840. [DOI] [PubMed] [Google Scholar]
- 9.Liu X-X, Li X-J, Zhang B, Liang Y-J, Zhou C-X, Cao D-X, et al. MicroRNA-26b is underexpressed in human breast cancer and induces cell apoptosis by targeting SLC7A11. FEBS Lett. 2011;585:1363–7. doi: 10.1016/j.febslet.2011.04.018. [DOI] [PubMed] [Google Scholar]
- 10.Rahbari R, Holloway AK, He M, Khanafshar E, Clark OH, Kebebew E. Identification of Differentially Expressed MicroRNAs in Parathyroid Tumors. Ann Surg Oncol. 2010;18:1158–65. doi: 10.1245/s10434-010-1359-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Danielsson K, Ebrahimi M, Wahlin YB, Nylander K, Boldrup L. Increased levels of COX-2 in oral lichen planus supports an autoimmune cause of the disease. JEADV. 2011 doi: 10.1111/j.1468-3083.2011.04306.x. DOI: 10.1111/j.1468-3083.2011.04306.x. [DOI] [PubMed] [Google Scholar]
- 12.Wu N, Zhao X, Liu M, Liu H, Yao W, Zhang Y, et al. Role of MicroRNA-26b in Glioma Development and Its Mediated Regulation on EphA2. Plos ONE. 2011;6:e16264. doi: 10.1371/journal.pone.0016264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kent OA, Mendell JT. A small piece in the cancer puzzle: microRNAs as tumors suppressors and oncogenes. Oncogene. 2006;25:6188–96. doi: 10.1038/sj.onc.1209913. [DOI] [PubMed] [Google Scholar]
- 14.Weidhaas JB, Babar I, Nallur SM, Trang P, Roush S, Boehm M, et al. MicroRNAs as Potential Agents to Alter Resistance to Cytotoxic Anticancer Therapy. Cancer Res. 2007;67:11111–6. doi: 10.1158/0008-5472.CAN-07-2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jeong SH, Wu HG, Park WY. LIN28B confers radio-resistance through the posttranscriptional control of KRAS. Exp Mol Med. 2009;41:912–8. doi: 10.3858/emm.2009.41.12.097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arora H, Qureshi R, Jin S, Park AK, Park WY. miR-9 and let-7g enhance the sensitivity to ionizing rediation by suppression of NFKB1. Exp Mol Med. 2011;43:298–304. doi: 10.3858/emm.2011.43.5.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ma YL, Zhang P, Wang F, Moyer MP, Yang JJ, Liu ZH, et al. Human embryonic stem cells and metastatic colorectal cancer cells shared the common endogenous human microRNA-26b. J Cell Mol Med. 2011;15:1941–54. doi: 10.1111/j.1582-4934.2010.01170.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moon RT, Kohn AD, De Ferrari GV, Kaykas A. Wnt and β-catenin signalling: diseases and therapies. Nature. 2004;5:689–99. doi: 10.1038/nrg1427. [DOI] [PubMed] [Google Scholar]
- 19.Paul S, Dey A. Wnt signaling and cancer development: therapeutic implication. Neoplasma. 2008;55:165–76. [PubMed] [Google Scholar]
- 20.Zhang Z, Florez S, Gutierrez-Hartmann A, Martin JF, Amendt BA. MicroRNAs regulate pituitary development, and microRNA 26b specifically targets lymphoid enhancer factor 1 (Lef-1), which modulates pituitary transcription factor 1 (Pit-1) expression. J Biol Chem. 2010;285:34718–28. doi: 10.1074/jbc.M110.126441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kriegl L, Horst D, Reiche JA, Engel J, Kirchner T, Jung A. LEF-1 and TCF4 expression correlate inversely with survival in colorectal cancer. J Translational Medicine. 2010;8:123–31. doi: 10.1186/1479-5876-8-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Niemann C, Owens DM, Schettina P, Watt FM. Dual Role of Inactivating Lef1 Mutations in Epidermis: Tumor Promotion and Specification of Tumor Type. Cancer Res. 2007;67:2916–21. doi: 10.1158/0008-5472.CAN-06-3427. [DOI] [PubMed] [Google Scholar]
- 23.Rubinson DA, Dillon CP, Kwiatkowski AV, Sievers C, Yang L, Kopinja J, et al. A lentivirus-based system to functionally silence genes in primary mammalian cells, stem cells and transgenic mice by RNA interference. Nat Genet. 2003;33:401–6. doi: 10.1038/ng1117. [DOI] [PubMed] [Google Scholar]
- 24.Lee S-O, Chintharlapalli S, Liu S, Papineni S, Cho SD, Yoon K, et al. p21 Expression Is Induced by Activation of Nuclear Nerve Growth Factor–Induced Bα (Nur77)in Pancreatic Cancer Cells. Mol Cancer Res. 2009;7:1169–78. doi: 10.1158/1541-7786.MCR-08-0473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Amendt BA, Sutherland LB, Russo AF. Multifunctional Role of the Pitx2 Homeodomain Protein C-Terminal Tail. Mol Cel Biol. 1999;19:7001–10. doi: 10.1128/mcb.19.10.7001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang Z, Wlodarczyk BJ, Niederreither K, Venugopalan S, Florez S, Finnell RH, et al. Fuz Regulates Craniofacial Development Through Tissue Specific Responses to Signaling Factors. PLoS ONE. 2011;6:e24608. doi: 10.1371/journal.pone.0024608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Balaguer F, Moreira L, Lozano JJ, Link A, Ramirez G, Shen Y, et al. Colorectal cancers with microsatellite instability display unique miRNA profiles. Clin Cancer Res. 2011;17:6239–49. doi: 10.1158/1078-0432.CCR-11-1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.He T-C, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, et al. Identification of c-MYC as a Target of the APC Pathway. Science. 1998;281:1509–12. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- 29.Shtutman M, Zhurinsky J, Simcha I, Albanese C, D'Amico M, Pestell R, et al. The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. Proc Natl Acad Sci USA. 1999;96:5522–7. doi: 10.1073/pnas.96.10.5522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tetsu O, McCormick F. β-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. 1999;398:422–6. doi: 10.1038/18884. [DOI] [PubMed] [Google Scholar]
- 31.Li TW-H, Ting J-HT, Yokoyama NN, Bernstein A, van de Wetering M, Waterman ML. Wnt Activation and Alternative Promoter Expression of LEF1 in Colon Cancer. Mol Cell Biol. 2006;26:5284–99. doi: 10.1128/MCB.00105-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Preisinger AC, Leppert M, et al. Genetic Alterations during colorectal-tumor develoment. N Eng J Med. 1988;319:525–32. doi: 10.1056/NEJM198809013190901. [DOI] [PubMed] [Google Scholar]
- 33.van de Wetering M, Sancho E, Verweij C, de Lau W, Oving I, Hurlstone A, et al. The β-catenin/TCF-4 Complex Imposes a Crypt Progenitor Phenotype on Colorectal Cancer Cells. Cell. 2002;111:241–50. doi: 10.1016/s0092-8674(02)01014-0. [DOI] [PubMed] [Google Scholar]
- 34.Merrill BJ, Gat U, DasGupta R, Fuchs E. Tcf3 and Lef1 regulate lineage differentiation of multipotent stem cells in skin. Genes Dev. 2001;15:1688–705. doi: 10.1101/gad.891401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van Genderen C, Okamura RM, Farinas I, Quo R-G, Parslow TG, Bruhn L, et al. Development of several organs that require inductive epithelial-mesenchymal interactions is impared in LEF-1-deficient mice. Genes Dev. 1994;8:2691–703. doi: 10.1101/gad.8.22.2691. [DOI] [PubMed] [Google Scholar]
- 36.Kim K, Lu Z, Hay ED. Direct evidence for the role pf beta-catenin/LEF-1 signaling pathway in induction of EMT. Cell Biol Int. 2002;26:463–76. doi: 10.1006/cbir.2002.0901. [DOI] [PubMed] [Google Scholar]
- 37.Hovanes k, Li TWH, Waterman ML. The human LEF-1 gene contains a promoter preferentially active in lymphocytes and encodes multiple isoforms derived from alternative splicing. Nuc Acids Res. 2000;28:1994–2003. doi: 10.1093/nar/28.9.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.ODonnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT. c-Myc-regulated microRNAs modulate E2F1 expression. Nature. 2005;435:839–43. doi: 10.1038/nature03677. [DOI] [PubMed] [Google Scholar]
- 39.Mestdagh P, Fredlund E, Pattyn F, Schulte JH, Muth D, Vermeulen J, et al. MYCN/c-MYC-induced microRNAs repress coding gene networks associated with poor outcome in MYCN/c-MYC-activated tumors. Oncogene. 2010;29:1394–404. doi: 10.1038/onc.2009.429. [DOI] [PubMed] [Google Scholar]
- 40.Ma L, Young J, Prabhala H, Pan E, Mestdagh P, Muth D, et al. miR-9, a MYC/MYCN-activated microRNA, regulates E-cadherin and cancer metastasis. Nat Cell Biol. 2010;12:247–56. doi: 10.1038/ncb2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bui TV, Mendell JT. Myc: Maestro of MicroRNAs. Genes & Cancer. 2010;1:568–75. doi: 10.1177/1947601910377491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Frenzel A, Loven J, Henriksson MA. Targeting MYC-Regulated miRNAs to Combat Cancer. Genes & Cancer. 2011;1:660–7. doi: 10.1177/1947601910377488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chang T-C, Yu D, Lee Y-S, Wentzel EA, Arking DE, West KM, et al. Widespread microRNA repression by Myc contributes to tumorigenesis. Nat Genet. 2008;40:43–50. doi: 10.1038/ng.2007.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kota J, Chivukula RR, O'Donnell KA, Wentzel EA, Montgomery CL, Hwang H-W, et al. Therapeutic microRNA Delivery Suppresses Tumorigenesis in a Murine Liver Cancer Model. Cell. 2009;137:1005–17. doi: 10.1016/j.cell.2009.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]