

Abstract
A persistent inflammatory and oxidative stress is a hallmark of most chronic CNS pathologies (Alzheimer's (ALS)) as well as the aging CNS orchestrated by the proinflammatory cytokines tumor necrosis factor alpha (TNFα) and interleukin-1 beta (IL-1β). Loss of the integrity and plasticity of neuronal morphology and connectivity comprises an early step in neuronal degeneration and ultimate decline of cognitive function. We examined in vitro whether TNFα or IL-1β impaired morphology and motility of growth cones in spinal cord neuron cultures. TNFα and IL-1β paralyzed growth cone motility and induced growth cone collapse in a dose-dependent manner reflected by complete attenuation of neurite outgrowth. Scavenging reactive oxygen species (ROS) or inhibiting NADPH oxidase activity rescued loss of neuronal motility and morphology. TNFα and IL-1β provoked rapid, NOX-mediated generation of ROS in advancing growth cones, which preceded paralysis of motility and collapse of morphology. Increases in ROS intermediates were accompanied by an aberrant, nonproductive reorganization of actin filaments. These findings suggest that NADPH oxidase serves as a pivotal source of oxidative stress in neurons and together with disruption of actin filament reorganization contributes to the progressive degeneration of neuronal morphology in the diseased or aging CNS.
1. Introduction
A spreading inflammatory reaction accompanied by oxidative stress is prevalent in most chronic CNS diseases and acute CNS trauma as well as in the aging CNS [1–3]. The proinflammatory cytokines TNFα (tumor necrosis factor α) and IL-1β (interleukin-1β) exert pleiotropic functions both in CNS development and CNS pathogenesis [4–6]. Persistent high-level expression of TNFα and IL-1β is important to the progressive degeneration of neuronal connectivity and loss of neuronal plasticity ultimately leading to cognitive decline. The role of TNFα and IL-1β as inducers of apoptotic events is well documented, whereas the recognition of their morphogenetic function is more recent [7]. TNFα and IL-1β released from microglia cells inhibited neurite outgrowth, reduced branching, and caused neurite retraction in cultures of Neuro2A cells or primary hippocampal neurons [8, 9]. The intricate pattern of neuronal connectivity innate to cognitive function rests as much on the integrity and stability of the axonal and dendritic architecture as on the plasticity of motile structures to maintain, form, or regenerate connections, which is intimately linked to the dynamic reorganization of the actin cytoskeleton [10, 11]. ROS intermediates greatly affect both the dynamics and organization of actin filaments during oxidative stress of physiological redox signaling [12, 13]. TNFα paralyzed actin filament reorganization in neuroblastoma cells due to oxidative damage, whereas physiological levels of ROS intermediates seem to be necessary for proper growth cone motility [14, 15]. TNFα and IL-1β potently stimulate NADPH oxidase (NOX) activities in neurons and glia cells often localized in coalescing lipid rafts [16, 17]. The members of the NADPH oxidase (NOX) family (NOX1–5, DUOX1/2), defined by the large membrane flavoprotein gp91phox of the phagocyte NAPDH oxidase (NOX2), are ubiquitously expressed in all cell types and have emerged as principal ROS sources both in cellular signaling and disease progression in response to cytokines, growth factors, and hormones [18, 19]. Functional NOX requires an intricate assembly between two membrane proteins (a NOX isoform, gp22phox) and several cytosolic factors (p47phox, p40phox, p67phox) under the regulation of the small GTPases Rac1 or Rac2 [20]. Rho GTPases harbor a dual role in cytokine signaling as regulators of both NOX assembly and the reorganization of actin filament structures [21–23]. In light of these reports, we examined whether ROS intermediates generated by NOX activities in neuronal growth cones are implicated in mediating the neurotoxic effects of TNFα or IL-1β on neurite outgrowth. A mechanistic understanding of the detrimental consequences of TNFα and IL-1β on neuronal connectivity in the CNS neurons is vital to intervene with progressive neurodegeneration in the aging or diseased CNS.
2. Materials and Methods
2.1. Reagents
Unless state otherwise, all reagents were purchased from Sigma (St. Louis, MO). Dulbecco's Modified Eagle Medium (high glucose DMEM), Leibovitz's L-15, and isopropyl β-D-thiogalactopyranoside were obtained from Invitrogen Corporation (Carlsbad, CA). Characterized fetal bovine serum (FBS) was from Hyclone (Logan, UT) and it was heat-inactivated according to the manufacturer. Laminin was from Roche Diagnostics Corporation (Indianapolis, IN). The redox sensitive fluorescent indicators carboxymethyl-2′,7′-dihydrodichlorofluorescein diacetate, 2′,7′-dihydrodichlorofluorescein diacetate, and Calcein Blue were obtained from Molecular Probes (Eugene, OR). Human recombinant tumor necrosis factor α (TNFα) and interleukin-1β (IL-1β), Diphenylene iodonium (DPI), N-acetyl-L-cysteine (NAC), and Manganese(III) tetrakis(4-benzoic acid) porphyrin chloride (MnTBAP) were purchased from Calbiochem (San Diego, CA). Polyclonal rabbit anti-TNF receptor I, polyclonal rabbit anti-TNF receptor II, polyclonal rabbit anti IL-1 receptor type I, polyclonal goat anti-NOX2, polyclonal goat anti-p47phox including respective blocking peptides were all from St. Cruz Biotechnology (St. Cruz, CA). A monoclonal mouse anti-p44/42 MAPK, a monoclonal mouse anti-phospho p44/42 MAPK, and a monoclonal mouse anti-phospho JNK were from Cell Signaling Technologies (Danvers, MA). Fluorescein or rhodamine-labeled goat anti-rabbit and goat anti-mouse IgG (resp.) were from Chemicon (Temecula, CA), whereas rhodamine-conjugated phalloidin was obtained from Cytoskeleton Inc. (Denver, CO). The polyclonal rabbit anti-p67phox was purchased from Abcam (Cambridge, MA).
2.2. Spinal Cord Neuron Cultures
Dissociated, low-density cultures of spinal cord (SC) neurons were established as described [24]. Briefly, spinal cord tissue was dissected from 7 day-old chick embryos (E7) and a single cell suspension obtained after enzymatic digestion (0.5 mg/mL trypsin, 2 mM EDTA, 10 min, 37°C) followed by trituration. After preplating (1 h, 37°C, 5% CO2 atmosphere, high glucose DMEM, pH 7.3/10% FBS), nonadherent cells (predominantly SC neurons) were collected (3 min, 200 ×gmax) and resuspended in SC medium (high glucose DMEM pH 7.3, 10% FBS, 12 nM fluorodeoxyuridine, 30 nM uridine, and 1% N3 nutrient supplement) [25]. SC explants were prepared by pushing freshly dissected E7 spinal cord tissue through a wire-mesh (50–75 μm). Dissociated SC neurons (75,000 cells/mL) or SC explants (2 per culture) were plated onto glass cover slips (22 × 22 mm2, number 1, Carolina Biological Supply Company, Burlington, NC) mounted over a 1.5 cm hole drilled into the bottom of a 35 mm culture dish. Glass cover slips were treated with poly-D-lysine (100 μg/mL, borate buffer pH 8.4, 30 min, RT) prior to coating with laminin (5 μg/cm2, 30 min, RT). These SC neuron cultures contained less than 5% nonneuronal cells with no immunoreactivity against microglia markers.
2.3. Growth Cone Particle Preparations
Preparations of highly enriched growth cone particles were obtained from freshly dissected whole chick embryo brains (E10-12) [26, 27]. After homogenizing (Dounce homogenizer, ice-cold 5 mM HEPES pH 7.3, 1 mM MgCl2, 0.32 M Sucrose, 6–8 volumes per wet weight), homogenates were centrifuged (15 min, 1600 ×gmax, 4°C). Resulting supernatants were overlaid onto 5 mM HEPES pH 7.3, 1 mM MgCl2, and 0.75 M sucrose and then centrifuged (150,000 ×gmax, 1 h, 4°C), and material at the interface was collected. After dilution (6-7 times in low sucrose buffer), the suspension was overlaid onto MaxiDense (4-5 sample volumes per volume Maxidense) and centrifuged at 40,000 ×gmax (1 h, 4°C), and growth cone particles (GCPs) were collected on top of the MaxiDense cushion. GCPs were resuspended in Kreb's buffer (145 mM NaCl, 5 mM KCl, 1.2 mM NaH2PO4 × H2O, 1.2 mM MgCl2, and 5 mM HEPES pH 7.3) and protein concentration determined (BCA assay, Thermo Scientific, Rockford, IL, USA).
2.4. Measurement of Neurite Lengths
Dissociated SC neurons were grown after the onset of neurite outgrowth indicated by the majority of cells extending at least one process longer than 2 cell body diameters. Cultures were incubated (1 h) with pharmacological agents, followed by bath application (6–8 h) of 100 ng/mL TNFα, 100 ng/mL IL-1β, or 10 μg/mL ovalbumin. After fixation (2% glutaraldehyde), the length of the longest neurite per neuron was measured of at least 50 randomly selected SC neurons only considering processes adhering to the following criteria: (i) emerging from an isolated cell body, (ii) longer than two cell diameters, and (iii) no contact to other neuronal processes or cell bodies. The distribution of neurite length in a population of SC neurons was obtained by plotting the percentage of neurons with neurites longer than a given length against neurite length [28]. As a characteristic for neurite outgrowth under a given condition, the neurite length reached by 50% of neurite-bearing SC neurons (NL50) was calculated as criteria for statistical significance [29]. Purified, recombinant Rac1V12-GST, Rac1N17-GST, or GST were introduced into freshly dissociated SC neurons (7–10 mg/mL, 500,000 cells, 200 μL 50 mM Tris-Cl pH 7.5, 100 mM NaCl, and 5 mM MgCl2) by trituration loading at the time of plating [26]. Following trituration, SC neurons were immediately transferred in SC medium and plated as described above. For each condition, measurements were performed in duplicate cultures from two independent dissections.
2.5. Measurement of Growth Cone Advance
Dissociated SC neurons (12 to 16 h in culture) were transferred into observation medium (Leibovitz'L15 without phenol red, pH 7.4, 5 mg/mL ovalbumin, 1% N3), overlaid with light mineral oil to avoid evaporation, and placed onto the microscope stage equilibrated at 37 ± 0.2°C (Nikon TE2000 U). After 15-minute recovery, images of advancing growth cones (20x, phase contrast) were acquired at 3-minute time intervals for a 30 min time period (Coolsnapfx, Photometrics, Tuscon, AZ). Cytokines (100 ng/mL TNFα, 100 ng/mL IL-1β) or 10 μg/mL Ovalbumin were bath-applied at t = 8 min. The extension of the growth cone/neurite boundary (μm) was measured as a function of time (min) (Metamorph Software, Meridian Instrument Co, Kent, WA). At least 20 growth cones were monitored for each condition in duplicate cultures from at least two different dissections.
2.6. Recombinant Mutant Rac1 Protein
Recombinant Rac1V12-GST or Rac1N17-GST protein was induced in E. coli DH5α with 0.5 mM isopropyl β-D-thiogalacto-pyranoside (4 h) [30]. After cell lysis (50 mM Tris-Cl pH 7.5, 50 mM NaCl, 5 mM MgCl2, and 1 mM dithiothreitol), a soluble protein fraction was obtained (20,000 ×gmax, 20 min, 4°C) and subjected to affinity chromatography on glutathione-conjugated agarose. Bound proteins were eluted (5 mM glutathione in lysis buffer), concentrated to 5–7 mg/mL in 50 mM Tris-Cl pH 7.5, 100 mM NaCl, and 5 mM MgCl2 (Centrifugal Devices, Millipore, Bedford, MA), and stored in 100 μL aliquots at −80°C after snap-freezing. Protein preparations showed single bands on Coomassie blue-stained 10% SDS polyacrylamide gels.
2.7. Adenoviral Expression of Rac1 Mutants in SC Neurons
Recombinant, replication deficient adenovirus carrying genes for constitutively active Rac1 (Rac1V12 with N-terminal FLAG tag), dominant negative Rac1 (Rac1N17 with N-terminal FLAG tag), or lacZ were expressed in E7 chick SC neurons as described previously [31]. At the time of plating, dissociated SC neurons were infected with recombinant adenovirus at 200 moi (multiplicity of infection) in 300 μL SC medium. Cultures were replenished with 200 μL fresh SC medium after 12 h and grown for additional 48 h. At three days after infection, yields of neuronal infections generally exceeded 70% [31, 32]. Amplification of viral stocks was performed in 293 HEK cells and titers greater than 5 × 108 plaque forming units per mL were routinely obtained. Viral stocks were stored at –80°C.
2.8. Quantitative ROS Imaging in SC Neuron Cell Bodies
Dissociated SC neurons were loaded with 10 μM 2′,7′-dihydrodichlorofluorescein diacetate (DCF) or 5 μM dihyroethidium (DMEM/10% FBS) for 30 min (37°C, 5% CO2 atmosphere), washed, and allowed to recover (15 min, DMEM/10% FBS). Cultures were switched to observation medium, overlaid with light mineral oil, transferred to the heated microscope stage (Zeiss Axiovert125S), and allowed to adapt for 15 min. Images of dissociated SC neurons (random fields of view) were acquired under phase contrast (20x Plan-Apo objective) and FITC illumination (Ex 465–495 nm, DM 505, Em 515–555) using a Peltier cooled CCD camera (Sensys, Photometrics, Tuscon, AZ) after the recovery phase (defined as basal condition), after pharmacological treatments (30 min), and after cytokine exposure (100 ng/mL TNFα or 100 ng/mL IL-1β). Hydrogen peroxide was added to all cultures following treatments to ensure proper loading. As our criteria for SC neurons, we analyzed only cells displaying a large round cell body and one neuronal process at least longer than three cell diameters. Maximum DCF fluorescence intensity per neuronal cell body was determined on a pixel-by-pixel basis following background subtraction (average background of all images at t = 0 min for each condition) followed by erosion (2 pixels) and overlay with the original image (Zeiss imaging analysis software KS 300). All values were normalized to the average DCF fluorescence intensity in control (initial conditions). No morphological changes of neuronal cell bodies were detectable in our assay conditions indicated by constant cell body areas. ROS measurements were performed in duplicate cultures obtained from 3 to 5 independent dissections. At least 50 SC neurons were measured in duplicate cultures of three independent dissections.
2.9. Quantitative ROS Imaging in Advancing SC Neuron Growth Cones
SC explants were incubated (30 min, DMEM/10% FBS) with 20 μM DCF (5′-(and 6′)chloromethyl-dichlorodihydrofluorescein diacetate) and 4 μM Calcein Blue (CB), an oxidation-inert fluorescence indicator. After recovery (15 min, DMEM/10% FBS), cultures were switched to observation medium and overlaid with light mineral oil. Images of advancing growth cones were acquired under phase contrast (40x Plan-Apo), FITC illumination (DCF fluorescence), and DAPI illumination (CB fluorescence) (Coolsnapfx) before (pre-stimulus images at t = 0, 2, 4 min) and after (after stimulus images, 2 min time intervals, 16 min time period) bath application of 100 ng/mL TNFα, 100 ng/mL IL-1β, or 10 μg/mL ovalbumin. As our positive loading control, following the observation period, all growth cones were exposed to 100 μM hydrogen peroxide. For image analysis, DCF and CB fluorescence intensities (F DCF and F CB) were integrated (pixel-by-pixel basis) over the growth cone area for each growth cone observed at each time point (Int F DCF,t and Int F CalcB,t with t = time interval) after background subtraction (t = 0 image) and the ratio of integrated DCF and CB fluorescence intensities for each growth cone at each time point was calculated (R t = Int F DCF,t/Int F with t = time interval). Next, the average ratio of integrated DCF and CB fluorescence intensities of all growth cones observed at t = 0 min (pre-stimulus) was calculated (av R 0) followed by normalization of all ratios for at t = 0 min (R t n/av R 0, n = growth cone 1, 2, to n for each condition). At least 15 growth cones were analyzed per condition from three different dissections to provide statistical significance (∗P < 0.05).
2.10. ROS Quantification in Growth Cone Particle Preparations
Freshly prepared GCPs (100 μg in Kreb's buffer) were loaded with 20 μM DCF (30 min, 4°C) in the presence of pharmacological reagents (10 μM DPI, 500 μM NAC), washed (14,000 ×gmax), allowed to recover (10 min, 4°C) with pharmacological reagents present, and then exposed to 100 or 200 ng/mL TNFα (45 min, 4°C). For lysis, GCPs were resuspended in 2% SDS, 10 mM Tris-Cl pH 7.5, 10 mM NaF, 5 mM dithiothreitol, and 2 mM EGTA; sonicated; and cleared by centrifugation (14,000 ×gmax, 5 min). Total DCF fluorescence intensity was measured (100 μL aliquots, black 96 well plates) using a Beckman Coulter Multimode DTX 880 microplate reader (495 nm excitation filter, 525 emission filter). All data were adjusted to total soluble protein concentration (BCA assay) and normalized to control condition to account for unspecific fluorescence and/or autofluorescence artifact. For all conditions, measurements were obtained in duplicates from three different GCP preparations.
2.11. Indirect Immunocytochemistry
Dissociated SC neurons were fixed (4% paraformaldehyde, 10 mM MES pH 6.1, 138 mM KCl, 3 mM MgCl2, 2 mM EGTA, 15 min, RT) followed by three washes with TBS (20 mM Tris-Cl pH 7.5, 150 mM NaCl). After blocking (30 min), cultures were incubated (2 h, RT) with primary antibodies (4 μg/mL, 2 mg/mL BSA in TBS) against cytokine receptors or MAP kinases, rinsed with TBS (3 times, 15 min each), and incubated with respective secondary antibodies. Cultures were transferred to 60% glycerol/PBS. Images were acquired under FITC illumination on a Radiance 2000 confocal microscope (Bio Rad).
2.12. Actin Filament Quantification
To visualize actin filaments, SC neurons were fixed and permeabilized (0.5% Triton X-100, 15 min). After rinsing (0.1% Triton X-100 in TBS), cultures were incubated (20 min) with rhodamine-conjugated phalloidin (1 : 10 in 1% Triton X-100 in TBS, Cytoskeleton Inc., Denver, CO), washed, and stored in 60% glycerol (4°C) until inspection. Images were acquired (40x oil, Plan Fluor) using a Zeiss LSM 510 confocal microscope equipped with a HeNe laser and an Argon laser. For each condition, 60 randomly selected SC neuron growth cones were scored for the presence of at least one large lamellipodia-like structure (three dissections, n = 180) and the percentage of responding growth cones determined.
2.13. Plasma Membrane Translocation of p67phox
Freshly prepared GCPs (100 μg per sample) were treated with methyl-β-cyclodextrin (0.1%) or buffer (30 min, 4°C) and then exposed to 200 ng/mL TNFα (1 h, 4°C), whereas cultures of E10 forebrain neurons were subjected to 200 nM PMA (1 h, 37°C). GCPs (centrifugation) or forebrain neurons (scraping) were transferred into a 0.33 M sucrose buffer (20 mM Tris-HCL pH 8.0, 2 mM EDTA, 0.5 EGTA, 2 mM AEBSF, and 25 μg/mL Leupeptin), lysed by sonication, and centrifuged (25,000 ×gmax, 15 min) to obtain an enriched plasma membrane fraction (pellet). Pellets were resuspended in sucrose buffer containing 1% Triton X-100 and 0.01% saponin (4°C, 15 min) or in 20 mM Tris-HCL pH 8.0, 2 mM EDTA, 0.5 mM EGTA, and 2 mM AEBSF with regard to gel electrophoresis or ELISA analysis, respectively. Suspensions were centrifuged (25,000 ×gmax, 15 min, 4°C), membrane protein was recovered as the supernatant, and total protein concentration was determined. For ELISA, aliquots of plasma membrane protein (20 μg/mL, TBS/1% Triton-X-100) were incubated (12 h, 25°C) in 96-well high protein absorbent plates (Falcon). After blocking with BSA (5% w/v, TBS, 1% Trition-X-100, 1 h), wells were incubated (overnight, 4°C) with a polyclonal rabbit anti-p67phox (3 μg/mL), rinsed (TBS/1% Triton-X-100), and incubated with a goat anti-rabbit IgG antibody conjugated to HRP (1 : 2000, 45 min, 25°C). After three consecutive washes, wells were supplemented with 100 μL TMB (tetramethylbenzidine) and absorbance at 620 nm was measured 10 min later (Beckman Coulter Multimode DTX 880 microplate reader. All values were adjusted to total membrane protein and normalized to control conditions (duplicates, three independent experiments).
2.14. Gel Electrophoresis and Western Blotting
SC tissue, SC neuron cultures, or GDPs were solubilized in 2% SDS, 10 mM Tris-Cl pH 7.5, 10 mM NaF, 5 mM dithiothreitol, and 2 mM EGTA and sonicated, and a total soluble protein fraction was obtained by centrifugation (supernatant, 14,000 ×gmax, 5 min). Fractions of total plasma membrane proteins were obtained as described above. Samples containing 20 μg of total soluble protein in Laemmli buffer were subjected to 10% SDS gel electrophoresis followed by western blotting onto PVDF membranes (Bio-Rad, Hercules, CA) [33, 34]. Membranes were blocked (2% nonfat dry milk, TBS pH 8.5, overnight, 4°C), washed (TBS/0.05% Tween 20/2% BSA), and incubated with polyclonal antibodies against cytokine receptor, MAP kinase, phospho MAP kinase, NOX2, p47phox, or p67phox (1 h, 4 μg/mL, in TBS/0.05% Tween 20/2% BSA). Membranes were washed (TBS/0.05% Tween 20/2% BSA) and incubated (1 h, RT) with the respective secondary, alkaline phosphatase-conjugated IgG (1 : 10,000, Sigma, St. Louis, MO) followed by colorimetric development (NBT/BCIP one step, Thermo Scientific, Rockford, IL). The specificity of antibodies was verified in lysates of HeLa cells, RAW264 cells, and SH-SY5Y cells (including blocking peptides).
2.15. Statistical Analysis
One-way ANOVA analysis and a Kruskal-Wallis test were employed for comparisons among multiple conditions. Dunnett's t-test was used when comparing the means of multiple conditions with a single control. All statistical values are given as SEM with a significance of *P < 0.05 unless indicated otherwise. Measurements were obtained in duplicates from 3 to 5 separate experiments unless stated otherwise.
3. Results
3.1. TNFα and IL-1β Paralyze Growth Cone Motility and Induce Growth Cone Collapse
Recent reports detailed that TNFα exerts morphogenetic functions (rearrangements of the actin cytoskeleton) without induction of apoptosis [7]. We examined whether TNFα or IL-1β has the potency to alter motility of neuronal growth cones, an actin-cytoskeleton driven mechanism. Live-video, phase contrast microscopy of advancing SC neuron growth cones revealed that an acute exposure to TNFα or IL-1β paralyzed growth cone motility and also caused the collapse of growth cone morphology accompanied by neurite beading and retraction compared to control (ovalbumin) (Figure 1(a)). To demonstrate a direct effect of cytokines on growth cones, we utilized polystyrene beads (4 μm in diameter) covalently coated with TNFα, IL-1β, or ovalbumin (control) to restrict contact between neurons and cytokines exclusively to advancing growth cones [35]. Growth cones encountering cytokine-coated beads displayed a series of stereotypic changes in behavior ultimately resulting in growth cone collapse (Figure 1(b)). After establishing long-lasting adhesion contacts with filopodia, growth cones entered a phase characterized by highly mobile, lamellipodia-like structures, which were nonproductive for advance, followed by a collapse of morphology (TNFα: 78 ± 4%, n = 22 and IL-1β: 83 ± 7%, n = 18 of observed growth cones, resp.). None of these growth cone responses occurred upon contact with ovalbumin-coated beads. Cytokine receptors were expressed on SC neurons and their processes (Figure 1(c)). Whole SC tissue extracts (western blotting) exhibited immunoreactivity against TNF receptor 1 (TNF-R1, apparent MW = 48 kDa), TNF receptor 2 (TNF-R2, apparent MW = 70 kDa), and IL-1β receptor type 1 (IL-1R, apparent MW = 76 kDa) in accordance with previous reports [36]. Expression of TNF-R1 and IL-1R was found on neuronal cell bodies, neurites, as well as growth cones; however, TNF-R2 expression was predominantly localized to neuronal cell bodies with virtually no expression on neurites or growth cones.
Figure 1.
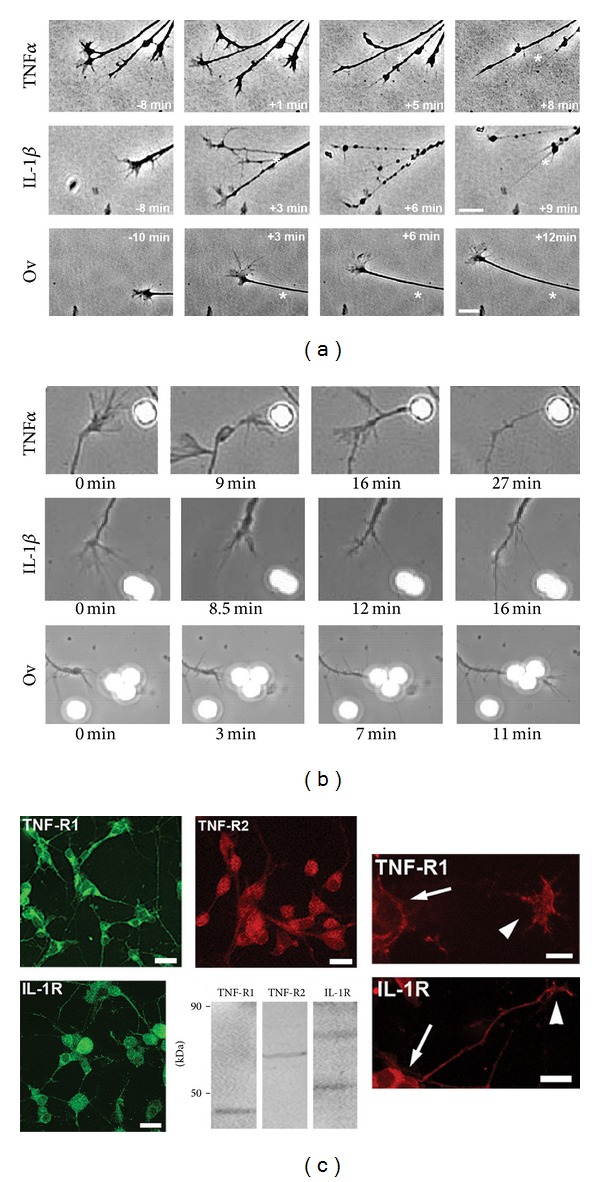
TNFα and IL-1β impair motility and morphology of neuronal growth cones. (a) Acute exposure of dissociated E7 SC neurons (laminin) to 100 ng/mL TNFα (top panel) or IL-1β (middle panel) provoked paralysis of growth cone advance and rapid degeneration of the morphology of growth cones and neurites as opposed to 10 μg/mL ovalbumin (bottom panel). Stars indicate location of the growth cone/neurite border with respect to first image in each panel, respectively (scale bar = 10 μm). (b) To restrict cytokine exposure exclusively to advancing growth cones, polystyrene beads (2.5 × 105 beads/mL, 4 μm in diameter) coated with TNFα or IL-1β were applied to SC neuron cultures and growth cone-bead encounters observed under phase contrast (63x oil, phase contrast). Following physical contact of growth cones to cytokine-coated beads (TNFα—top panel, IL-1β—middle panel), growth cone motility ceased followed by the progressive degeneration of growth cone morphology upon reaching complete collapse. Contact with ovalbumin-coated beads had no influence on growth cone morphology and advance (bottom panel). (c) E7 SC neurons grown on laminin for 2 days were fixed in paraformaldehyde. Cytokine receptors were revealed by indirect immunocytochemistry and analyzed by confocal microscopy (Zeiss LSM510, 40x oil, NA 1.30). TNFα receptor 1 (TNF-R1) and IL-1β receptor (IL-1R) were expressed on cell bodies (arrows) and neurites were expressed as well as on growth cones and filopodia (arrowheads). In contrast, TNFα receptor 2 (TNF-R2) expression was restricted to cell bodies. (Scale bars: upper panel, 20 μm; lower panel 10 μm). Western blots of whole spinal cord (E7 chick) extracts revealed immunoreactivity (stars) against avian cytokine receptors TNF-R1 (48 kDa), TNF-R2 (70 kDa), and IL-1R (76 kDa) as determined in spinal cord lysates (50 μg total protein per lane).
Next, we quantified growth cones advance (the spatial displacement of the growth cone/neurite boundary over time) in SC explant cultures upon acute exposure to TNFα, IL-1β, or ovalbumin (Ov, control) (Figure 2). Both TNFα and IL-1β caused a rapid, dose-dependent paralysis of growth cone advance and even neurite retraction within less than 10 minutes of addition compared to Ov (Figures 2(a) and 2(b)). Acute bath application of 100 ng/mL TNFα or IL-1β reduced the percentage of advancing growth cones by 83 ± 6% (n = 48, *P < 0.05) compared to bath application of 10 μg/mL Ov. In contrast, a considerable fraction of growth cones resumed advance following bath application of 50 ng/mL TNFα or IL-1β (TNFα: 42 ± 14%, n = 24, *P < 0.05 and IL-1β: 30 ± 8% n = 20, *P < 0.05) however at much slower growth rates (31 ± 6 μm/h and 21 ± 5 μm/h, resp.) compared to growth rates prior to cytokines application (TNFα: 79 ± 8 μm/h, n = 22 and IL-1β: 89 ± 9 μm/h, n = 26). As expected, application of 10 μg/mL Ov had no effect on growth cone advance (before application: 108 ± 10 μm/h and after application: 113 ± 6 μm/h). According to reports and our own findings, we tested whether ROS intermediates were implicated in cytokine-mediated growth cone paralysis and collapse. Scavenging ROS with 10 μM MnTBAP or inhibition of NADPH oxidase (NOX) activities with 2 μM DPI both protected growth cone advance upon exposure to 100 ng/mL TNFα (69 ± 1%, n = 26 and 71 ± 10%, n = 38 of advancing growth cones, resp.) albeit with decreased growth rates (96 ± 9 μm/h, 17% reduction and 82 ± 9 μm/h, 26% reduction, resp.) compared to 10 μg/mL Ov (Figure 2(c)). Growth cone advance was not affected by an addition of MnTBAP (124 ± 10 μm/h) or DPI (98 ± 13 μm/h) in the presence of ovalbumin during the period of observation. Growth cone advance in the presence of 100 ng/mL IL-1β was also rescued by 10 μM MnTBAP or 2 μM DPI (77 ± 12%, n = 38 and 69 ± 6%, n = 50, resp.) (Figure 2(d)). Exogenous addition of ROS to growth cones (100 μM hydrogen peroxide) also reduced the percentage of advancing growth cones (44 ± 2%, n = 42, *P < 0.05).
Figure 2.
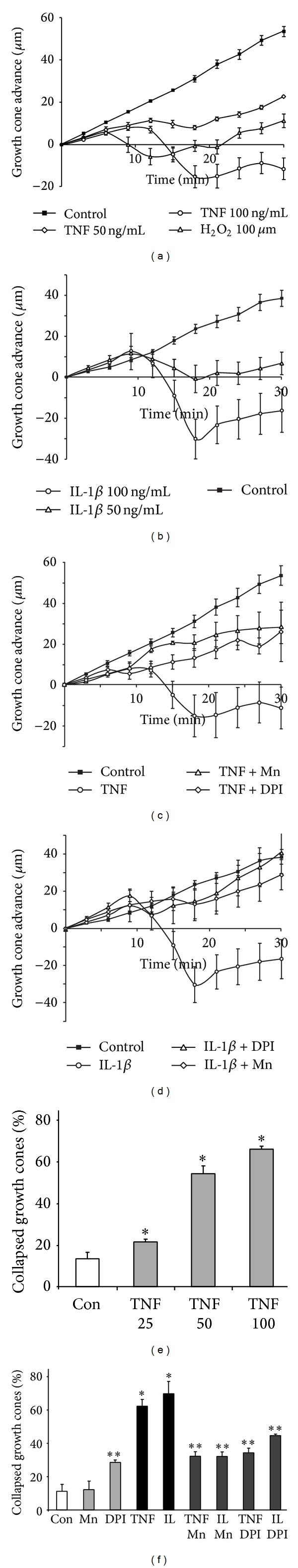
TNFα and IL-1β provoke a redox-sensitive collapse of growth cone motility and morphology. Advancing growth cones in SC neurons cultures (laminin) were randomly selected and images were acquired at 3 min time intervals (20x magnification, phase contrast) in the presence of 10 μM MnTBAP, 2 μM DPI, or PBS (equal volume) before and after acute exposure (t = 6 min) to TNFα (50 or 100 ng/mL), IL-1β (50 or 100 ng/mL), or ovalbumin (10 μg/mL). Growth cone advance was measured as the extension of the growth cone/neurite boundary (μm) per time interval and plotted against time (min) with slopes indicating growth rates. (a and b) Growth cones ceased motility and advance within minutes upon exposure to cytokines (open diamonds, open circles) compared to a steady growth cone advance under control (ovalbumin, closed squares). Growth cones exposed to 100 μM H2O2 (open triangles) mostly responded with paralysis, yet slow recovery was measured. In the presence of 50 ng/mL cytokines (open diamonds), growth cones resumed advance after a lag phase however at much slower growth rates, whereas no recovery was detected at concentrations of 100 ng/mL TNFα or IL-1β (open circles). (c and d) ROS scavenging with 5 μM MnTBAP (open triangles) or NOX inhibition with 2 μM DPI (open diamonds) rescued growth cone advance upon acute exposure to 100 ng/mL TNFα (c) or 100 ng/mL IL-1β (d), respectively (open circles). All data were obtained from at least three different dissections (duplicate cultures each, >30 growth cones total) with error bars representing SEM. (e) TNFα elicited a dose-dependent growth cone collapse at concentrations higher than 50 ng/mL. Growth cones with collapsed morphology were quantified (random fields of view) 30 min after application to allow for possible recovery of morphology. (f) Preincubation of SC neuron culture either with 10 μM MnTBAP or with 2 μM DPI provided significant protection against growth cone collapse in the presence of 100 ng/mL TNFα or 100 ng/mL IL-1β (dark grey bars; **P < 0.05) as opposed to cytokines alone (black bars), which caused substantial growth cone collapse (dark bar, *P < 0.05) compared to control (open bar). A presence of 10 μM MnTBAP (light grey bar, Mn) had no effect on basal levels of collapsed growth cones, whereas 2 μM DPI increased the percentage of collapse growth cones. All data (e and f) were obtained from at least three different dissections (duplicate cultures each). Error bars represent ±SEM.
Both cytokines also elicited a dose-dependent growth cone collapse quantified 30 min after addition as shown for TNFα (25 ng/mL: 16 ± 3%, n = 135; 50 ng/mL: 59 ± 4%, ∗P < 0.05, n = 125; 100 ng/mL: 71 ± 8%, *P < 0.05, n = 122) compared to 10 μg/mL Ov (13 ± 3%, n = 69) (Figure 2(e)). Moreover, scavenging ROS (10 μM MnTBAP) or inhibiting NOX (2 μM DPI) (32 ± 4%, n = 100 and 34 ± 4%, n = 99, resp., ∗∗P < 0.05) protected growth cone morphology from the presence of 100 ng/mL TNFα as opposed to the absence of pharmacological agents (63 ± 7%, n = 87, ∗P < 0.05) (Figure 2(f)). Similarly, MnTBAP (31 ± 3%, n = 156) and DPI (44 ± 1%, n = 186) also negated IL-1β-induced growth cone collapse (69 ± 8%, n = 196, *P < 0.05). The percentage of collapsed growth cone remained unaltered by the presence of 10 μM MnTBAP (11 ± 6%, n = 99), yet it did significantly increase with 2 μM DPI (27 ± 1%, n = 63). Taken together, these data revealed that TNFα and IL-1β caused rapid degeneration of growth cone morphology and complete loss of motility through a redox-dependent mechanism. The rapid response of growth cones to cytokines together with the expression of cytokines receptors on growth cones strongly suggested a direct mechanism as opposed to indirect effects through neurodegenerative processes originating in neuronal cell bodies.
3.2. A Cytokine-Activated NADPH Oxidase Generates ROS in Growth Cones and Cell Bodies of SC Neurons
To determine the generation of intracellular ROS in advancing growth cones exposed to cytokines, we employed quantitative ratiometric fluorescence analysis utilizing the oxidation-sensitive fluorescent indicator 2′,7′-dihydrodichlorofluorescein (DCF, 10 μM) and the redox inert fluorescence indicator Calcein Blue (CB, 4 μM). Ratiometric analysis distinguishes changes in DCF fluorescence due to ROS formation from dilution/concentration effects simply due to rapid changes in growth cone morphology. Bath application of 100 ng/mL TNFα or 100 ng/mL IL-1β elicited a rise in the relative maximum DCF/CB fluorescence ratio indicative of the formation of ROS (Figure 3). As shown in Figure 3(a) (false colored DCF/CB ratio images), ROS formation was sustained and preceded loss of morphology of the advancing growth cone reflected by atrophy and beading of filopodia, condensation of growth cone body, and retraction. Quantitative analysis demonstrated a significant and sustained ROS formation in advancing growth cones upon exposure to TNFα (100 ng/mL, n = 14), IL-1β (100 ng/mL, n = 10), or 100 μM hydrogen peroxide (positive control, n = 11) compared to 10 μg/mL ovalbumin (negative control, n = 22) (Figure 3(b)). Neither loading with DCF (131 ± 18 μm/h, n > 15) nor loading with CB (118 ± 14 μm/h, n > 15) at the concentrations used affected growth cone advance per se compared to unloaded control (113 ± 9 μm/h).
Figure 3.
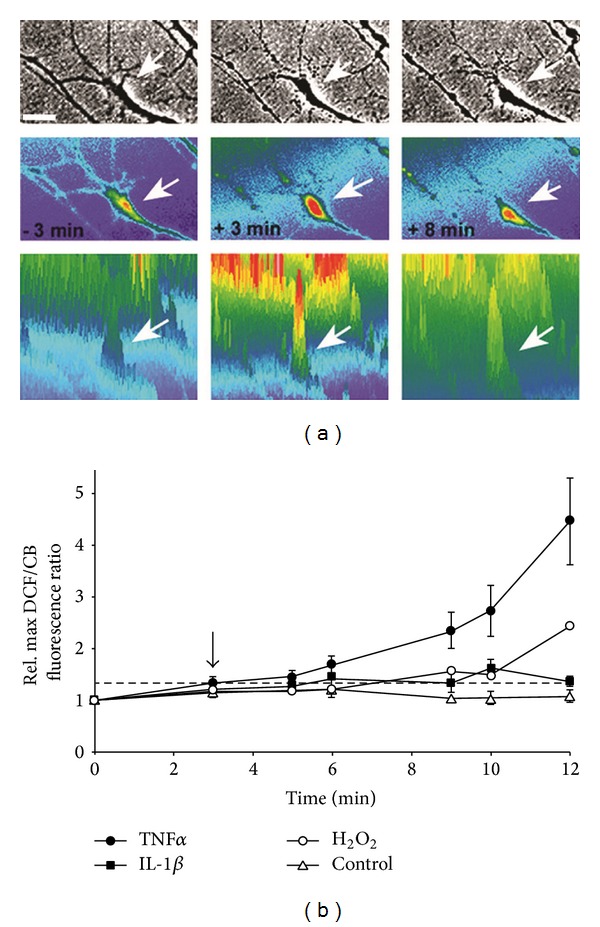
TNFα stimulates ROS formation in advancing growth cones. ROS formation in advancing SC neuron growth cones was revealed by ratiometric fluorescence imaging. Dissociated E7 chick SC neurons (laminin) were loaded (30 min) with the oxidation-sensitive fluorescent indicator 2′,7′-dihydrodichlorofluorescein (10 μM DCF) and the oxidation-inert fluorescent indicator Calcein blue (4 μM CB). Images of randomly selected growth cones were acquired under FITC fluorescence illumination (DCF), DAPI fluorescence illumination (CB), and phase contrast at short time intervals before and after addition of stimuli (40x, oil, identical parameters). (a) Intracellular ROS formation is represented by a heat spectrum ranging from blue (basal ROS levels) to red (increased ROS levels). Growth cone morphology (arrow) disintegrated over a time period of 11 minutes upon acute exposure to 100 ng/mL TNFα (phase images, upper panel). Ratio imaging (middle panel) revealed a sharp increase in ROS production (t = +3 min) shortly after TNFα exposure that persisted (t = +8 min) compared to basal levels (t = −3 min) further illustrated by profile images of ratiometric images (lower panel) (Scale bar = 5 μm). Note the loss of filopodia and lamellipodia and the contraction of the growth cone body (last panel) were preceded by an increase in ROS. (b) ROS formation in advancing SC neuron growth cones was quantified by ratiometric fluorescence imaging. Maximum DCF and CB fluorescence intensities per growth cone area were determined on a pixel-by-pixel basis and DCF/CB ratios calculated for each condition and time point. All DCF/CB ratios were normalized (average DCF/CB ratio at t = 0 min) and plotted against time. Exposure to 100 ng/mL TNFα (filled circles, n = 14), 100 ng/mL IL-1β (filled squares, n = 10), or 100 μM hydrogen peroxide (positive control, open circles, n = 11) at t = 3 min (arrow) elicited a significant ROS formation (stippled line = significance threshold) in advancing growth cones within less than 5 min upon addition compared to 10 μg/mL ovalbumin (negative control, open triangles, n = 22).
To examine NOX-dependent ROS production in neuronal growth cones, we utilized freshly isolated growth cone particle preparations (GCPs) obtained from chick E10 forebrain neurons, which are capable of showing complex responses to extrinsic stimuli [37–39]. As shown in Figure 4(a), addition of 100 ng/mL TNFα to laminin-adherent GCPs resulted in MAP kinase and JNK activation, which was corroborated in cultured forebrain neurons. No basal MAP kinase activity (phosphor MAP kinase) was detectable in GCPs under control conditions possibly due a lack of growth factors (serum free plating conditions). Indicative for the formation of a functional NOX multiprotein complex, the NOX activator PMA (200 ng/mL) significantly increased the relative plasma membrane association of p67phox in chick forebrain neurons (Figure 4(b)). Moreover, native gel electrophoresis and NBT staining revealed several NADPH oxidoreductase activities in GCPs, one colocalizing with NOX2 immunoreactivity. The large membrane subunit NOX2 and the cytosolic subunits p67phox and p47phox were also detected in lysates of SC neurons (Figure 4(b)). Addition of TNFα (100 ng/mL) to DCF-loaded GCPs stimulated a significant, dose-dependent increase in ROS formation, which was negated by the NOX inhibitor DPI (5 μM) or the ROS scavenger NAC (1 mM) (Figure 4(c)). The small spatial dimensions of plated GCPs prevented ROS quantification utilizing microscopic imaging. Therefore, we resorted to a protocol involving lysis as previously established [14]. Moreover, plasma membrane association of p67phox in GCPs significantly increased in response to 100 ng/mL TNFα, which was abolished by the lipid raft disrupter methyl-β-cyclodextrin, suggesting that the assembly of NOX occurred predominantly in lipid rafts (Figure 4(d)). The insignificant increase of p67phox in plasma membrane fractions from methyl-β-cyclodextrin-treated cells could result from unspecific adsorption due to the dramatic change in membrane lipid composition. TNFα-coated polystyrene beads or bath application of 100 ng/mL TNFα to DCF-loaded SC neurons stimulated a transient increase in DCF fluorescence intensity in neuronal cell bodies (Figures 5(a) and 5(b)). Cytokine-stimulated ROS formation was dose-dependent with significant stimulation at concentrations higher than 50 ng/mL and saturation at 100 ng/mL (data not shown). Maximum ROS formation (1.62 ± 0.1, *P < 0.01) in SC neuron cell bodies occurred 5.3 ± 0.8 min following bath application of 100 ng/mL TNFα lasting for 6.4 ± 0.6 min (16 ± 2 cells per time interval) (Figure 5(c)). 100 ng/mL IL-1β also increased ROS formation (1.39 ± 0.02, *P < 0.01) within 5 ± 0.7 min upon application and lasted for 7.8 ± 1.4 min (n = 20 ± 2 cells per time interval). Scavenging ROS with 2 mM NAC or 10 μM MnTBAP both suppressed TNFα-stimulated ROS formation (0.75 ± 0.11; n = 29 and 0.99 ± 0.05; n = 65, resp.) to levels indistinguishable from control (10 μg/mL ovalbumin, 0.99 ± 0.05, n = 123) in contrast to TNFα alone (1.53 ± 0.05, *P < 0.01, n = 184) (Figure 5(d)). Inhibiting NOX activity (5 μM DPI) also negated ROS formation (1.03 ± 0.07; n = 70, *P < 0.01) upon exposure to TNFα. Similar data were obtained upon acute addition of 100 ng/mL IL-1β to SC neurons (data not shown). As our positive control, 200 μM hydrogen peroxide greatly increased relative maximum DCF fluorescence intensity (1.75 ± 0.02; n = 109, *P < 0.01), which was abolished in the presence of 10 μM MnTBAP (0.97 ± 0.02; n = 109) (Figure 5(d)). Depleting Rac1 activity using adenoviral-mediated expression of FLAG-tagged, dominant-negative Rac1N17 completely suppressed TNFα or IL-1β-stimulated ROS formation (1.13 ± 0.05, n = 93 and 0.84 ± 0.05, n = 71, resp.) as opposed to lacZ expression (TNFα: 1.53 ± 0.05, *P < 0.05, n = 184, and IL-1β: 1.30 ± 0.05, *P < 0.05, n = 117, resp.) (Figure 6(a)) [31]. In contrast, expression of constitutively active Rac1V12 in SC neurons was sufficient to increase ROS formation (1.65 ± 0.04, n = 450, *P < 0.05) compared to 200 μM hydrogen peroxide (1.98 ± 0.16, n = 60, *P < 0.05) (Figures 6(b) and 6(c)). ROS scavenging with 2 mM NAC or 10 μM MnTBAP as well as NOX inhibition with 10 μM DPI abolished Rac1V12-stimulated ROS formation (1.11 ± 0.09, n = 62; 0.96 ± 0.09, n = 109; and 1.05 ± 0.07, n = 138, resp.). Similar results were obtained using purified recombinant GST chimeras of Rac1V12 and Rac1N17 transfected into E7 SC neurons by trituration loading (data not shown). Expressing Rac1N17 alone (1.03 ± 0.04, n = 247) or lacZ (0.99 ± 0.03, n = 105), our control, had no effect on basal levels of ROS (Figure 6(c)). However addition of 2 mM NAC or 10 μM DPI to Rac1N17-expressing SC neurons both reduced ROS formation significantly below basal levels (0.76 ± 0.04, n = 94 and 0.55 ± 0.02, n = 106, resp., *P < 0.05). These findings revealed that TNFα and IL-1β stimulate a functional assembly of a Rac1-regulated NOX2 complex in the plasma membrane of neuronal growth cones resulting in a transient increase in ROS formation.
Figure 4.
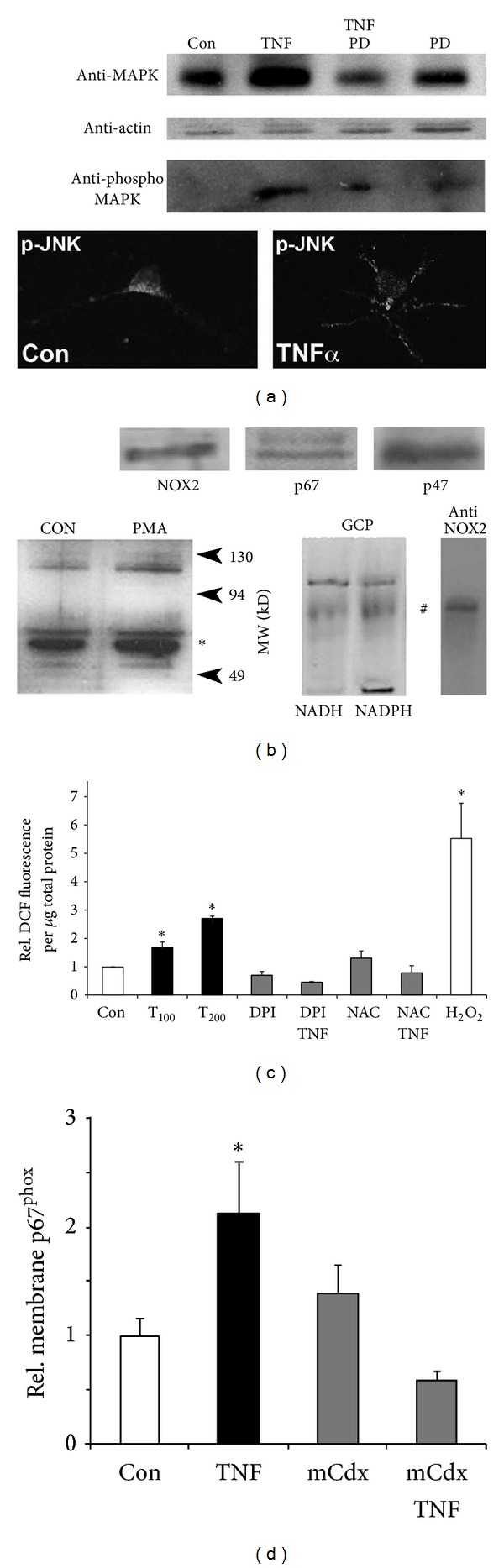
NADPH oxidase mediates ROS formation in growth cones exposed to TNFα. (a) Exposure of freshly isolated growth cone particles (GCPs) to TNFα activates MAP kinase. GCPs were plated on laminin in Krebs buffer (2 h), washed, and incubated with the MAP kinase inhibitors PD98059 (25 μM, 20 min) or PBS (equivalent volume) prior to an addition of 100 ng/mL TNFα. Attached GCPs were lysed and equal amounts of total protein were subjected to SDS gel electrophoresis (10%) followed by western blotting and detection (chemiluminescence) of MAP kinase (top panel) and phospho-MAP kinase immunoreactivity (bottom panel). Actin served as a loading control (middle panel). Exposure of GCPs to TNFα stimulated MAP kinase activity (increased phospho-MAP kinase), which was negated by PD98059. Also, TNFα treatment enhanced expression of MAP kinase (increases in total MAP kinase), a response apparently diminished by a presence of PD98059. Confocal images acquired (63x, oil) of cultured chick forebrain neurons (laminin, 24 h) exposed to 100 ng/mL of TNFα (TNFα) revealed phospho-JNK immunoreactivity (white signal) as a discrete, punctate pattern in cell bodies and neuronal process in contrast to the homogenous appearance in neuronal soma under control conditions (PBS, equivalent volume). Cultures were fixed with 4% paraformaldehyde and immunostained against phospho-JNK. (b) Neuronal growth cones contain a functional NADPH oxidase activity. Lysates obtained from SC neuron cultures revealed immunoreactivity against the large membrane subunit NOX2 and the cytosolic subunits p67phox and p47phox. Incubation of chick (E7) forebrain neurons with the NOX activator PMA (400 ng/mL) increased plasma membrane association of the cytosolic subunit p67phox (∗) compared to controls (Con) indicative for the formation of the NOX multiprotein complex. Native gel electrophoresis, combined with in-gel NBT staining, of freshly isolated growth cone particles (GCPs) obtained from E10–12 chick forebrain demonstrated one NADPH oxidoreductases activity (#) colocalizing with NOX2 immunoreactivity. (c) GCPs were loaded with 10 μM DCF (30 min) in the presence of 2 μM DPI (NOX inhibitor), 500 μM NAC (antioxidant), or PBS (mock, equivalent volume) prior to addition of TNFα (45 min). DCF fluorescence was determined in GCP lysates, adjusted to total soluble protein, and normalized to control (Con, open bar). TNFα stimulated a significant, dose-dependent ROS formation in GCPs (T100 = 100 ng/mL, T200 = 200 ng/mL, *P < 0.05), which was abolished by DPI or NAC. Peroxide (H2O2) served as a positive control. (d) Plasma membrane association of p67phox normalized to control conditions was quantified (western blotting) using plasma membrane-enriched fractions. Exposure of GCPs to 200 ng/mL TNFα significantly increased the relative plasma membrane association of p67phox (TNF, *P < 0.05) compared to control (Con) indicating the formation of a functional NOX complex. Preincubation of GCPs with methyl-β-cyclodextrin (0.1%) negated plasma membrane translocation of p67phox in the presence of TNFα (mCdx-TNF). All data represent SEM from at least two experiments (triplicate conditions each) with *P < 0.05.
Figure 5.
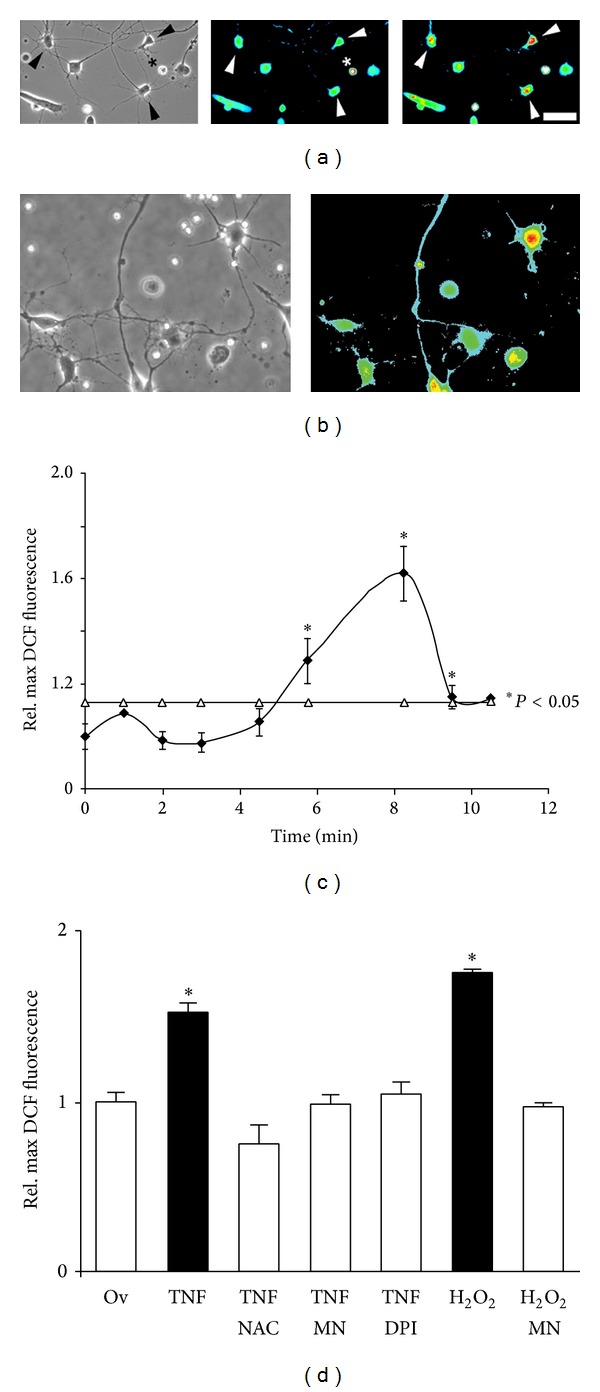
Cytokines elicit ROS formation in cell bodies of SC neurons. Dissociated E7 SC neurons were loaded with 10 μM DCF (30 min), allowed to recover for 15 min, and then incubated (45 min) with NAC, MnTBAP, DPI, or PBS (equal volume) prior to cytokines exposure. Images were acquired at identical parameters (20x). (a) DCF-loaded SC neurons exhibited a distinct neuronal morphology (arrowheads) extending one long and several shorter neuronal processes (left panel, phase contrast image). Note cultures are virtually free of nonneuronal cells. Under control conditions, SC neuron cell bodies displayed basal levels of ROS formation (middle panel, arrowheads). Degenerating cells (star) exhibit very high fluorescence intensity in the absence of any stimuli. Addition of 100 ng/mL TNFα stimulated a robust increase in intracellular ROS (right panel, arrowheads, 8 min after addition). Fluorescence images were false colored (heat spectrum) with fluorescence intensities increasing from blue (basal ROS levels) to red (high ROS levels). (b) TNFα-coated polystyrene beads increased DCF fluorescence intensity in SC neurons indicative of ROS formation. Although qualitative in nature, SC neurons with multiple bead contacts (arrow head) as opposed to single bead contact (asterisk) displayed more intense DCF fluorescence. (c) Maximum DCF fluorescence intensities per neuronal cell body were determined on a pixel-by-pixel basis after background subtraction over time and all values normalized to the average maximum DCF fluorescence intensity under control conditions at t = 0 min (relative maximum DCF fluorescence intensity). SC neurons responded with a transient increase in ROS formation in cell bodies (16 ± 2 cells per time interval) upon exposure to 100 ng/mL TNFα (2.5) min (threshold of statistical significance *P < 0.01 shown as open triangles). (d) 100 ng/mL TNFα stimulated a significant increase in ROS formation (*P < 0.01), which was negated by the presence of ROS scavengers (TNFα-NAC 2 mM or TNFα-MnTBAP 10 μM, resp.) or NOX inhibitor (TNFα-DPI 5 μM) to levels indistinguishable from 10 μg/mL ovalbumin (Ov), our control. As a control for DCF loading, 200 μM hydrogen peroxide (H2O2) greatly increased relative maximum DCF fluorescence (*P < 0.01), which was suppressed in the presence of 10 μM MnTBAP (H2O2-Mn). All measurements were obtained from at least three independent dissections (duplicate cultures each).
Figure 6.
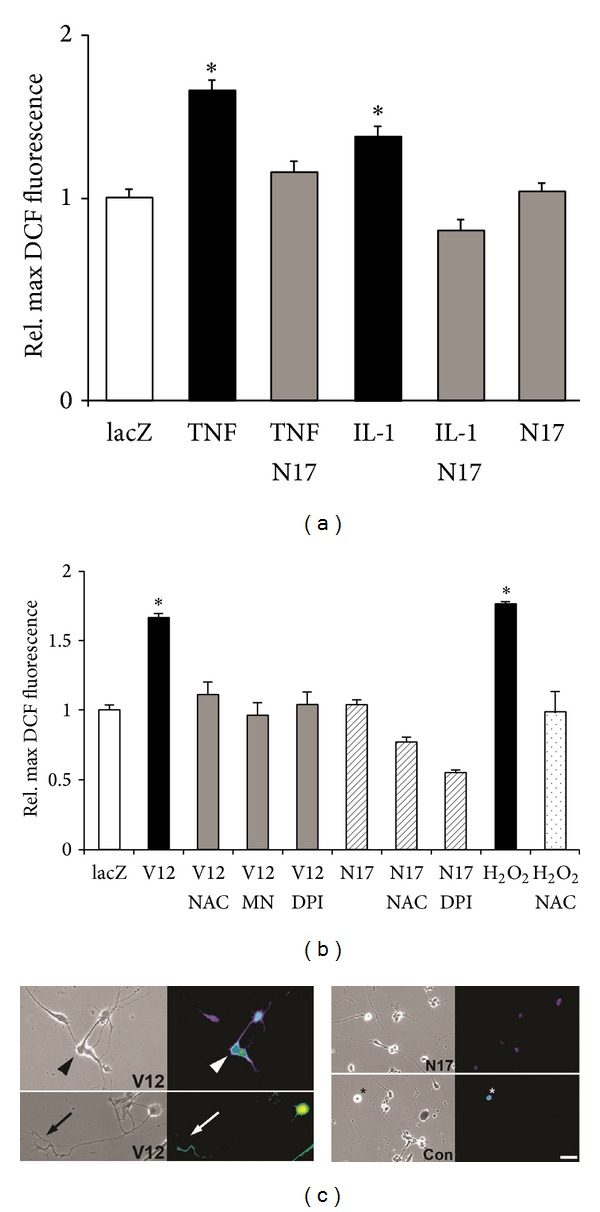
TNFα and IL-1β stimulate a Rac1-mediated ROS formation in SC neurons. Dissociated SC neurons grown on laminin were infected with recombinant, replication deficient adenovirus (200 moi, infection at the time of plating) carrying FLAG-tagged constitutively active Rac1V12, FLAG-tagged dominant negative Rac1N17, or lacZ. Three days after infection, SC neurons were loaded with 10 μM DCF (30 min), incubated with NAC, MnTBAP, or DPI, and exposed to cytokines (100 ng/mL). Random images were acquired (20x) under FITC fluorescence illumination, relative maximum DCF fluorescence intensity was quantified, and all values normalized to lacZ-expressing SC neurons exposed to 10 μg/mL Ovalbumin (lacZ), our control. (a) Expression of RacN17 completely abolished ROS formation in SC neurons upon exposure to 100 ng/mL TNFα or IL-1β (TNFα-N17 and IL-1β-N17, resp.) compared to lacZ-expressing SC neurons (TNFα and IL-1β, resp., *P < 0.01) without altering basal levels of ROS formation (N17). (b) Expression of Rac1V12 was sufficient to induce ROS formation in SC neurons, which was negated by 2 mM NAC, 10 μM MnTBAP, or 10 μM DPI. Peroxide (200 μMol/L H2O2) served as a DCF loading control. (c) DCF-loaded SC neurons expressing RacV12 (V12) revealed increases in ROS formation both in cell bodies (arrowhead) as well as in neuronal growth cones (arrow) and distal neurites (left panel). In contrast, SC neurons expressing RacN17 (N17) exhibited basal DCF fluorescence intensity compared to lacZ (Con) expressing SC neurons (right panel). Note the intense DCF fluorescence in a degenerating cell (asterisk). A heat spectrum ranging from blue (basal ROS levels) to red (increased ROS levels) indicates ROS production (scale bar = 40 μm).
3.3. TNFα and IL-1β Attenuate Neurite Outgrowth of SC Neurons in a Redox-Dependent Manner
Next, we examined whether the redox-dependent impairment of growth cone advance is of consequence for neurite outgrowth. Bath application of 100 ng/mL TNFα or IL-1β attenuated neurite outgrowth in SC neuron cultures in a dose-dependent manner indicated by a shift in the distribution to shorter neurite lengths compared to 10 μg/mL ovalbumin (Ov), our control (Figures 7(a) and 7(b), Table 1). Scavenging ROS (10 μM MnTBAP) rescued neurite outgrowth in the presence of TNFα and IL-1β, whereas the NOX inhibitor (2 μM DPI) provided only partial protection of neurite outgrowth (Figures 7(c) and 7(d)). Notably, neurite outgrowth of SC neurons on laminin exhibited inherent redox dependence even in the absence of TNFα or IL-1β (Figures 7(e) and 7(f)). In accordance with previous reports, increasing concentrations of the ROS scavenger N-acetyl-L-cysteine (2 mM NAC, 50% reduction) or 20 μM MnTBAP (10% reduction) inhibited neurite outgrowth compared to control, whereas 10 μM MnTBAP was ineffective [15]. However, we measured a significant increase in neurite outgrowth with 5 μM MnTBAP (25% increase).
Figure 7.
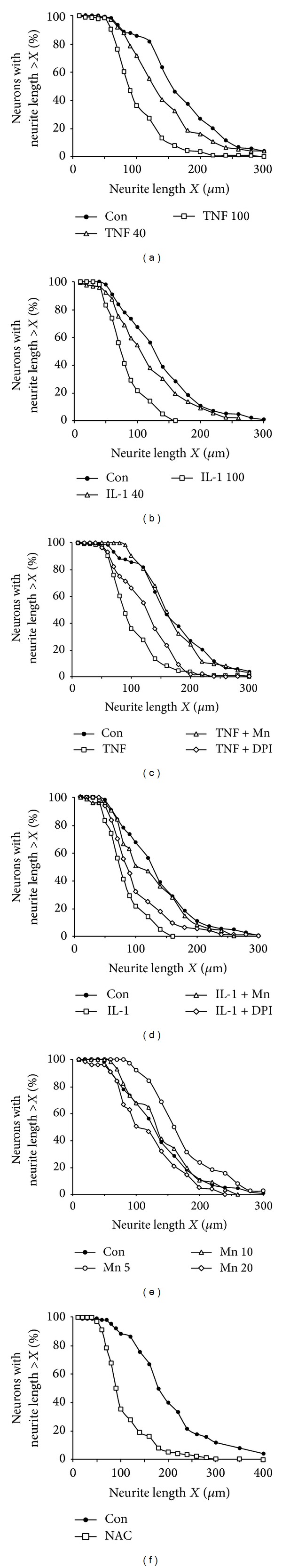
TNFα and IL-1β impede neurite outgrowth in a redox-sensitive manner. Dissociated SC neurons were grown on laminin past the onset of neurite initiation and then incubated with 10 μM MnTBAP, 2 μM DPI, or PBS (10 μL) for 1 h prior to bath application of cytokines or ovalbumin (6 to 8 h) followed by fixation (2% glutaraldehyde). The longest neurite per neuron was measured of randomly selected neurons and the percentage of neurons with a given neurite length was plotted against neurite length. (a and b) Persistent presence of TNFα (a) or IL-1β (b) significantly reduced neurite outgrowth in a dose-dependent manner (40 ng/mL, open triangles; and 100 ng/mL, open squares) indicated by the shift of the neurite length distribution to shorter neurites compared to control (10 μg/mL ovalbumin, filled circles). (c and d) Despite a continuous presence of 100 ng/mL TNFα ((c), open squares) or 100 ng/mL IL-1β ((d), open squares), scavenging ROS with 10 μM MnTBAP (open triangles) rescued neurite outgrowth compared to controls (10 μg/mL ovalbumin, filled circles), whereas inhibiting NOX activity with 2 μM DPI (open diamonds) was only partially protective. (Neurite number measured ≥ 60 for each condition, two experiments, and duplicate cultures). (e) In the presence of 20 μM MnTBAP (open diamonds), neurite outgrowth was significantly decreased compared to control (PBS, filled circles). However, concentrations of 10 μM MnTBAP (open triangles) did not significantly alter neurite outgrowth and 5 μM MnTBAP (open circles) on the contrary causes a significant increase in neurite length. (Neurite number measured > 75 for each condition, two experiments, and duplicate cultures). (f) Lastly, an overabundance of the radical scavenger NAC (2 mM, open squares) dramatically reduced neurite outgrowth compared to control (PBS, filled circles) indicated by the shift towards shorter neurites.
Table 1.
Redox-sensitive inhibition of neurite outgrowth in response to TNFα and IL-1β.
| Condition | NL50 ± SEM | n |
|---|---|---|
| Control (10 mg/mL Ov) | 147 ± 16 μm | 104 |
| TNFα 40 ng/mL | 118 ± 6 μm∗ | 74 |
| TNFα 100 ng/mL | 86 ± 13 μm∗ | 173 |
| TNFα 100 ng/mL + 10 mM MnTBAP | 145 ± 13 μm | 62 |
| TNFα 100 ng/mL + 2 mM DPI | 113 ± 12 μm∗∗ | 56 |
| Control (10 mg/mL Ov) | 128 ± 7 μm | 61 |
| IL-1β 40 ng/mL | 109 ± 4 μm∗ | 92 |
| IL-1β 100 ng/mL | 80 ± 3 μm∗ | 107 |
| IL-1β 100 ng/mL + 10 mM MnTBAP | 112 ± 9 μm∗∗ | 113 |
| IL-1β 100 ng/mL + 2 mM DPI | 87 ± 8 μm∗ | 78 |
| PBS | 128 ± 12 μm | 156 |
| MnTABP 5 mM | 161 ± 10 μm∗ | 82 |
| MnTABP 10 mM | 133 ± 7 μm | 75 |
| MnTABP 20 mM | 115 ± 7 μm∗ | 56 |
| PBS | 179 ± 7 μm | 103 |
| NAC 2 mM | 89 ± 7 μm∗ | 102 |
SC neuron cultures were incubated with pharmacological inhibitors (MnTBAP, DPI) or PBS prior to addition of cytokines for 6–8 hours. After fixation, the average neurite length of the longest neurite per neuron reached by 50% of neurons (NL50) was quantified for each condition. ∗Significant differences from controls. ∗∗Significant difference from cytokine only at P < 0.05 by one-way ANOVA and Dunnett's t-test.
To assess the role of Rac1 on neurite outgrowth, Rac1V12 (constitutively active Rac1) or Rac1N17 (dominant negative Rac1) was introduced as purified recombinant GST chimera into freshly dissected SC neurons by trituration loading [26] (Figure 8). Depletion of Rac1 activity (Rac1N17-GST) in SC neurons protected neurite outgrowth upon addition of 100 ng/mL TNFα (NL50 = 81 ± 5 μm, n = 85, **P < 0.05) or 100 ng/mL IL-1β (NL50 = 79 ± 6 μm, n = 68, **P < 0.05) compared to GST-loaded SC neurons (NL50 = 64 ± 3 μm, n = 75 and NL50 = 69 ± 2 μm, n = 51), respectively (Figure 8(a)). However, introduction of Rac1N17-GST diminished neurite outgrowth even in the absence of adverse stimuli (NL50 = 84 ± 4 μm, n = 132, *P < 0.05) compared to GST-loaded SC neurons (NL50 = 91 ± 3 μm, n = 99) in accordance with previous findings [26, 40]. Constitutive activation of Rac1 (Rac1V12-GST) in SC neurons also resulted in reduction of neurite length (NL50 = 83 ± 5 μm, n = 195, *P < 0.05) compared to GST-loaded SC neurons (NL50 = 98 ± 5 μm, n = 112) (Figure 8(b)). Interestingly, addition of 5 μM MnTBAP restored neurite outgrowth of RacV12-GST-loaded SC neurons (NL50 = 108 ± 6, n = 136, **P < 0.05) to levels indistinguishable from GST-loaded SC neurons (NL50 = 103 ± 7 μm, n = 103, **P < 0.05). This finding implied a role for Rac1 as a regulator of TNFα or IL-1β-stimulated oxidative stress. These findings provided evidence that attenuation of neurite outgrowth in the presence of TNFα or IL-1β required a persistent, Rac1-regulated formation of ROS. Moreover, neurite outgrowth on laminin exhibited an innate redox-sensitivity.
Figure 8.
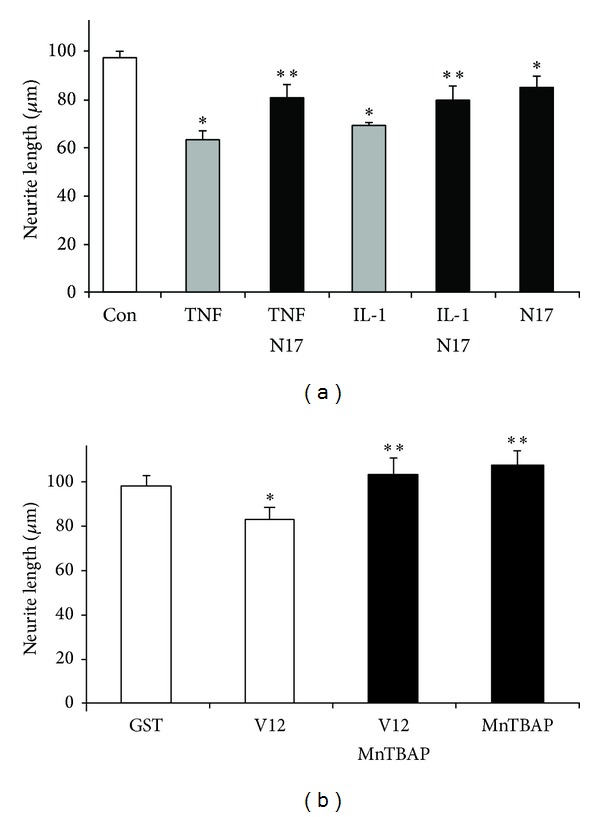
The small GTPase Rac1 mediates redox-dependent neurite outgrowth. Freshly dissected SC neurons were trituration-loaded with purified recombinant Rac1V12-GST, Rac1N17-GST, or GST only (7 mg/mL each) and grown (laminin) after the onset of neurite initiation. Cultures were supplemented with 100 ng/mL TNFα, 100 ng/mL IL-1β, or 10 μg/mL ovalbumin (8 h) and the average neurite length of the longest neurite reached by 50% of SC neurons (NL50) for each condition quantified (n > 50). (a) Introduction of Rac1N17-GST (dominant negative mutation, black bars) significantly protected neurite outgrowth in the presence of TNFα (TNFα-N17, **P < 0.05) or IL-1β (IL-1β-N17, **P < 0.05) compared to SC neurons loaded with GST, which exhibited a great reduction in neurite length in the presence of cytokines (grey bars *P > 0.05) compared to control (ovalbumin, open bar). Not unexpected, Rac1N17-GST even in the absence of cytokines reduced neurite outgrowth (N17, *P < 0.05) compared to control. (b) Similarly, introduction of Rac1V12-GST (constitutively active mutation) significantly reduced neurite lengths (V12, *P < 0.05) compared to GST-loaded SC neurons (GST). Notably, the presence of 5 μM MnTBAP increased neurite outgrowth of Rac1V12-GST-loaded SC neurons (V12-MnTBAP, **P < 0.05) to levels indistinguishable from neurite outgrowth of GST-loaded SC neurons treated with 5 μM MnTBAP (MnTBAP, **P < 0.05) compared to control. All data were obtained from at least three different dissections (duplicate cultures each) with error bars representing.
3.4. Cytokines Elicit a Redox-Dependent Reorganization of Actin Filaments in Neuronal Growth Cones
Earlier studies in neuroblastoma cells revealed oxidative damage to actin due to TNFα exposure [14]. We found that growth cones responded rapidly to cytokine exposure with changes in morphology prior to collapse suggesting reorganization of actin filaments. We quantified the percentage of growth cones responding to cytokines with at least one distinct actin filament-rich, lamellipodial structure as a function of both time and pharmacological treatment (Figure 9). Growth cones exhibited a rapid, yet transient increase in actin filament-rich structures within 15 min upon exposure to 100 ng/mL TNFα (65 ± 5%, n = 180, *P < 0.05) or 100 ng/mL IL-1β (84 ± 5%, n = 180, *P < 0.05) compared to control (23 ± 5%, n = 180, ∗P < 0.05) and persisted up to 30 min (TNFα: 67 ± 5%, n = 180, *P < 0.05, and IL-1β: 58 ± 5%, n = 180, *P < 0.05) (Figures 9(a) and 9(d)). However, the percentage of growth cones with actin filament-rich structures greatly subsided 45 min after addition (TNFα: 40 ± 4%, n = 180, **P < 0.05, and IL-1β: 37 ± 5%, n = 180, **P < 0.05) compared to the initial response. Inhibiting NOX activity (5 μM DPI) greatly diminished the percentage of responding growth cones upon bath application of cytokines (DPI + TNFα: 32 ± 6%, n = 180, and DPI + IL-1β: 49 ± 6%, n = 180, **P < 0.05) compared to absence of DPI (TNFα: 60 ± 6%, n = 180, *P < 0.05, and IL-1β: 76 ± 6%, n = 180, *P < 0.05) measured 15 min after addition of cytokines (Figures 9(b) and 9(e)). The percentage of responding growth cones was not altered by DPI treatment alone (18 ± 6%, n = 180) compared to control (23 ± 6%, n = 180). Similarly, scavenging ROS with 20 μM MnTBAP also negated the formation of actin filament-rich structures in growth cones exposed to cytokines (Mn + TNFα: 24 ± 8%, n = 180, and Mn + IL-1β: 18 ± 8%, n = 180) to levels indistinguishable from control (18 ± 4%, n = 180) compared to cytokines alone (TNFα: 66 ± 8%, n = 180, *P < 0.05, and IL-1β: 84 ± 8%, n = 180, *P < 0.05) (Figures 9(c) and 9(f)). A presence of MnTBAP did not alter the percentage of growth cones with actin filament-rich structures (18 ± 3%, n = 180). Taken together, these data suggested that TNFα and IL-1β stimulated a redox-dependent, yet transient reorganization of actin filaments in neuronal growth cones prior to collapse of morphology.
Figure 9.
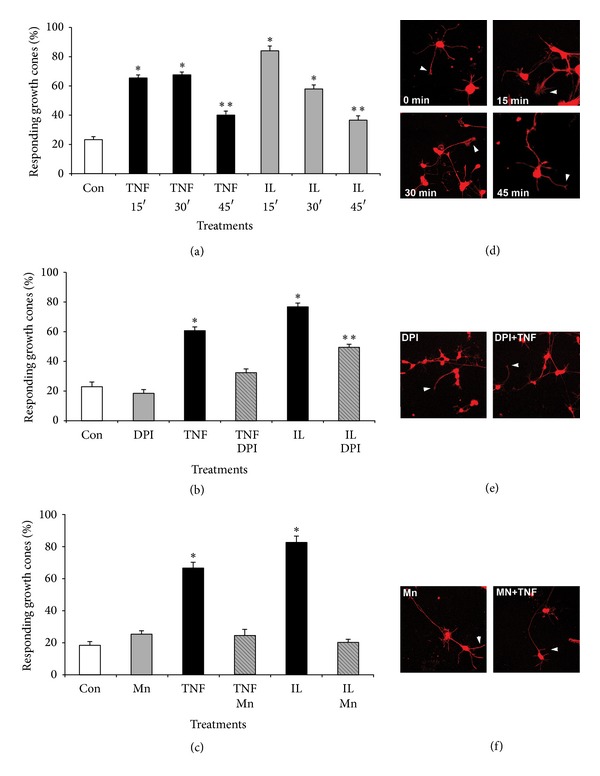
Cytokines elicit redox-dependent reorganization of actin filaments in neuronal growth cones. SC neurons grown on laminin (2 days) were incubated with 40 μM MnTBAP, 5 μM DPI or left untreated prior to addition of TNFα or IL-1β (200 ng/mL) for increasing time periods (15 min, 30 min, and 45 min). Cultures were fixed, permeabilized (Triton-X-100), and stained with rhodamine phalloidin to reveal filamentous actin. Random images were acquired (40x, confocal microscope), growth cones were scored for the presence of at least one, distinct actin filament-rich structure, and all values normalized to control conditions (% responding growth cones). (a) Both TNFα and IL-1β significantly increased the percentage of growth cones with actin-filament rich structures (*P < 0.01) at 15 min and 30 min after exposure followed by a significant decrease at 45 min after exposure (**P < 0.01 compared to t = 15 min) as opposed to control. (b) Inhibition of NOX activity with 2 μM DPI largely negated the formation of actin-filament rich structures upon exposure to TNFα (TNF-DPI) or IL-1β (IL-DPI, **P < 0.01) as opposed to TNFα only (TNFα, *P < 0.01) or IL-1β only (IL, *P < 0.01), respectively. Large actin filament-rich structures were not affected by DPI in the absence of cytokines (DPI) compared to control (Con). (c) Scavenging ROS with 10 μM MnTBAP abolished the formation of actin filament-rich structures in response to TNFα (TNFα-Mn) or IL-1β (IL-Mn) when compared to TNFα only (TNF, *P < 0.01) or IL-1β only (IL, *P < 0.01). The degree of actin filament-rich structures in the presence of MnTBAP alone (Mn) was indistinguishable from control (Con). ∗Significant difference from control and ∗∗significant difference from respective cytokine treatment at P < 0.05 by Kruskal Wallis test and Dunnett's t-test. (d–f) Cultured SC neurons were treated with pharmacological inhibitors prior to bath application of cytokines, fixed with 4% paraformaldehyde, and stained for actin filaments with rhodamine phalloidin (red signal). (d) Representative images of SC neurons with increasing exposure time to TNFα. Actin filament-rich structures in growth cones (lamellipodia) are indicated with arrowheads. (e) Incubation with DPI largely abolished the formation of actin filament-rich structures in SC neurons cultures. (f) Scavenging ROS with MnTBAP effectively negated appearance of actin filament-rich structures in growth cones of SC neurons.
4. Discussion
We demonstrated that long-term exposure of SC neurons to the proinflammatory cytokines TNFα and IL-1β provoked the loss of growth cone motility and the subsequent degeneration of growth cone morphology (collapse). These changes in growth cone advance and behavior translated into the impairment of neurite outgrowth and disruption of process architecture of SC neurons, which was rescued either by scavenging ROS, inhibiting NOX activity, or depleting Rac1 activity despite a presence of TNFα or IL-1β. Both cytokines stimulated the formation of ROS intermediates and a transient phase of actin filament organization in advancing growth cones. Importantly, ROS intermediates and actin filament reorganization preceded paralysis of growth cone motility and collapse of morphology implying a causative action of this signaling mechanism. Taking into account that exhaustive ROS scavenging (excess of antioxidants) also impaired neurite outgrowth, it is feasible that productive growth cone motility could demand an optimal level of ROS intermediates. We propose that a redox rheostat under the regulation of the Rac1 shapes growth cone motility and hence neurite outgrowth, in response to many extrinsic stimuli other than cytokines (Figure 10).
Figure 10.
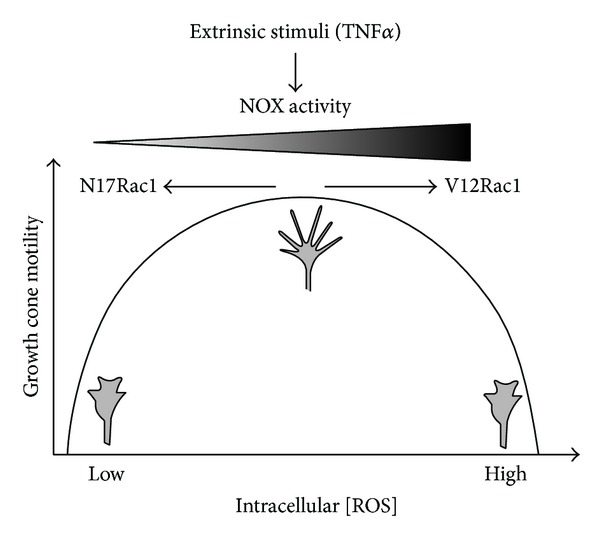
A Rac1-dependent redox rheostat regulates growth cone motility in response to extrinsic stimuli. Exposure of neuronal growth cones either to TNFα or to IL-1β rapidly stimulates ROS generation through NOX activity under the regulation of the small GTPase Rac1 and concomitant paralysis and degeneration of morphology of growth cones. Moreover, growth cone motility is not only sensitive to the overabundance of ROS either via NOX activation or via constitutive Rac1 activity (Rac1V12) but is also impaired by a substantial depletion of ROS (antioxidants) or inhibition of Rac1 activity (Rac1N17). Consequently, productive growth cone motility requires an optimal concentration of intracellular ROS generated by a Rac1-regulated NOX activity. It is plausible that this Rac1/NOX-redox rheostat is responsive to many extrinsic stimuli including cytokines, growth factors, hormones, and cell adhesion molecules for which redox signaling mechanism has been demonstrated.
Prolonged bath application of TNFα or IL-1β (6–8 h) to dissociated SC neurons or SC explants stunted neurite outgrowth in a dose-dependent manner (Figure 7, Table 1) in accordance with previous findings in hippocampal neurons or neuroblastoma cells [8, 9]. In lieu of the well-documented apoptotic potency of TNFα and IL-1β, it was imperative to determine whether inhibition of neurite outgrowth had its origin in neuronal cell bodies (apoptosis, necrosis) or directly in the distal compartment of growth cones. Foremost, cytokines were applied to SC neurons after the onset of neurite outgrowth with the majority of processes longer than two cell diameters. Since the number of neurites per neuron (a measure of neurite initiation) remained unaltered and no measureable neuronal cell death occurred over the time period of cytokine exposure (6–8 h) as determined by a live/dead fluorescence assay (calcein green/propidium iodide counterstaining), neither a decrease in neurite initiation nor a decrease in neuronal cell death could account for the observed reduction in neurite outgrowth. Significant neuronal cell death (>15%) was however apparent 24 hours after cytokine addition and increased to over 40% after 2 days. Evidence for direct action of cytokines on growth cone motility and morphology was obtained by live-video phase microscopy (Figures 1 and 2). Acute exposure to TNFα or IL-1β (bath applied) impaired growth cone motility in a dose-dependent manner within 10 to 15 minutes upon application. Initial paralysis of growth cone advance was followed by degeneration of morphology and subsequent collapse. Restricting cytokine exposure exclusively to advancing growth cones in our bead assay corroborated these findings. In addition, bead assays revealed a transient increase in lamellipodia-like structures or in the vicinity of neuron-bead contacts, which were nonproductive for growth cone advance. This transient increase in actin filaments (Figure 9) was reminiscent of our findings in SH-SY5Y neuroblastoma, yet in the case of primary neurons it revealed a physiological consequence [14]. Direct effects of cytokines on growth cones were further supported by the expression of IL-R1 (IL-1β receptor) and TNF-R1 (high affinity p55 TNFα receptor) on the entire neuronal surface including cell bodies and growth cones (Figure 1). Together these findings provided strong evidence that TNFα and IL-1β directly impaired growth cone motility and morphology as the underlying cause for the attenuation of neurite outgrowth over longer time periods of exposure.
Initial evidence for a role of ROS intermediates was suggested by studies demonstrating that antioxidants (ROS scavenging) or DPI (inhibiting NOX-like activities) largely rescued neurite outgrowth, growth cone motility, growth cone morphology, and lastly actin filament organization in the presence of cytokines. Interestingly, neurite outgrowth of SC neurons on laminin exhibited innate redox dependence. Whereas moderate MnTBAP concentrations (5 μM) significantly increased neurite outgrowth, concentration higher than 10 μM stunted neurite outgrowth corroborating studies in Aplysia neurons [15]. It is plausible that moderate MnTBAP concentration (5 μM) scavenges excessive amounts of superoxide derived from mitochondrial respiration and as byproduct of several enzymatic reactions. Under basal culture conditions, this excessive superoxide might compromise optimal growth cone motility (actin dynamics, tubulin dynamics) and hence the observed enhancement of neurite outgrowth by moderate concentrations of scavenger [41–43]. In contrast, higher MnTBAP concentrations could impair vital redox-dependent mechanism necessary for proper neurite outgrowth [44]. Using the oxidation-sensitive fluorescence indicator DCF, we demonstrated substantial yet transient generation of ROS (likely superoxide) in growth cones and cell bodies of SC neurons upon exposure to TNFα and IL-1β (Figures 3 and 5). Similar results were obtained using the superoxide-specific fluorescence indicator dihydroethidium, yet investigations suffered from considerable neurotoxicity of dihydroethidium (data not shown). Previous studies in chick cortical neurons and human SH-SY5Y neuroblastoma cells demonstrated superoxide production in response to TNFα utilizing dihydroethidium oxidation or a SOD-inhibitable cytochrome C oxidation as demonstrated [14, 45]. A significant contribution from nonneuronal cells (microglia) was unlikely since (i) SC neuron cultures contained less than 3% nonneuronal cells, (ii) analyzed cells all exhibited distinct neuronal characteristics (i.e., round cell body, one process longer than 2 cell diameters), and (iii) microglia invade and populate the developing CNS at late embryonic or even postnatal stages [46]. Ratiometric imaging demonstrated significant ROS production in advancing growth cones within 3 to 5 min upon exposure to inflammatory stress as opposed to changes in motility and morphology observed 10 to 15 min following exposure. Several findings strongly suggested a NOX2-like activity as the source of ROS in SC neurons, in particular superoxide [14, 47]. SC neurons exhibited immunoreactivity against NOX2 and the cytosolic subunits p47phox and p67phox. A presence of other NOX isoforms can however not be excluded, yet antibody quality against NOX1 or NOX3 was insufficient to produce a conclusive finding. TNFα stimulated a dose-dependent increase in ROS intermediates in freshly isolated growth cone particle preparations in conjunction with a presence of NADPH oxidoreductases activity. The phorbol ester PMA as well as TNFα induced translocation of the cytosolic subunit p67phox to plasma membranes. Parallel studies on the translocation of the cytosolic subunit p47phox to plasma membranes were hampered by the considerable variability of immunoreactivity. Expression of NOX-like activities was reported in both primary PNS and CNS neurons [48–51]. Lastly, the antioxidant MnTBAP inhibits the TNFα or IL-1 β-stimulated ROS formation. Also, pharmacological inhibition with DPI was effective in blocking TNFα or IL-1β-mediated responses of SC neurons including ROS formation and partial rescue of growth motility, neurite outgrowth, growth cone collapse, and actin filament reorganization (Figures 2, 4, 5, 7, and 9). Notably, all these experiments revealed considerable toxicity of DPI and thus, depending on the duration of DPI application, put restrictions on the usable concentrations of DPI. Mechanistically, DPI blocks single electron transport reactions such as NOX-mediated generation of superoxide. Nevertheless, numerous other enzymatic reactions are also compromised foremost electron transport of complex I in mitochondria, which could account for the observed toxicity. Lastly, depleting Rac1 activity negated cytokine-mediated ROS formation in SC neurons, whereas Rac1 overexpression alone was sufficient to increase ROS formation. The small GTPase Rac1 emerged as a pivotal regulator of cytokine-stimulated ROS formation in SC neurons in accordance with previous reports [22, 52, 53]. In our study, depleting Rac1 activity abolished ROS formation in the presence of TNFα or IL-1β yet without affecting basal ROS levels (Figures 6(b)–6(d)). In contrast, overexpression of constitutively active Rac1 alone was sufficient to stimulated ROS generation in SC neurons and also reduced neurite outgrowth, which was not further exacerbated upon addition of TNFα or IL-1β (data not shown). Effects of Rac1 depletion or overexpression were addressed by trituration loading of purified recombinant Rac1-GST mutant chimeras since adenoviral-mediated expression of Rac1 mutants required a 3-day expression period incompatible with neurite outgrowth analysis [26, 31]. Not unexpectedly, introduction of Rac1N17 or Rac1V12 resulted in a measurable reduction in neurite outgrowth on laminin even in the absence of cytokines [11, 26, 54, 55]. In support of our findings, numerous reports link Rac1 as a regulator of ROS intermediates in cellular signal transduction. Neurite extension in PC12 cells upon NGF stimulation encompasses increases in Rac1 activity and H2O2 formation [56]. A Rac1 mutant lacking residues 124–135 of the insert region blocking ROS generation disrupted membrane ruffling and mitogenesis in fibroblasts and caused a downregulation of RhoA [44, 57–59]. With respect to actin cytoskeleton dynamics, TNFα and IL-1β induced a transient phase of actin filament reorganization (increases in actin filament density) in neuronal growth cones, which was however nonproductive for motility. Interestingly, actin filament reorganization was not repeatable with another addition of cytokines even after wash-out and extensive recover time suggesting permanent or persistent damage to actin filament reorganization by cytokines. Irreversible oxidative damage (carbonylation) to actin was detectable in neuroblastoma cells exposed to TNFα [14]. These findings provided evidence that NOX activation in neuronal growth cones serves as source of ROS intermediates in response to cytokines and that ROS intermediates are likely causative for cytokine-mediated degeneration of neuronal growth cones.
Inflammatory and oxidative stress is a hallmark of neurodegeneration in most chronic, acute, and even some psychiatric CNS pathologies [3, 60, 61]. The proinflammatory cytokines TNFα and IL-1β are important to orchestrate inflammatory and oxidative stress in the disease and aging CNS although pleiotropic actions in the adult and developing CNS are known, hence establishing a complex and delicate balance between neuroprotection and neurotoxicity. Members of the Nox/Duox family have emerged as [46, 62] key sources of oxidative stress in aging and the progression of many pathologies beyond the CNS and have been recognized as key therapeutic targets [63–66]. Our findings provide support for a role of NOX activity in primary neurons as a principal source of oxidative stress triggered by TNFα or IL-1β [16]. Overabundance of ROS intermediates is likely to disrupt proper actin filament dynamics, which is vital for the plasticity and morphology of distal neuronal processes including neurite outgrowth, sprouting, and spine dynamics. Strategies to block NOX activities could thus proof beneficial to blunt inflammatory stress in the diseased and aging CNS and to halt cognitive decline [66, 67].
Acknowledgments
I would like to extend my appreciation to Drs. Richard Bridges and Mike Kavanaugh (University of Montana) and Dr. Paul Letourneau (University of Minnesota, Minneapolis) for many helpful discussions that were vital in shaping this research. Thanks to Shellie Stewart-Smeets for her contribution on actin filament experiments and Dianne Cabrie for technical assistance. I am indebted to Dr. Mark Wright (University of Alaska, Fairbanks) for his support with native gel electrophoresis and Dr. Kelly Auer (University of Alaska, Fairbanks) for her support with MAP kinase analysis. Support for this work was provided by the Christopher Reeve Paralysis Foundation (KA1-0004-2), NIH/NINDS grant 1U54NS41069, and COBRE The University of Montana.
Abbreviations
- DCF:
2′,7′-dichlorofluorescein
- DMEM:
Dulbecco's Modified Eagle Medium
- DPI:
Diphenylene iodonium
- FBS:
Fetal bovine serum
- GST:
Glutathione-S-transferase
- IL-1β:
Interleukin-1 beta
- MnTBAP:
manganese(III) tetrakis(4-benzoic acid) porphyrin chloride
- NAC:
N-acetyl-L-cysteine
- ROS:
Reactive oxygen species
- SC:
Spinal cord
- TBS:
Tris-buffered saline
- TNFα:
Tumor necrosis factor alpha.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Amor S, Puentes F, Baker D, van der Valk P. Inflammation in neurodegenerative diseases. Immunology. 2010;129(2):154–169. doi: 10.1111/j.1365-2567.2009.03225.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cacquevel M, Lebeurrier N, Chéenne S, Vivien D. Cytokines in neuroinflammation and Alzheimer’s disease. Current Drug Targets. 2004;5(6):529–534. doi: 10.2174/1389450043345308. [DOI] [PubMed] [Google Scholar]
- 3.Lucas S-M, Rothwell NJ, Gibson RM. The role of inflammation in CNS injury and disease. British Journal of Pharmacology. 2006;147(supplement 1):S232–S240. doi: 10.1038/sj.bjp.0706400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McCoy MK, Tansey MG. TNF signaling inhibition in the CNS: implications for normal brain function and neurodegenerative disease. Journal of Neuroinflammation. 2008;5, article 45 doi: 10.1186/1742-2094-5-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mrak RE, Griffin WST. Glia and their cytokines in progression of neurodegeneration. Neurobiology of Aging. 2005;26(3):349–354. doi: 10.1016/j.neurobiolaging.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 6.Viviani B, Bartesaghi S, Corsini E, Galli CL, Marinovich M. Cytokines role in neurodegenerative events. Toxicology Letters. 2004;149(1–3):85–89. doi: 10.1016/j.toxlet.2003.12.022. [DOI] [PubMed] [Google Scholar]
- 7.Mathew SJ, Haubert D, Krönke M, Leptin M. Looking beyond death: a morphogenetic role for the TNF signalling pathway. Journal of Cell Science. 2009;122(12):1939–1946. doi: 10.1242/jcs.044487. [DOI] [PubMed] [Google Scholar]
- 8.Münch G, Gasic-Milenkovic J, Dukic-Stefanovic S, et al. Microglial activation induces cell death, inhibits neurite outgrowth and causes neurite retraction of differentiated neuroblastoma cells. Experimental Brain Research. 2003;150(1):1–8. doi: 10.1007/s00221-003-1389-5. [DOI] [PubMed] [Google Scholar]
- 9.Neumann H, Schweigreiter R, Yamashita T, Rosenkranz K, Wekerle H, Barde Y-A. Tumor necrosis factor inhibits neurite outgrowth and branching of hippocampal neurons by a Rho-dependent mechanism. The Journal of Neuroscience. 2002;22(3):854–862. doi: 10.1523/JNEUROSCI.22-03-00854.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gungabissoon RA, Bamburg JR. Regulation of growth cone actin dynamics by ADF/cofilin. Journal of Histochemistry and Cytochemistry. 2003;51(4):411–420. doi: 10.1177/002215540305100402. [DOI] [PubMed] [Google Scholar]
- 11.Luo L. Rho GTPases in neuronal morphogenesis. Nature Reviews Neuroscience. 2000;1(3):173–180. doi: 10.1038/35044547. [DOI] [PubMed] [Google Scholar]
- 12.Moldovan L, Irani K, Moldovan NI, Finkel T, Goldschmidt-Clermont PJ. The actin cytoskeleton reorganization induced by Rac1 requires the production of superoxide. Antioxidants and Redox Signaling. 1999;1(1):29–43. doi: 10.1089/ars.1999.1.1-29. [DOI] [PubMed] [Google Scholar]
- 13.Thannickal VJ, Fanburg BL. Reactive oxygen species in cell signaling. American Journal of Physiology: Lung Cellular and Molecular Physiology. 2000;279(6):L1005–L1028. doi: 10.1152/ajplung.2000.279.6.L1005. [DOI] [PubMed] [Google Scholar]
- 14.Barth BM, Stewart-Smeets S, Kuhn TB. Proinflammatory cytokines provoke oxidative damage to actin in neuronal cells mediated by Rac1 and NADPH oxidase. Molecular and Cellular Neuroscience. 2009;41(2):274–285. doi: 10.1016/j.mcn.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Munnamalai V, Suter DM. Reactive oxygen species regulate F-actin dynamics in neuronal growth cones and neurite outgrowth. Journal of Neurochemistry. 2009;108(3):644–661. doi: 10.1111/j.1471-4159.2008.05787.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chakraborty S, Kaushik DK, Gupta M, Basu A. Inflammasome signaling at the heart of central nervous system pathology. Journal of Neuroscience Research. 2010;88(8):1615–1631. doi: 10.1002/jnr.22343. [DOI] [PubMed] [Google Scholar]
- 17.Morgan MJ, Kim Y-S, Liu Z. Lipid rafts and oxidative stress-induced cell death. Antioxidants and Redox Signaling. 2007;9(9):1471–1483. doi: 10.1089/ars.2007.1658. [DOI] [PubMed] [Google Scholar]
- 18.Kawahara BT, Quinn MT, Lambeth JD. Molecular evolution of the reactive oxygen-generating NADPH oxidase (Nox/Duox) family of enzymes. BMC Evolutionary Biology. 2007;7, article 109 doi: 10.1186/1471-2148-7-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nature Reviews Immunology. 2004;4(3):181–189. doi: 10.1038/nri1312. [DOI] [PubMed] [Google Scholar]
- 20.Groemping Y, Rittinger K. Activation and assembly of the NADPH oxidase: a structural perspective. Biochemical Journal. 2005;386(3):401–416. doi: 10.1042/BJ20041835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leonardi A, Ellinger-Ziegelbauer H, Franzoso G, Brown K, Siebenlist U. Physical and functional interaction of filamin (actin-binding protein-280) and tumor necrosis factor receptor-associated factor 2. The Journal of Biological Chemistry. 2000;275(1):271–278. doi: 10.1074/jbc.275.1.271. [DOI] [PubMed] [Google Scholar]
- 22.Peppelenbosch M, Boone E, Jones GE, et al. Multiple signal transduction pathways regulate TNF-induced actin reorganization in macrophages: inhibition of Cdc42-mediated filopodium formation by TNF. Journal of Immunology. 1999;162(2):837–845. [PubMed] [Google Scholar]
- 23.Singh R, Wang B, Shirvaikar A, et al. The IL-1 receptor and rho directly associate to drive cell activation in inflammation. Journal of Clinical Investigation. 1999;103(11):1561–1570. doi: 10.1172/JCI5754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuhn TB. Growing and working with spinal motor neurons. Methods in Cell Biology. 2003;71:67–87. doi: 10.1016/s0091-679x(03)01005-7. [DOI] [PubMed] [Google Scholar]
- 25.Bottenstein JE, Sato GH. Growth of a rat neuroblastoma cell line in serum free supplemented medium. Proceedings of the National Academy of Sciences of the United States of America. 1979;76(1):514–517. doi: 10.1073/pnas.76.1.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kuhn TB, Brown MD, Bamburg JR. Rac1-dependent actin filament organization in growth cones is necessary for b1-integrin-mediated advance but not for growth on poly-D-lysine. Journal of Neurobiology. 1998;37:524–540. doi: 10.1002/(sici)1097-4695(199812)37:4<524::aid-neu3>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 27.Owen Lockerbie R, Miller VE, Pfenninger KH. Regulated plasmalemmal expansion in nerve growth cones. The Journal of Cell Biology. 1991;112(6):1215–1227. doi: 10.1083/jcb.112.6.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chang S, Rathjen FG, Raper JA. Extension of neurites on axons is impaired by antibodies against specific neural cell surface glycoproteins. The Journal of Cell Biology. 1987;104(2):355–362. doi: 10.1083/jcb.104.2.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stoeckli ET, Kuhn TB, Duc CO, Ruegg MA, Sonderegger P. The axonally secreted protein axonin-1 is a potent substratum for neurite growth. The Journal of Cell Biology. 1991;112(3):449–455. doi: 10.1083/jcb.112.3.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hall A. Rho GTpases and the actin cytoskeleton. Science. 1998;279(5350):509–514. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- 31.Kuhn TB, Brown MD, Wilcox CL, Raper JA, Bamburg JR. Myelin and collapsin-1 induce motor neuron growth cone collapse through different pathways: inhibition of collapse by opposing mutants of rac1. The Journal of Neuroscience. 1999;19(6):1965–1975. doi: 10.1523/JNEUROSCI.19-06-01965.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brown M, Cornejo B, Kuhn T, Bamburg J. Cdc42 stimulates neurite outgrowth and formation of growth cone filopdia and lamellipodia. Journal of Neurobiology. 2000;43:352–364. doi: 10.1002/1097-4695(20000615)43:4<352::aid-neu4>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 33.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227(5259):680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 34.Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proceedings of the National Academy of Sciences of the United States of America. 1979;76(9):4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kuhn TB, Schmidt MF, Kater SB. Laminin and fibronectin guideposts signal sustained but opposite effects to passing growth cones. Neuron. 1995;14(2):275–285. doi: 10.1016/0896-6273(95)90285-6. [DOI] [PubMed] [Google Scholar]
- 36.Wride MA, Sanders EJ. Expression of tumor necrosis factor-α (TNFα)-cross-reactive proteins during early chick embryo development. Developmental Dynamics. 1993;198(3):225–239. doi: 10.1002/aja.1001980308. [DOI] [PubMed] [Google Scholar]
- 37.Cypher C, Letourneau PC. Identification of cytoskeletal, focal adhesion, and cell adhesion proteins in growth cone particles isolated from developing chick brain. Journal of Neuroscience Research. 1991;30(1):259–265. doi: 10.1002/jnr.490300126. [DOI] [PubMed] [Google Scholar]
- 38.Helmke S, Pfenninger KH. Growth cone enrichment and cytoskeletal association of non-receptor tyrosine kinases. Cell Motility and the Cytoskeleton. 1995;30(3):194–207. doi: 10.1002/cm.970300304. [DOI] [PubMed] [Google Scholar]
- 39.Negre-Aminou P, Pfenninger KH. Arachidonic acid turnover and phospholipase A2 activity in neuronal growth cones. Journal of Neurochemistry. 1993;60(3):1126–1136. doi: 10.1111/j.1471-4159.1993.tb03263.x. [DOI] [PubMed] [Google Scholar]
- 40.Jin Z, Strittmatter SM. Rac1 mediates collapsin-1-induced growth cone collapse. The Journal of Neuroscience. 1997;17(16):6256–6263. doi: 10.1523/JNEUROSCI.17-16-06256.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cooper CE, Patel RP, Brookes PS, Darley-Usmar VM. Nanotransducers in cellular redox signaling: modification of thiols by reactive oxygen and nitrogen species. Trends in Biochemical Sciences. 2002;27(10):489–492. doi: 10.1016/s0968-0004(02)02191-6. [DOI] [PubMed] [Google Scholar]
- 42.Dalle-Donne I, Rossi R, Milzani A, di Simplicio P, Colombo R. The actin cytoskeleton response to oxidants: from small heat shock protein phosphorylation to changes in the redox state of actin itself. Free Radical Biology and Medicine. 2001;31(12):1624–1632. doi: 10.1016/s0891-5849(01)00749-3. [DOI] [PubMed] [Google Scholar]
- 43.Lassing I, Schmitzberger F, Björnstedt M, et al. Molecular and structural basis for redox regulation of β-actin. Journal of Molecular Biology. 2007;370(2):331–348. doi: 10.1016/j.jmb.2007.04.056. [DOI] [PubMed] [Google Scholar]
- 44.Nimnual AS, Taylor LJ, Bar-Sagi D. Redox-dependent downregulation of Rho by Rac. Nature Cell Biology. 2003;5(3):236–241. doi: 10.1038/ncb938. [DOI] [PubMed] [Google Scholar]
- 45.Gustafson SJ, Dunlap KL, McGill CM, Kuhn TB. A nonpolar blueberry fraction blunts NADPH oxidase activation in neuronal cells exposed to tumor necrosis factor-α . Oxidative Medicine and Cellular Longevity. 2012;2012:12 pages. doi: 10.1155/2012/768101.768101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Merrill JE. Tumor necrosis factor alpha, interleukin 1 and related cytokines in brain development: normal and pathological. Developmental Neuroscience. 1992;14(1):1–10. doi: 10.1159/000111642. [DOI] [PubMed] [Google Scholar]
- 47.Hordijk PL. Regulation of NADPH oxidases: the role of Rac proteins. Circulation Research. 2006;98(4):453–462. doi: 10.1161/01.RES.0000204727.46710.5e. [DOI] [PubMed] [Google Scholar]
- 48.Noh KM, Koh JY. Induction and activation by zinc of NADPH oxidase in cultured cortical neurons and astrocytes. The Journal of Neuroscience. 2000;20(23, article RC111) doi: 10.1523/JNEUROSCI.20-23-j0001.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vallet P, Charnay Y, Steger K, et al. Neuronal expression of the NADPH oxidase NOX4, and its regulation in mouse experimental brain ischemia. Neuroscience. 2005;132(2):233–238. doi: 10.1016/j.neuroscience.2004.12.038. [DOI] [PubMed] [Google Scholar]
- 50.Dvorakova M, Höhler B, Richter E, Burritt JB, Kummer W. Rat sensory neurons contain cytochrome b558 large subunit immunoreactivity. NeuroReport. 1999;10(12):2615–2617. doi: 10.1097/00001756-199908200-00032. [DOI] [PubMed] [Google Scholar]
- 51.Tammariello SP, Quinn MT, Estus S. NADPH oxidase contributes directly to oxidative stress and apoptosis in nerve growth factor-deprived sympathetic neurons. The Journal of Neuroscience. 2000;20(1, article RC53) doi: 10.1523/JNEUROSCI.20-01-j0006.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Puls A, Eliopoulos AG, Nobes CD, Bridges T, Young LS, Hall A. Activation of the small GTPase Cdc42 by the inflammatory cytokines TNFα and IL-1, and by the Epstein-Barr virus transforming protein LMP1. Journal of Cell Science. 1999;112(17):2983–2992. doi: 10.1242/jcs.112.17.2983. [DOI] [PubMed] [Google Scholar]
- 53.Wojciak-Stothard B, Entwistle A, Garg R, Ridley A. Regulation of TNF alpha-induced reorganization of the actin cytoskeleton and cell-cell junctions by rho, rac, and cdc42 in human endothelial cells. Journal of Cell Physiology. 1998;176:150–165. doi: 10.1002/(SICI)1097-4652(199807)176:1<150::AID-JCP17>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 54.Hall A, Nobes CD. Rho GTPases: molecular switches that control the organization and dynamics of the actin cytoskeleton. Philosophical Transactions of the Royal Society B: Biological Sciences. 2000;355(1399):965–970. doi: 10.1098/rstb.2000.0632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Meyer G, Feldman EL. Signaling mechanisms that regulate actin-based motility processes in the nervous system. Journal of Neurochemistry. 2002;83(3):490–503. doi: 10.1046/j.1471-4159.2002.01185.x. [DOI] [PubMed] [Google Scholar]
- 56.Suzukawa K, Miura K, Mitsushita J, et al. Nerve growth factor-induced neuronal differentiation requires generation of rac1-regulated reactive oxygen species. The Journal of Biological Chemistry. 2000;275(18):13175–13178. doi: 10.1074/jbc.275.18.13175. [DOI] [PubMed] [Google Scholar]
- 57.Freeman JL, Abo A, Lambeth JD. Rac “insert region” is a novel effector region that is implicated in the activation of NADPH oxidase, but not PAK65. The Journal of Biological Chemistry. 1996;271(33):19794–19801. doi: 10.1074/jbc.271.33.19794. [DOI] [PubMed] [Google Scholar]
- 58.Joneson T, Bar-Sagi D. A Rac1 effector site controlling mitogenesis through superoxide production. The Journal of Biological Chemistry. 1998;273(29):17991–17994. doi: 10.1074/jbc.273.29.17991. [DOI] [PubMed] [Google Scholar]
- 59.Karnoub AE, Der CJ, Campbell SL. The insert region of Rac1 is essential for membrane ruffling but not cellular transformation. Molecular and Cellular Biology. 2001;21(8):2847–2857. doi: 10.1128/MCB.21.8.2847-2857.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Allan SM, Rothwell NJ. Cytokines and acute neurodegeneration. Nature Reviews Neuroscience. 2001;2(10):734–744. doi: 10.1038/35094583. [DOI] [PubMed] [Google Scholar]
- 61.Sei Y, Vitkovic L, Yokoyama MM. Cytokines in the central nervous system: regulatory roles in neuronal function, cell death and repair. Neuroimmunomodulation. 1995;2(3):121–133. doi: 10.1159/000096881. [DOI] [PubMed] [Google Scholar]
- 62.Vitkovic L, Bockaert J, Jacque C. “Inflammatory” cytokines: neuromodulators in normal brain? Journal of Neurochemistry. 2000;74(2):457–471. doi: 10.1046/j.1471-4159.2000.740457.x. [DOI] [PubMed] [Google Scholar]
- 63.Bedard K, Krause K-H. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiological Reviews. 2007;87(1):245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 64.Kawahara T, Lambeth JD. Molecular evolution of Phox-related regulatory subunits for NADPH oxidase enzymes. BMC Evolutionary Biology. 2007;7, article 178 doi: 10.1186/1471-2148-7-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Krause K-H, Lambeth D, Krönke M. NOX enzymes as drug targets. Cellular and Molecular Life Sciences. 2012;69(14):2279–2282. doi: 10.1007/s00018-012-1006-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lambeth JD, Krause K-H, Clark RA. NOX enzymes as novel targets for drug development. Seminars in Immunopathology. 2008;30(3):339–363. doi: 10.1007/s00281-008-0123-6. [DOI] [PubMed] [Google Scholar]
- 67.Gao H-M, Zhou H, Hong J-S. NADPH oxidases: novel therapeutic targets for neurodegenerative diseases. Trends in Pharmacological Sciences. 2012;33(6):295–303. doi: 10.1016/j.tips.2012.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]