

Abstract
After severe burn injury and other major traumas, glucose tolerance tests have demonstrated delayed glucose disposal. This “diabetes of injury” could be explained by insulin deficiency, and several studies have shown that soon after trauma (“ebb phase”) insulin concentrations are reduced in the face of hyperglycemia. After resuscitation of trauma patients (“flow phase”), βcell responsiveness normalizes and plasma insulin levels are appropriate or even higher than expected however, glucose intolerance and hyperglycemia persist. In the acute care setting, several approaches have been used for treating insulin resistance, including: insulin infusion, propranolol, and glucagon-like-peptide-1 (GLP-1). Recently, it was demonstrated that a tetrapeptide with anti-oxidant properties (D-Arg-Dmt-Lys-Phe-NH2, SS31), but not its inactive analogue (Phe-D-Arg-Phe-Lys-NH2, SS20) attenuates insulin resistance in mice maintained on a high fat diet. In this report the effects of SS31 and SS20 on burn induced insulin resistance was studied in mice. Oral glucose tolerance tests (OGTT) were performed in 4 groups of 6 mice with thermal injury with or without pretreatment with SS31 or SS20 and sham controls. In addition, biodistribution of 18FDG was measured in burned mice with and without SS31 treatment and shams (subsets of these animals were also studied by μPET); for comparison purposes groups of 6 cold-stressed mice with and without SS31 treatment were also studied. The results of these studies demonstrate that SS31 but not SS20 ameliorated burn induced insulin resistance. In addition, SS31 treatment resulted in marked reduction in the increased 18FDG uptake by brown adipose tissue (BAT) in burned but not cold-stressed animals; suggesting that the stressors act by different mechanisms. Overall, these studies confirmed that SS31 can be used to reverse burn induced insulin resistance and provide a firm pre-clinical basis for future clinical trials of SS-31 for the treatment of insulin resistance in patients with burn injury.
Keywords: Burn-injury, trauma, insulin-resistance, cold-stress, anti-oxidant oxidative stress, OGTT, μPET, 18FDG, SS31, SS20, ROS
INTRODUCTION
Maintenance of blood glucose levels is one of the most tightly regulated systems in the body, and although all cells require glucose, it is only available from exogenous sources as well as hepatic and to a lesser extent renal cortical gluconeogenisis (1,2). Since glucose cannot be stored in significant amounts except as glycogen in liver and muscle, transport into the cell by specific glucose transporting proteins is essential for normal cellular function (3).
Insulin plays a critical role in maintaining normal levels of plasma glucose. Insulin secretion is stimulated when blood glucose levels rise, which stimulates the uptake of glucose by skeletal muscle and other tissues via specific glucose transporter proteins and decreases glucose production by the liver (4). There are several key proteins in the insulin/glucose regulatory pathway cascade, including insulin receptor substrate 1 (IRS-1) and Akt1/Protein kinase Bα (Akt1/PKBα) (5). When levels of insulin and glucose are abnormally high in the fasting state, in conditions such as Type 2 Diabetes, severe trauma and burn injury, a condition called insulin resistance exists (6).
In the acute care setting, several approaches have been used for treating insulin resistance, including: insulin infusion (7), propranolol (8) and more recently, glucagon-like-peptide-1 (GLP-1) (9). Very recently, we demonstrated that simvastatin can also attenuate burn induced insulin resistance (10). The use of insulin infusion is the most straight forward treatment, however, it can produce severe and potentially life threatening hypoglycemia. Propranolol acts by inhibiting the effect of epinephrine (11,12). The mechanism (s) by which GLP-1 reverses insulin resistance is more complex and involves both insulinotropic effects and direct inhibition of glucagon (13,14). The mechanism for statin effects on insulin resistance are poorly defined but could be related to their anti inflammatory actions. It was demonstrated that a tetrapeptide (D-Arg-Dmt-Lys-Phe-NH2, SS31) with anti-oxidant properties (scavenger of reactive oxygen species, ROS) but not its inactive analogue (Phe-D-Arg-Phe-Lys-NH2, SS20) can reverse the insulin resistance that occurs in mice maintained on a high fat diet (15). In this report the effects of SS31 and SS20 on burn induced insulin resistance was studied with a murine model of burn injury.
Oxidative injury has been implicated a wide variety of clinical disorders including: diabetes and burn injury. However, currently available antioxidants have not proven to be particularly effective for treating these conditions, possibly due to their inability to reach the relevant sites of free radical generation, especially if mitochondria are the primary source of reactive oxygen species (ROS). Oxidative damage to mitochondria has been shown to impair their function and lead to cell death via apoptosis and necrosis. Because dysfunctional mitochondria produce more ROS, a feed-forward loop is set up whereby ROS-mediated oxidative damage to mitochondria favors more ROS generation, resulting in a vicious cycle (16).
The recently developed SS (Szeto-Schiller) peptide antioxidants represent a novel approach for targeted delivery of antioxidants to the inner mitochondrial membrane (17). The most common feature of these peptides centers on alternating aromatic and basic amino acid residues (aromatic-cationic peptides). These peptides can scavenge hydrogen peroxide and peroxynitrite and inhibit lipid peroxidation. Their antioxidant action can be attributed to the presence of a dimethyltyrosine residue. SS31 (D-Arg-Dmt-Lys-Phe-NH2) is among the most potent ROS scavengers in this family of peptides. Since cells are highly permeable to SS31 (with a 3+ charge) and mitochondria are the most electronegative cellular organelles, the peptide accumulates >1,000-fold at the inner mitochondrial membrane (18) where it scavenges ROS, and protects mitochondria from permeability transition, swelling, and cytochrome c release (18). It is believed that the ROS scavenging effect of SS31 is mediated via the dimethyl tyrosine residue in its sequence. Since the dimethyl tyrosine residue in SS31 is not present in SS20 (Phe-D-Arg-Phe-Lys-NH2) this peptide dose not display ROS scavenging properties.
Among the SS peptides, SS31 has been most extensively studied and appears to be highly potent. Studies with isolated mitochondrial preparations and cell cultures have shown that SS31 can scavenge ROS, reduce mitochondrial ROS production, and inhibit mitochondrial permeability transition (19). Excellent efficacy has also been demonstrated with animal models of myocardial ischemia-reperfusion injury (20), ischemic brain injury (21), neurodegeneration (22), islet isolation for transplantation (23), renal fibrosis (24) 1-methyl-4-phenyl-1,2,3,6- tetrahydropyridine (MPTP) neurotoxicity (25) and the insulin resistance that develops in mice maintained on a high fat diet (26).
In this report, the effects of SS31 and SS20 on burn induced insulin resistance were studied with a murine model of burn injury.
MATERIALS AND METHODS
Materials
18F labeled FDG was prepared by routine methods (27). D-Arg-Dmt-Lys-Phe-NH2 (SS31) and Phe-D-Arg-Phe-Lys-NH2 (SS20) were purchased from Peptide 2.0, Herndon, VA. Ethyl ether was purchased from Fisher Scientific Co, Valley Stream Parkway, Melvern PA.
Burn Injury in Mice
Male CD1 mice weighing 25 to 28 g were purchased from Charles River Laboratories (Boston, MA). Full-thickness, non-lethal thermal injuries (30 % total body surface area [TBSA]) were produced, as described previously (28)(29) using a protocol approved by the Subcommittee on Research Animal Care of the Massachusetts General Hospital. Briefly the mice were anesthetized with ether and their dorsal and ventral areas were shaved with animal hair clippers. Under ether anesthesia, the animals were placed in molds (acrylic plastic ¼ inch thick) exposing 30% TBSA (dorsal plus ventral areas) followed by emersion of the dorsal area in a water bath at 90°C for 9 seconds and emersion of the ventral area in the same water bath for 4 seconds. The animals were immediately resuscitated with saline (50 ml/kg) by intraperitoneal (I.P.) injection. The % TBSA was confirmed at necropsy by removal of the skin and measurement of the area burned vs. the total area of the pelt. Sham (control) mice, were anesthetized with ether, shaven, placed in molds, exposed to room temperature water and injected I.P. with saline. After the procedure, the animals were caged individually for the duration of the study without food, but with water provided ab libitum. Animals were monitored for post-treatment complications, including infection and failure of the normal healing process (granulation tissue formation and sloughing of injured skin); animals with either infection or abnormal healing were excluded from the study.
Cold-Stress in Mice
To produce cold stress, the mice were placed in a cold room at 4°C for 24 hours with overnight fasting and access to water ab libitum. The mice were housed three to a cage and the radiopharmaceutical (18FDG) was administered on the following morning.
Effect of SS31 on Oral Glucose Tolerance Tests (OGTT) Performed in Burn Injured and Sham Treated Mice
Burned mice treated with SS31 or SS20 and sham controls were studied at 24 hrs after injury. The doses of SS31 and SS20 were 40.0 mg/kg, injected IP in the resuscitation fluid immediately after injury and 12 h later by IP injection. After fasting overnight a 2 g/kg glucose solution was administered via gavage. Glucose levels were determined on serial whole blood samples with a glucometer (Ascencia® Contour®, Bayer Healthcare LLC Mishawaka IN). Areas under the plasma glucose curves (AUC’s) were calculated using the trapazoidal rule. Initial glucose levels in the sham mice used in these studies never exceeded 100 mg/dl.
Effect of SS31 and SS20 Treatment on Regional Glucose Metabolism
The biodistribution of 18FDG was measured in groups of burned mice, burned mice treated with SS31 of SS20, and sham treated controls. One day after burn injury as described above, SS31 and SS20 was injected IP (40 mg/kg) followed by fasting overnight. On the following morning, the unanesthetized mice were injected with 18FDG (50 μCi) via tail vein. Approximately 60 minutes after tracer injection, the animals were sacrificed by cervical dislocation and complete biodistribution was measured. The results were calculated as the percentage of the injected dose per gram wet weight of tissue (%ID/g). All results were expressed as mean ± SEM. A sub-set of animals was injected with a larger dose of 18FDG (~500 μCi) and the distribution of tracer was evaluated by μPET.
μPET Imaging
Groups of burn injured mice, burn injured mice treated with SS31 or SS20 (40 mg/kg) and sham treated controls were studied by 18FDG-μPET. μPET imaging was performed with a P4 μPET camera (Concord Microsystems Inc. Knoxville, TN). One hour after intravenous injection of 18FDG (~500 μCi) via tail vein (without anesthesia,) the mice were anesthetized, positioned and stabilized in the gantry of the PET camera and a 10 min image was acquired in list mode. The primary imaging characteristics of the P4 camera are in-plane and axial resolutions of ~2 mm FWHM, 63 contiguous slices of 1.27 mm separation and a sensitivity of ~650 cps/μCi. In all animals, the region from the head to the base of the tail was included in the 8.0 cm field of view of the camera. The PET images were reconstructed using an iterative algorithm (Ordered Subset Expectation Maximization, OSEM) and a ramp filter with a cut-off of 0.5. Data for attenuation correction was measured with a rotating point source containing 68Ge. All projection data were corrected for nonuniformity of detector response, dead time, random coincidences, and scattered radiation. The PET camera was cross-calibrated to a well scintillation counter by comparing the camera response from a uniform distribution of an 18F solution in a 5.0 cm cylindrical phantom with the response of a well counter to an aliquot of the same solution.
Statistical Analysis
Statistical analyses were performed using 1-way or 2-way ANOVA (as appropriate) and individual means were compared using Duncan’s New Multiple range test. All results were expressed as mean ± SEM. P-values of <0.05 were considered to be statistically significant.
RESULTS
Figure 1 illustrates glucose time-plasma concentration curves from OGTTs performed on mice with burn injury, burned mice treated with SS31 or SS20 and sham treated controls. Two-way ANOVA demonstrated highly significant main effects of treatment, F3,160= 48.06 p<0.00001 and time, F7,160= 9.90; p<0.00001. Compared with sham treated control animals, burn injury produced marked elevation and delayed normalization of blood glucose levels; baseline (p<0.0001), 5 min (p<0.005), 10 min (p<0.05), 15 min (p<0.05), 30 min (p<0.005), 60 min (p<0.005), 120 min (p<0.005) and 180 min (p<0.05). This elevation was significantly (p<0.05) reduced at 30, 60 and 120 min. by treatment of the burned animals with SS31. In contrast, treatment with SS20 did not reduce burn induced elevations in glucose levels at any time point. In fact, although not statistically significant, at the early times (baseline to 15 min) glucose levels tended to be higher in burned animals treated with SS20 compared with untreated burned animals.
Figure 1.

Effect of thermal injury and SS31 or SS20 treatment on oral glucose tolerance tests in mice; time dependence of blood glucose levels. Each value represents the mean ± sem for 6–12 animals; ***p< 0.005 and *p<0.05 for burn vs sham, ++p<0.005 and +p<0.05 for burn + SS-31 vs burn alone.
Figure 2 shows the AUCs calculated from the blood glucose curves in Figure 1. One-way ANOVA demonstrated a highly significant main effect of treatment on AUC, F3,29= 20.15; p<0.00001. AUCs for burned animals and burned animals treated with SS20 were significantly greater than the values for SS31 treated animals and sham treated controls (p<0.0001). The AUC of the SS31 treated group was significantly reduced compared to burned animals and burned animals treated with SS-20. Overall, the results of these studies clearly demonstrate that burn injury markedly elevates plasma levels of glucose (consistent with the results of previous studies) and that this effect is reduced by treatment with SS31 but not SS20.
Figure 2.

Areas under the plasma glucose curves (AUC’s) in Figure 1. Each value represents the mean ± sem for 6–12 animals; ***p < 0.0001 vs sham, ++p<0.001 vs burn alone and burn + SS-31 and SS-20 treated animals and sham controls.
Figure 3 illustrates the effects treatment with SS-31 on burn injury and cold stress induced changes in 18FDG uptake by brown adipose tissue (BAT) in mice. ANOVA demonstrated a highly significant main effect of treatment, F4,45 = 30.65; p<0.0001. Compared with sham treated mice, both burn injury and cold-stress produced highly significant increases in 18FDG uptake by BAT (p<0.0001). Cold-stress produced a greater increase in 18FDG uptake by BAT that burn injury (p<0.01). Treatment with SS31 produced a significant (p<0.05) reduction in 18FDG uptake by in burned animals. Surprisingly, peptide treatment had no effect on the increase in 18FDG uptake by BAT that was produced by cold stress. In addition, there were no effects of SS31 treatment on 18FDG uptake in any of the other tissues that were sampled.
Figure 3.

Effects treatment with SS31 on burn injury and cold stress induced changes in 18FDG uptake by brown adipose tissue (BAT) in mice. Each value represents the mean ± sem for 7 animals; ***p < 0.0001 vs sham, ++p<0.01 vs burn injured animals, *p < 0.05 vs burned animals that were not treated with peptide.
μPET Imaging
Representative 18FDG μPET images (sagittal slices) of a sham control mouse (left panel), a mouse with burn injury (middle panel) and a burn injured mouse treated with SS31 (right panel) are illustrated in Figure 4. 18F-FDG μPET imaging demonstrated intense focal uptake at sites of BAT after burn injury. Uptake in BAT was so intense that it was associated with significant reductions in uptake by all other tissues, including brain. No such areas of intense 18FDG uptake were observed in sham treated control mice. In the mouse treated with SS31, there was reduced 18FDG in BAT, and a partial normalization of 18FDG uptake in brain.
Figure 4.
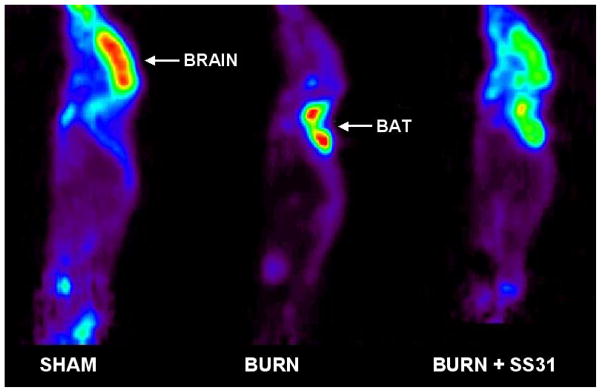
Representative 18FDG-μPET images of a sham treated mouse (Left panel), a mouse with burn injury (Middle panel) and a mouse with burn injury that was treated with SS31 (Right panel). 18F-FDG μPET imaging demonstrated intense focal uptake at sites of BAT after burn injury. Uptake in BAT was so intense that it was associated with significant reductions in uptake by all other tissues, including brain. In the mouse treated with SS-31, there was reduced 18FDG in BAT, and a partial normalization of 18FDG uptake in brain.
DISCUSSION
Oxidative stress has been shown to have a role in the insulin resistance associated with type 2 diabetes; based in part on the established role that ROS play in the endothelial, renal, and neural complications associated with hyperglycemia in late-stage diabetes (30). Numerous studies using nonspecific general antioxidant treatments have provided indirect evidence for a potential link between oxidative stress and insulin resistance (31,32,33,34,35,36). Recently, more direct evidence was provided by studies with cultured adipocytes using a mitochondrial-targeted strategy in which ROS were shown to play a causal role in the development of both TNF-α and glucocorticoid induced insulin resistance (37). However, the nature and molecular source(s) of ROS, the mechanisms governing their production, and their relevance to high-fat diet-induced insulin resistance (the most prevalent form of the disease) remain unknown.
In addition to providing energy to the cell, mitochondria are recognized as a site for the generation, dispensation, and removal of a number of intracellular signaling effectors, including hydrogen peroxide (H202), calcium, and nitric oxide. The emission rate of H202 from mitochondria, which reflects the balance between the rate of electron leak/superoxide formation from the respiratory system and scavenging of H202 in the matrix, varies over a remarkably consistent range (38). Once in the cytosol, H202 can alter the redox state by either reacting directly with thiol residues in redox-sensitive proteins or shifting the ratio of reduced to oxidized glutathione (GSH/GSSG); the main redox buffer of the cell. Thus, the rate at which H202 is emitted from mitochondria is considered an important index of mitochondrial function and modulator of the overall cellular redox environment (39). Two recent investigations have indicated that the rate of mitochondrial H202 emission is significantly greater when basal respiration is supported by fatty acid versus carbohydrate-based substrates (40,41), raising the possibility that mitochondrial H202 emission may be a primary factor in the etiology of insulin resistance. Anderson et al demonstrated that hydrogen peroxide production by mitochondria is linked to high-fat-diet induced insulin resistance in both rodents and humans (15).
Burn injury is associated with oxidative stress (42,43,44) as well as insulin resistance (8, 9, 28, 29). Using the euglycemic insulin clamp technique it has been demonstrated that: (i) maximal rate of glucose disposal is reduced in trauma patients; (ii) the metabolic clearance rate of insulin is almost twice normal in these patients; and (iii) post-trauma insulin resistance appears to occur in peripheral tissues, probably skeletal muscle, and is consistent with a post-receptor effect (31,45). Ikezy et al (46) demonstrated that burn injury results in impaired insulin-stimulated transport of [3H] 2-deoxyglucose into soleus muscle strips in vitro in rats. These investigations, as well as other studies from our laboratory (47,) have also demonstrated that insulin stimulated phosphoinositide 3-kinase (PI 3-kinase) activity, which is pivotal for glucose transport in muscle by glucose transporter 4 (GLUT 4), is decreased by burn injury to rats as measured by its IRS-1 associated activity. These data are consistent with alterations in post receptor signaling following burn injury, which results in burn induced insulin resistance. The present study was designed to determine whether SS31, which has been show to have protective effects at the mitochondrial level, effects glucose clearance from the blood after burn injury or uptake of the glucose analogue 18FDG. The mechanism(s) by which burn injury in this mouse model alters these two parameters of glucose metabolism is not understood at this point. However, it is interesting to note that treatment with SS31 but not SS20 seemed to ameliorate the changes produced by the burn injury. Whether this is related to changes in mitochondrial function in the burned mouse under these conditions is not clear. We have observed, however, that chronic treatment of burned rats with SS31 reduces the increased oxygen consumption produced by the burn injury (48, 49). One possibility is that the increased oxygen consumption produced by burn injury to rats is related to altered mitochondrial metabolism, which might be effected by SS31.
CONCLUSIONS
The results of these studies demonstrate that SS31, but not SS20, significantly attenuated burn induced insulin resistance in mice. In addition, SS31 had major effects on the biodistribution of 18FDG. Most significantly, SS31 treatment resulted in marked reduction of 18FDG uptake by brown adipose tissue (BAT). These findings provide a firm pre-clinical basis for future clinical trials of SS31 for the treatment of insulin resistance in patients with burn injury.
References
- 1.Nordlie R, Foster J, Lange A. Regulation of glucose production by the liver. Ann Rev Nutr. 1999;19:379–406. doi: 10.1146/annurev.nutr.19.1.379. [DOI] [PubMed] [Google Scholar]
- 2.Gerich J, Meyer C, Woerle H, Stumvoll M. Renal gluconeogenesis: its importance in human glucose homeostatis. Diabetes Care. 2001;24(2):381–391. doi: 10.2337/diacare.24.2.382. [DOI] [PubMed] [Google Scholar]
- 3.Joost H, Thorens B. The extended GLUT-family of sugar/polyol transport facilitators: nomenclature, sequence characteristics, and potential function of its novel members (Review) Mol Memb Biol. 2001;18:247–256. doi: 10.1080/09687680110090456. [DOI] [PubMed] [Google Scholar]
- 4.Saltiel A, Kahn R. Insulin signaling and the regulation of glucose and lipid metabolism. Nature. 2001;414:799–806. doi: 10.1038/414799a. [DOI] [PubMed] [Google Scholar]
- 5.Jiang Z, Zhou Q, Coleman K, Chouinard M, Boese Q, Czech M. Insulin signaling through Akt/protein kinase B analyzed by small interfering RNA-mediated gene silencing. Proc Natl Acad Sci U S A. 2003;100:7569–7574. doi: 10.1073/pnas.1332633100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reaven G. The insulin resistance syndrome: definition and dietary approaches to treatment. Annu Rev Nutr. 2005;25:391–406. doi: 10.1146/annurev.nutr.24.012003.132155. [DOI] [PubMed] [Google Scholar]
- 7.Pereira C, Herndon D. the pharmacologic modulation of the hypermetabolic response to burn. Adv Surg. 2005;39:245–261. doi: 10.1016/j.yasu.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 8.Herndon D, Dasu M, Wolfe R, Barrow R. Gene expression profiles and protein balance in skeletal muscle after beta-adrenergic blockage. Am J Physiol Endocrinol Metab. 2003;285(4):E783–789. doi: 10.1152/ajpendo.00508.2002. [DOI] [PubMed] [Google Scholar]
- 9.Gleeson J, Berenbeim D, Gilkin R. Incretin mimetics: promising new therapeutic options in the treatment of type 2 diabetes. 2005. J Manag Care Pharm. 2005;11(7 Suppl):S2–13. doi: 10.18553/jmcp.2005.11.s7.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bonab AA, Carter EA, Paul K, Kaneki M, Yu YM, Tompkins RG, Fischman AJ. Effect of simvastatin on burn-induced alterations in tissue specific glucose metabolism: implications for burn associated insulin resistance. Int J Mol Med. 2010;26(3):311–316. [PMC free article] [PubMed] [Google Scholar]
- 11.Deibert DC, Defronzo RA. Epinephrine-induced Insulin Resistance in Man. J Clin Invest. 1980;65(3):717–721. doi: 10.1172/JCI109718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Influence of sympatho-adrenal system on insulin sensitivity using the euglycemic clamp technique. Kusunoki M, Oshida Y, Iguchi A, Iida T, Suga T, Funado T, Sato Y, Kato K, Sakamoto N. Diabetes Res Clin Pract. 1992;17(2):125–131. doi: 10.1016/0168-8227(92)90157-m. [DOI] [PubMed] [Google Scholar]
- 13.Nauck MA, Kleine N, Orskov C, Hoist JJ, Willms B, Creutzfeldt W. Normalization of fasting hyperglycaemia by exogenous glucagon-like peptide 1 (7–36 amide) in Type 2 (non-insulin-dependent) diabetic patients 3. Diabetologia. 1993;36:741–744. doi: 10.1007/BF00401145. [DOI] [PubMed] [Google Scholar]
- 14.Nicolaus M, Bridle DL, Linke R, Woerle HJ, Schirra J. Endogenous GLP-1 regulates postprandial glycemia in humans: relative contributions of insulin, glucagon, and gastric emptying. J Clin Endocrinol Metab. 2011;96(1):229–236. doi: 10.1210/jc.2010-0841. [DOI] [PubMed] [Google Scholar]
- 15.Anderson EJ, Lustig ME, Boyle KE, Woodlief TL, Kane DA, Lin CT, Price JW, Kang L, Rabinovitch PS, Szeto HH, Houmard JA, Cortright RN, Wasserman DH, Neufer PD. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J Clin Invest. 2009;119:573–58. doi: 10.1172/JCI37048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Szeto HH. Mitochondria-targeted peptide antioxidants: novel neuroprotective agents. AAPS J. 2006;8(3):E521–31. doi: 10.1208/aapsj080362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Szeto HH. Cell-permeable, mitochondrial-targeted, peptide antioxidants. AAPS J. 2008;8(2):E277–283. doi: 10.1007/BF02854898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhao K, Zhao GM, Wu D, Soong Y, Birk AV, Schiller PW, Szeto HH. Cell-permeable peptide antioxidants targeted to inner mitochondrial membrane inhibit mitochondrial swelling, oxidative cell death, and reperfusion injury. J Biol Chem. 2004;279(33):34682–34690. doi: 10.1074/jbc.M402999200. [DOI] [PubMed] [Google Scholar]
- 19.Szeto HH. Mitochondria-targeted cytoprotective peptides for ischemia- reperfusion injury. Antioxid Redox Signal. 2008;10(3):601–619. doi: 10.1089/ars.2007.1892. [DOI] [PubMed] [Google Scholar]
- 20.Cho J, Won K, Wu D, Soong Y, Liu S, Szeto HH, Hong MK. Potent mitochondria-targeted peptides reduce myocardial infarction in rats. Coron Artery Dis. 2007;18(3):215–220. doi: 10.1097/01.mca.0000236285.71683.b6. [DOI] [PubMed] [Google Scholar]
- 21.Cho S, Szeto HH, Kim E, Kim H, Tolhurst AT, Pinto JT. A novel cell-permeable antioxidant peptide, SS31, attenuates ischemic brain injury by down-regulating CD36. J Biol Chem. 2007;282(7):4634–42. doi: 10.1074/jbc.M609388200. [DOI] [PubMed] [Google Scholar]
- 22.Petri S, Kiaei M, Damiano M, Hiller A, Wille E, Manfredi G, Calingasan NY, Szeto HH, Beal MF. Cell- permeable peptide antioxidants as a novel therapeutic approach in a mouse model of amyotrophic lateral sclerosis. J Neurochem. 2006;98(4):1141–1148. doi: 10.1111/j.1471-4159.2006.04018.x. [DOI] [PubMed] [Google Scholar]
- 23.Thomas DA, Stauffer C, Zhao K, Yang H, Sharma VK, Szeto HH, Suthanthiran M. Mitochondrial targeting with antioxidant peptide SS-31 prevents mitochondrial depolarization, reduces islet cell apoptosis, increases islet cell yield, and improves posttransplantation function. J Am Soc Nephrol. 2007;18(1):213–222. doi: 10.1681/ASN.2006080825. [DOI] [PubMed] [Google Scholar]
- 24.Mizuguchi Y, Chen J, Seshan SV, Poppas DP, Szeto HH, Felsen D. A novel cell-permeable antioxidant peptide decreases renal tubular apoptosis and damage in unilateral ureteral obstruction. Am J Physiol Renal Physiol. 2008;295(5):F1545–F1553. doi: 10.1152/ajprenal.00395.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang L, Zhao K, Calingasan NY, Luo G, Szeto HH, Beal MF. Mitochondria targeted peptides protect against 1-methyl-4-phenyl- 1,2,3,6-tetrahydropyridine neurotoxicity. Antioxid Redox Signal. 2009;11(9):2095–2104. doi: 10.1089/ars.2009.2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Anderson EJ, Lustig ME, Boyle KE, Woodlief TL, Kane DA, Lin CT, Price JW, Kang L, Rabinovitch PS, Szeto HH, Houmard JA, Cortright RN, Wasserman DH, Neufer PD. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodants and humans. J Clin Invest. 2009;119:573–581. doi: 10.1172/JCI37048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hamacher K, Coenen HH, Stocklin G. Efficient stereospecific synthesis of no-carrier-added 2-[18F]-fluoro- 2-deoxy-D-glucose using aminopolyether supported nucleophilic substitution. J Nucl Med. 1986;27(2):235–238. [PubMed] [Google Scholar]
- 28.Pawlik TM, Carter EA, Bode BP, Fischman AJ, Tompkins RG. Central Role of IL-6 in burn induced stimulation of hepatic amino acid transport. Int J Mol Med. 2003;12:541–548. [PubMed] [Google Scholar]
- 30.Evans JL, Goldfine ID, Maddux BA, Grodsky GM. Oxidative stress and stress-activated signaling pathways: a unifying hypothesis of type 2 diabetes. Endocr Rev. 2002;23:599–622. doi: 10.1210/er.2001-0039. [DOI] [PubMed] [Google Scholar]
- 31.Bonnard C, Durand A, Peyrol S, Chanseaume E, Chauvin MA, Morio B, Vidal H, Rieusset J. Mitochondrial dysfunction results from oxidative stress in the skeletal muscle of diet-induced insulin-resistant mice. J Clin Invest. 2008;118:789–800. doi: 10.1172/JCI32601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Evans JL, Maddux BA, Goldfine ID. The molecular basis for oxidative stress-induced insulin resistances. Antioxid Redox Signal. 2005;7:1040–1052. doi: 10.1089/ars.2005.7.1040. [DOI] [PubMed] [Google Scholar]
- 33.Maddux BA, See W, Lawrence JC, Goldfine AL, Goldfine ID, Evans JL. Protection against oxidative stress - induced insulin resistance in rat L6 muscle cells by micromolar concentrations of α-lipoic acid. Diabetes. 2001;50:404–410. doi: 10.2337/diabetes.50.2.404. [DOI] [PubMed] [Google Scholar]
- 34.Rudich A, Tirosh A, Potashnik R, Khamaisi M, Bashan N. Lipoic acid protects against oxidative stress induced impairment in insulin stimulation of protein kinase B and glucose transport in 3T3-L1 adipocytes. Diabetologia. 1999;42:949–957. doi: 10.1007/s001250051253. [DOI] [PubMed] [Google Scholar]
- 35.Saengsirisuwan V, Kinnick TR, Schmit MB, Henriksen EJ. Interactions of exercise training and lipoic acid on skeletal muscle glucose transport in obese Zucker rats. J Appl Physiol. 2001;91:145–153. doi: 10.1152/jappl.2001.91.1.145. [DOI] [PubMed] [Google Scholar]
- 36.Saengsirisuwan V, Perez FR, Sloniger JA, Maier T, Henriksen EJ. Interactions of exercise training and {alpha}-lipoic acid on insulin signaling in skeletal muscle of obese Zucker rats. Am J Physiol Endocrinol Metab. 2004;287:E529–E536. doi: 10.1152/ajpendo.00013.2004. [DOI] [PubMed] [Google Scholar]
- 37.Houstis N, Rosen E, Lander ES. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature. 2006;440:944–948. doi: 10.1038/nature04634. [DOI] [PubMed] [Google Scholar]
- 38.Stone JR, Yang S. Hydrogen peroxide: a signaling messenger. Antioxid Redox Signal. 2006;8:243–270. doi: 10.1089/ars.2006.8.243. [DOI] [PubMed] [Google Scholar]
- 39.Schafer FQ, Buettner GR. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic Biol Med. 2001;30:1191–1212. doi: 10.1016/s0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- 40.Anderson EJ, Yamazaki H, Neufer PD. Induction of endogenous uncoupling protein 3 suppresses mitochondrial oxidant emission during fatty acid-supported respiration. J Biol Chem. 2007;282:31257–31266. doi: 10.1074/jbc.M706129200. [DOI] [PubMed] [Google Scholar]
- 41.St-Pierre J, Buckingham JA, Roebuck SJ, Brand MD. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J Biol Chem. 2002;277:44784–44790. doi: 10.1074/jbc.M207217200. [DOI] [PubMed] [Google Scholar]
- 42.Gurer A, Ozdogan M, Gokakin AK, Gomceli I, Gulbahar O, Arikok AT, Kulacoglu H, Aydin R. Tissue oxidative stress level and remote organ injury in two-hit trauma model of sequential burn injury and peritoneal sepsis are attenuated with N-acetylcysteine treatment in rats. Ulus Travma Acil Cerrahi Derg. 2009;15:1–6. [PubMed] [Google Scholar]
- 43.Foldi V, Csontos C, Bogar L, Roth E, Lantos J. Effects of fluid resuscitation methods on burn trauma induced oxidative stress. J Burn Care Res. 2009;30:957–966. doi: 10.1097/BCR.0b013e3181bfb75e. [DOI] [PubMed] [Google Scholar]
- 44.Parihar A, Parihar MS, Milner S, Bhat S. Oxidative stress and anti-oxidative mobilization in burn injury. Burns. 2008;34:6–17. doi: 10.1016/j.burns.2007.04.009. [DOI] [PubMed] [Google Scholar]
- 45.Black PR, Brooks DC, Bessey PQ, Wolfe RR, Wilmore DW. Mechanism of insulin resistance following injury. Ann Surg. 1982;196:420–435. doi: 10.1097/00000658-198210000-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ikezu T, Okamoto T, Yonezawa K, Tompkins RG, Martyn JA. Analysis of thermal injury-induced insulin resistance in rodents, Implications of postreceptor mechanisms. J Biol Chem. 1997;272(40):25289–25295. doi: 10.1074/jbc.272.40.25289. [DOI] [PubMed] [Google Scholar]
- 47.Carter EA, Burks D, Fischman A, White M, Tompkins R. Insulin resistance in thermally-injured rats is associated with post-receptor alterations in skeletal muscle, liver and adipose tissue. Int J Mol Med. 2004;14(4):653–658. [PubMed] [Google Scholar]
- 48.Yoh K, Carter EA, Lin F, Fischman AJ, Aikawa N, Tompkins RG, Yu YM. Mitochondria-targeted peptide attenuates the hypermetabolism after burn injury. J Burn Care. 2009;30:S106. [Google Scholar]
- 49.Yoh K, Carter EA, Lin F, Fischman AJ, Aikawa N, Tompkins RG, Yu YM. Mitochondria-targeted peptide attenulates the burn induced hypermetabolism by the down regulated UDP-1 expression in rat bown adipose tissue. J Burn Care. 2010;31:S104. [Google Scholar]