

Abstract
Mimetics of conformational protein epitopes have broad applications, but have been difficult to identify using conventional peptide phage display. The tenth type III domain of human fibronectin (FNfn10) has two extended, randomizable surface-exposed loops and might be more amenable to the identification of such mimetics. We therefore selected a library of FNfn10 clones, randomized in both loops (15 residues in all), for binding to monoclonal antibodies (MAb) that recognize the HIV-1 envelope glycoprotein. Anti-idiotypic monobodies (αIMs) mimicking both “linear” epitopes (2F5 and 4E10 MAbs) and conformational epitopes (b12 and VRC01 MAbs) were generated. αIMs selected against 2F5 and 4E10 frequently displayed sequence homology to the corresponding linear native epitopes. In the case of b12 and VRC01, we expected that the two constrained loop domains of FNfn10 would both contribute to complex conformational interactions with target antibodies. However, mutagenesis studies revealed differences from this simple model An αIM selected against b12 was found to bind its cognate antibody via only a few residues within the BC loop of FNfn10, with minimal contribution from the FG loop. Unexpectedly, this was sufficient to generate a protein that engaged its cognate antibody in a manner very similar to HIV-1 Env, and with a strong KD (43 nM). In contrast, an αIM selected against VRC01 engaged its cognate antibody in a manner that was dependent on both BC and FG loop sequences. Overall, these data suggest that the FNfn10 scaffold can be used to identify complex structures that mimic conformational protein epitopes.
Keywords: Anti-Idiotype, HIV-1, Phage Display, Monobody, Fibronectin
Anti-idiotype antibodies provide powerful tools for studies of molecular mimicry and epitope topology [1, 2], as well as for biotechnology applications such as the specific detection and quantitation of therapeutic proteins in clinical settings [3, 4]. The large physical size of antibody molecules, combined with constraints on their structure and diversity, has led to an interest in alternative approaches to developing anti-idiotypic reagents.
Conventional peptide phage display technology involving the display of short linear peptides on the surface of filamentous phage Scott and Smith [5] has been successfully used to derive peptide mimics of linear virus-neutralizing antibody epitopes [6, 7]. However, this approach has been less successful in developing mimics of non-linear, conformational structures [8] – including mimics of the well-characterized HIV-1 broadly neutralizing antibody (bNAb), b12 [9, 10]. Constraining linear peptide sequences by flanking them with cysteine residues to generate a loop structure or embedding a loop structure within a stable scaffold has been used to improve the affinity of the peptides to their target antibody, presumably by reducing peptide flexibility reducing peptide flexibility [11–13]. This suggests that a protein scaffold with several randomizable loop domains might be proficient at making complex, conformationally-dependent, interactions with target antibodies – and thereby facilitate the identification of conformational epitope mimetics.
To test this hypothesis, we performed a series of proof-of-concept experiments, using an alternative display scaffold based on the tenth fibronectin type III domain (FNfn10) of human fibronectin. FNfn10 is a small β-sandwich protein domain with a folding pattern similar to immunoglobulin [14, 15] (Figure 1A); it has also been shown to exhibit a high level of physical stability and the ability to permit selection of ligand-binding proteins with nanomolar affinity [15–17]. The FNfn10 scaffold offers potential advantages over both the conventional, linear peptide display and antibody platforms. First, FNfn10 contains three adjacent structural loops (BC, DE, FG) that can be diversified either alone or in combination – thereby creating the potential for the generation of both linear and discontinuous structures on the scaffold surface. Second, the suggested preference for monobodies to bind to residues found in protein-protein interacting surfaces coupled with their smaller size and more efficient expression would offer significant advantages relative to antibody-based reagents [15, 17].
Fig 1.

FN structure and construction of randomized libraries. (A.) Structural representation of the fibronectin scaffold. β strands A–G of the FNfn10 molecular scaffold are shown (yellow arrows), along with the 3 adjacent surface exposed loops (BC, FG and DE; turquoise). The structure was generated using CN3D, from the source data file MMDB IB 57520. (B.) Table of the randomized molecular libraries used in screening experiments.
In the present work, we randomized the BC and FG loops of FNfn10 creating large peptide display libraries from which we selected anti-idiotypic monobodies (αIMs) capable of binding to a panel of well-characterized HIV-1 monoclonal antibodies (MAb) including 4E10, 2F5, Z13e1, 447-52D, VRC-01, and b12. In this manner, we generated αIMs that mimicked both “linear” MAb epitopes (2F5, 4E10, Z13e1, and 447-52D) and conformational MAb epitopes (b12 and VRC-01). αIMs that mimicked the linear MAb epitopes frequently demonstrated sequence homology to the corresponding MAb epitopes within HIV-1 Env. In contrast, and as expected, αIMs that mimicked the conformational b12 and VRC-01 epitopes did not show sequence homology with HIV-1 Env. However, these αIMs specific for b12 were shown to engage the same contact residues that mediate binding of the b12 antibody to HIV-1 Env. This is in striking contrast to the previously described B2.1 peptide, a b12-binding peptide derived by conventional peptide display technology, which bound to the b12 antibody in a manner quite different from that employed by HIV-1 Env [9, 10, 18]. Additionally, αIMs that mimicked both discontinuous (b12) and “linear” (4E10 and 2F5) antibody epitopes were found to efficiently compete with native epitopes for binding to their cognate MAb.
Mutagenesis studies of two αIMs selected against conformation-dependent MAbs revealed distinct modes of interaction between αIMs and their cognate antibodies. In one case, binding was mediated by a small number of residues within the BC loop of FNfn10, with minimal contribution from the FG loop. In the other case, binding of the αIM to its cognate antibody was dependent on both BC and FG loop sequences. Overall, these data suggest that the FNfn10 scaffold can be used to facilitate the discovery of complex structures that mimic conformational protein epitopes.
Experimental Procedure
Generation of the Monobody Library
The FNfn10 scaffold was cloned into pAP-III6FL, a derivative of pAP-III6 containing a full-length gene III of M13, downstream of a FLAG epitope sequence. This plasmid was introduced into CJ236, an E. coli strain which incorporates deoxyuracil rather than thymine, and single-stranded DNA (ssDNA) containing uracil was produced by infection with VCS M13 helper phage (Agilent); ssDNA from the phagemid particles was then purified using a QiaPrep Spin M13 kit (Qiagen). A derivative containing Ase I restriction sites and in-frame stop codons (ATTAAT) within the BC and FG loops was generated by site-directed mutagenesis [19] and confirmed by sequence analysis. For NHK library construction, the template was annealed to mutagenic oligonucleotides targeting the BC (aa 23–29) and FG (aa 77–84) loops in large-scale reactions as described [20]. The library oligonucleotides (BC: 5’-CGTGATACGGTAATAACGMDNMDNMDNMDNMDNMDNMDNCCAGCTGATCAGCAGG CT and FG: 5’-AATCGAGATTGGCTTGGAMDNMDNMDNMDNMDNMDNMDNMDNAGTAACAGCATAT ACAGTGATGGT) partially randomized seven positions in the BC loop and eight positions in the FG loop using NHK (N=A+C+G+T; H=A+C+T; K=G+T) codons. All amino acids except Arg, Trp, Cys and Gly are encoded by using the NHK scheme. The template mix was electroporated into TG-1 cells (Agilent) and ~5×108 ampicillin resistant transformants were obtained. Colony PCR followed by restriction digestion of the FNfn10 insert with Ase I revealed that about 40% of the colonies obtained had incorporated both oligonucleotides, yielding a functional library size of about 2×108. The remaining unmutated or singly mutated clones will not display a monobody upon infection with helper phage, and therefore are eliminated during the panning process.
The colonies were resuspended by scraping the plates with LB broth and an aliquot containing >10 times the number of transformants in the library was subcultured and grown in LB medium at 37°C for two hours. Ten mLs of this culture were infected with either VCS M13 helper phage (for monovalent display) for two hours and diluted into 200 mL of LB containing ampicillin (100 µg/mL) and kanamycin (70 µg/mL). The culture was grown overnight at 30°C and phage from the supernatant was harvested by precipitation with polyethylene glycol, resuspended in 50 mM Tris-HCl, pH7.5, 150 mM NaCl (TBS) containing 0.5% casein and 15% glycerol, and frozen in aliquots at −80°C.
Libraries generated using triphosphoramidites (Glen Research, 19 codon mix, no cysteine) were prepared using a derivative of pAP-III6 containing the Fnfn10 scaffold with the phoA promoter and FNfn10- truncated gene III fusion protein region inverted to generate ssDNA complementary to the sense strand trimer-containing oligonucleotides (Tri BC: 5’-CTGATCAGCTGG(Tri)7CGTTATTACCGT and Tri-FG: 5’-TACGCTGTTACT(Tri)8TCCAAGCCAATC) where Tri indicates the 19 codon mix. The trimer library oligonucleotides were obtained from the W.M. Keck Oligonucleotide Synthesis Facility at Yale University. Large-scale mutagenesis and library generation were as described above. A total of ~1×109 doubly mutated clones were obtained from 5 large scale electroporations.
Library Selection on MAbs
Target MAb (b12, 447-52D, Z13e1, 4E10, 2F5, VRC-01 were obtained from the AIDS Reagent Repository [21–36]; Rtx (Rituxan; specific for the B cell surface protein, CD20; [37]), 1F1 (specific for Dengue virus; kindly provided by Dr. Jacob J. Schlesinger, University of Rochester; [38]) and F10 (specific for influenza virus hemagglutinin; [39]), were used as irrelevant controls. Note that the final F10 antibody used for our FN selections corresponded to a pairing of the heavy chain from the F10 antibody [39], with an arbitrary λ chain - and is therefore designated as F10λ
Antibodies were immobilized in microtiter dish wells at 50 µg/mL in TBS overnight, blocked with TBS containing 0.5% casein. Aliquots of the libraries diluted 1:1 with TBS + casein were added to two wells (50 µL/well) and the plate was shaken for 2 hours at room temperature. Phage were removed and the wells were washed with TBS+0.5% Tween 20 seven times over 20 minutes followed by one wash with water. Bound phage were eluted with 0.1 M glycine HCl, pH 2 + 0.1% bovine serum albumin for 15 minutes. The eluate was removed, neutralized with Tris base, transduced into mid-log TG-1 cells and plated on LB plates containing ampicillin. The next day, colonies were scraped from plates in 5 mL of LB and subcultured into fresh medium for production of the next round of phage. One mL aliquots of mid-log culture were infected with VCS M1 3 helper and grown as described above. Second and third rounds of phage were prepared from 30 mL cultures as described above and 50 mL aliquots were applied to single wells coated with the target MAb. Individual clones after the second or third round of enrichment were tested by phage ELISA to confirm binding to the target MAb and lack of reactivity to an irrelevant myeloma IgG1 protein. The FNfn10 inserts of positive clones were PCR amplified using flanking vector primers and sequenced.
Production and Purification of αIMs
Selected FNfn10 variants were sub-cloned into a pET22 vector (Novagen) by introducing a methionine codon in front of the FLAG epitope as part of an NdeI restriction enzyme site. The carboxy -terminus of the FNfn10 was modified to add a birA [13] site and the modified FNfn10 was cloned between the NdeI and XhoI sites of the vector, adding a His6 purification tag immediately following the birA site. Cultures were grown in LB to mid-exponential phase and protein expression was induced for 2 hours by addition of 1 mM IPTG and 50 µM biotin. Cells were harvested and lysed with Bugbuster® reagent (Novagen) and purified by nickel affinity chromatography using Ni2+ magnetic beads and a Thermo Kingfisher instrument. Proteins were eluted from the beads with PBS containing 250 mM imidazole and stored at 4°C. The purity of the proteins was determined by SDS-PAGE and Simply Blue staining (Invitrogen). For some experiments, the protein was dialyzed into PBS before use. We verified the functionality of the biotinylated αIMs by comparing the titer of the proteins after capture on streptavidin-coated wells using both anti-Flag (Sigma) and the cognate antibody to the αIM.
Generation of b12 MAb Mutants
The b12 MAb and alanine substitution mutants were generated by cloning synthetic fragments encoding the variable regions of b12 (GeneArt) into vectors for mammalian cell expression described previously [40]. The wild type variable heavy chain (VH) and Y53A and Y98A mutants were directly cloned into the heavy chain expression vector from the synthetic genes. The W100A mutant was generated from a single chain antibody fragment variable (scFv) construct by site-directed mutagenesis as described above and the mutant VH region was PCR amplified and cloned into the mammalian expression vector. Each VH variant along with the wild type b12 VH was cotransfected with the b12 variable light chain (VL) plasmid into HEK293 cells using FuGene (Roche) and supernatant was harvested three and six days post-transfection. Antibodies were purified from culture supernatant using protein G magnetic beads (Thermo) and processed on a Thermo Kingfisher instrument. Antibodies were eluted from the beads in 0.1 M glycine HCl, pH 3, neutralized with Tris base and dialyzed into PBS.
Production of the b12 Fab was accomplished by modifying the heavy chain expression vector by deleting the hinge, CH2 and CH3 domains and replacing them with a hexa-histidine tag. Five days post-transfection the cell culture supernatants were collected and centrifuged at 3500×g to remove cell debris. Fab protein was purified by metal affinity chromatography using Ni-NTA resin (Qiagen). Columns were washed with increasing concentrations of imidazole (20mM and 40mM) followed by elution in the presence of 250mM imidazole. Fractions containing purified protein were pooled, dialyzed against PBS.
Alanine Scanning Mutagenesis
A representative b12 MAb-binding αIM, b12 3–5 αIM, was chosen for further characterization. Single alanine mutations at all positions of the b12 3–5 αIM BC and FG loops (except for position 26 which is Ala in wild type b12 3–5 αIM) were generated using site-directed mutagenesis of the b12 3–5 αIM sequence as described above. Individual biotinylated variant proteins were prepared from pET clones as described and immobilized on streptavidin-coated wells using a saturating amount of bio-αIM. A non-saturating dilution of the b12 MAb was prepared and added to all wells in an ELISA assay. After incubation with anti-human IgG-HRP conjugate, bound antibody was detected after incubation with TMB substrate. In addition to alanine substitution mutants, two hybrid FNfn10 proteins were also evaluated. Each retained the b12 3–5 αIM BC loop sequence, but we exchanged the FG loop for those found in the Rtx and the 1F1 αIMs. These hybrid clones were generated by swapping the C-terminal half of the genes using the unique EcoRI restriction site present between the BC and FG loops of the scaffold. For confirmation of specificity of the b12 MAb against our b12 αIM, an ELISA (described below) was performed after passive coating wells with b12 3–5 αIM, 4E10 αIM, 1F1 αIM, and RTX αIM. b12 MAb was added at 150 µg/mL followed by goat anti-human IgG conjugated to HRP at 1:1000. After washing, HRP substrate (TMB) was added and color was allowed to develop and the plate was photographed. The reaction was stopped by the addition of 50 µL of 1 M phosphoric acid and the optical density was read at 420 nm.
Generation of VRC-01 and 447-52D Mutants
Loop swap hybrid αIMs of VRC-01 3-2, VRC-01 3–11, VRC-01 3–13, 447-52D J2, and 447-52D J3 were generated as described above. Hybrids either retained the original wild-type BC or FG loop, while the reciprocal loop was swapped for an all serine substitute. The BC loop hybrids, FG loop hybrids, and parental αIMs were analyzed by phage ELISA (described below).
The VRC-01 3-2 αIM was mutated by a variant of combinatorial alanine scanning mutagenesis [41] whereby all non-serine residues within the BC and FG loops were restricted to either the wild type amino acid or serine, with the exception of positions encoding Met, His or Arg where it was necessary to have 4 amino acids represented. The oligonucleotides used to generate the serine scan library were: BC: 5’-CTGCTGATCAGCTGGTYGCGMRTKCGYCTAKSKCTCGTTATTACCGTATC and FG: 5’- GTATACGCTGTTACTYCGTYTTMTAGTTYTYCTMRTAKSTCCAAGCCAATCTCG, where K = G or T, M = A or C, R = A or G, Y = C or T, and S = C or G. The annealed and extended template mixture was electroporated into TG1 and ~1×108 transformants were obtained. After infection with helper phage, the displayed phage library was panned against anti-FLAG or VRC-01 MAb.
ELISA Analysis
Phage ELISA: Phage were prepared by PEG precipitation from 1.2 mL cultures using VCS M13 helper, re-suspended in 0.3 mL in TBS+0.5% casein and 50 µL was added to the wells. After 1 hour incubation at room temperature, the wells were washed 10 times and a 1:2000 dilution of anti-M13-HRP conjugate was added for 1 hour incubation. After washing, HRP substrate (TMB) was added and color was allowed to develop and the plate was photographed. The reactions were stopped by addition of 50 µL of 1 M phosphoric acid and the optical density was read at 420 nm.
Antibody ELISA: Purified αIMs were diluted in TBS+casein to ~1 µg/mL and captured on streptavidin coated wells. After washing with TBS, serial half-log dilutions of selected MAbs were added to each well and incubated for one hour at room temperature. Addition of secondary goatanti-human IgG-HRP antibody (KPL, Gaithersburg MD) followed by HRP substrate (TMB) as described above.
b12 Competition ELISA: Purified, biotinylated b12 3–5 αIM was bound to streptavidin coated wells and washed with PBS + 0.05% Tween (PBS-T). Serial 2-fold dilutions of oligomeric YU2gp140 [42] or YU2gp140 D368R mutant, from 0.4 to 25 µg/mL, were preincubated with a fixed amount of IgG1 b12 MAb (1 µg/mL) at 37°C for 30 min, before being added to b12 3–5 αIM coated wells for 2 hours at room temperature. After washes with PBS-T, a secondary antihuman-IgG-HRP antibody was added at 1:5000 dilution for 1 hour at room temperature. Following five washes, the ELISAs were developed with 100 µL of TMB substrate. The reactions were stopped by adding 100 µL 1 M sulfuric acid to each well and the optical density at 450 µnm was read. Each condition was repeated in duplicate.
2F5 and 4E10 Competition ELISA: Peptides corresponding to the 2F5 and 4E10 epitopes from HIV-1 gp41 were synthesized and conjugated to BSA (2F5: CNEQELLELDKWASLWSGGRGGL and 4E10: CSLWNWFDITNWLWRRK) [25–28]. The BSA-conjugated peptides were then coated at 5 µg/mL overnight onto polystyrene wells. A serial half log titration of the respective MAb was added sequentially to each well and developed as described in the Antibody ELISA section. A titration curve was created to determine the optimal concentration of MAb for the competition ELISA. 2F5/4E10 peptide conjugated to BSA was coated overnight at 5µg/mL in polystyrene wells. After washing five times with TBS, the plates were blocked with Blocker® TM for two hours at room temperature. 2F5 MAb (50 ng/mL) was pre-incubated with half log dilutions of 2F5 αIM, MPER peptide, or F10Aλ αIM protein. 4E10 MAb (15.8 ng/mL) was pre-incubated with half log dilutions of 4E10 5–2 αIM, 4E10 5–5 αIM, MPER peptide, or F10λ αIM protein. Alternatively, a 1:400 dilution of sera from either an HIV-1 infected person (subject 026) or a HIV-1 negative person (subject 1609) was pre-incubated with half log dilutions of 4E10 5–2 αIM, 4E10 5–5 αIM, MPER peptide, or F10Aλ αIM before addition to the coated plates. After extensive washing with TBS, MAb bound to the αIMs or peptide was detected with goat anti-human IgG-HRP conjugate at a 1:2000 dilution. HRP substrate (TMB) was used for the development of the assay and the reactions were stopped by addition of 50 µL of 1 M phosphoric acid to each well after an image was acquired. At this point, the optical density was read at 420 nm.
Detection of Rituximab in pooled normal sera: Biotinylated Rtx αIM or b12 3–5 αIM was coupled to neutravidin microtiter wells at 1 µg/mL in triplicate. Rituximab was serially diluted in half log increments in blocking solution containing 1% pooled normal sera, starting at a concentration of 10 µg/mL. After extensive washing, goat anti-human IgG conjugated to HRP was added at a 1:1000 dilution. HRP substrate (TMB) was added and color was allowed to develop.The wells were photographed and the reaction was stopped by the addition of 50 µL of 1 M phosphoric acid to each well. The optical density was recorded at 420 nm.
Collection of Human Serum Samples
Peripheral blood samples were obtained from HIV-1 infected persons at the University of Rochester Medical Center and the University of Washington HIV clinics between 2004 and 2010. Samples from healthy control subjects were obtained at the University of Rochester. All subjects provided signed written informed consent. All procedures and methods were approved by the University of Rochester Research Subjects Review Board and the University of Washington Institutional Review Board.
SPR Kinetic Binding Analysis
The binding affinity of b12 3–5 αIM protein to the b12 Fab was determined by surface plasmon resonance using a Reichert SR7500DC SPR instrument. Streptavidin was immobilized via standard amine coupling onto a planar mixed self assembled monolayer consisting of 10% carboxyl groups in a polyethylene glycol background. The surface was first activated with a mix of 0.04 mg/mL 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimide HCl (EDC) (Pierce) and 0.01 mg/mL N-hydroxysuccinimide (NHS) (Sigma)) to form NHS esters on the surface. Streptavidin at 200 µg/mL in 10 mM sodium acetate was injected over both the sample and reference channels. 2000 micro refractive index units of streptavidin were immobilized. The surface was then blocked by injecting 1 M Ethanolamine HCl pH 8.5 for 10 minutes. 470 micro refractive index units of biotinylated αIM was then captured on the sample channel only. Association and dissociation data were collected by injecting dilutions of the Fab (in PBS, 0.05% Tween 20) over the sample and reference channels at 50 µl/min in duplicate. Data overlays were performed using Scrubber 2.0a (Biologic Software Pty., Australia). The data were double referenced using the reference channel along with buffer blank injections. Overlays were exported to a SPR data analysis program [43] that used Bayesian analysis for determination of binding constants. The association phase of each sample injection was fit to a single exponential growth equation using GraphPad Prism (GraphPad Software Incorporated) to determine the equilibrium binding level. The calculated equilibrium for each injection was plotted versus concentration and fit to a Langmuir binding isotherm (Response = Equilibrium Response * ((KA*[ b12 Fab])/(KA*[ b12 Fab]+1)) to determine the equilibrium binding constants.
Results
Generation of Anti-Idiotypic Monobodies
We generated two distinct libraries of randomized BC and FG loops displayed on the FNfn10 scaffold (Figure 1). These libraries were then selected against a variety of target monoclonal antibodies (MAb), including several with broadly neutralizing activity against HIV-1, one murine MAb and a murine-derived therapeutic MAb. Selections against the MAbs were generally completed after three iterative cycles of panning, and the majority of the selected population consisted of clones that were positive for MAb binding by phage ELISA (Table 1). We found no αIMs that cross-reacted with other MAbs, as might have been expected since all of the human MAb targets were of the IgG1 isotype. However this result is consistent with the observations of Koide and co-workers [44] who have suggested that the FNfn10 monobodies have a predilection for binding to surfaces involved in protein-protein interactions (such as antibody combining sites).
Table 1. Summary of Anti-Idiotypic Monobody Selections on Monoclonal Antibodies.
All enrichments were performed for 3 rounds on the indicated target MAb (coated at 50 µg/mL in microtiter plate wells) except for the 447-52D enrichment, which was stopped after 2 rounds when high level enrichment was already evident.
| Target mAb | Library1 | Rounds | ELISA positive |
Number Unique Sequences |
|---|---|---|---|---|
| b12 | NHK, Tri | 3 | 20/23 | 11 |
| 4E10 | Tri | 3 | 33/34 | 9 |
| 1F1 | Comb NHK + Tri | 3 | 6/6 | 3 |
| 2F5 | Comb NHK + Tri | 4 | 8/24 | 2 |
| Z13e1 | Tri | 3 | 12/15 | 9 |
| 447-52D | NHK-HP, Tri | 2 | 16/16 | 16 |
| VRC-01 | Comb NHK | 3 | 8/16 | 7 |
| Rituxan | Tri | 3 | 18/20 | 7 |
The libraries used were either the NHK: produced by infection with VCS helper; Tri: trimer library produced by infection with VCS helper; Comb NHK+Tri: an equal volume mixture of the two VCS-produced libraries; NHK-HP +Tri: the NHK library was infected with helper phage and combined with an equal volume of the trimer library. Individual binding clones after 2 or 3 rounds of enrichment were identified by phage ELISA and the αIM inserts were amplified by colony PCR and sequenced.
Individual clones were identified as binders by phage ELISA and the sequences of their BC and FG loops were then determined. In nearly all enrichments we did not screen exhaustedly, so the number of clones reported in Table 1 represents a minimum number, particularly for those enrichments that yielded a large number of binding clones. We also evaluated a subset of the selected phage for their reactivity with non-selecting MAbs. As shown in Figure 2, individual clones reacted only with their cognate MAb and had no reactivity with any non-selecting antibody. We determined the sequence of all positive clones and examples of the results are shown in Table 2. As might be anticipated, some of the αIMs selected against target antibodies that recognize mainly linear epitopes (2F5, 4E10, 447-52D, Z13e1) had partial amino acid homology to the reported epitopes recognized by these MAbs.
Fig 2.

ELISA analysis of the binding specificity of epitope mimics displayed on phage. The indicated antibodies (top axis) were immobilized in triplicate in plate wells, and phage clones displaying the indicated αIMs (left axis) were then added to the wells. After washing, bound phage was detected using anti-M13-HRP conjugate. It can be readily appreciated that each of the phage clones reacted specifically with its cognate antibody (top axis), but not with irrelevant/unrelated antibodies.
Table 2. Summary of Selections – Sequences of Clones by Target MAb.
Sequence analysis of representative anti-idiotypic monobodies. Clones designated “Tri-XX-YY” correspond to clones derived from the Trimer library. All other clones were derived from the NHK library. All clones were isolated and sequenced after three iterative cycles of panning against the target antibody except for the 447-52D αIMs, which were characterized after 2 rounds. Bold residues indicate partial homology to the reported linear epitopes for 4E10: (NWFNIT), Z13e1: (WNWFDITN), 447-52D: (SIHXGPXX), and 2F5: (ELDKWAS). For Rituximab binders, bold residues indicate partial homology with a mimotope sequence previously isolated from a cyclic peptide display library (WWEWS/T) [2]. The * indicates a BC loop sequence containing only 6 amino acids in the variegated region that probably arose from a shortened mutagenic oligonucleotide produced using tri-phosphoramidites.
| Clone | BC Loop | FG Loop | Clone | BC Loop | FG Loop |
|---|---|---|---|---|---|
| b12 | Z13 | ||||
| FN3-2 | VHFALPV | TNHYMV | Tri-Z13-3-1 | QPTFMPE | MIPWMVPG |
| FN3-5 | VHFALPV | HISHQHIL | Tri-Z13-3-2 | EWQVDAE | DWWWDTIE |
| Tri-3-1 | VHFAWTV | NWGDTHQH | Tri-Z13-3-3 | MEYSWQY | PWNWVDLT |
| Tri-3-6 | VHWALPV | WWSGQWMP | Tri-Z13-3-4 | ERWAMWY | HPWIKWYW |
| Tri-3–8 | VHFAYPA | DITMGYMY | Tri-Z13-3-5 | AHAWLPE | WHPWFQYE |
| Tri-3–9 | MDHTWLP | QWDQNVWP | Tri-Z13-3-7 | QEASWLS | WVPEQFDQ |
| Tri-Z13-3–9 | YDEWFWT | DPWLMPPA | |||
| 2F5 | Tri-Z13-3-11 | RRFWPPF | TATKSFWW | ||
| Tri-2F5-15 | MWDKWSY | WWIGEFPV | Tri-Z13-3-14 | SSTEWFV | DIPKQWGK |
| Tri-2F5-17 | PYDKWAY | RWYWVPHY | |||
| 447-52D | |||||
| 4E10 | Hp1 | TVHAVPT | LYPLDQSS | ||
| Tri-5-2 | TEWPEQY | RHLFEYAE | Hp2 | SANFPSV | DLHFYVVS |
| Tri-5-5 | RWDPFTH | QNWFNQTS | Hp3 | ANAPVAT | LYPMQASL |
| Tri-2–6 | DAHSFTA | WNWFEQTN | Hp4 | SKSTFHV | TSYYKKFP |
| 4E10-3-3 | DAPAVTV | HNFFAQSA | Hp5 | LAPLSVV | LYPYPVAS |
| 4E10-3–5 | DAPAVTV | VLTHNFNN | Hp6 | QFQYPVV | STYPLLIF |
| 4E10-3–6 | SPLTFPA | FNFFLQTA | Hp7 | APPLNVV | LYPKPEPH |
| Hp8 | APSLPVV | LYPFPTQD | |||
| VRC01 | J1 | FPSSLPV | LYNPQYMY | ||
| 3-1 | SPPTFPM | SLLDFPIV | J2 | STSPLPT | LYPSHLSS |
| 3-2 | LPHAPMA | PFYSFPHM | J3 | PPVMSPV | LYPPAKVH |
| 3–5 | MPFVFTP | SYVPFAD | J4 | LPPPHPL | LYPYPPIS |
| 3–10 | LHHSPSA | LTLLPNYV | J5 | YVTLHPV | LYPYPTHL |
| 3–11 | HISFPYT | SLFYPVPS | J6 | TLPMLPT | LYPYPYEY |
| 3–13 | ATSLSTL | FPNYSYPL | Tri-J7 | NWRWVMED | SIHMGPRG |
| 3–16 | ASSLPSL | FAPQLHTL | J8 | PATQHPV | LYPLPPLL |
| Rituxan | PKLGVNK | VWEWDQPQ | 1F1 | MHQMPWV | YWRTTPFM |
Characterization of Anti-Idiotypic Monobodies for Critical Binding Residues
The b12 MAb is a well-characterized broadly neutralizing antibody that recognizes a discontinuous epitope within the CD4 binding site of the HIV-1 Env [21–24]. Previous studies have shown that the b12 MAb residues Y53, Y98, and W100 are essential for binding to HIV-1 gp140 [10]. We therefore examined the reactivity of our b12-binding phage clones with wild-type b12 MAb and with Y53G, Y98A, and W100A mutants of b12. As a positive control for these experiments, we used the B2.1 peptide phage clone described by Scott and co-workers (kind gift of Dr. J.K. Scott, Simon Fraser University, British Columbia, Canada). This phage clone was selected from a conventional peptide display library [36] and does not bind to the b12 MAb in the site where gp120 binds [9, 45]. As a consequence, some mutations in the b12 MAb can differentiate this peptide from gp120 - including mutations at Y53, Y98 and W100 [9, 18]. We therefore used these mutations to assess whether our b12-reactive monobodies bind in the gp120 binding site, as opposed to the B2.1 peptide binding site (which is on the other side of CDR-H3 from the gp120 binding site). As shown in Figure 3, all of the b12-specific αIM clones tested bound to the wild-type b12 MAb, but failed to bind to the Y53G, Y98A, W100A b12 mutants. Thus the b12 αIMs bind in a distinctly different mode to the MAb than the B2.1 peptide, and require the same residues of the b12 paratope that are necessary for binding to gp120.
Fig 3.

ELISA analysis of the specificity of selected b12 reactive phage clones. The indicated b12 antibodies (wild-type or mutant; top axis) were immobilized in triplicate in plate wells, and phage clones displaying the indicated αIMs (left axis) were then added to the wells. After washing, bound phage was detected using anti-M13-HRP conjugate. B2.1 is a positive control phage encoding a peptide mimotope that binds wild type b12 MAb as well as the three mutant forms of the b12 MAb [9, 18]. CRPII is an irrelevant phage clone, not selected against the b12 MAb.
We next produced soluble recombinant protein corresponding to a subset of the phage clones. αIM-encoding sequences from phage clones of interest were sub-cloned into a prokaryotic expression vector, and expressed in E. coli BL21 host cells with terminal His, biotin, and FLAG tags. Proteins were then purified using metal affinity chromatography to bind the protein His tag. Samples of our purified b12 3–5 αIMs were then analyzed by SDS-PAGE and visualized by staining with Coomassie Blue (Simply Blue™) dye (Figure 4A). A single protein species of the expected molecular weight (15 – 17 kDa) was detected. Subsequently, we tested the ability of this protein to bind the b12 MAb. As shown in Figure 4B, the b12 3–5 αIM bound efficiently to immobilized b12 MAb, as did HIV-1 gp140 [42], the positive control for the assay. We further examined the ability of HIV-1 gp140 to competitively inhibit binding of the b12 MAb to immobilized b12 3–5 αIM. An HIV-1 gp140 mutant that is unable to bind to the b12 MAb was used as a control (D368R) [46]. The results (Figure 4C) show that wild-type gp140, but not the D368R mutant, competitively inhibited binding of the b12 MAb to our b12 3–5 αIM protein.
Fig 4.

Binding of purified b12 αIMs to the b12 MAb is competitively inhibited by HIV-1 Env. (A) The b12 3–5 αIM was expressed in E coli BL21 DE3 cells. A 4–12% Bis-Tris gel was loaded with samples of the uninduced culture (Lane 2), induced culture (Lane 3), and the purified eluate collected after column purification of the b12 αIM (Lane 4); Lane 1 contains molecular weight markers. (B) ELISA analysis of the binding of serially diluted b12 MAb to biotinylated b12 3–5 αIM (5µg/mL) and HIV-1 Env gp140 oligomers [42]. (C) b12 MAb (1 µg/ml) was pre-incubated with varying amounts of wild type HIV-1 Env gp140 oligomers or mutant gp140 oligomers (D368R; this lacks the ability to bind to b12) and then added to the wells coated with b12 3–5 αIM. The b12 3–5 αIM competitively inhibits binding of the b12 MAb to HIV-1 Env gp140 oligomers.
To determine which residues of the αIM were important antibody binding, we performed a systematic alanine substitution mutagenesis of the BC and FG loop sequences and then tested each variant for its ability to bind the b12 MAb by ELISA. As shown in Figure 5A, BC loop residues H24 and F25 appear to play a critical role in recognition of the b12 MAb as they decreased binding by at least 10 fold. In contrast, Figure 5B shows only FG loop residue L84 contributed significantly to b12 MAb recognition, as reflected by the approximately 5-fold reduced binding by the L84A mutant. We conclude that the primary interaction between the b12 3–5 αIM and its cognate antibody is mediated through the engineered BC loop domain (notably, residues H24, F25), and that the FG loop contributes more modestly to MAb binding (notably, residue L84).
Fig 5.

Contribution of BC and FG loop residues to b12 MAb binding by the b12 3-5 αIM. αIMs, including wild type b12 3–5 αIM and mutated derivatives thereof (as indicated), were captured on ELISA plates. b12 MAb was then added, and antibody binding detected with anti-human IgG-HRP. (A) For the wild-type b12 3–5 αIM sequence, the BC loop sequence is VHFALPV and (B) the FG loop sequence is HISHQHIL. A library of single alanine mutants for each of the amino acids in both the BC and FG loops was created and analyzed for b12 binding. (C) The FG loop of wild-type b12 3–5 αIM was replaced with FG loops from irrelevant αIMs: 1F1 (FG loop = YWRTTPFM) or Rtx (FG loop = VWEWDQPQ). (D) Wells were coated with αIM as indicated and an ELISA was performed with b12 MAb; the data indicate that MAb binding was specific to the b12 3–5 αIM – and did not occur with irrelevant αIMs, selected against different antibody targets (1F1, Rtx, 4E10).
We also conducted an additional experiment to examine the role of the FG loop in contributing to b12 MAb binding by the b12 3–5 αIM. To do this, the FG loop of the b12 3–5 FN αIM was exchanged for FG loop sequences from two irrelevant control αIMs (1F1 and Rtx). When the FG loop of b12 3–5 αIM was exchanged for the corresponding loop from the Rtx αIM, binding to the b12 MAb was almost completely abrogated. However, binding was only modestly affected when the FG loop was exchanged for the corresponding domain from the 1F1 αIM; (Figure 5C). The profound inhibitory effect of the Rtx FG loop domain is consistent with a role for the FG loop in binding to the b12 MAb, but the modest effect of the 1F1 FG loop sequence raises the possibility of an alternative interpretation: that the Rtx FG loop may have introduced a new, disruptive or sterically hindering group. Further studies will be necessary to fully resolve this issue.
As an additional control, we examined the ability of αIMs selected against other MAbs, for their ability to bind to the b12 MAb. As expected, only the b12 3–5 αIM (and not other recombinant αIMs, selected on different antibody targets) was reactive with b12 (Figure 5D).
The analysis of the b12 3–5 αIM mutant revealed that, unexpectedly, most of the key residues were in the BC loop. To determine whether this was a generalizable finding, we conducted additional “loop exchange” experiments using αIMs reactive with two other Mabs: (i) the VRC-01 MAb (which recognizes a conformational epitope in the CD4-binding site of HIV-1 Env), and (ii) the 447-52D Mab (which recognizes a linear epitope in the V3 region of HIV-1 Env) (Table 2). We generated hybrid αIMs in which either the full BC or FG loop was replaced with a series of serine residues (equal in length to the wild-type BC or FG loop). These BC hybrids, FG hybrids, and parental αIMs were tested by phage ELISA. Figure 6 shows the VRC-01 αIM hybrids were uniformly unable to bind to the VRC-01 Mab, unlike their respective parental αIMs. In contrast, one of two the 447-52D αIM hybrids retained the ability to bind efficiently to its cognate antibody. Figure 6 shows that clone J2 (which contains a “Ser-replacement” BC loop) was able to bind to the 447-52D Mab, whereas clone J3 (which contains a “Ser-replacement” FG loop) was not. These data suggest that both the FG and BC loops can contribute to binding of FNfn10-based αIMs to targeted antibodies.
Fig 6.

Contribution of BC and FG loops to the binding of VRC01 and 447-52D IMs to their cognate antibodies. VRC-01 3-2, 3–11, 3–13 and 447-52D J2 and J3 αIMs were mutated, so as to replace either the full BC or FG loop with serine residues. Binding of the resulting hybrids to their cognate Mab was then assessed, and results compared to binding to the corresponding parental αIMs. In all cases, except 447-52D J2, substitution of either the BC or FG loop with serine residues resulted in abrogation of binding of the αIMs to their cognate antibodies.
To confirm this, we further characterized the VRC-01 3-2 αIM by using a variant form of combinatorial scanning mutagenesis. Specifically, we used a modified serine-scanning library to analyze the contribution of individual amino acid residues on the BC and FG loops, to the interaction between anti-idiotypic monobodies and their cognate antibodies. Serine was used to replace wild-type residues because it is a small, hydrophilic amino acid that may be less disruptive to the overall structure of solvent-exposed loops than the more commonly used, but hydrophobic, alanine side chain.
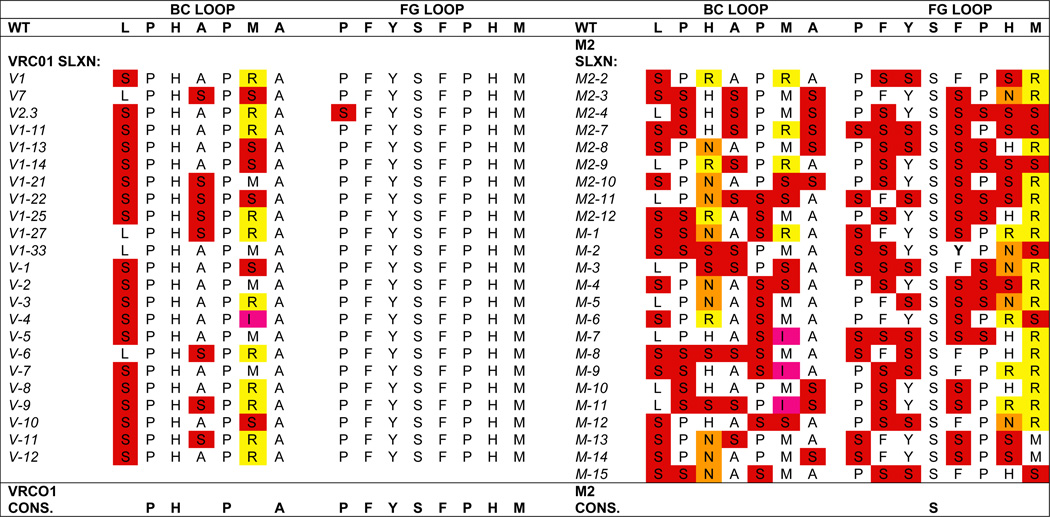
To perform this analysis, we generated a “mini-library” in which the amino acid residues in the BC and FG loops were either: (i) the wild-type sequence, or (ii) replaced with serine (except for methionine and histidine residues, which were replaced with a mixture of asparagine, arginine, and isoleucine; this was necessary because of the degeneracy of the genetic code). Phage prepared from this library were then separately selected for binding to the VRC-01 anti-Env MAb, or to the irrelevant M2 anti-FLAG MAb. Table 3 shows the frequencies of the wild-type amino acids found in the selected clones. Sequence data for clones selected against the irrelevant M2 anti-FLAG MAb illustrate the diversity of the modified serine-scanning “mini-library”. In contrast, sequences of clones selected for binding to the VRC-01 MAb demonstrate that the original sequence of the entire FG loop is important for VRC-01 MAb binding, along with 4/7 residues in the BC loop. Thus, for the VRC-01 3-2 αIM, important contact residues were present in both the BC and FG loops.
Table 3.
Mapping of VRC-01 3-2 αIM residues required for antibody binding by site-directed mutagenesis. Site-directed mutagenesis was used to create a “mini-library” in which each position in the BC and FG loops of VRC01 3-2 αIM was permitted to be either the wild-type sequence or a serine residue (except in the case of methionine and histidine, which were replaced by either serine, asparagine, arginine, and isoleucine). The resulting library was then selected for binding to either the VRC-01 Mab (left) or the irrelevant M2 anti-FLAG antibody (right). A selection of clones, 23 panned on VRC-01 and 24 panned on anti-FLAG, were sequenced; results are presented. Wild-type VRC-01 sequences are unshaded, while substitutions are color coded by amino acid (e.g., red denotes serine). Sequences of the clones selected against the VRC-01 3-2 αIM demonstrate the critically important role of the complete FG loop and 4 of 7 residues within the BC loop in contributing to the binding of the VRC-01 3-2 αIM to its cognate antibody. In contrast, sequences of the clones selected against the irrelevant M2 anti-FLAG antibody reveal the diversity of the parental “mini-library”.
 |
Characterization of Anti-Idiotypic Monobodies Reactive with MAbs 2F5 and 4E10
We also characterized αIMs reactive with MAbs that recognize linear antigenic epitopes. For this analysis, we chose 2F5 and 4E10 reactive αIMs that showed partial homology with the corresponding native MAb epitopes from HIV-1 gp41; we also selected a clone (4E10 5-2 αIM) that did not show homology to the 4E10 epitope. We assayed the ability of these αIMs, as well as an irrelevant αIM (F10λ; negative control) and the full length MPER peptide (ELLELDKWASLWNWFDITNWLWYIK; positive control) to bind to the 2F5 and 4E10 MAbs using a competition ELISA. Figure 7A shows that the 2F5–17 αIM was able to competitively inhibit the binding of the 2F5 MAb to its cognate peptide. Similar results were also obtained with both 4E10 αIMs (Figure 7B), including the αIM with no amino acid homology to the 4E10 peptide epitope (4E10 5-2 αIM). The results in Figure 7 also suggest that the affinity of the 2F5–17 αIM for its cognate antibody is about 10-fold lower than the affinity of the MPER peptide for the 2F5 MAb (Figure 7A). Similar results were obtained with the 4E10 specific αIMs, although the 4E10 5-2 protein appeared to bind its cognate antibody with only about 2–3 reduced affinity, when compared to the MPER peptide (Figure 7B).
Fig 7.

The 2F5 and 4E10 αIMs competitively inhibit the binding of their cognate antibodies to their target epitopes in HIV-1 Env. (A) 2F5 epitope peptide conjugated to BSA was immobilized in triplicate wells. A fixed concentration of 2F5 MAb was then pre-incubated with half log dilutions of 2F5 αIM, MPER peptide, or an irrelevant αIM (F10λ) before addition to the coated plate. After washing, bound 2F5 Mab was detected using anti-human IgG-HRP. (B) 4E10 epitope peptide conjugated to BSA was immobilized in triplicate wells. A fixed concentration of 4E10 was then pre-incubated with half log dilutions of 4E10 αIM (5-5 or 5-2), MPER peptide, or an irrelevant αIM (F10λ) before addition to the coated plate. After washing, bound 4E10 Mab was detected using anti-human IgG-HRP. (C, D) Sera from an HIV-positive person (026) (C), or from an HIV-negative person (1609) (D) was pre-incubated with half log dilutions of 4E10 5-2 αIM, 4E10 5-5 αIM, MPER peptide, or F10λ αIM before addition to a plate coated with 4E10 epitope peptide. After washing, bound 4E10 MAb detected with anti-human IgG-HRP.
Finally, we performed a competition binding experiment using sera from an HIV-positive person with high levels of serologic reactivity to the MPER peptide. In this case, the 4E10 αIMs competitively inhibited binding of human IgG antibodies to the MPER peptide, whereas an irrelevant αIM had no effect (Figure 7C). In contrast, the αIMs had no effect on the background MPER-binding activity of an HIV-negative control serum (Figure 7D).
Use of Anti-Idiotypic Monobodies to Detect a Therapeutic Monoclonal Antibody in Human Sera
To examine the utility of αIM as a tool to probe antibody specificities in human sera, we conducted an experiment in which the Rtx MAb was serially diluted in 1% pooled normal human sera, and then allowed to react with Rtx or b12 3–5 αIM coated ELISA plates. This analysis revealed that the Rtx αIM was able to detect Rituximab concentrations as low as 1 nM or less (Figure 8). This is considerably more sensitive than a conventional peptide ELISA assay for measurement of Rituximab concentration in human serum [41], suggesting that αIM proteins may have utility for pharmacokinetic studies of therapeutic monoclonal antibodies.
Fig 8.

Rtx αIM can be used as a sensitive probe to detect Rituximab in normal human sera. Rtx αIM or b12 3–5 αIM was immobilized in wells in triplicate. Rituximab was then serially diluted in 1% pooled normal human sera (itself diluted in blocking solution), and added to the wells. After extensive washing, bound Rituximab was detected with anti-human IgG-HRP.
Determination of the b12 bNAb Binding Affinity of the b12 3–5 αIM
We determined the affinity of the b12 3–5 αIM using SPR with the recombinant b12 Fab as the analyte and immobilized bio-αIM as the ligand. The results are shown in Figure 9. The calculated KD of 43 nM is in the range of affinities observed with similarly sized two loop libraries subjected to phage display-mediated enrichment [3]. While this affinity is relatively strong, it likely could be significantly improved using off-rate selections [1] to derive tighter binders that might function as more effective epitope mimics.
Fig 9.

SPR analysis of b12 3–5 αIM binding to the b12 Fab. (A) Association and dissociation phases of Fab binding to immobilized biotinylated b12 3–5 αIM captured on a streptavidin surface. Concentrations of Fab used were (top to bottom) 50 nM, 25 nM, 12.5 nM, 6.25 nM and 3.125 nM. (B) Langmuir isotherm of Fab binding. The Kd was determined by calculating the value of equilibrium Rmax for each injection, plotted versus concentration and fit to a Langmuir binding isotherm (Response = Equilibrium Response * ((KA*[ b12 Fab])/(KA*[ b12 Fab]+1)). (C) Distribution analysis of b12 Fab binding kinetics to surface-immobilized b12 3-5 αIM. Integration of the distribution inside a polygon drawn around the high affinity peak gives a binding capacity of 718.9 RU, an average koff = 0.0068/s, and an average KD = 43 nM.
Discussion
In this study we report the use of the tenth domain of human fibronectin (FNfn10) as a scaffold to generate anti-idiotypic monobodies recognized by a variety of monoclonal antibodies, including several HIV-1 Env-specific, broadly neutralizing antibodies (bNAbs). We successfully generated αIMs that mimic both linear (2F5, 4E10) and conformational (b12) epitopes. Our original goal was to explore the use of a more compact, easily manipulated and efficiently expressed display scaffold as an alternative to either peptide or antibody display to derive specific binding partners to antibodies. We believe such reagents could find use in a variety of applications such as protein purification, pharmacokinetic analysis of therapeutic antibodies and, potentially, vaccine development [3, 4, 47–49]. We have recently described their potential utility for immune response profiling in autoimmune disease [44].
We originally envisioned that two constrained loop domains on the FNfn10 scaffold protein would allow for complex, conformationally-dependent, interactions with target antibodies. Our analysis of the mechanism of αIM and antibody interaction has revealed unexpected differences from this simple model. In the case of the two antibodies with complex discontinuous epitopes, b12 and VRC01, we observe very different modes of interaction. For b12, the 3–5 αIM exhibits a strong requirement at only two positions within the BC loop, with the remaining BC positions and most of the FG loop being rather tolerant of substitutions while retaining the ability to bind to b12. Remarkably, this αIM nevertheless accurately mimics the actual gp120 epitope with respect to three key residues of the VH domain required for antigen binding.
The VRC01 3–2 αIM represents nearly the complete opposite situation. In this case, almost the entire original sequence selected from the library must be maintained to retain binding to the Mab. The complete FG loop and 4 of 7 residues within the BC loop were reselected in a combinatorial scanning of the two loops. Interestingly, the other VRC01-specific sequences selected have no similarity to each other. Presumably, these represent alternative conformations of the αIM lodged in the antibody paratope, each yielding a distinctive sequence. Further analysis using VRC01 CDR mutants will be necessary to fully define the mechanism of interaction.
For antibodies that recognize linear epitopes, exemplified by 4E10, 447-52D and 2F5, Z13e1, some of the selected αIMs bore sequences in one of the loops that were homologous to the reported linear epitopes. This suggests that these αIMs may be interacting with their cognate antibodies predominantly through a single loop. Other αIMs selected on these MAbs lack any obvious resemblance to the epitope and may interact in a manner that is more akin to the VRC01 specific αIMs. Despite the partial homology to the epitope found in the former type of αIMs, they were nevertheless found to compete with the identified linear peptide epitope for binding to the cognate antibodies and the 4E10 5-5 αIM could be used to detect serum antibodies in sera from an HIV patient with high reactivity to the linear MPER peptide.
Recent approaches to the generation of improved epitope mimics for HIV-1 bNAb include the grafting of both linear (2F5, 4E10) and discontinuous (b12) epitope sequences onto computationally selected or remodeled protein scaffolds that optimize epitope display [50–54]. Our monobody-based approach offers a complementary method that can be readily combined with mutagenesis and selection techniques, in order to isolate variants with altered specificity and affinity. This may facilitate the derivation of antigenic structures capable of reacting with germline precursors and/or early progenitors of somatically hypermutated bNAbs [47–49, 55]. Other potential future applications may include the use of combinations of FNfn10-based αIMs and native epitopes grafted onto optimized scaffolds in heterologous prime-boost immunization regimens intended to promote immuno-focusing on the targeted bNAb epitope [52]. Overall, the use of these robust, inexpensive and easy to purify alternatives to conventional antigens could have broad applications in diagnostics, pharmacokinetic analyses of therapeutic monoclonal antibodies, and vaccine development.
Acknowledgements
The following reagents were obtained through the NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH: HIV-1 gp120 Monoclonal Antibody (IgG1 b12) from Dr. Dennis Burton and Carlos Barbas; HIV-1 gp41 Monoclonal Antibodies (2F5, 4E10) from Dr. Hermann Katinger; IgG1 Z13e1 Monoclonal Antibody from Dr. Michael Zwick; and HIV-1 V3 Monoclonal Antibody (447-52D) from Dr. Susan Zolla-Pazner. We also thank Dr. Jacob J. Schlesinger (University of Rochester) for providing the Dengue virus specific 1F1 MAb, and Dr. Jamie K. Scott (Simon Fraser University, British Columbia, Canada) for giving us the B2.1 phage clone.
Funding Information: This work was supported by the National Institutes of Health [R21 AI087149 and P30 AI078498 to S.D.; R21 AI078459 and R01 AI084808 to I.S.; T32 DE007202 to J.M.; T32 GM068411 to L.R.B.; and P30 AI027757 and R37 AI049660 to I.S.].
Abbreviations (List the abbreviations used in an individual paragraph)
- HIV-1
Human Immunodeficiency Virus
- αIM
Anti-idiotypic monobody
- Env
Envelope surface glycoprotein of HIV-1
- ELISA
Enzyme linked Immunosorbant assay
- bNAb
Broadly eutralizing antibody
- Mab
Monoclonal antibody
References
- 1.Goletz S, et al. Selection of large diversities of antiidiotypic antibody fragments by phage display. J Mol Biol. 2002;315(5):1087–1097. doi: 10.1006/jmbi.2001.5314. [DOI] [PubMed] [Google Scholar]
- 2.Simmons DP, et al. Shark IgNAR antibody mimotopes target a murine immunoglobulin through extended CDR3 loop structures. Proteins. 2008;71(1):119–130. doi: 10.1002/prot.21663. [DOI] [PubMed] [Google Scholar]
- 3.Jiang G, et al. Evaluation of semi-homogeneous assay formats for dual-specificity antibodies. J Immunol Methods. 2013;387(1–2):51–56. doi: 10.1016/j.jim.2012.09.010. [DOI] [PubMed] [Google Scholar]
- 4.Tornetta M, et al. Isolation of human anti-idiotypic antibodies by phage display for clinical immune response assays. J Immunol Methods. 2007;328(1-2):34–44. doi: 10.1016/j.jim.2007.08.008. [DOI] [PubMed] [Google Scholar]
- 5.Scott JK, Smith GP. Searching for peptide ligands with an epitope library. Science. 1990;249(4967):386–390. doi: 10.1126/science.1696028. [DOI] [PubMed] [Google Scholar]
- 6.Larralde OG, et al. Identification of hepatitis A virus mimotopes by phage display, antigenicity and immunogenicity. J Virol Methods. 2007;140(1–2):49–58. doi: 10.1016/j.jviromet.2006.10.015. [DOI] [PubMed] [Google Scholar]
- 7.Houimel M, Dellagi K. Peptide mimotopes of rabies virus glycoprotein with immunogenic activity. Vaccine. 2009;27(34):4648–4655. doi: 10.1016/j.vaccine.2009.05.055. [DOI] [PubMed] [Google Scholar]
- 8.Matthews LJ, Davis R, Smith GP. Immunogenically fit subunit vaccine components via epitope discovery from natural peptide libraries. J Immunol. 2002;169(2):837–846. doi: 10.4049/jimmunol.169.2.837. [DOI] [PubMed] [Google Scholar]
- 9.Saphire EO, et al. Structure of a high-affinity “mimotope” peptide bound to HIV-1-neutralizing antibody b12 explains its inability to elicit gp120 cross-reactive antibodies. J Mol Biol. 2007;369(3):696–709. doi: 10.1016/j.jmb.2007.01.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zwick MB, et al. Identification and characterization of a peptide that specifically binds the human, broadly neutralizing anti-human immunodeficiency virus type 1 antibody b12. J Virol. 2001;75(14):6692–6699. doi: 10.1128/JVI.75.14.6692-6699.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McLafferty MA, et al. M13 bacteriophage displaying disulfide-constrained microproteins. Gene. 1993;128(1):29–36. doi: 10.1016/0378-1119(93)90149-w. [DOI] [PubMed] [Google Scholar]
- 12.O’Neil KT, et al. Identification of novel peptide antagonists for GPIIb/IIIa from a conformationally constrained phage peptide library. Proteins. 1992;14(4):509–515. doi: 10.1002/prot.340140411. [DOI] [PubMed] [Google Scholar]
- 13.Bonnycastle LL, et al. Probing the basis of antibody reactivity with a panel of constrained peptide libraries displayed by filamentous phage. J Mol Biol. 1996;258(5):747–762. doi: 10.1006/jmbi.1996.0284. [DOI] [PubMed] [Google Scholar]
- 14.Koide A, et al. The fibronectin type III domain as a scaffold for novel binding proteins. J Mol Biol. 1998;284(4):1141–1151. doi: 10.1006/jmbi.1998.2238. [DOI] [PubMed] [Google Scholar]
- 15.Koide A, Koide S. Monobodies: antibody mimics based on the scaffold of the fibronectin type III domain. Methods Mol Biol. 2007;352:95–109. doi: 10.1385/1-59745-187-8:95. [DOI] [PubMed] [Google Scholar]
- 16.Richards J, et al. Engineered fibronectin type III domain with a RGDWXE sequence binds with enhanced affinity and specificity to human alphavbeta3 integrin. J Mol Biol. 2003;326(5):1475–1488. doi: 10.1016/s0022-2836(03)00082-2. [DOI] [PubMed] [Google Scholar]
- 17.Lipovsek D. Adnectins: engineered target-binding protein therapeutics. Protein Eng Des Sel. 2010;24(1–2):3–9. doi: 10.1093/protein/gzq097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zwick MB, et al. Molecular features of the broadly neutralizing immunoglobulin G1 b12 required for recognition of human immunodeficiency virus type 1 gp120. J Virol. 2003;77(10):5863–5876. doi: 10.1128/JVI.77.10.5863-5876.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kunkel TA, Roberts JD, Zakour RA. Rapid and efficient site-specific mutagenesis without phenotypic selection. Methods Enzymol. 1987;154:367–382. doi: 10.1016/0076-6879(87)54085-x. [DOI] [PubMed] [Google Scholar]
- 20.Tonikian R, et al. Identifying specificity profiles for peptide recognition modules from phage-displayed peptide libraries. Nat Protoc. 2007;2(6):1368–1386. doi: 10.1038/nprot.2007.151. [DOI] [PubMed] [Google Scholar]
- 21.Barbas CF, 3rd, et al. Recombinant human Fab fragments neutralize human type 1 immunodeficiency virus in vitro. Proc Natl Acad Sci U S A. 1992;89(19):9339–9343. doi: 10.1073/pnas.89.19.9339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Burton DR, et al. A large array of human monoclonal antibodies to type 1 human immunodeficiency virus from combinatorial libraries of asymptomatic seropositive individuals. Proc Natl Acad Sci U S A. 1991;88(22):10134–10137. doi: 10.1073/pnas.88.22.10134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roben P, et al. Recognition properties of a panel of human recombinant Fab fragments to the CD4 binding site of gp120 that show differing abilities to neutralize human immunodeficiency virus type 1. J Virol. 1994;68(8):4821–4828. doi: 10.1128/jvi.68.8.4821-4828.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burton DR, et al. Efficient neutralization of primary isolates of HIV-1 by a recombinant human monoclonal antibody. Science. 1994;266(5187):1024–1027. doi: 10.1126/science.7973652. [DOI] [PubMed] [Google Scholar]
- 25.Buchacher A, et al. Generation of human monoclonal antibodies against HIV-1 proteins; electrofusion and Epstein-Barr virus transformation for peripheral blood lymphocyte immortalization. AIDS Res Hum Retroviruses. 1994;10(4):359–369. doi: 10.1089/aid.1994.10.359. [DOI] [PubMed] [Google Scholar]
- 26.Purtscher M, et al. Restricted antigenic variability of the epitope recognized by the neutralizing gp41 antibody 2F5. AIDS. 1996;10(6):587–593. doi: 10.1097/00002030-199606000-00003. [DOI] [PubMed] [Google Scholar]
- 27.Purtscher M, et al. A broadly neutralizing human monoclonal antibody against gp41 of human immunodeficiency virus type 1. AIDS Res Hum Retroviruses. 1994;10(12):1651–1658. doi: 10.1089/aid.1994.10.1651. [DOI] [PubMed] [Google Scholar]
- 28.Stiegler G, et al. A potent cross-clade neutralizing human monoclonal antibody against a novel epitope on gp41 of human immunodeficiency virus type 1. AIDS Res Hum Retroviruses. 2001;17(18):1757–1765. doi: 10.1089/08892220152741450. [DOI] [PubMed] [Google Scholar]
- 29.Conley AJ, et al. Neutralization of primary human immunodeficiency virus type 1 isolates by the broadly reactive anti-V3 monoclonal antibody, 447-52D. J Virol. 1994;68(11):6994–7000. doi: 10.1128/jvi.68.11.6994-7000.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gorny MK, et al. Neutralization of diverse human immunodeficiency virus type 1 variants by an anti-V3 human monoclonal antibody. J Virol. 1992;66(12):7538–7542. doi: 10.1128/jvi.66.12.7538-7542.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gorny MK, et al. Human monoclonal antibodies to the V3 loop of HIV-1 with intra- and interclade cross-reactivity. J Immunol. 1997;159(10):5114–5122. [PubMed] [Google Scholar]
- 32.Gorny MK, et al. Repertoire of neutralizing human monoclonal antibodies specific for the V3 domain of HIV-1 gp120. J Immunol. 1993;150(2):635–643. [PubMed] [Google Scholar]
- 33.Nyambi PN, et al. Mapping of epitopes exposed on intact human immunodeficiency virus type 1 (HIV-1) virions: a new strategy for studying the immunologic relatedness of HIV-1. J Virol. 1998;72(11):9384–9391. doi: 10.1128/jvi.72.11.9384-9391.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zolla-Pazner S, et al. Serotyping of primary human immunodeficiency virus type 1 isolates from diverse geographic locations by flow cytometry. J Virol. 1995;69(6):3807–3815. doi: 10.1128/jvi.69.6.3807-3815.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nelson JD, et al. An affinity-enhanced neutralizing antibody against the membrane-proximal external region of human immunodeficiency virus type 1 gp41 recognizes an epitope between those of 2F5 and 4E10. J Virol. 2007;81(8):4033–4043. doi: 10.1128/JVI.02588-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zwick MB, et al. Broadly neutralizing antibodies targeted to the membrane-proximal external region of human immunodeficiency virus type 1 glycoprotein gp41. J Virol. 2001;75(22):10892–10905. doi: 10.1128/JVI.75.22.10892-10905.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reff ME, et al. Depletion of B cells in vivo by a chimeric mouse human monoclonal antibody to CD20. Blood. 1994;83(2):435–445. [PubMed] [Google Scholar]
- 38.Megret F, et al. Use of recombinant fusion proteins and monoclonal antibodies to define linear and discontinuous antigenic sites on the dengue virus envelope glycoprotein. Virology. 1992;187(2):480–491. doi: 10.1016/0042-6822(92)90450-4. [DOI] [PubMed] [Google Scholar]
- 39.Sui J, et al. Structural and functional bases for broad-spectrum neutralization of avian and human influenza A viruses. Nat Struct Mol Biol. 2009;16(3):265–273. doi: 10.1038/nsmb.1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tiller T, et al. Efficient generation of monoclonal antibodies from single human B cells by single cell RT-PCR and expression vector cloning. J Immunol Methods. 2008;329(1-2):112–124. doi: 10.1016/j.jim.2007.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weiss GA, et al. Rapid mapping of protein functional epitopes by combinatorial alanine scanning. Proc Natl Acad Sci U S A. 2000;97(16):8950–8954. doi: 10.1073/pnas.160252097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mattiacio J, et al. Dense display of HIV-1 envelope spikes on the lambda phage scaffold does not result in the generation of improved antibody responses to HIV-1 Env. Vaccine. 2011;29(14):2637–2647. doi: 10.1016/j.vaccine.2011.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moos PJ, Edes K, Fitzpatrick FA. Inactivation of wild-type p53 tumor suppressor by electrophilic prostaglandins. Proc Natl Acad Sci U S A. 2000;97(16):9215–9220. doi: 10.1073/pnas.160241897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sullivan MA, et al. Anti-idiotypic monobodies for immune response profiling. Methods. 2012;58(1):62–68. doi: 10.1016/j.ymeth.2012.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhou T, et al. Structural definition of a conserved neutralization epitope on HIV-1 gp120. Nature. 2007;445(7129):732–737. doi: 10.1038/nature05580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li Y, et al. Broad HIV-1 neutralization mediated by CD4-binding site antibodies. Nat Med. 2007;13(9):1032–1034. doi: 10.1038/nm1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.de la Cruz VF, Lal AA, McCutchan TF. Immunogenicity and epitope mapping of foreign sequences via genetically engineered filamentous phage. J Biol Chem. 1988;263(9):4318–4322. [PubMed] [Google Scholar]
- 48.Greenwood J, Willis AE, Perham RN. Multiple display of foreign peptides on a filamentous bacteriophage. Peptides from Plasmodium falciparum circumsporozoite protein as antigens. J Mol Biol. 1991;220(4):821–827. doi: 10.1016/0022-2836(91)90354-9. [DOI] [PubMed] [Google Scholar]
- 49.Parmley SF, Smith Gp. Filamentous fusion phage cloning vectors for the study of epitopes and design of vaccines. Adv Exp Med Biol. 1989;251:215–218. doi: 10.1007/978-1-4757-2046-4_21. [DOI] [PubMed] [Google Scholar]
- 50.Correia BE, et al. Computational protein design using flexible backbone remodeling and resurfacing: case studies in structure-based antigen design. J Mol Biol. 2011;405(1):284–297. doi: 10.1016/j.jmb.2010.09.061. [DOI] [PubMed] [Google Scholar]
- 51.Correia BE, et al. Computational design of epitope-scaffolds allows induction of antibodies specific for a poorly immunogenic HIV vaccine epitope. Structure. 2010;18(9):1116–1126. doi: 10.1016/j.str.2010.06.010. [DOI] [PubMed] [Google Scholar]
- 52.Guenaga J, et al. Heterologous epitope-scaffold prime:boosting immuno-focuses B cell responses to the HIV-1 gp41 2F5 neutralization determinant. PLoS One. 2011;6(1):e16074. doi: 10.1371/journal.pone.0016074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ofek G, et al. Elicitation of structure-specific antibodies by epitope scaffolds. Proc Natl Acad Sci U S A. 2010;107(42):17880–17887. doi: 10.1073/pnas.1004728107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Azoitei ML, et al. Computation-guided backbone grafting of a discontinuous motif onto a protein scaffold. Science. 2011;334(6054):373–376. doi: 10.1126/science.1209368. [DOI] [PubMed] [Google Scholar]
- 55.Zhou T, et al. Structural basis for broad and potent neutralization of HIV-1 by antibody VRC01. Science. 2010;329(5993):811–817. doi: 10.1126/science.1192819. [DOI] [PMC free article] [PubMed] [Google Scholar]