

Abstract
Established and emerging data demonstrate that a ‘preclinical’ period of disease precedes the onset of clinical rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE), as well as other autoimmune rheumatic diseases (ARDs).This preclinical stage of development of disease is characterized by abnormalities in disease-related biomarkers before the onset of the clinically apparent signs and symptoms. Numerous genetic and environmental risk factors for ARDs have also been identified, and many of these factors are likely to act before the clinical appearance of tissue injury to initiate and/or propagate autoimmunity and autoimmune disease. Thus, biomarkers representative of these autoimmune processes could potentially be used in conjunction with other clinical parameters during the preclinical period of ARDs to predict the future development of clinically apparent disease. This Review focuses on the preclinical stages of RA and SLE, as our current understanding of these diseases can be used to present an overall model of the development of ARDs that might ultimately be used to develop screening programmes and preventive strategies. Important considerations for the future development of such approaches, in particular, the issues that require additional research and how they might be addressed, are also discussed.
Introduction
Autoimmune rheumatic diseases (ARDs) encompass a wide variety of illnesses in which innate and adaptive immune responses lead to autoimmune-mediated tissue damage. In total, ARDs affect approximately 5% of the population and result in substantial morbidity, increased mortality and high financial costs.1–5 As such, measures to prevent ARDs would lead to marked improvements in public health.
Increasing evidence suggest that many ARDs, in particular, rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE)—the ARDs for which the natural history in humans is best understood—have a ‘pre-clinical’ period of development (Figure 1; Table 1).6–13 During this preclinical stage of disease, genetic and environ mental risk factors interact, probably sequentially, to initiate and propagate the development of autoimmunity, ultimately culminating in detectable tissue inflammation and injury. Furthermore, disease-related biomarkers, particularly autoantibodies, develop and evolve, initially in the absence of clinical signs and symptoms of tissue injury.13 These findings suggest that combined analysis of such biomarkers and other risk factors in asympto matic (or minimally symptomatic) individuals could identify individuals at high risk of future rheumatic disease, which might ultimately enable early therapeutic intervention to prevent progression of disease to a clinically meaningful state. Herein, we describe an overall model of ARD development based on the extensive data that are available on preclinical disease in RA and SLE. We also highlight certain features of pre-clinical disease development and, potentially, prevention that could, with further study, be applied to a broad range of ARDs that have preclinical stage.
Figure 1.

Overall model of the development of autoimmune rheumatic disease. Autoimmunity is probably initiated owing to a combination of a | genetic, environmental and stochastic factors, and b | at an anatomic site, which might not be the main target of the subsequent autoimmune response. c | Initially, autoimmunity can be present in absence of clinically apparent tissue injury, but might be detectable through analysis of disease biomarkers. d | Over time, further pathogenic changes in autoimmune responses occur, mediated by ongoing genetic, environmental and stochastic factors. e | Eventually, clinically apparent tissue injury occurs and the affected individual subsequently presents to a health-care provider. Processes a–d are considered to represent the ‘preclinical’ phases of disease development. Abbreviations: ACPA, anti-citrullinated peptide antibody; ANA, antinuclear antibody; RF, rheumatoid factor; SLE, systemic lupus erythematosus.
Table 1.
| Examples of autoimmune diseases with a known preclinical period of disease development
| Disease | Preclinical (auto)antibodies | Details |
|---|---|---|
| T1DM | Autoantibodies targeting variety of autoantigens involved in insulin production (for example, anti-insulin antibodies, anti-islet cell antibodies) | The presence of two or more of these types of autoantibodies is almost 100% predictive of future T1DM144 |
| Inflammatory bowel disease | Anti-Saccharomyces-cerevisiae-mannan antibodies; perinuclear ANCAs | Anti-S.-cerevsiae-mannan antibodies: present before clinical onset of Crohn disease in 10/32 cases versus 0/95 controls (P <0.01)146 Perinuclear ANCAs: present before the clinical onset of ulcerative colitis in 2/8 cases and 0/24 controls (P = 0.01)146 |
| Granulomatosis with polyangiitis | ANCAs | Present before clinical onset of vasculitis147-149 Levels of ANCAs were increased before diagnosis of vasculitis in 17/27 cases and 0/27 controls (P <0.01) based on samples stored in the US Department of Defence Biobank149 |
| Rheumatic fever | Anti-streptococcal-GlcNAc antibodies that also recognize the heart valve endothelium, laminin and laminar basement membrane | Pharyngeal infection preceded onset of autoimmune-mediated injury to various tissues (for example, cardiac, joint and skin tissues)127 |
| Autoimmune myositis | Various myositis-related autoantibodies | Production might precede clinical manifestations of disease150 |
| APS | aPL antibodies | Shown to be present before embolic events116 In some patients, aPL antibodies preceded the onset of clinically apparent SLE, with Coomb's test positivity in patients with APS most strongly associated with future SLE (OR = 66; P = 0.027)151 |
| Rheumatoid arthritis | RF and ACPAs | Levels increased before clinical appearance of inflammatory arthritis6-11,72,140,152,153 |
| SLE | ANAs | Present before the appearance of clinical features of disease12,154,155 |
| Sjogren's syndrome | Anti-Ro/SSA antibodies or anti-La/SSB antibodies | 8/11 initially asymptomatic women who were found to be positive for these antibodies after they delivered children with neonatal lupus developed symptomatic Sjogren’s within 4.75 years156 |
| Coeliac disease | Anti-gliadin antibodies and anti-endomesial antibodies | Levels were increased before the clinical onset of coeliac disease157 |
| Autoimmune thyroid disease | Anti-thyroglobulin and anti-thyroid-peroxidase antibodies | Present before the onset of hypothyroidism;158-161 in a longitudinal study with 20 years of follow-up, anti-thyroid antibodies were strongly associated with future hypothyroidism (OR=8 [95°% CI = 5–15] in women, and 25 [95°% CI = 10–63] in men)158 |
| Autoimmune biliary disease | Antibodies targeting mitochondria antibodies and/or the pyruvate dehydrogenase complex | 2/3 patients with Sjogren's syndrome and increased anti-mitochondrial antibodies concentrations developed symptomatic primary biliary cirrhosis with 5 years162 In ~1,400 samples from an Estonian Biobank, 3/8 subjects with antibodies to pyruvate dehydrogenase complex developed abnormal liver function tests within 9 years; higher levels of autoantibodies and autoantibodies of multiple immunoglobulin classes were more strongly associated with liver disease163 |
| Autoimmune adrenal disease | Autoantibodies to adrenal cortex cells or 21-hydroxylase | In patients with established T1DM, these antibodies preceded the development of clinically apparent adrenal insufficiency164,165 |
Abbreviations: ACPA, anti-cirullinated peptide antibody; ANA, antinuclear antibody; ANCA, antineutrophil cytoplasmic antibody; aPL, antiphospholipid (antibody); APS, antiphospholipid antibody syndrome; CI, confident interval; GlcNAc, N-acetyl-β-D-glucosamine; OR, odds ratio; RF, rheumatoid factor; SLE, systemic lupus erythematosus; T1DM, type 1 diabetes mellitus.
Defining preclinical rheumatic disease
An overall model of the development of ARDs is presented in Figure 1. In this model, and throughout this manuscript, the term ‘preclinical’ is defined as a period of detectable autoimmunity and/or inflammation predating the onset of clinically apparent tissue inflammation and injury. Currently, the definition of ‘clinically apparent’ is primarily based on widely used clinical parameters that can clearly be identified and attributed to an ARD, such as signs and symptoms of synovitis in the case of RA, and injury of the kidneys, skin, nervous system and haematological system in SLE. Indeed, classification systems incorporating such clinical parameters have been developed for many rheumatic diseases; however, these classification schemes might change over time as new developments, particularly regarding biomarkers and imaging modalities, enable the routine detection of earlier clinical stages of disease.
In fact, efforts have already been made to define terminology and definitions pertaining to the early natural history of both RA and SLE, in particular, before disease that is classifiable by existing schemes. Specifically, as part of European League Against Rheumatism (EULAR) Study Group for Risk Factors for RA, Gerlag et al.13 have recommended terminology for certain phases of the development of RA that include the following: genetic risk factors for RA; environmental risk factors; systemic autoimmunity; symptoms without clinical arthritis; unclassified arthritis; and classifiable RA. This study group13 also noted that these phases could be used in combination; for example, genetic and environmental risk factors for RA, and systemic autoimmunity might be detected in an individual without clinical arthritis, unclassified arthritis or RA. Notably, in an effort to avoid stigmatizing such individuals who have risk factors and even autoimmunity in the absence of classifiable disease, the EULAR recommendations13 also noted that an individual should not be classified as having ‘preclinical RA’ unless they later develop clinical disease. In SLE, the term ‘incomplete lupus erythematosus’ (ILE) has been used to define early signs and symptoms of disease, or a potentially milder form of the disease that might not ever meet classification criteria for definite SLE.14–18
Central to the issue of defining a preclinical period of disease is distinction of the characteristics that indicate autoimmunity, as well as those indicative of tissue injury and clinically apparent disease. The presence of a highly disease-specific autoantibody might be generally accepted as an example of a measure of autoimmunity that could define a preclinical disease state. For example, in many case–control studies, anti-citrullinated peptide anti bodies (ACPAs) are highly specific (>90% in most studies) for established RA,19 as well as highly predictive (positive predictive values [PPV] of >90% in most studies) of future development of RA.9,10 However, the relationship with preclinical autoimmunity remains uncertain for auto antibodies less specifically associated with ARDs, such as antinuclear antibodies (ANAs), which have been shown to be present at titres of >1:40 in up to 27% of co mmunity-based subjects,20 most of whom will never develop a clinically apparent rheumatic disease.20,21
The measures of tissue injury that should be used to define the onset of clinically apparent disease are another important consideration in characterizing the preclinical period. Whether this distinction should continue to be based on established measures, such as physical examination and routine biomedical tests, requires clarification because, as sensitive imaging measures and other markers of tissue injury are developed and applied to the study of preclinical ARD, the definition of clinically apparent disease might need to change. In addition, many current classification criteria for rheumatic diseases were developed to identify defined patient populations for inclusion in clinical trials and, in many cases, their applicability to clinical practice remains unclear. In fact, clinical manifestations that a clinician could potentially label as a specific disease, and indeed subsequently initiate treatment for, might not be classifiable by existing, consensus classification schemes.22 For example, a patient who has one swollen joint and elevated levels of an RA-related autoantibody might be diagnosed with RA by their health-care provider and treated with disease-modifying therapy, even though disease in this individual does not meet formal, established classification criteria for RA.23 Similarly, the tools commonly used to measure and assess the activity of various rheumatic diseases, such as the Disease Activity Score (DAS) for RA24 and SLE Disease Activity Index (SLEDAI),25 have not been well studied in patients who are in the early phases of disease before fulfilling classification criteria.
These issues attest that additional work is needed to develop valid means to classify and assess the pre clinical phases of ARDs. Importantly, the phases of ARD development will need to be related to classic public health schemes, in which the term ‘screening’ typically describes approaches aimed at identification of individuals who are in an asymptomatic or minimally symptomatic phase of disease, and who can undergo interventions to prevent progression to future disease (Figure 2).26 Clear, standardized definitions and classification schemes, which enable appropriate stratification of patients in studies of the natural history of disease, clinical trials and clinical practice, will lead to advances that ultimately facilitate clinical care of individuals with preclinical ARD.
Figure 2.
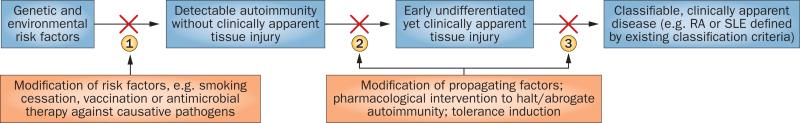
Points in the natural history of autoimmune rheumatic disease development that might represent therapeutic windows to prevention of initiation or progression of disease. In this model, intervention at point 1, probably involving modification of risk factors, might prevent the initial development of autoimmunity (primary prevention). Intervention at point 2, potentially by modification of potentiating risk factors or by targeting immune processes underlying the development of autoimmunity, might halt the progression of ‘benign’ (preclinical) autoimmunity to a more pathogenic state, and perhaps ‘reset’ the immune system and restore immune tolerance (secondary prevention). Therapeutic intervention at point 3 could, potentially, block or abrogate the progression of early symptomatic disease to fully differentiated disease, or prevent substantial organ injury and other complications (tertiary prevention), but might be unlikely to reverse clinically relevant autoimmune responses and thus prevent the development of persistent clinical disease. Of note, defining intervention at point 2 as ‘secondary prevention’ could be controversial, as autoimmunity in absence of obvious tissue injury might indicate to some that true disease has not yet occurred; however, intervention at this point in individuals at risk of developing clinical disease could conceivably hold more promise for preventing chronic disease than initiation of treatment at a point when tissue damage is already evident. Abbreviations: RA, rheumatoid arthritis; SLE, systemic lupus erythematosus.
Genetic and environmental risk factors
Multiple genetic, epigenetic and environmental risk factors for ARDs, particularly RA and SLE, have been identified, and some examples are presented in Table 2. In the context of a model of preclinical disease development, many of these genetic and environmental factors probably act before clinically apparent manifestations of RA, SLE or other ARDs to initiate and/or propagate disease. Furthermore, such associations raise the possibility that modification of certain environmental risk factors could lead to prevention of ARDs.
Table 2.
| Examples of genetic and environmental factors associated with ARDs
| Risk factor | Examples of associated ARDs | References |
|---|---|---|
| Genetic factors | ||
| MHC alleles | Multiple ARDs Including RA and SLE | 166,167 |
| PTPN22 | Multiple ARDs including RA and SLE, as well as multiple sclerosis and type 1 diabetes mellitus | 168 |
| Complement genes | Multiple ARDs including SLE | 169,170 |
| Epigenetic factors (for example, DNA methylation) | Multiple ARDs including RA and SLE | 171,172 |
| Environmental factors | ||
| Socioeconomic status | Multiple ARDs including RA and SLE | 126,173–175 |
| Sunlight or ultraviolet light | SLE | 176 |
| Tobacco smoke | RA | 177 |
| Occupational dust | RA, SLE and SSc | 32 |
| Dietary/nutritional factors | RA and SLE | 174 |
| Microbes | Rheumatic fever, RA, SLE (EBV), possibly giant cell arteritis | 36,103,105,127,128,178 |
| Hormonal factors | RA, SLE and SSc | 174,179 |
| Alcohol consumption | Protective in RA | 180,181 |
| Other factors | ||
| Gender | Most ARDs are more prevalent in women; unclear if this is due to hormonal or other factors (such as gene-dose effects) | 182 |
| Adulterated rapeseed oil | SSc-like disease | 183 |
| Organic solvents | SLE and SSc | 176 |
| Life stress | RA and SLE | 184 |
| Pharmaceuticals | Procainamide and hydralazine have been associated with an SLE-like disorder; thionamides used to treat hyperthyroidism can result in ANCA+ disease | 185 |
| Race or ethnicity | High rates of RA and SLE are seen in certain ethnic/racial groups; whether genetic and/or environmental differences explain this relationship is unclear | 54,55,83,179,186 |
| In individuals without current ARDs | ||
| Tobacco | Exposure to tobacco smoke is associated with RF positivity in the absence of RA | 33,34,187 |
| Hormones | Oral contraceptive use is associated with decreased risk of RF positivity in the absence of RA | 34 |
| Microbes | Serological evidence of EBV infection can precede a clinical diagnosis of SLE Serological evidence of infection with oral microbes is associated with RA-related autoantibody positivity in subjects without RA | 37,188,189 |
| Gene–environment interactions | Smoking combined with certain MHC alleles was associated with increased risk of future RA in the Nurses’ Health Study | 190 |
Abbreviations: ANCA, antineutrophil cytoplasmic antibody; ARD, autoimmune rheumatic disease; EBV Epstein–Barr virus; RA, rheumatoid arthritis; RF, rheumatoid factor; SLE, systemic lupus erythematosus; SSc, systemic sclerosis.
An important caveat, however, is that the majority of the known genetic and environmental risk factors for diseases such as RA and SLE have been identified through case–control studies, in which patients with established disease were compared with those without disease. Moreover, most of these studies could have been affected, to some extent, by recall bias due to patients’ incorrect recollection and reporting of the duration and timing of environmental exposures that might have occurred years before the clinically apparent onset of disease.27 Whether particular risk factors are involved in the initiation or propagation of the disease, or both, also remains unclear at present. For example, exposure to tobacco smoke has been identified as a strong risk factor for RA in multiple studies;28–32 however, because this relationship has largely been studied retrospectively in patients with established disease, whether smoking is an initial trigger for autoimmunity and/or a propagating factor, or even perhaps a permissive factor for some other aetiologic agent such as a bacterial organism, remains unclear. Likewise, clarification is needed regarding the precise roles that genetic factors, such as HLA-DRB1 alleles in RA, have in initiating autoimmunity and/or propagating disease to a clinically apparent state once autoimmunity has developed.
Nevertheless, some studies have evaluated risk factors for ARDs that might be relevant to the develop ment of preclinical disease (Table 2). For example, a strong association between smoking and increased levels of rheumatoid factor (RF) in individuals without current RA has been reported.33,34 In addition, data from the prospective Nurses’ Health Study35 have demonstrated that combined exposure to tobacco smoke and expression of certain HLA molecules considerably increases the risk of future RA. This finding suggesting that gene– environment interactions, at least those involving these factors, play an important part in the develop ment of future RA, although the issue of whether smoking is a triggering versus a propagating factor remains unaddressed.
Of note, multiple sources of evidence can strengthen the association of an ARD with a specific risk factor. For example, the result of serological studies in patients with established SLE and individuals with preclinical SLE (often identified retrospectively after development of clinical SLE), and data from animal models of lupus, all combine to suggest that Epstein–Barr virus (EBV) infection precedes the development of SLE-related auto antibodies.36–39 In particular, these data suggest that, in susceptible individuals, EBV antigens can promote the generation of initial autoimmune responses to nuclear antigens, such as Ro/SSA, through molecular mimicry, which are subsequently amplified through processes such as epitope spreading.37 Together, these findings indicate that specific risk factors can influence develop ment of disease during the preclinical period, and might thus represent true triggers of disease. Further study of the preclinical period of ARDs will be important to understand the precise role of such risk factors in the aetiology of ARDs, particularly if we are to develop pr eventive measures that target these factors.
Preclinical studies in RA and SLE
Although multiple ARDs seem to have a preclinical period of development (Table 1), studies of the preclinical stages of SLE and RA have provided us with key data regarding the evolution of autoimmunity before the onset of clinically detectable, immune-mediated tissue inflammation; therefore, these diseases can be used to model the development of ARDs. Datasets in indivi duals with preclinical SLE and RA have been generated using both retrospective and prospective approaches. Retrospective approaches take advantage of available ‘convenient’ biological samples, usually large biobanks of stored serum from which cases and controls can be identified for comparative studies. In particular, in Europe, retrospective studies have utilized large serum biobanks in Sweden, Finland, and the Netherlands, whereas studies in the USA have used the large Department of Defence repository of serum samples obtained from military personnel at regular intervals during their service. These studies have collectively focused on dissecting the repertoire of auto-antibodies and cytokines that are detectable in serum or plasma from individuals who eventually developed SLE or RA; the availability of serial preclinical samples from the same individuals has been particularly important for defining the evolution of the autoantibody repertoire and the associated changes in levels of circulating cytokines. As a result of these studies, a clear picture is emerging regarding the preclinical expansion and amplification of disease specific autoantibodies and inflammation.
Such retrospective approaches are practical and cost-effective, but suffer from some inherent biases related to the composition and assembly of the biobanks, most of which are not established to test specific scientific hypotheses. By contrast, prospective approaches comprising longitudinal studies of at-risk populations are designed to test a specific hypothesis, and can largely eliminate the compositional biases associated with biobanks. Furthermore, prospective studies can collect high-quality data from questionnaires, clinical examination and other methodologies, together with biological samples, providing additional information of potential importance. The major disadvantage of long-term prospective studies, however, is that such studies are expensive and difficult to sustain over the timeframe that is required to accumulate an appropriate number of incident cases. Furthermore, prospective approaches that are not specifically designed to evaluate a particular ARD might not record key data relevant to that disease, such as first onset of joints symptoms in RA. Never theless, as summarized below, the data gathered from both the retrospective and prospective studies to date have been remarkably consistent, and have led to some important conclusions regarding the evolution of the autoimmune phenomenology during the preclinical stages of SLE and RA, and probably other related au toimmune diseases.
Studies of preclinical SLE
Preclinical autoantibodies in SLE
Seminal studies by researchers at the University of Oklahoma, USA,12,40,41 examined preclinical samples from 130 US military personnel who developed SLE. 36% of the individuals in this SLE cohort were male and 62% were African-American;12,36,37 these frequencies are higher than those observed in most SLE studies, a trend that probably reflects the composition of the military population from which cases were drawn. The mean age of SLE-onset was 30 years, and approximately five preclinical samples, on average, were analysed for each of the individuals included in these studies.12,36,37 Analysis of this population clearly demonstrated a high prevalence of preclinical autoantibody positivity, with ANA positivity at a titre of ≥1:120 in 78% of the SLE-cohort a mean of approximately 3 years before clinical diagnosis, compared with 0% ANA positivity at this level in matched military control samples.12 However, in many cases the earliest available sample was positive, therefore, this duration of preclinical autoimmunity might be an underestimation.12 Indeed, SLE-associated autoantibodies were detectable >9 years before diagnosis of classifiable SLE in some individiuals.12 Moreover, a key observation was that, overall, certain autoantibodies were detected earlier before onset of SLE than others: ANAs, anti- phospholipid antibodies, and anti-Ro/SSA and anti-La/SSB antibodies were all detected substantially earlier than antibodies targeting double-stranded DNA (dsDNA), the Smith (Sm) antigen and ribonucleo-proteins (RNP).12 In the case of anti-dsDNA antibodies, anti-Sm antibodies and anti-RNP autoantibodies, which were rare in the control samples (≤3% postivity), the proportion of indivi duals in the SLE cohort who became positive increased considerably in the year immediately preceding clinical diagnosis.12
Similar findings have been demonstrated by Eriksson and colleagues who studied 38 patients from northern Sweden in whom stored serum samples from before a diagnosis of SLE were available.42 Specifically, they found that positivity for any nuclear antigen (using indirect immunofluorescence to detect ANAs and a multiplex assay for antibodies to extractable nuclear antigens [ENAs]) was observed in 63% of patients a mean of 8.7 years before diagnosis of SLE, with auto antibodies targeting Ro/SSA being the earliest of the anti-ENA antibodies to appear; antibodies to other ENAs (such as dsDNA and Sm) were detected closer to diagnosis.
Overall, findings from these studies have led to the concept that individuals who develop SLE have an initial preclinical stage of ‘benign autoimmunity’ that develops into a more ominous stage of ‘pathogenic auto-immunity’ that in turn rapidly evolves into clinically apparent disease and tissue inflammation. According to this concept, the autoantibodies closely associated with the pathogenesis of SLE, such as anti-Sm antibodies, are hypothesized to be produced as a result of maturation and amplification of the autoimmune response, and epitope spreading in the preclinical period.
Preclinical inflammation in SLE
In addition to autoantibodies, established and emerging data have identified abnormalities in a variety of immune-related and inflammation-related pathways, including the complement system, cytokines and chemokines, and the more recently described ‘microparticles’ detected in association with classified SLE and also ILE.43,44 In particular, dysregulation of IFN-α seems to be an important aspect of SLE-related autoimmunity in both classified and incomplete forms of disease.45 Furthermore, IFN-α might be related to the presence of SLE-related autoantibodies rather than clinical manifestations of disease,45,46 and therefore IFN-α might be speci fically related to development of auto immunity. These same processes might also play a part in the develop ment of SLE in the preclinical period of dis ease development, and thus could ultimately be potential tar gets for prevention of progression to clinical dis ease. However, this possibility has not been well studied; therefore, going forward, the role of these processes in preclinical SLE should be an area of active investigation.
Studies of preclinical RA
Preclinical rheumatoid factor positivity
The seminal observations regarding preclinical autoimmunity in SLE are echoed by similar observations in preclinical RA. The earliest studies, dating as far back as the 1980s, demonstrated that RF was present in serum samples many years before disease onset in indivi duals who ultimately developed RA. For example, epidemio-logical studies in a large Finnish community-based cohort demonstrated that the majority of indivi duals who developed seropositive RA during the study period were RF-positive in the years immediately preceding disease onset.47–50 Moreover, a number of these indivi duals were also positive for anti-keratin antibodies (AKA) and anti- perinuclear factor (APF), which are more specific ally associated with RA;51 furthermore, these latter two auto-antibodies have subsequently been shown to re cognize citrullinat ed epitopes, and thus represent ACPAs.52,53
In the USA, important NIH-funded prospective longitudinal epidemiological studies in the Pima Indians of Arizona, a population that—similarly to other American Indian populations—is known to have a high prevalence of RA,54 also demonstrated preclinical autoimmunity involving RF.7,55 In these studies, medical history, physical examination of the joints, radiographs and serum levels of RF were assessed biennially for up to 19 years in more than 2,700 individuals, initially without RA.7,55 During the study period, 70 new cases of RA developed, with the data demonstrating that the incidence of RA increased with progressively higher titres of RF, reaching a peak of 48.3 cases per 1,000 person-years for RF titres >1:256.7 These findings highlight the risk of future disease associated with high RF titres and also the high incidence of RA in this American Indian population.
Preclinical ACPA positivity
Subsequent to the discovery of ACPAs and the demonstration of their high degree of specificity for RA,52,53 studies were initiated to evaluate the potential presence of these autoantibodies during the preclinical period of disease. Two landmark European retrospective studies, one in Sweden9 and the other in the Netherlands,10 analysed stored serum samples—from a public health study and derived from a blood bank, respectively—isolated from individuals who ultimately developed RA and control individuals who did not. Both studies demonstrated that ACPAs and RF were detectable months or even years before the development of RA.9,10 Indeed the proportion of ACPA-positive and/or RF-positive indivi duals who later developed RA increased progressively until clinical onset of disease, and most individuals were seropositive for both autoantibodies in the months immediately before diagnosis. Preclinical auto-immunity in RA has also been examine retrospectively in the Studies of the Etiology of Rheumatoid Arthritis (SERA) study,11 which used the large Department of Defence serum repository that was also utilized in the studies of development of autoimmunity in SLE.12,36,37 In addition to confirming the observations regarding pre-clinical autoimmunity made in the European studies,9,10 this US study11 demonstrated that the period of preclinical autoantibody seropositivity increased with age at RA onset, a finding that was later validated in the Dutch sample set mentioned above.11,56 This observation might provide important insights into the effect of age on the evolution of RA-associated autoimmunity, suggesting that aetiological risk factors that determine the temporal relationship between RA-related autoantibodies and clinically apparent onset of disease might differ with age. Furthermore, this variation in the duration of preclinical autoimmunity represents an important consideration for any proposed screening protocols.
Evidence of epitope spreading
Studies in these retrospective cohorts have subsequently been extended to examine other autoantibody bio-markers relevant to RA, and these efforts have collectively provided important mechanistic information regarding the immunological events that precede the onset of clinical disease. Echoing the observations made in preclinical SLE, a key observation in RA was the demon stration of epitope spreading in the ACPA response during the pre-clinical stage of RA.57–59 Methodo logies based on arrays of citrullinated auto antigens have revealed that the breadth of ACPA responses increased in most individuals as the clinical diagnosis of RA became apparent.58 Furthermore, a wide spectrum of citrullinated autoantigens seems to be recognized by ACPAs, and the epitopes targeted are not restricted to the previously well-characterized auto-antigens fibrinogen, vimentin and enolase.60,61 Although no single initial (auto)antigenic target of RA-associated autoimmunity has been demonstrated, Brink et al.59 found that certain citrullinated peptides (citrullinated fibrinogen and vimentin, for example) were some of the earliest targets of autoantibodies in patients with RA; autoimmunity targeting other epitopes (such as those derived from citrullinated enolase and filaggrin) developed closer to disease onset, and autoantibodies recognizing citrullinated collagen increased most prominently after onset of clinical RA. These findings need confirmation; however, they echo findings in SLE, and suggest that certain antigens contribute to an initial break in immune tolerance, with subsequent epitope spreading resulting in autoimmune responses to other antigens, which causes a transition to clinically apparent disease. Furthermore, such expansion of the repertoire of recognized ACPAs is reflected by increasing levels of anti-cyclic citrullinated peptide (anti-CCP) antibodies,58 which represent ACPAs detectable using a widely available clinical assay.62 Thus, an individual with rising anti-CCP antibody titres, representing a broadening of the ACPA response, is probably at considerable risk of imminent RA onset.58
Other important characteristics of the autoantibody responses during the preclinical period of RA also seem to occur, including abnormal galactosylation that might render these autoantibodies more pathogenic.63 Furthermore, increased avidity of ACPAs for citrullinated autoantigens,64 as well as expansion of antibody isotype usage and class switching, have been noted.65 The latter processes suggest an important role for T cells in the maturation and amplification of autoimmune responses.66 Indeed, the RA-predisposing HLA-DRB1 alleles comprising the ‘shared-epitope’ have been proposed to play a key part in facilitating the maturation of the ACPA response by efficiently presenting citrullinated peptides to T cells.60,67,68 In support of this hypothesis, a prospective multicentre Dutch study69 demonstrated an association between the shared epitope and the breadth of citrullinated peptides recognized by ACPAs present in the sera of anti-CCP-antibody-positive and/or IgMRF-positive individuals with ‘arthralgia’ but no obvious arthritis (that is, patients with joint symptoms but no clinically detectable synovitis); however, no association between the range of ACPA responses and the development of clinical RA was found in this study,69 probably due to statistical limitations.
Preclinical inflammation in RA
The availability of multiplexing technologies capable of simultaneously quantifying multiple biomarkers has been particularly helpful in dissecting changes in soluble cytokine and chemokine networks in preclinical samples. Retrospective studies utilizing such methodologies have shown that, in parallel with the evolution of the auto antibody responses described above, the level of multiple soluble cytokines and chemokines progressively increased before onset of clinical RA.70 Indeed, an increase in the range of cytokines and chemokines that are expressed at abnormal levels predicts imminent onset of RA.71 These cytokines and chemokines include those targeted with current therapeutic agents (such as TNF, IL-1 and IL-6), as well as numerous others, suggesting that multiple pathways of inflammation are affected in preclinical RA.70–74 Interestingly, expression levels of both proinflammatory and anti-inflammatory cytokines are increased, and the available data point to a general perturbation of the soluble cytokine networks, as opposed to a predictable rise in one or several cytokines, before onset of classifiable disease.70–72 However, some data suggest that certain cytokines and chemokine abnormalities could precede the appearance of autoantibodies, and the inflammatory pathways that these abnormalities affect might have a mechanistic role in the earliest generation of autoimmunity. In particular, Deane and colleagues71 found that elevated levels of CXC-motif chemokine 10 (CXCL10; also known as 10 kDa IFN-γ-induced protein [IP-10]) and IL-1α preceded the appearance of ACPAs. Furthermore, El-Gabalawy et al.75 reported that elevations of CC-motif chemokine 2 (CCL2; also known as monocyte chemoattractant protein-1 [MCP-1]) were present even in the absence of autoantibodies in arthritis-free first-degree relatives of American Indian probands with RA; this group is currently undergoing longitudinal follow-up to determine if these abnormalities are associated with incident autoantibodies and ultimately clinically apparent RA. As technologies used to study a broad range of cytokines, chemokines and other inflammatory factors improve, and high-quality sample sets from individuals with preclinical RA are obtained, the relationship between certain inflammatory pathways and the initiation and propagation of autoimmunity can be elucidated to identify targets for abrogation or prevention of future RA.
Studies in high-risk populations
A considerable proportion of ACPA-positive and/or RF-positive individuals with arthralgia, such as those included in a Dutch study of patients with ‘arthralgia’ but no clinically apparent inflammatory arthritis,76 have been shown to subsequently develop inflammatory arthritis and classifiable RA. Furthermore, the severity and distribution of symptoms in such individuals have been found to be predictive of the development of swollen joints and classifiable RA.77 Importantly, the North American SERA study,78 which evaluated first-degree relatives of primarily white patients with RA, reported an association between anti-CCP antibody positivity, as well as expansion of the ACPA repertoire according to array testing, and joint tender ness in the absence of swelling; thus, such changes in autoantibody responses could potentially cause underlying tissue injury in some individuals before onset of clinically classified RA. In general, these findings suggest that complex interplay exists between the development of autoimmune responses and tissue injury, and therefore clearly distinguishing between preclinical autoimmunity and clinically apparent disease is difficult. This issue will require careful analysis in real-time prospective studies in individuals at risk of RA.
A prospective longitudinal study64 has demonstrated a substantially higher prevalence of ACPA and RF in the first-degree relatives of American Indian patients with RA from Central Canada (Cree and Ojibway) and Alaskan Natives (Tlingit) compared with the prevalence of these antibodies reported in the primarily white first-degree relatives who were followed in the SERA study.78,79 Moreover, data from incident cases of inflammatory arthritis drawn from these American Indian or Alaskan Native populations have so far confirmed the high risk of imminent RA associated with positivity for both ACPAs and RF, while providing further evidence of epitope spreading of the ACPA response in association with increasing cytokine levels.80 Interestingly, the cytokine profile of the unaffected American Indian first-degree relatives more closely resembled that observed in their relatives with RA than the profile found in a control population of American Indian individuals with no family history of autoimmune disease.75 Studies in these populations have also revealed clustering of genetic and environmental risk factors for RA in the population as a whole, and particularly in specific high-risk multi-case families.75,81–86 Furthermore, ‘RA-like’ joint symptoms and arthralgia were markedly more common in first-degree relatives compared with the control population, a phenomenon that was not fully explained by either the presence of ACPA and/or RF, or a specific cytokine profile, suggesting that such clustering of other genetic and environmental risk factors might underlie this susceptibility to joint symptoms.87
In addition to the well-established studies discussed, a number of other prospective studies in groups of individuals at high risk of RA are being undertaken to address complementary questions.88–90 In parallel, new biomarkers are becoming available with which to further dissect the immune and inflammatory mechanisms that are operative in the preclinical period of RA, SLE and other ARDs. For example, antibodies targeting protein-arginine deiminase type-4 (PAD4) have been shown to be elevated in preclinical RA.91 In addition, an analysis of anti-PAD4 antibodies in American Indian patients with RA and their first-degree relatives revealed that, in contrast with anti-CCP antibodies, these RA-associated autoantibodies were virtually undetectable in the first-degree relatives but were common in patients with established RA.92 Furthermore, the presence of anti-carbamylated antibodies has been detected in serum samples collected before the onset of RA,93 and emerging data in patients with established RA suggests that neutrophil extracellular trap (NET) formation are a source of citrullinated autoantigens in RA.94 In addition, although emerging data is available regarding T-cell reactivity to specific citrullinated proteins and other antigens in patients with established RA,95,96 no studies to date have directly evaluated the antigen specificity of T-cell and/or B-cell subsets during the preclinical stage of RA and compared this with the ACPA responses. Hope fully, well-designed prospective studies that methodically isolate and analyse peripheral blood mononuclear cells, including utilization of emerging technologies such as single-cell analyses that can identify antigen reactivity of specific immune cells,97 will address this important question in the near future.
An overall model of ARD development
On the basis of the currently available data regarding the natural history of SLE and RA, as well as several other diseases (Table 1), many ARDs probably initially develop as a result of combined genetic, environmental and perhaps stochastic factors that initiate inflammation and autoimmunity (Figure 1). Once manifest, autoimmunity evolves over time under the influence of the same, or perhaps additional similar or disparate genetic and environ mental factors, to a more pathogenic stage through multiple processes: expansion of auto reactive T cells and B cells; epitope spreading; increases in inflammation; upregulation of signalling molecules; inflammation-related antigen production and presentation; and alterations of autoantibodies, such as glyco sylation, that render them more capable of inducing disease. Data regarding the specific type of autoantibody (that is, ACPAs or RF) initially produced in preclinical RA are conflicting, with some finding that ACPAs precede RF,69 whereas other have demonstrated that these autoantibodies appear almost simultaneously;11 nevertheless, a combined elevation of ACPA and RF levels is highly specific for imminent onset of RA,9,10,71 and therefore suggests that synergy between these two autoantibody responses could promote the development of clinically apparent arthritis. Ultimately, autoimmunity progresses to a point at which tissue injury occurs, and the clinical symptoms and signs of ARD develop.
Importantly, the precise mechanisms by which individuals transitions from a preclinical state to clinically apparent disease remain unknown. In SLE and RA, epitope spreading might progress from reactivity to a non pathogenic target to a point where tissue proteins that are relevant to disease states (for example, joint pro teins in RA and renal proteins in SLE) are targeted, result ing in injury.98 In RA, a combination of auto-antibodies, such as ACPAs and RF, could form immune com plexes that trigger joint inflammation.99 Of note, in some studies in RA and SLE, the expansion in the repertoire of ACPA and SLE-related autoantibodies, respectively, seems to diminish or halt after diag nosis.12,58 Further more, patients who are seronegative at the time of onset of clinically apparent synovitis rarely convert to seropositivity for RF and/or anti-CCP antibodies after the clinical onset of inflammatory arthritis, with <9% seronegative patients converting to seropositivity in one meta-analysis of 12 publications.100 Whether these findings reflect the effects of treatment with immunomodulatory therapies or some threshold of expansion that is related to the onset of clinically apparent disease remains unclear. Also of note, the prolonged period of preclinical autoimmunity seen in RA and SLE suggests that these diseases, as well as other ARDs, might be initiated at an anatomical site distal to the organs eventually injured in clinically apparent disease (Figure 1).101 Mucosal surfaces represent an attractive potential site of initiation of ARDs, given their exposure to environmental insults and their dedicated immunologic machinery (mucosal associated lymphatic tissue, for example) that can mount auto immune responses.102 In fact, mucosal inflammation and/or organisms associated with mucosal sites have been associated with the develop ment of a number of ARDs.103–107 For example, several studies have demonstrated the presence of lung inflammation before the onset of symptomatic articular RA, and this site has been implicated in the initial generation of RA-related autoantibodies.108–111 Certainly, in established RA, autoantibodies are generated in the joint112 and ACPA-producing B cells have also been identified in the circulation,113 and autoantibody generation within the kidney has been demonstrated in SLE;114 however, auto antibodies might be generated at these sites after initiation of auto immunity at another anatomical site.
These issues, in particular the mechanisms that initially trigger autoimmunity and enable the transition between preclinical autoimmunity and clinically apparent disease, will need further exploration. Nevertheless, these data raise the possibility that, if an individual could be identified before their immune response has progressed to a more pathogenic state, intervention to block expansion of autoimmunity could abrogate or even halt future disease. Importantly, many indivi duals develop autoimmunity but never develop a clinically apparent ARD, or only have modest manifestations of disease that are difficult to classify as a specific ARD; autoimmunity might even resolve in some indivi duals, as evidenced in some studies that reported the dis appearance of detectable circulating autoantibodies in individuals who did not develop RA.71 Therefore, identification of factors that accurately predict pathogenic autoimmunity will be important to avoid potential overtreatment of ‘benign’ autoimmunity. Nonetheless, findings in individuals who have risk factors for an ARD or exhibit asymptomatic autoimmunity but who do not develop a classifiable disease might prove invaluable in understanding the natural history of ARDs, including the genetic and environ mental triggers that can lead to disease. In addition, as knowledge of the roles of autoimmunity in health and disease grows, asymptomatic or benign autoimmunity might be recognized as being more relevant to health or disease than once thought. In particular, emerging data demonstrating that the presence of antiphospholipid antibodies, ANAs and RF is associated with cardiovascular disease (CVD), even in absence of an overt ARD.115–117 As a result of such findings, autoimmunity could potentially be classified as a pathogenic condition that would benefit from intervention in the absence of a fully classifiable ARD or symptoms of disease, such as fatigue and arthralgias, that can be attributable to autoimmune-mediated injury.
Predicting future onset of ARDs
An important step in screening for and preventing ARDs is to develop a test, or panel of tests, that accurately identifies individuals who are at risk of future disease, at a time when they are in a phase of disease development that might not otherwise warrant intervention (Figure 2). Such instruments will be especially important if ARDs are to be the subject of population-based screening and prevention programmes, similar to those that have been developed for CVD and certain cancers.
Examples of studies that have used preclinical bio-markers or genetic and epidemiologic factors to predict future onset of SLE and RA are presented in Table 3. In particular, in a case–control study of preclinical SLE-related autoantibodies in military personnel, Arbuckle and colleagues12 found that ANA titres of >1:120 were 100% specific for future onset of SLE, although given the frequency of ANA positivity in the general population,18 the specificity of this test is likely to be much lower if used in a broader population; indeed, as found in other studies, a high prevalence of ANA positivity at titres >1:120 in the general population would make prediction of future SLE based on serum levels of ANAs alone impractical,18 although using more specific tests for certain types of ANA (anti-dsDNA antibodies and/or anti-Sm antibodies, for example) might improve biomarker-based predictions of future SLE.
Table 3.
| Examples of studies of autoantibodies and other biomarkers in preclinical SLE and RA
| Study | Biomarkers analysed | Findings |
|---|---|---|
| Arbuckle et al. (2003)12 | ANAs (and anti-ENA antibodies) | ANAs at a titre of 1:120 are highly specific for SLE based on analysis of stored samples (collected before onset of SLE) in case–control fashion* |
| Rantapaa-Dahlqvist et al. (2003)9 | RF and ACPAs | PPV of 100% for a diagnosis of RA within 1.5 years in individuals positive for both IgA RF and ACPAs (based on case–control data)* |
| Nielen et al. (2004)10 | RF (IgM) and ACPAs | PPV of up to 100% for a diagnosis of RA within 5 years based on 5-year incidence rates of 0.001 (general population) or 3.9% (estimated for high-risk individuals from families with a multicase history of RA)* |
| Deane et al. (2010)191 | RF, ACPAs, CRP, and multiple cytokines and chemokines | ACPAs and/or two or more RF isotypes >96% specific for future RA; highest levels of autoantibodies <3 years before diagnosis |
| Bos et al. (2010)76 | RF and ACPAs | 27% ACPA+ individuals developed inflammatory arthritis after a median of 11 months of follow-up; rates of over 50% within ~1 year were seen in patients with highest ACPA titres |
| van de Stadt et al. (2012)77 | Multiple environmental factors, symptoms and biomarkers | In individuals with joint symptoms (‘arthralgia’) in the absence of inflammatory arthritis on examination, factors including gender, lack of alcohol consumption, and symptom duration and distribution were predictive of developing RA; however, the strongest risk factors were increased levels of RF and ACPAs |
| Karlson et al. (2013)118 | Multiple genetic and environmental factors | Genetic and environmental factors ascertained before the onset of RA in the Nurses’ Health Studies were used to predict future RA with an AUC of 0.716 |
A caveat of these findings is that the high specificities of the biomarkers was determined in case–control studies and when these tests are applied to a general population, in which the estimated prevalence disease might be low, the PPVs could fall substantially. Abbreviations: ACPA, anti-citrullinated peptide antibody; ANA, antinuclear antibody; AUC, area under the curve; CRP, C-reactive protein; ENA, extractable nuclear antigen; PPV, positive predictive value; RA, rheumatoid arthritis; RF, rheumatoid factor; SLE, systemic lupus erythematosus.
Perhaps the best data regarding prediction of a future onset of an ARD relate to RA. In this disease, elevated levels of both ACPAs and RF in combination have been shown to be highly predictive of future RA (Table 3), with some case–control studies reporting estimated positive predictive values (PPV) for future disease of 100%.9–11 Again, the PPVs of these tests are likely to be lower when applied to the general population, considering the prevalence rates of the disease. For instance, in a case–control study, Rantapaa-Dahlqvist and colleges9 found that positivity for ACPAs and IgM RF has a 100% PPV for future RA, although in a population with a 1% frequency of RA the PPV fell to 16%. Nevertheless, these results suggest that positivity for multiple auto antibodies can identify individuals with pathogenic forms of autoimmunity who are at highest risk of future disease. Further more, in a Dutch study of ACPA-positive and/or RF-positive individuals with arthralgia but no clinically detectable synovitis, which has followed 374 individuals for several years, 131 individuals have developed inflammatory arthritis.77 In this group, the risk of developing RA was associated with positivity for both ACPA and RF, and up to 40% of individuals with arthralgia who were positive for both ACPA and RF developed RA within approximately two years, with higher rates of progression and earlier onset of disease observed in indivi duals with high-titre ACPA positivity (at least a threefold increase over normal titres). In addition to autoantibody status, other reported predictors of future RA in these Dutch patients with arthralgia include: family history of RA (that is, having a first-degree family member with the disease); no or limited alcohol consumption; duration, severity and location of joint symptoms (including morning stiffness); and a patient-reported history of swollen joints.77 Furthermore, work by Karlson and colleagues,118 and others,119 that have used genetic factors to predict the likelihood of future RA suggest that ultimately a combined assessment of symptoms, and genetic, environmental, serologic and inflammatory factors could be a reasonable approach to identify individuals at high risk of a future ARD.
Predicting the overall likelihood of a future ARD is important. However, the ability to predict the timing of future onset of clinically apparent disease is also important to inform individuals of their personal risk for a future ARD within a defined time period, as well as for designing prevention trials, which have a limited period of follow-up, that are adequately powered. Predictive tools in CVD exemplify this approach, as indivi duals are currently evaluated for risk of a cardiovascular event within a defined period using models such as the Framingham Risk Score.121 The ‘timing’ of ARD develop ment has been evaluated in RA to some extent in the Dutch study discussed earlier, in which indivi duals with the highest risk scores had the highest incidence of RA develop ment (80% with RA within 60 months), and furthermore had the quickest rate of onset of RA (60% had developed RA within 24 months).77 In addition, a study in US Military personnel by Sokolove and colleges58 found that elevated expression of a certain combination of ACPAs, cytokines and chemokines was approximately 58% sensitive and 87% specific for onset of RA within 2 years. Furthermore, indirect evidence indicates that certain bio-markers in SLE, such as elevated anti-dsDNA and anti-Smith antibodies, are elevated relatively shortly before the onset of clinical symptoms; therefore, elevations of these auto antibodies in individuals who are at-risk for future SLE might signal ‘imminent’ clinically apparent disease. Overall, these findings suggest that predictive approaches can identify both the likelihood and timing of future ARDs, although this needs further exploration.
ARD screening
In 1968, the WHO presented recommendations regarding the features that make a disease appropriate for screening and prevention programmes (Box 1).122 ARDs, and particularly RA and SLE, could be considered to meet several of the conditions stipulated by the WHO; however, not all of the criteria have been satisfied with regard to RA and SLE (or most other ARDs), at present. Specifically, the true predictive value of auto antibodies for future disease in large-scale population-based studies remains largely unknown. Given that most of these studies did not performed detailed clinical evaluations in control individuals, the association between auto-immunity detected in control individuals and preclinical symptoms of disease cannot be determined precisely; therefore, disease-related autoantibodies might be more specific and thus more predictive than is currently believed. Furthermore, the characteristics of the population or populations that it would best to target for screening are not clear. Screening individuals with a potentially increased baseline risk of future ARD, such as the relatives of patients with established disease,123,124 individuals from high-risk populations (such as American Indians in RA),54 or women—who have a higher prevalence of most ARDs than men125—could potentially improve the diagnostic accuracy of screening tests, but might be of limited utility in the general population. Indeed, many ARD cases are sporadic, and for RA and SLE in particular, the majority of cases occur in absence of a known family history of disease.124,126 Therefore, the type of screening strategy would provide the greatest overall benefit to those at risk of development of ARDs requires clarification, but ultimately, screening will probably rely on inexpensive, readily available, accurate tests that could be used in broad population-based approaches. Importantly, any such approaches will need to be designed based on high-quality natural history studies of ARD development, with input from experts in public health and cost-effective approaches to screening for, and prevention of, disease.
ARD prevention
Although extensive data regarding optimal screening strategies are not currently available for ARDs, several examples of potential preventive approaches to ARDs are presented in Figure 2. To some extent, effective primary preventive strategies have already been developed for some ARDs. For instance, an understanding of the natural history of disease, and the subsequent development of antibiotic therapy and vaccinations have resulted in substantial declines in the incidences of in rheumatic fever and hepatitis-B-associated polyarteritis nodosa, respectively, at least in developed countries.127,128 Studies have also provided insights into the potential prevention strategies that might be of benefit in other ARDs. For example, increasing evidence suggests that early treatment of classifiable RA leads to improved long-term outcomes, and perhaps increased rates of drug-free remission.129 In addition, in palindromic rheumatism, which has been likened to a ‘preclinical’ state of RA,130 case–control studies have demonstrated that the use of antimalarial agents, such as chloroquine, can slow or halt progression to persistent disease, including the development of RA.131,132 Furthermore, in patients with ILE, antimalarial therapy has been shown to delay the onset of classifiable SLE, and also decrease the repertoire and expression levels of autoantibodies present when full diagnostic criteria for the disease were met.40 These findings suggest that the underlying SLE-associated auto immunity can be modulated if treated early in the course of disease, before fully classifiable clinical manifestation of the disease. Similarly, trials in early, undifferen tiated clinically apparent inflammatory arthritis have demon strated that methotrexate, repeated doses of cortico steroids, or abatacept-mediated blockade of interactions between antigen presenting cells and T cells can delay or halt the development of classifiable disease.133–135 By contrast, in a cohort of patients with arthralgia and positivity for RA-related autoantibodies, two doses of dexamethasone (100 mg intramuscularly at baseline and at 6 months) did not seem to halt progression to clinically apparent arthritis after a median follow-up of 26 months.136
These ‘clues’ to preventative strategies for ARDs notwithstanding, multiple issues need to be addressed to enable the development of feasible preventive strategies for most ARDs. Some of these issues relate to the items highlighted by the WHO (Box 1) and discussed above, whereas additional ARD-specific issues are listed in Box 2, with additional discussion presented in Supplementary Table 1. In particular, the mechanisms by which intervening in preclinical ARDs might lead to prevention of progression to clinical disease are not clear. Certainly eliminating a key factor that initially triggers autoimmunity could lead to disease prevention, but whether clinical onset of the disease can be prevented by intervention after an individual has developed auto-immunity is unknown. On the basis of the findings in SLE and RA discussed, early intervention could potentially block epitope spreading, which seems to represent a process important to a transition from initial auto immunity to clinically apparent disease. Although effective treatments are available for most clinically apparent ARDs, the efficacy and safety of these treatments as implemented in a preclinical period with the intent to prevent the future onset of clinically apparent disease is, however, not well understood. Notably, therapies effective in established disease might not be necessary or sufficient to abrogate preclinical autoimmunity. For example, although TNF inhibitors are highly efficacious in the treatment active RA,137 such therapies might not be effective in preclinical RA; elevations in TNF levels have been detected in preclinical RA, but this pathway might not be crucial for disease development and thus targeting this cytokine might not provide adequate prevention of disease onset. As discussed above, additional studies and further longitudinal follow-up is needed to identify the association between elevations in IL-1α, CXCL10 and CCL2 levels and future autoimmunity; however, given that these bio markers might portent risk of future auto-immunity, perhaps targeting these pathways would prove effective in preventing disease progression in individuals with preclinical RA.75 In addition, although established and emerging data implicate IFN-α in SLE and even in ILE,45 whether this pathway is also important in the very early pathogenesis of the disease remains unclear.
Developing a sufficient understanding of ARDs to support the implementation of effective screening and prevention programmes is a daunting task; however, a complete understanding of all ARDs might not be necessary to initiate preventive measures in certain diseases. For example, a fair understanding of the role of biomarkers in predicting risk of future RA already exists. Furthermore, although the specific factors that initiate and propagate most ARDs remain unknown, the use of interventions that have broad beneficial effects on health and could also be of benefit in reducing the risk of ARDs might be a reason able approach to prevention of disease in individuals identified based on expression levels of these bio-markers or from high-risk populations. For example, smoking cessation has a variety of health benefits, and could potentially lead to reduction in the incidence of RA of up to a 35%, based on some available evidence.28 In addition, use of a pharmaco logical agent already known to be effective in clinically apparent disease might be a reason able approach to prevention of ARDs while we await a more detailed understanding of the natural history of disease, which might in fact be obtained in a prevention trial using such currently available therapies. For example, treatment of an indivi dual deemed to be at-risk of RA or SLE based on biomarker profiling with a relatively safe and well-tolerated therapy, such as hydroxychloroquine, might prevent the future onset of clinically apparent disease, similar to the effect seen in studies of palindromic rheumatism and ILE.40,131,138
Importantly, in asymptomatic individuals with risk factors for ARD, many of whom will not develop future disease, the use of approaches such as lifestyle modification, vaccinations and antimicrobial or anti- inflammatory therapies with excellent safety records, is likely to be much more acceptable than prophylactic therapy with expensive drugs that are potentially associated with greater risks. However, the use of targeted disease-modifying therapies might be considered justified in individuals with early signs or symptoms of disease and/or a high risk of future ARD according to accurate predictive instruments, although efforts are needed to clearly define this ‘high-risk’ population. Notably in this regard, an ongoing clinical trial is investigating rituximab for the prevention of future RA in individuals with no evidence of inflammatory arthritis upon physical examination but who have elevated levels of RF and ACPAs plus one or more of the following measures: increased levels of C-reactive protein or evidence of subclinical synovitis obtained using ultra-sonography or MRI.139 In this trial, which began in the Netherlands in 2009, the individuals are being treated with a single dose of either rituximab or placebo, and the primary outcome is a decrease in the number of individuals who develop classifiable RA at 4 years. The results of this trial should be become available in the next few years and will probably provide important data regarding preclinic al RA and prevention of RA.
Future directions
The greatest challenge to prevention of ARDs is obtaining sufficient knowledge of the mechanisms underlying development of disease that operate during the pre clinical period. Understanding of this stage of the natural history of these diseases is essential to enable accurate identification of autoimmunity, prediction of clinically meaningful disease and to provide a rationale for preventive interventions can be applied at an individual level using an evidence-based approach that appropriately balances risks of interventions with benefits of prevention. As we have described, retrospective analyses of stored samples, together with a growing number of prospective studies in high-risk populations, including ongoing studies in American Indian populations,83 nurses,140 and first-degree relatives of patients with RA and SLE,141–143 as well as studies of individuals who have presented to clinical care with disease that does not meet the current classification criteria (such as patients with arthralgia or ILE),17,77 have provided much important information regarding the mechanisms of SLE and RA development in the preclinical period. Similar future studies will continue to provide additional information, hopefully by taking advantage of established and emerging technologies (genetic and epigenetic testing, microbiome analyses, array-based testing, and single-cell analyses, for example) to assess the relationship between genetic and environmental factors and the development of autoimmunity and inflammation. In addition, partnering studies of ARDs with other large-scale natural history studies of CVD or cancer could enable pooling of resources to optimize the understanding of ARDs. Mechanistic studies using relevant animal models will also be important to understanding the natural history of ARDs. Nevertheless, well-designed prospective clinical studies of both the mechanisms of disease development and interventions to abrogate or halt the future develop ment of ARDs will be necessary to develop preventive strategies for ARDs that can be broadly applied. Furthermore, the analyses of the cost-effectiveness of such approaches will also be required. How such studies will be funded and implemented is an open question; however, the rheumatologic community, as well as the wider medical and general community, and funding sources including governmental agencies should be responsible for determining the value of such approaches to society as a whole. Perhaps approaches such as those used in the autoimmune disease type 1 diabetes mellitus (T1DM), which follows a similar model of development as RA and SLE with the presence of autoantibodies that are highly specific for future disease preceding clinically apparent disease onset, could be used.144 Specifically, much has been learned about the natural history of T1DM through large-scale collaborative projects, such as TrialNet,145 which involved multiple clinical and research centres worldwide that united to develop and perform natural history studies and im plement clinical prevention trials.
Conclusions
We have focused herein on RA and SLE as model ARDs with a preclinical period of development, although most if not all ARDs are likely to have a preclinical stage of disease characterized by initiation and propagation of autoimmunity. Obtaining a clearer understanding of pre-clinical ARDs, including the genetic and environmental aetiological factors, and biomarkers that characterize the early stages of pathogenesis, would facilitate the develop predictive tools that ultimately enable screening and prevention strategies. Such screening and preventative initiatives could substantially reduce the burden that these diseases place on public health. Although further studies are needed to develop effective preventive approaches for all ARDs, if increased focus is placed on these issues by the biomedical community, other important organizations (including funding agencies) and society in general, we could reveal sufficient information on some ARDs to implement preventive strategies in the near future.
Supplementary Material
Key points.
■ Preclinical autoimmune rheumatic disease (ARD) can be defined as the presence of abnormalities in immune function and responses in the absence of clinically manifest tissue injury
■ Increasing data support the existence of preclinical phase of rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE) that can be identified using biomarkers of autoimmunity and inflammation
■ RA and SLE could serve as models for understanding the mechanisms of disease development and, ultimately, prevention of other ARDs that in aggregate affect a substantial portion of the population
■ Understanding the preclinical phases of ARD might enable accurate identification of at-risk individuals and the development of preventive interventions that might modify risk factors or target immune pathways underlying disease
■ Identifying both the overall likelihood of future development of an ARD and the timing of onset of clinically apparent disease will be important for ‘personalized’ medicine and designing prevention trials
■ A range of studies focused on preclinical ARD are needed to clarify the natural history of ARD, and thus enable the development of screening programmes and early, potentially preventive, interventions
Box 1 WHO recommendations for disease screening122.
■ The disease should represent an important health problem
■ A treatment should be available for the disease
■ Facilities for diagnosis and treatment of the disorder should be available
■ A latent (preclinical) stage of the disease should be detectable
■ A test or examination for the condition (such as analysis of an autoantibody that defines a preclinical state) should exist
■ The screening test should be acceptable to the general population
■ The natural history of the disease should be adequately understood
■ An agreed policy on whom to treat is required
■ The total cost of identifying a case among the population should be economically balanced in relation to medical expenditure as a whole
■ Case-finding should be a continuous process, necessitating regular repeat testing, not just a ‘once and for all’ project
Box 2 Potential issues regarding prevention of ARDs*.
■ Understanding of the natural history of ARDs must be sufficient to enable accurate prediction of future disease in each individual and to identify therapeutic targets for prevention of disease development
■ The populations that should be included in prediction models for disease must be identified
■ Appropriate public health efforts that identify individuals at-risk of ARDs in whom disease prevention would be reasonable approach must be determined; the cost-effectiveness of preventing ARDs also need to be evaluated
■ How high the likelihood of future ARD should be before initiating preventive therapy needs to be established
■ What individuals who are at-risk of a future ARD will be willing to undergo for prevention must be considered
■ Efforts are needed to determine whether intervention is worthwhile in individuals with autoimmunity, even if they have low risk of progression to a clinically apparent ARD
■ Studies are needed to determine at what point in preclinical autoimmunity intervention is most reasonable
■ Which pharmacologic agents or other intervention that will adequately abrogate disease in its preclinical phase are appropriate to use in individuals without clinical disease remains unclear
■ Clarification is required as to what we hope to achieve with preventive interventions and adequate tools (such as symptom assessments, imaging technologies and biomarker testing) must be in place to measure such responses to a preventive intervention
*See Supplementary Table 1 online for further discussion of how these issues could be addressed. Abbreviation: ARDs, autoimmune rheumatic diseases.
Review criteria.
PubMed was searched for publications using key words that included “preclinical”, “autoantibodies”, “natural history”, and/or phrases including “before the clinical onset”, with each of these terms/phrases in turn linked to terms defining a variety of autoimmune rheumatic diseases (ARDs). No restrictions were applied with regard to year of publication or article type. The reference lists of identified articles related to preclinical ARDs were also searched for additional publications. Articles for inclusion were selected based on the authors’ opinion of their relevance to the topic.
Acknowledgements
K. D. Deane has received research support from the NIH (grant: AI103023), the American College of Rheumatology Research and Education Foundation, and the Walter S. and Lucienne Driskill Foundation. H. El-Gabalawy has received research from the Canadian Institutes of Health Research (grant: MOP7770).
Footnotes
Competing interests K. D. Deane declares that he has submitted a patent application for the use of biomarkers to predict actionable outcomes in rheumatoid arthritis. H. El-Gabalaway declares no competing interests.
Author contributions Both authors made substantial contributions all stages of the preparation of this manuscript for submission.
Supplementary information is linked to the online version of the paper at www.nature.com/nrrheum.
References
- 1.Lawrence RC, et al. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States: Part II. Arthritis Rheum. 2007;58:26–35. doi: 10.1002/art.23176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Helmick CG, et al. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States: Part I. Arthritis Rheum. 2007;58:15–25. doi: 10.1002/art.23177. [DOI] [PubMed] [Google Scholar]
- 3.Jacobson DL, Gange SJ, Rose NR, Graham NM. Epidemiology and estimated population burden of selected autoimmune diseases in the United States. Clin. Immunol. Immunopathol. 1997;84:223–243. doi: 10.1006/clin.1997.4412. [DOI] [PubMed] [Google Scholar]
- 4.Birnbaum H, et al. Societal cost of rheumatoid arthritis patients in the US. Curr. Med. Res. Opin. 2010;26:77–90. doi: 10.1185/03007990903422307. [DOI] [PubMed] [Google Scholar]
- 5.Proceedings. Isr. J. Med. Sci. Vol. 26. Tel-Hashomer, Israel: Jan 25, 1990. Fourth autoimmunity meeting—Noel Rose day. pp. 661–718. [No authors listed] 1990. [PubMed] [Google Scholar]
- 6.Aho K, Palosuo T, Heliovaara M. Predictive significance of rheumatoid factor. J. Rheumatol. 1995;22:2186–2187. [PubMed] [Google Scholar]
- 7.del Puente A, Knowler WC, Pettitt DJ, Bennett PH. The incidence of rheumatoid arthritis is predicted by rheumatoid factor titer in a longitudinal population study. Arthritis Rheum. 1988;31:1239–1244. doi: 10.1002/art.1780311004. [DOI] [PubMed] [Google Scholar]
- 8.Silman AJ, Hennessy E, Ollier B. Incidence of rheumatoid arthritis in a genetically predisposed population. Br. J. Rheumatol. 1992;31:365–368. doi: 10.1093/rheumatology/31.6.365. [DOI] [PubMed] [Google Scholar]
- 9.Rantapaa-Dahlqvist S, et al. Antibodies against cyclic citrullinated peptide and IgA rheumatoid factor predict the development of rheumatoid arthritis. Arthritis Rheum. 2003;48:2741–2749. doi: 10.1002/art.11223. [DOI] [PubMed] [Google Scholar]
- 10.Nielen MM, et al. Specific autoantibodies precede the symptoms of rheumatoid arthritis: a study of serial measurements in blood donors. Arthritis Rheum. 2004;50:380–386. doi: 10.1002/art.20018. [DOI] [PubMed] [Google Scholar]
- 11.Majka DS, et al. Duration of preclinical rheumatoid arthritis-related autoantibody positivity increases in subjects with older age at time of disease diagnosis. Ann. Rheum. Dis. 2008;67:801–807. doi: 10.1136/ard.2007.076679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arbuckle MR, et al. Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N. Engl. J. Med. 2003;349:1526–1533. doi: 10.1056/NEJMoa021933. [DOI] [PubMed] [Google Scholar]
- 13.Gerlag DM, et al. EULAR recommendations for terminology and research in individuals at risk of rheumatoid arthritis: report from the Study Group for Risk Factors for Rheumatoid Arthritis. Ann. Rheum. Dis. 2012;71:638–641. doi: 10.1136/annrheumdis-2011-200990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Asherson RA, Cervera R, Lahita RG. Latent, incomplete or lupus at all? J. Rheumatol. 1991;18:1783–1786. [PubMed] [Google Scholar]
- 15.Ball EM, Bell AL. Lupus arthritis—do we have a clinically useful classification? Rheumatology (Oxford) 2012;51:771–779. doi: 10.1093/rheumatology/ker381. [DOI] [PubMed] [Google Scholar]
- 16.Greer JM, Panush RS. Incomplete lupus erythematosus. Arch. Intern. Med. 1989;149:2473–2476. [PubMed] [Google Scholar]
- 17.Stahl Hallengren C, Nived O, Sturfelt G. Outcome of incomplete systemic lupus erythematosus after 10 years. Lupus. 2004;13:85–88. doi: 10.1191/0961203304lu477oa. [DOI] [PubMed] [Google Scholar]
- 18.Vila LM, Mayor AM, Valentin AH, Garcia-Soberal M, Vila S. Clinical outcome and predictors of disease evolution in patients with incomplete lupus erythematosus. Lupus. 2000;9:110–115. doi: 10.1191/096120300678828073. [DOI] [PubMed] [Google Scholar]
- 19.Whiting PF, et al. Systematic review: accuracy of anti-citrullinated peptide antibodies for diagnosing rheumatoid arthritis. Ann. Intern. Med. 2010;152:456–464. doi: 10.7326/0003-4819-152-7-201004060-00010. [DOI] [PubMed] [Google Scholar]
- 20.Wandstrat AE, et al. Autoantibody profiling to identify individuals at risk for systemic lupus erythematosus. J. Autoimmun. 2006;27:153–160. doi: 10.1016/j.jaut.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 21.Satoh M, et al. Prevalence and sociodemographic correlates of antinuclear antibodies in the United States. Arthritis Rheum. 2012;64:2319–2327. doi: 10.1002/art.34380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dougados M, Gossec L. Classification criteria for rheumatic diseases: why and how? Arthritis Rheum. 2007;57:1112–1115. doi: 10.1002/art.23015. [DOI] [PubMed] [Google Scholar]
- 23.Arnett FC, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988;31:315–324. doi: 10.1002/art.1780310302. [DOI] [PubMed] [Google Scholar]
- 24.Dougados M, Aletaha D, van Riel P. Disease activity measures for rheumatoid arthritis. Clin. Exp. Rheumatol. 2007;25:S22–S29. [PubMed] [Google Scholar]
- 25.Guzman J, Cardiel MH, Arce-Salinas A, Sanchez-Guerrero J, Alarcon-Segovia D. Measurement of disease activity in systemic lupus erythematosus. Prospective validation of 3 clinical indices. J. Rheumatol. 1992;19:1551–1558. [PubMed] [Google Scholar]
- 26.Peckham CS, Dezateux C. Issues underlying the evaluation of screening programmes. Br. Med. Bull. 1998;54:767–778. doi: 10.1093/oxfordjournals.bmb.a011728. [DOI] [PubMed] [Google Scholar]
- 27.Koren G, Nickel S. Sources of bias in signals of pharmaceutical safety in pregnancy. Clin. Invest. Med. 2010;33:E349–E355. doi: 10.25011/cim.v33i6.14585. [DOI] [PubMed] [Google Scholar]
- 28.Klareskog L, Gregersen PK, Huizinga TW. Prevention of autoimmune rheumatic disease: state of the art and future perspectives. Ann. Rheum. Dis. 2010;69:2062–2066. doi: 10.1136/ard.2010.142109. [DOI] [PubMed] [Google Scholar]
- 29.Verpoort KN, et al. Association of smoking with the constitution of the anti-cyclic citrullinated peptide response in the absence of HLA-DRB1 shared epitope alleles. Arthritis Rheum. 2007;56:2913–2918. doi: 10.1002/art.22845. [DOI] [PubMed] [Google Scholar]
- 30.Sugiyama D, et al. Impact of smoking as a risk factor for developing rheumatoid arthritis: a meta-analysis of observational studies. Ann. Rheum. Dis. 2010;69:70–81. doi: 10.1136/ard.2008.096487. [DOI] [PubMed] [Google Scholar]
- 31.Padyukov L, Silva C, Stolt P, Alfredsson L, Klareskog L. A gene–environment interaction between smoking and shared epitope genes in HLA-DR provides a high risk of seropositive rheumatoid arthritis. Arthritis Rheum. 2004;50:3085–3092. doi: 10.1002/art.20553. [DOI] [PubMed] [Google Scholar]
- 32.Oliver JE, Silman AJ. Risk factors for the development of rheumatoid arthritis. Scand. J. Rheumatol. 2006;35:169–174. doi: 10.1080/03009740600718080. [DOI] [PubMed] [Google Scholar]
- 33.Tuomi T, Heliovaara M, Palosuo T, Aho K. Smoking, lung function, and rheumatoid factors. Ann. Rheum. Dis. 1990;49:753–756. doi: 10.1136/ard.49.10.753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bhatia SS, et al. Rheumatoid factor seropositivity is inversely associated with oral contraceptive use in women without rheumatoid arthritis. Ann. Rheum. Dis. 2007;66:267–269. doi: 10.1136/ard.2006.060004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Keenan BT, et al. Effect of interactions of glutathione S-transferase T1, M1, P1 and HMOX1 gene promoter polymorphisms with heavy smoking on the risk of rheumatoid arthritis. Arthritis Rheum. 2010;62:3196–3210. doi: 10.1002/art.27639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.James JA, Robertson JM. Lupus and Epstein–Barr. Curr. Opin. Rheumatol. 2012;24:383–388. doi: 10.1097/BOR.0b013e3283535801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McClain MT, et al. Early events in lupus humoral autoimmunity suggest initiation through molecular mimicry. Nat. Med. 2005;11:85–89. doi: 10.1038/nm1167. [DOI] [PubMed] [Google Scholar]
- 38.James JA, et al. Systemic lupus erythematosus in adults is associated with previous Epstein–Barr virus exposure. Arthritis Rheum. 2001;44:1122–1126. doi: 10.1002/1529-0131(200105)44:5<1122::AID-ANR193>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 39.Cooper G, et al. Recent advances and opportunities in research on lupus: environmental influences and mechanisms of disease. Cien. Saude Colet. 2009;14:1865–1876. doi: 10.1590/s1413-81232009000500028. [DOI] [PubMed] [Google Scholar]
- 40.James JA, et al. Hydroxychloroquine sulfate treatment is associated with later onset of systemic lupus erythematosus. Lupus. 2007;16:401–409. doi: 10.1177/0961203307078579. [DOI] [PubMed] [Google Scholar]
- 41.McClain MT, et al. The prevalence, onset, and clinical significance of antiphospholipid antibodies prior to diagnosis of systemic lupus erythematosus. Arthritis Rheum. 2004;50:1226–1232. doi: 10.1002/art.20120. [DOI] [PubMed] [Google Scholar]
- 42.Eriksson C, et al. Autoantibodies predate the onset of systemic lupus erythematosus in northern Sweden. Arthritis Res. Ther. 2011;13:R30. doi: 10.1186/ar3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wahren-Herlenius M, Dorner T. Immunopathogenic mechanisms of systemic autoimmune disease. Lancet. 2013;382:819–831. doi: 10.1016/S0140-6736(13)60954-X. [DOI] [PubMed] [Google Scholar]
- 44.Dye JR, Ullal AJ, Pisetsky DS. The role of microparticles in the pathogenesis of rheumatoid arthritis and systemic lupus erythematosus. Scand. J. Immunol. 2013;78:140–148. doi: 10.1111/sji.12068. [DOI] [PubMed] [Google Scholar]
- 45.Li QZ, et al. Interferon signature gene expression is correlated with autoantibody profiles in patients with incomplete lupus syndromes. Clin. Exp. Immunol. 2010;159:281–291. doi: 10.1111/j.1365-2249.2009.04057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Weckerle CE, et al. Network analysis of associations between serum interferon-α activity, autoantibodies, and clinical features in systemic lupus erythematosus. Arthritis Rheum. 2011;63:1044–1053. doi: 10.1002/art.30187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tuomi T, Palosuo T, Aho K. The distribution of class-specific rheumatoid factors is similar in rheumatoid and pre-illness sera. Scand. J. Immunol. 1986;24:751–754. doi: 10.1111/j.1365-3083.1986.tb02196.x. [DOI] [PubMed] [Google Scholar]
- 48.Aho K, Tuomi T, Heliovaara M, Palosuo T. Rheumatoid factors and rheumatoid arthritis. Scand. J. Rheumatol. Suppl. 1988;74:41–44. doi: 10.3109/03009748809102938. [DOI] [PubMed] [Google Scholar]
- 49.Aho K, Palosuo T, Raunio V, Tuomi T. The timing of rheumatoid factor seroconversions. Arthritis Rheum. 1987;30:719–720. doi: 10.1002/art.1780300623. [DOI] [PubMed] [Google Scholar]
- 50.Aho K, Heliovaara M, Maatela J, Tuomi T, Palosuo T. Rheumatoid factors antedating clinical rheumatoid arthritis. J. Rheumatol. 1991;18:1282–1284. [PubMed] [Google Scholar]
- 51.Aho K, von Essen R, Kurki P, Palosuo T, Heliovaara M. Antikeratin antibody and antiperinuclear factor as markers for subclinical rheumatoid disease process. J. Rheumatol. 1993;20:1278–1281. [PubMed] [Google Scholar]
- 52.Schellekens GA, de Jong BA, van den Hoogen FH, van de Putte LB, van Venrooij WJ. Citrulline is an essential constituent of antigenic determinants recognized by rheumatoid arthritis-specific autoantibodies. J. Clin. Invest. 1998;101:273–281. doi: 10.1172/JCI1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schellekens GA, et al. The diagnostic properties of rheumatoid arthritis antibodies recognizing a cyclic citrullinated peptide. Arthritis Rheum. 2000;43:155–163. doi: 10.1002/1529-0131(200001)43:1<155::AID-ANR20>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 54.Ferucci ED, Templin DW, Lanier AP. Rheumatoid arthritis in American Indians and Alaska Natives: a review of the literature. Semin. Arthritis Rheum. 2005;34:662–667. doi: 10.1016/j.semarthrit.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 55.Del Puente A, Knowler WC, Pettitt DJ, Bennett PH. High incidence and prevalence of rheumatoid arthritis in Pima Indians. Am. J. Epidemiol. 1989;129:1170–1178. doi: 10.1093/oxfordjournals.aje.a115238. [DOI] [PubMed] [Google Scholar]
- 56.Bos WH, Nielen MM, Dijkmans BA, van Schaardenburg D. Duration of pre-rheumatoid arthritis anti-cyclic citrullinated peptide positivity is positively associated with age at seroconversion. Ann. Rheum. Dis. 2008;67:1642. doi: 10.1136/ard.2007.085456. [DOI] [PubMed] [Google Scholar]
- 57.van der Woude D, et al. Epitope spreading of the anti-citrullinated protein antibody response occurs before disease onset and is associated with the disease course of early arthritis. Ann. Rheum. Dis. 2010;69:1554–1561. doi: 10.1136/ard.2009.124537. [DOI] [PubMed] [Google Scholar]
- 58.Sokolove J, et al. Autoantibody epitope spreading in the pre-clinical phase predicts progression to rheumatoid arthritis. PLoS ONE. 2012;7:e35296. doi: 10.1371/journal.pone.0035296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brink M, et al. Multiplex analyses of antibodies against citrullinated peptides in individuals prior to development of rheumatoid arthritis. Arthritis Rheum. 2013;65:899–910. doi: 10.1002/art.37835. [DOI] [PubMed] [Google Scholar]
- 60.Verpoort KN, et al. Fine specificity of the anti-citrullinated protein antibody response is influenced by the shared epitope alleles. Arthritis Rheum. 2007;56:3949–3952. doi: 10.1002/art.23127. [DOI] [PubMed] [Google Scholar]
- 61.Mathsson L, et al. Antibodies against citrullinated vimentin in rheumatoid arthritis: higher sensitivity and extended prognostic value concerning future radiographic progression as compared with antibodies against cyclic citrullinated peptides. Arthritis Rheum. 2008;58:36–45. doi: 10.1002/art.23188. [DOI] [PubMed] [Google Scholar]
- 62.Demoruelle MK, Deane K. Antibodies to citrullinated protein antigens (ACPAs): clinical and pathophysiologic significance. Curr. Rheumatol. Rep. 2011;13:421–430. doi: 10.1007/s11926-011-0193-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ercan A, et al. Aberrant IgG galactosylation precedes disease onset, correlates with disease activity, and is prevalent in autoantibodies in rheumatoid arthritis. Arthritis Rheum. 2010;62:2239–2248. doi: 10.1002/art.27533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Suwannalai P, et al. Avidity maturation of anti-citrullinated protein antibodies in rheumatoid arthritis. Arthritis Rheum. 2012;64:1323–1328. doi: 10.1002/art.33489. [DOI] [PubMed] [Google Scholar]
- 65.Kokkonen H, et al. Antibodies of IgG, IgA and IgM isotypes against cyclic citrullinated peptide precede the development of rheumatoid arthritis. Arthritis Res. Ther. 2011;13:R13. doi: 10.1186/ar3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hill JA, et al. Arthritis induced by posttranslationally modified (citrullinated) fibrinogen in DR4-IE transgenic mice. J. Exp. Med. 2008;205:967–979. doi: 10.1084/jem.20072051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hill JA, et al. Cutting edge: the conversion of arginine to citrulline allows for a high-affinity peptide interaction with the rheumatoid arthritis-associated HLA-DRB1*0401 MHC class II molecule. J. Immunol. 2003;171:538–541. doi: 10.4049/jimmunol.171.2.538. [DOI] [PubMed] [Google Scholar]
- 68.Klareskog L, Ronnelid J, Lundberg K, Padyukov L, Alfredsson L. Immunity to citrullinated proteins in rheumatoid arthritis. Annu. Rev. Immunol. 2008;26:651–675. doi: 10.1146/annurev.immunol.26.021607.090244. [DOI] [PubMed] [Google Scholar]
- 69.van de Stadt LA, et al. The extent of the anti-citrullinated protein antibody repertoire is associated with arthritis development in patients with seropositive arthralgia. Ann. Rheum. Dis. 2011;70:128–133. doi: 10.1136/ard.2010.132662. [DOI] [PubMed] [Google Scholar]
- 70.Kokkonen H, et al. Up-regulation of cytokines and chemokines predates the onset of rheumatoid arthritis. Arthritis Rheum. 2010;62:383–391. doi: 10.1002/art.27186. [DOI] [PubMed] [Google Scholar]
- 71.Deane KD, et al. The number of elevated cytokines and chemokines in preclinical seropositive rheumatoid arthritis predicts time to diagnosis in an age-dependent manner. Arthritis Rheum. 2010;62:3161–3172. doi: 10.1002/art.27638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jorgensen KT, et al. Cytokines, autoantibodies and viral antibodies in premorbid and postdiagnostic sera from patients with rheumatoid arthritis: case–control study nested in a cohort of Norwegian blood donors. Ann. Rheum. Dis. 2008;67:860–866. doi: 10.1136/ard.2007.073825. [DOI] [PubMed] [Google Scholar]
- 73.Lubbers J, et al. The type I IFN signature as a biomarker of preclinical rheumatoid arthritis. Ann. Rheum. Dis. 2013;72:776–780. doi: 10.1136/annrheumdis-2012-202753. [DOI] [PubMed] [Google Scholar]
- 74.van Baarsen LG, et al. Gene expression profiling in autoantibody-positive patients with arthralgia predicts development of arthritis. Arthritis Rheum. 2010;62:694–704. doi: 10.1002/art.27294. [DOI] [PubMed] [Google Scholar]
- 75.El-Gabalawy HS, et al. Familial clustering of the serum cytokine profile in the relatives of rheumatoid arthritis patients. Arthritis Rheum. 2012;64:1720–1729. doi: 10.1002/art.34449. [DOI] [PubMed] [Google Scholar]
- 76.Bos WH, et al. Arthritis development in patients with arthralgia is strongly associated with anti-citrullinated protein antibody status: a prospective cohort study. Ann. Rheum. Dis. 2010;69:490–494. doi: 10.1136/ard.2008.105759. [DOI] [PubMed] [Google Scholar]
- 77.van de Stadt LA, Witte BI, Bos WH, van Schaardenburg D. A prediction rule for the development of arthritis in seropositive arthralgia patients. Ann. Rheum. Dis. 2013;72:1920–1926. doi: 10.1136/annrheumdis-2012-202127. [DOI] [PubMed] [Google Scholar]
- 78.Young KA, et al. Relatives without rheumatoid arthritis show reactivity to anti-citrullinated protein/peptide antibodies which are associated with arthritis-related traits: Studies of the Etiology of Rheumatoid Arthritis. Arthritis Rheum. 2013;65:1995–2004. doi: 10.1002/art.38022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ioan-Facsinay A, et al. Marked differences in fine specificity and isotype usage of the anti-citrullinated protein antibody in health and disease. Arthritis Rheum. 2008;58:3000–3008. doi: 10.1002/art.23763. [DOI] [PubMed] [Google Scholar]
- 80.El Gabalawy H, et al. Evolution of preclinical autoimmunity in individuals at risk for development of rheumatoid arthritis [abstract 1688]. Arthritis Rheum. 2012;64(Suppl. 10):S722. [Google Scholar]
- 81.Blanchard AK, et al. Oral health in a First Nations and a non-Aboriginal population in Manitoba. Int. J. Circumpolar Health. 2012;71:17394. doi: 10.3402/ijch.v71i0.17394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.El-Gabalawy HS, et al. Non-HLA genes modulate the risk of rheumatoid arthritis associated with HLA-DRB1 in a susceptible North American Native population. Genes Immun. 2011;12:568–574. doi: 10.1038/gene.2011.30. [DOI] [PubMed] [Google Scholar]
- 83.El-Gabalawy HS, et al. Immunogenetic risks of anti-cyclical citrullinated peptide antibodies in a North American Native population with rheumatoid arthritis and their first-degree relatives. J. Rheumatol. 2009;36:1130–1135. doi: 10.3899/jrheum.080855. [DOI] [PubMed] [Google Scholar]
- 84.Hitchon CA, et al. Antibodies to porphyromonas gingivalis are associated with anticitrullinated protein antibodies in patients with rheumatoid arthritis and their relatives. J. Rheumatol. 2010;37:1105–1112. doi: 10.3899/jrheum.091323. [DOI] [PubMed] [Google Scholar]
- 85.Oen K, et al. Familial seropositive rheumatoid arthritis in North American Native families: effects of shared epitope and cytokine genotypes. J. Rheumatol. 2005;32:983–991. [PubMed] [Google Scholar]
- 86.Oen K, et al. HLA associations of seropositive rheumatoid arthritis in a Cree and Ojibway population. J. Rheumatol. 1998;25:2319–2323. [PubMed] [Google Scholar]
- 87.Smolik I, Robinson DB, Bernstein CN, El-Gabalawy HS. First-degree relatives of patients with rheumatoid arthritis exhibit high prevalence of joint symptoms. J. Rheumatol. 2013;40:818–824. doi: 10.3899/jrheum.121016. [DOI] [PubMed] [Google Scholar]
- 88.Barra L, et al. Anti-citrullinated protein antibodies in unaffected first-degree relatives of rheumatoid arthritis patients. Arthritis Rheum. 2013;65:1439–1447. doi: 10.1002/art.37911. [DOI] [PubMed] [Google Scholar]
- 89.Kim SK, et al. Greater prevalence of seropositivity for anti-cyclic citrullinated peptide antibody in unaffected first-degree relatives in multicase rheumatoid arthritis-affected families. Korean J. Intern. Med. 2013;28:45–53. doi: 10.3904/kjim.2013.28.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Arlestig L, et al. Antibodies against cyclic citrullinated peptides of IgG, IgA and IgM isotype and rheumatoid factor of IgM and IgA isotype are increased in unaffected members of multicase rheumatoid arthritis families from northern Sweden. Ann. Rheum. Dis. 2012;71:825–829. doi: 10.1136/annrheumdis-2011-200668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kolfenbach JR, et al. Autoimmunity to peptidyl arginine deiminase type 4 precedes clinical onset of rheumatoid arthritis. Arthritis Rheum. 2010;62:2633–2639. doi: 10.1002/art.27570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ferucci ED, et al. Prevalence of anti-peptidylarginine deiminase type 4 antibodies in rheumatoid arthritis and unaffected first-degree relatives in indigenous North American populations. J. Rheumatol. 2013;40:1523–1528. doi: 10.3899/jrheum.130293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Shi J, et al. Anti-carbamylated protein antibodies (anti-CarP) are present in arthralgia patients and predict the development of rheumatoid arthritis. Arthritis Rheum. 2013;65:911–915. doi: 10.1002/art.37830. [DOI] [PubMed] [Google Scholar]
- 94.Khandpur R, et al. NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci. Transl. Med. 2013;5:178ra40. doi: 10.1126/scitranslmed.3005580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Snir O, et al. Multifunctional T cell reactivity to native and glycosylated type-II collagen in rheumatoid arthritis. Arthritis Rheum. 2012;64:2482–2488. doi: 10.1002/art.34459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Snir O, et al. Identification and functional characterization of T cells reactive to citrullinated vimentin in HLA-DRB1*0401-positive humanized mice and rheumatoid arthritis patients. Arthritis Rheum. 2011;63:2873–2883. doi: 10.1002/art.30445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nguyen CQ, Ogunniyi AO, Karabiyik A, Love JC. Single-cell analysis reveals isotype-specific autoreactive B cell repertoires in Sjögren's syndrome. PLoS ONE. 2013;8:e58127. doi: 10.1371/journal.pone.0058127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Arend WP, Firestein GS. Pre-rheumatoid arthritis: predisposition and transition to clinical synovitis. Nat. Rev. Rheumatol. 2012;8:573–586. doi: 10.1038/nrrheum.2012.134. [DOI] [PubMed] [Google Scholar]
- 99.Sokolove J, Zhao X, Chandra PE, Robinson WH. Immune complexes containing citrullinated fibrinogen costimulate macrophages via Toll-like receptor 4 and Fcγ receptor. Arthritis Rheum. 2011;63:53–62. doi: 10.1002/art.30081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Barra L, Pope J, Bessette L, Haraoui B, Bykerk V. Lack of seroconversion of rheumatoid factor and anti-cyclic citrullinated peptide in patients with early inflammatory arthritis: a systematic literature review. Rheumatology (Oxford) 2011;50:311–316. doi: 10.1093/rheumatology/keq190. [DOI] [PubMed] [Google Scholar]
- 101.van de Sande MG, et al. Different stages of rheumatoid arthritis: features of the synovium in the preclinical phase. Ann. Rheum. Dis. 2011;70:772–777. doi: 10.1136/ard.2010.139527. [DOI] [PubMed] [Google Scholar]
- 102.Rangel-Moreno J, et al. Inducible bronchus-associated lymphoid tissue (iBALT) in patients with pulmonary complications of rheumatoid arthritis. J. Clin. Invest. 2006;116:3183–3194. doi: 10.1172/JCI28756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Morris D, Inman RD. Reactive arthritis: developments and challenges in diagnosis and treatment. Curr. Rheumatol. Rep. 2012;14:390–394. doi: 10.1007/s11926-012-0280-4. [DOI] [PubMed] [Google Scholar]
- 104.Fasano A. Leaky gut and autoimmune diseases. Clin. Rev. Allergy Immunol. 2012;42:71–78. doi: 10.1007/s12016-011-8291-x. [DOI] [PubMed] [Google Scholar]
- 105.Wegner N, et al. Autoimmunity to specific citrullinated proteins gives the first clues to the etiology of rheumatoid arthritis. Immunol. Rev. 2010;233:34–54. doi: 10.1111/j.0105-2896.2009.00850.x. [DOI] [PubMed] [Google Scholar]
- 106.Scher JU, Abramson SB. Periodontal disease, Porphyromonas gingivalis, and rheumatoid arthritis: what triggers autoimmunity and clinical disease? Arthritis Res. Ther. 2013;15:122. doi: 10.1186/ar4360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Scher JU, et al. Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. Elife. 2013;2:e01202. doi: 10.7554/eLife.01202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gizinski AM, et al. Rheumatoid arthritis (RA)-specific autoantibodies in patients with interstitial lung disease and absence of clinically apparent articular RA. Clin. Rheumatol. 2009;28:611–613. doi: 10.1007/s10067-009-1128-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Demoruelle MK, et al. Airways abnormalities and rheumatoid arthritis-related autoantibodies in subjects without arthritis: early injury or initiating site of autoimmunity? Arthritis Rheum. 2012;64:1756–1761. doi: 10.1002/art.34344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Fischer A, et al. Lung disease with anti-CCP antibodies but not rheumatoid arthritis or connective tissue disease. Respir. Med. 2012;106:1040–1047. doi: 10.1016/j.rmed.2012.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Willis VC, et al. Sputum autoantibodies in patients with established rheumatoid arthritis and subjects at risk of future clinically apparent disease. Arthritis Rheum. 2013;65:2545–2554. doi: 10.1002/art.38066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Snir O, et al. Antibodies to several citrullinated antigens are enriched in the joints of rheumatoid arthritis patients. Arthritis Rheum. 2010;62:44–52. doi: 10.1002/art.25036. [DOI] [PubMed] [Google Scholar]
- 113.Kerkman PF, et al. Circulating plasmablasts/plasmacells as a source of anticitrullinated protein antibodies in patients with rheumatoid arthritis. Ann. Rheum. Dis. 2013;72:1259–1263. doi: 10.1136/annrheumdis-2012-202893. [DOI] [PubMed] [Google Scholar]
- 114.Chang A, et al. In situ B cell-mediated immune responses and tubulointerstitial inflammation in human lupus nephritis. J. Immunol. 2011;186:1849–1860. doi: 10.4049/jimmunol.1001983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Meroni PL, Raschi E, Testoni C, Tincani A, Balestrieri G. Antiphospholipid antibodies and the endothelium. Rheum. Dis. Clin. North Am. 2001;27:587–602. doi: 10.1016/s0889-857x(05)70222-2. [DOI] [PubMed] [Google Scholar]
- 116.Devreese KM. Antiphospholipid antibodies: evaluation of the thrombotic risk. Thromb. Res. 2012;130(Suppl. 1):S37–S40. doi: 10.1016/j.thromres.2012.08.270. [DOI] [PubMed] [Google Scholar]
- 117.Liang KP, et al. Autoantibodies and the risk of cardiovascular events. J. Rheumatol. 2009;36:2462–2469. doi: 10.3899/jrheum.090188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Karlson EW, et al. Association of environmental and genetic factors and gene–environment interactions with risk of developing rheumatoid arthritis. Arthritis Care Res. (Hoboken) 2013;65:1147–1156. doi: 10.1002/acr.22005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Feitsma AL, et al. Risk of progression from undifferentiated arthritis to rheumatoid arthritis: the effect of the PTPN22 1858T-allele in anti-citrullinated peptide antibody positive patients. Rheumatology (Oxford) 2007;46:1092–1095. doi: 10.1093/rheumatology/kem006. [DOI] [PubMed] [Google Scholar]
- 120.Johansson M, Arlestig L, Hallmans G, Rantapaa-Dahlqvist S. PTPN22 polymorphism and anti-cyclic citrullinated peptide antibodies in combination strongly predicts future onset of rheumatoid arthritis and has a specificity of 100% for the disease. Arthritis Res. Ther. 2006;8:R19. doi: 10.1186/ar1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Dent TH. Predicting the risk of coronary heart disease I. The use of conventional risk markers. Atherosclerosis. 2010;213:345–351. doi: 10.1016/j.atherosclerosis.2010.06.019. [DOI] [PubMed] [Google Scholar]
- 122.Wilson JM, Jungner G. Principles and practice of screening for disease. WHO Public Health Papers. 1968;34:1–163. [Google Scholar]
- 123.Hemminki K, Li X, Sundquist J, Sundquist K. Familial associations of rheumatoid arthritis with autoimmune diseases and related conditions. Arthritis Rheum. 2009;60:661–668. doi: 10.1002/art.24328. [DOI] [PubMed] [Google Scholar]
- 124.Cardenas-Roldan J, Rojas-Villarraga A, Anaya JM. How do autoimmune diseases cluster in families? A systematic review and meta-analysis. BMC Med. 2013;11:73. doi: 10.1186/1741-7015-11-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Fairweather D, Frisancho-Kiss S, Rose NR. Sex differences in autoimmune disease from a pathological perspective. Am. J. Pathol. 2008;173:600–609. doi: 10.2353/ajpath.2008.071008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Silman AJ, Pearson JE. Epidemiology and genetics of rheumatoid arthritis. Arthritis Res. 2002;4(Suppl. 3):S265–S272. doi: 10.1186/ar578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Cunningham MW. Streptococcus and rheumatic fever. Curr. Opin. Rheumatol. 2012;24:408–416. doi: 10.1097/BOR.0b013e32835461d3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Guillevin L. Infections in vasculitis. Best Pract. Res. Clin. Rheumatol. 2013;27:19–31. doi: 10.1016/j.berh.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 129.Demoruelle MK, Deane KD. Treatment strategies in early rheumatoid arthritis and prevention of rheumatoid arthritis. Curr. Rheumatol. Rep. 2012;14:472–480. doi: 10.1007/s11926-012-0275-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Katz SJ, Russell AS. Palindromic rheumatism: a pre-rheumatoid arthritis state? J. Rheumatol. 2012;39:1912–1913. doi: 10.3899/jrheum.120995. [DOI] [PubMed] [Google Scholar]
- 131.Gonzalez-Lopez L, Gamez-Nava JI, Jhangri G, Russell AS, Suarez-Almazor ME. Decreased progression to rheumatoid arthritis or other connective tissue diseases in patients with palindromic rheumatism treated with antimalarials. J. Rheumatol. 2000;27:41–46. [PubMed] [Google Scholar]
- 132.Hanonen P, Mottonen T, Oka M. Treatment of palindromic rheumatism with chloroquine. BMJ. 1987;294:1289. doi: 10.1136/bmj.294.6582.1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.van Dongen H, et al. Efficacy of methotrexate treatment in patients with probable rheumatoid arthritis: a double-blind, randomized, placebo-controlled trial. Arthritis Rheum. 2007;56:1424–1432. doi: 10.1002/art.22525. [DOI] [PubMed] [Google Scholar]
- 134.Emery P, et al. The impact of T-cell co-stimulation modulation in patients with undifferentiated inflammatory arthritis or very early rheumatoid arthritis: a clinical and imaging study of abatacept. Ann. Rheum. Dis. 2009;69:510–516. doi: 10.1136/ard.2009.119016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Verstappen SM, et al. Beneficial effects of a 3-week course of intramuscular glucocorticoid injections in patients with very early inflammatory polyarthritis: results of the STIVEA trial. Ann. Rheum. Dis. 2010;69:503–509. doi: 10.1136/ard.2009.119149. [DOI] [PubMed] [Google Scholar]
- 136.Bos WH, Dijkmans BA, Boers M, van de Stadt RJ, van Schaardenburg D. Effect of dexamethasone on autoantibody levels and arthritis development in patients with arthralgia: a randomised trial. Ann. Rheum. Dis. 2010;69:571–574. doi: 10.1136/ard.2008.105767. [DOI] [PubMed] [Google Scholar]
- 137.Agarwal SK. Biologic agents in rheumatoid arthritis: an update for managed care professionals. J. Manag. Care Pharm. 2011;17:S14–S18. doi: 10.18553/jmcp.2011.17.s9-b.S14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Abraham RR. Palindromic rheumatism: strategies to prevent evolution to rheumatoid arthritis. South Med. J. 2012;105:322. doi: 10.1097/SMJ.0b013e318257c53e. author reply 3. [DOI] [PubMed] [Google Scholar]
- 139.Nederlands Trials Register Prevention of clinically manifest rheumatoid arthritis by B cell directed therapy in the earliest phase of the disease (PRAIRI) [online] 2010 http://www.trialregister.nl/trialreg/admin/rctview.asp?TC=2442. [Google Scholar]
- 140.Karlson EW, et al. Biomarkers of inflammation and development of rheumatoid arthritis in women from two prospective cohort studies. Arthritis Rheum. 2009;60:641–652. doi: 10.1002/art.24350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kolfenbach JR, et al. A prospective approach to investigating the natural history of preclinical rheumatoid arthritis (RA) using first-degree relatives of probands with RA. Arthritis Care Res. (Hoboken) 2009;61:1735–1742. doi: 10.1002/art.24833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Rasmussen A, et al. The lupus family registry and repository. Rheumatology (Oxford) 2011;50:47–59. doi: 10.1093/rheumatology/keq302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Navarra SV, et al. Studies of Filipino patients with systemic lupus erythematosus: autoantibody profile of first-degree relatives. Lupus. 2011;20:537–543. doi: 10.1177/0961203310385164. [DOI] [PubMed] [Google Scholar]
- 144.Eisenbarth GS, et al. Insulin autoimmunity: prediction/precipitation/prevention type 1A diabetes. Autoimmun. Rev. 2002;1:139–145. doi: 10.1016/s1568-9972(02)00035-6. [DOI] [PubMed] [Google Scholar]
- 145.Skyler JS, et al. Type 1 diabetes TrialNet—an international collaborative clinical trials network. Ann. NY Acad. Sci. 2008;1150:14–24. doi: 10.1196/annals.1447.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Israeli E, et al. Anti-Saccharomyces cerevisiae and antineutrophil cytoplasmic antibodies as predictors of inflammatory bowel disease. Gut. 2005;54:1232–1236. doi: 10.1136/gut.2004.060228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.McAdoo SP, Hall A, Levy J, Salama AD, Pusey CD. Proteinase-3 antineutrophil cytoplasm antibody positivity in patients without primary systemic vasculitis. J. Clin. Rheumatol. 2012;18:336–340. doi: 10.1097/RHU.0b013e31826d2005. [DOI] [PubMed] [Google Scholar]
- 148.Olson SW, et al. Asymptomatic autoantibodies associate with future anti-glomerular basement membrane disease. J. Am. Soc. Nephrol. 2011;22:1946–1952. doi: 10.1681/ASN.2010090928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Olson SW, et al. Relation between asymptomatic proteinase 3 antibodies and future granulomatosis with polyangiitis. Clin. J. Am. Soc. Nephrol. 2013;8:1312–1318. doi: 10.2215/CJN.10411012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Suber TL, Casciola-Rosen L, Rosen A. Mechanisms of disease: autoantigens as clues to the pathogenesis of myositis. Nat. Clin. Pract. Rheumatol. 2008;4:201–209. doi: 10.1038/ncprheum0760. [DOI] [PubMed] [Google Scholar]
- 151.Gomez-Puerta JA, et al. Long-term follow-up in 128 patients with primary antiphospholipid syndrome: do they develop lupus? Medicine (Baltimore) 2005;84:225–230. doi: 10.1097/01.md.0000172074.53583.ea. [DOI] [PubMed] [Google Scholar]
- 152.Chibnik LB, Mandl LA, Costenbader KH, Schur PH, Karlson EW. Comparison of threshold cutpoints and continuous measures of anti-cyclic citrullinated peptide antibodies in predicting future rheumatoid arthritis. J. Rheumatol. 2009;36:706–711. doi: 10.3899/jrheum.080895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Iijima T, et al. Prediction of postpartum onset of rheumatoid arthritis. Ann. Rheum. Dis. 1998;57:460–463. doi: 10.1136/ard.57.8.460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Olson SW, et al. Elevated subclinical double-stranded DNA antibodies and future proliferative lupus nephritis. Clin. J. Am. Soc. Nephrol. 2013;8:1702–1708. doi: 10.2215/CJN.01910213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Waltuck J, Buyon JP. Autoantibody-associated congenital heart block: outcome in mothers and children. Ann. Intern. Med. 1994;120:544–551. doi: 10.7326/0003-4819-120-7-199404010-00003. [DOI] [PubMed] [Google Scholar]
- 156.McCune AB, Weston WL, Lee LA. Maternal and fetal outcome in neonatal lupus erythematosus. Ann. Intern. Med. 1987;106:518–523. doi: 10.7326/0003-4819-106-4-518. [DOI] [PubMed] [Google Scholar]
- 157.Johnston SD, Watson RG, McMillan SA, Evans AE, Love AH. Serological markers for coeliac disease: changes with time and relationship to enteropathy. Eur. J. Gastroenterol. Hepat. 1998;10:259–264. doi: 10.1097/00042737-199803000-00013. [DOI] [PubMed] [Google Scholar]
- 158.Vanderpump MP, et al. The incidence of thyroid disorders in the community: a twenty-year follow-up of the Whickham Survey. Clin. Endocrinol. 1995;43:55–68. doi: 10.1111/j.1365-2265.1995.tb01894.x. [DOI] [PubMed] [Google Scholar]
- 159.Huber G, et al. Prospective study of the spontaneous course of subclinical hypothyroidism: prognostic value of thyrotropin, thyroid reserve, and thyroid antibodies. J. Clin. Endocrinol. Metab. 2002;87:3221–3226. doi: 10.1210/jcem.87.7.8678. [DOI] [PubMed] [Google Scholar]
- 160.Loviselli A, et al. The Sardinian Autoimmunity Study: 3. Studies on circulating antithyroid antibodies in Sardinian schoolchildren: relationship to goiter prevalence and thyroid function. Thyroid. 2001;11:849–857. doi: 10.1089/105072501316973109. [DOI] [PubMed] [Google Scholar]
- 161.Lazarus JH, Kokandi A. Thyroid disease in relation to pregnancy: a decade of change. Clin. Endocrinol. 2000;53:265–278. doi: 10.1046/j.1365-2265.2000.01087.x. [DOI] [PubMed] [Google Scholar]
- 162.Csepregi A, Szodoray P, Zeher M. Do autoantibodies predict autoimmune liver disease in primary Sjögren's syndrome? Data of 180 patients upon a 5 year follow-up. Scand. J. Immunol. 2002;56:623–629. doi: 10.1046/j.1365-3083.2002.01165.x. [DOI] [PubMed] [Google Scholar]
- 163.Kisand KE, et al. The follow-up of asymptomatic persons with antibodies to pyruvate dehydrogenase in adult population samples. J. Gastroenterol. 2001;36:248–254. doi: 10.1007/s005350170111. [DOI] [PubMed] [Google Scholar]
- 164.Marzotti S, Falorni A. Addison's disease. Autoimmunity. 2004;37:333–336. doi: 10.1080/08916930410001705466. [DOI] [PubMed] [Google Scholar]
- 165.Husebye E, Løvås K. Pathogenesis of primary adrenal insufficiency. Best Pract. Res. Clin. Endocrinol. Metab. 2009;23:147–157. doi: 10.1016/j.beem.2008.09.004. [DOI] [PubMed] [Google Scholar]
- 166.Gregersen PK, Silver J, Winchester RJ. The shared epitope hypothesis. An approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum. 1987;30:1205–1213. doi: 10.1002/art.1780301102. [DOI] [PubMed] [Google Scholar]
- 167.Rullo OJ, Tsao BP. Recent insights into the genetic basis of systemic lupus erythematosus. Ann. Rheum. Dis. 2013;72(Suppl. 2):ii56–ii61. doi: 10.1136/annrheumdis-2012-202351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Zikherman J, Weiss A. Unraveling the functional implications of GWAS: how T cell protein tyrosine phosphatase drives autoimmune disease. J. Clin. Invest. 2011;121:4618–4621. doi: 10.1172/JCI60001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Moulds JM, et al. Genetics of the complement system and rheumatic diseases. Rheum. Dis. Clin. North Am. 1992;18:893–914. [PubMed] [Google Scholar]
- 170.Cui Y, Sheng Y, Zhang X. Genetic susceptibility to SLE: recent progress from GWAS. J. Autoimmun. 2013;41:25–33. doi: 10.1016/j.jaut.2013.01.008. [DOI] [PubMed] [Google Scholar]
- 171.Nakano K, Whitaker JW, Boyle DL, Wang W, Firestein GS. DNA methylome signature in rheumatoid arthritis. Ann. Rheum. Dis. 2013;72:110–117. doi: 10.1136/annrheumdis-2012-201526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.De Santis M, Selmi C. The therapeutic potential of epigenetics in autoimmune diseases. Clin. Rev. Allergy Immunol. 2012;42:92–101. doi: 10.1007/s12016-011-8293-8. [DOI] [PubMed] [Google Scholar]
- 173.Aho K, Heliovaara M. Risk factors for rheumatoid arthritis. Ann. Med. 2004;36:242–251. doi: 10.1080/07853890410026025. [DOI] [PubMed] [Google Scholar]
- 174.Karlson EW, Deane K. Environmental and gene–environment interactions and risk of rheumatoid arthritis. Rheum. Dis. Clin. North Am. 2012;38:405–426. doi: 10.1016/j.rdc.2012.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.O'Neill S, Cervera R. Systemic lupus erythematosus. Best Pract. Res. Clin. Rheumatol. 2010;24:841–855. doi: 10.1016/j.berh.2010.10.006. [DOI] [PubMed] [Google Scholar]
- 176.Cooper GS, et al. Occupational risk factors for the development of systemic lupus erythematosus. J. Rheumatol. 2004;31:1928–1933. [PubMed] [Google Scholar]
- 177.Klareskog L, Padyukov L, Alfredsson L. Smoking as a trigger for inflammatory rheumatic diseases. Curr. Opin. Rheumatol. 2007;19:49–54. doi: 10.1097/BOR.0b013e32801127c8. [DOI] [PubMed] [Google Scholar]
- 178.Reveille JD. Genetics of spondyloarthritis—beyond the MHC. Nat. Rev. Rheumatol. 2012;8:296–304. doi: 10.1038/nrrheum.2012.41. [DOI] [PubMed] [Google Scholar]
- 179.Dooley MA, Hogan SL. Environmental epidemiology and risk factors for autoimmune disease. Curr. Opin. Rheumatol. 2003;15:99–103. doi: 10.1097/00002281-200303000-00002. [DOI] [PubMed] [Google Scholar]
- 180.Lu B, et al. Alcohol consumption and markers of inflammation in women with pre-clinical rheumatoid arthritis. Arthritis Rheum. 2010;62:3554–3559. doi: 10.1002/art.27739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Kallberg H, et al. Alcohol consumption is associated with decreased risk of rheumatoid arthritis: results from two Scandinavian case–control studies. Ann. Rheum. Dis. 2009;68:222–227. doi: 10.1136/ard.2007.086314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Hughes T, et al. Analysis of autosomal genes reveals gene–sex interactions and higher total genetic risk in men with systemic lupus erythematosus. Ann. Rheum. Dis. 2012;71:694–699. doi: 10.1136/annrheumdis-2011-200385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Patterson R, Germolec D. Review article toxic oil syndrome: review of immune aspects of the disease. J. Immunotoxicol. 2005;2:51–58. doi: 10.1080/15476910590960143. [DOI] [PubMed] [Google Scholar]
- 184.Rampton DS. The influence of stress on the development and severity of immune-mediated diseases. J. Rheumatol. Suppl. 2011;88:43–47. doi: 10.3899/jrheum.110904. [DOI] [PubMed] [Google Scholar]
- 185.Bonaci-Nikolic B, Nikolic MM, Andrejevic S, Zoric S, Bukilica M. Antineutrophil cytoplasmic antibody (ANCA)-associated autoimmune diseases induced by antithyroid drugs: comparison with idiopathic ANCA vasculitides. Arthritis Res. Ther. 2005;7:R1072–R1081. doi: 10.1186/ar1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Cooper GS, et al. Differences by race, sex and age in the clinical and immunologic features of recently diagnosed systemic lupus erythematosus patients in the southeastern United States. Lupus. 2002;11:161–167. doi: 10.1191/0961203302lu161oa. [DOI] [PubMed] [Google Scholar]
- 187.Korpilahde T, et al. Smoking history and serum cotinine and thiocyanate concentrations as determinants of rheumatoid factor in non-rheumatoid subjects. Rheumatology (Oxford) 2004;43:1424–1428. doi: 10.1093/rheumatology/keh365. [DOI] [PubMed] [Google Scholar]
- 188.Harley JB, James JA. Epstein–Barr virus infection induces lupus autoimmunity. Bull. NYU Hosp. Jt Dis. 2006;64:45–50. [PubMed] [Google Scholar]
- 189.Mikuls TR, et al. Porphyromonas gingivalis and disease-related autoantibodies in individuals at increased risk of rheumatoid arthritis. Arthritis Rheum. 2012;64:3522–3530. doi: 10.1002/art.34595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Karlson EW, et al. Gene–environment interaction between HLA-DRB1 shared epitope and heavy cigarette smoking in predicting incident rheumatoid arthritis. Ann. Rheum. Dis. 2010;69:54–60. doi: 10.1136/ard.2008.102962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Deane KD, et al. The number of elevated cytokines/chemokines in pre-clinical seropositive rheumatoid arthritis predicts time to diagnosis in an age-dependent manner. Arthritis Rheum. 2010;62:3161–3172. doi: 10.1002/art.27638. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.