

Abstract
Design of an efficient fingerprint that detects homologous proteins at distant sequence identity has been a great challenge. This paper proposes a strategy to extract an ideal-like fingerprint with high specificity and sensitivity from a group of sequences related to a fold. The approach is devised based on the assumptions that the critical residues for a protein fold may be conserved in three aspects, i.e. sequence, structure, and intramolecular interaction, and embedded in secondary structures. We hypothesized that the residues satisfying such conditions simultaneously may work as an efficient fingerprint. This idea was tested on protein folds of various classes, such as beta-strand rich, alpha + beta proteins and alpha/beta proteins with discrete sequence similarities. The fingerprint for each fold was generated by selecting the overlapped conserved residues (OCR) from the conserved residues obtained using independent three alignment methods, i.e. multiple sequence alignment, structure-based alignment, and alignment based on the interstrand hydrogen-bonds. The OCR fingerprints showed more than 90% detection efficiency for all the folds tested and were identified to be almost the minimal fingerprints composed of only critical residues. This study is expected to provide an important conceptual improvement in the identification or design of ideal fingerprints for a protein fold.
An exponential growth of protein sequence database motivated the development of various computational approaches for the recognition of structural/functional features and classification of uncharacterized protein sequences1,2,3,4. The methods basically utilize the protein sequence patterns or fingerprints that represent the proteins with specific structures or functions5,6,7. The patterns are generally generated by the alignment of a group of sequences with similar structure, function or family relationship. Three kinds of sequence patterns have been representatively used to tackle the relationship of protein sequences, structures and functions: (i) small motifs (e.g. identified by PROSITE, Pratt, TRIOLOGY, etc.) are the group of conserved residues identified from the short conserved sequences in the region well-known for substantial biological activity such as catalytic sites and metal ion binding sites8,9,10,11; (ii) multiple motifs or blocks (e.g. identified by PRINTS, InterPro, etc.) are the group of independent, sequentially or spatially distinct motifs that usually occur together and suggest a putative function12,13 and; (iii) profiles or family signatures are generated using the level of amino acid conservation at different positions in the alignment of complete protein domain. PROSITE, HHpred, PSI-BLAST, etc. are the tools used to identify such patterns14,15,16,17. These all patterns work as the signatures to identify similar features in uncharacterized sequences.
An ideal fingerprint for a given fold might be one that can detect all the homologous proteins with perfect sensitivity and exclude any non-homologous proteins with perfect specificity. Such a fingerprint should include the critical residues, which can detect all the homologous proteins, and not include any non-essential residues that can decrease the sensitivity. As mentioned above, many strategies were devised to identify such efficient sequence patterns and they were evaluated to be somewhat successful to characterize the protein sequences and structures. However, there are still some limitations in the fingerprints18,19,20. For instance, small motifs for substantial biological activity generally show high sensitivity, but low specificity in the detection of homologous sequences. On the other hand, the fingerprints such as blocks and profiles show high specificity, but relatively low sensitivity. In particular, the sensitivities of most sequence patterns are not satisfactory when finding remote protein homologs. Further intensive studies need to be executed to produce a lot more effective schemes to evoke a fingerprint close to ideality.
We propose a new approach to generate an efficient fingerprint for the detection of protein homologs. The approach was devised on the basis of following assumptions. First, the crucial residues for a protein fold might be conserved in three aspects, i.e. sequence, structure, and intramolecular interaction. Second, structurally important residues may be embedded in the secondary structure elements, such as α-helices and β-strands, rather than in the loop regions. Finally, the residues satisfying such conditions simultaneously might be the critical residues for a protein fold, and work as an efficient fingerprint for the detection of homologous sequences. To evaluate these hypotheses, this study attempts to identify the residues based on the above assumptions for various protein folds and examined their efficiencies as a fingerprint.
We begin by describing the general scheme of the design of fingerprints using the devised approach. The approach is first implemented on Immunoglobulin V-set domain (IgV) as a model system to present the detailed procedure. Next, the method is benchmarked by applying on various protein folds such as beta-strand rich, alpha + beta, and alpha/beta protein folds with a range of sequence similarities. These studies demonstrate that the proposed approach is effective to extract an efficient fingerprint with high specificity and sensitivity. The implications of our results for the protein homology detection are also discussed.
Results
Design of OCR-based fingerprints
Figure 1 shows the scheme of protein fingerprint mining based on the devised strategy. In the first step, the conserved residues in the three aspects, i.e. sequence, structure, and intramolecular interaction, were identified independently from a group of homologous sequences for a specific fold. To identify the residues conserved at sequence level, a general multiple sequence alignment (MSA) was performed using ClustalW21. Structure based alignment (SBA) was applied to the target sequences to identify structurally conserved residues using Dali server22. For the intramolecular interactions, this study focused on the non-local hydrogen bonds between beta-strands because they are considered as one of the most important factors to determine a protein fold and stability23. In addition, their patterns can be identified more clearly compared to other intramolecular interactions. To select the conserved residues for the hydrogen bond patterns of the beta-strands, the method to align the beta-strand sequences based on the inter-strand hydrogen bond patterns of the β-sheet was employed (This method will be referred to as SSS-based approach because this approach was devised to find the supersecondary structure(SSS)-determining residues)24. In this study, the hydrophobicity and hydrophilicity were used as the criteria of conservedness of a position to maximize the number of conserved positions in the alignments. In the second step, the amino acid positions found to be commonly conserved among the three different alignments were selected. The residues were called “Overlapped Conserved Residues” (OCR) and used to create the OCR fingerprint for the fold detection process. In addition, the OCR embedded in the beta strand region was used to generate the OCRS fingerprint. Further, OCRMIN fingerprint was produced by eliminating the conserved positions in the OCRS fingerprint one by one. The OCR-based fingerprints such as OCR, OCRS, and OCRMIN were used to detect the homologous proteins for a target fold, and their fold detection efficiencies were compared with the fingerprints obtained by MSA, SBA and SSS-based approaches.
Figure 1. Scheme of protein fingerprint mining.

Flow chart shows the steps to extract the various OCR fingerprints. First, three independent alignment methods, i.e. MSA, SBA and SSS-based method, were applied to the target folds using hydrophobicity and hydrophilicity as conservedness criteria and the conserved fingerprint from each method was obtained. Second, overlapped conserved residues in three alignments are identified to generate the OCR fingerprints. Further, elimination of the non-essential residues in OCR fingerprint generates the OCRS and OCRMIN fingerprints.
Implementation of OCR-based approach on Immunoglobulin V-set domain
In the first phase of this study, the OCR-based approach was implemented as a model system on the antibody variable domain-like proteins (IgV-set domain). The “IgV-set domain proteins” have a beta sandwich structure where ten strands are arranged in two β-sheets in a Greek-key fashion25, where the lowest sequence identity between the two structural homologous is ~23%. Protein Databank contains approximately 558 IgV-set domains, where the sequence length of the structural varies from 110 to 130 amino acid residues. This study illustrates how to identify the critical residues embedded in the beta-strands of the IgV-set domain using the OCR-based approach, and their efficiency as a protein signature to detect remote protein homologous was examined. The fold detection efficiency is a term to consider both detection sensitivity and specificity, and their exact definitions are described in Method section.
i) Homology detection efficiencies of MSA, SBA and SSS-based fingerprints
To create a protein sequence pattern for IgV-set domain, 10 distantly related protein sequences of IgV-set domains were selected (Supplementary Table S1 online). The conserved sequence patterns were created using three independent different alignment methods, i.e. MSA, SBA and SSS-based method. Figure 2 shows the sequence patterns generated from each sequence alignment method. The sequence patterns consisted of 43, 40 and 32% of the total residue numbers for MSA, SBA and SSS-based methods, respectively. The sequence patterns were tested to detect the homologous protein structures against the protein structure database, PDB, as the target database. Table 1 lists the homology detection efficiencies of the MSA, SBA and SSS-based fingerprints to 44, 51 and 76%, respectively. The conserved sequence patterns determined by these three methods were highly specific in nature with zero false positives. These results suggest that the specificities of the fingerprints are perfect, but there is a limitation in the sensitivities of the identified conserved sequence patterns.
Figure 2. Conserved sequence residues obtained by MSA, SBA and SSS methods.

Protein sequence patterns for Immunoglobulin-V set domain were obtained by MSA, SBA, SSS and OCR approach. Distribution of conserved positions in secondary structure elements (SSEs) is shown for each alignment method. Sequence pattern is PROSITE-like pattern. Here, the expression “x(d,r)” indicates the “d” as the minimum number of residues between two consecutive conserved positions and the distance “r” is the maximum number of residues between two consecutive conserved positions. Similarly, expression “x” is used if the minimum and maximum distance between two consecutive conserved positions is same.
Table 1. Database Scan results for Immunoglobulin V-set domain Proteins.
| Pattern Length | Fold Detection | ||||||
|---|---|---|---|---|---|---|---|
| Sr. No. | Sequence Pattern | #res | %res | #Hits | TP | FP | EFF |
| 1 | MSA | 54 | 43 | 246 | 246 | 0 | 44 |
| 2 | SBA | 50 | 40 | 285 | 285 | 0 | 51 |
| 3 | SSS | 40 | 32 | 424 | 424 | 0 | 76 |
| 4 | OCR | 29 | 23 | 435 | 435 | 0 | 80 |
| 5 | OCRS | 15 | 12 | 486 | 486 | 0 | 87 |
| 6 | OCRMIN | 14 | 11 | 554 | 542 | 12 | 95 |
Here, table lists the percentage of the sequence residues involved in the generated fingerprints as well as the detection efficiencies of the respective fingerprints for the IgV-set domain. Here #res indicates total conserved positions and %res indicates percentage of the conserved positions for each fold. Similarly, #Hits, TP, FP and EFF indicates total structural hits, true positive hits, false positive hits and fold detection efficiency, respectively.
ii) Homology detection efficiency of OCR-based approach
The common positions among the identified conserved positions in the three sequence alignments were used to develop the OCR fingerprint. The OCR fingerprint, shown in Figure 2, consists of 23% of the total residue numbers, which was almost 25 to 50% shorter in length than the previous three fingerprints. The fold detection efficiency of the OCR fingerprint was 80%, higher than the fold detection efficiencies of the MSA, SBA and SSS-based fingerprints, and there were no false positives (Table 1). These results suggest that the sensitivity of the OCR-based fingerprint for homology detection can be higher than the three individual methods by maintaining the perfect specificity despite the significant decrease in fingerprint size. This also provides an important insight that some non-essential residues in the MSA, SBA and SSS-based fingerprints can be eliminated, but the critical residues can be maintained during the extraction of the overlapped conserved residues.
iii) Homology detection efficiency of the OCR-fingerprint in beta-strands
To test the importance and efficiency of the fingerprints in the secondary structures, a new fingerprint was generated by selecting the conserved residues in the beta-strands of the IgV-set domain. The new fingerprint, designated OCRS, consisted of just 12% of the sequence residues, and its pattern length was just half of the OCR fingerprint. As shown in Table 1, the fold detection efficiency of the OCRS fingerprint was improved to 87% compared to the 80% efficiency of the original OCR fingerprint. The specificity of this fingerprint was also perfect. These results suggest that the OCR residues in the loop regions may be mostly non-essential residues that are mainly responsible for the decrease in the fold scan sensitivity. Therefore, the removal of these non-essential residues can improve the fold detection efficiency. This also suggests that the OCR residues in the beta-strands include the critical residues to detect the homologous proteins efficiently. Overall, the beta-strand embedded amino acids that are conserved in terms of the sequence, structure, and hydrogen bond pattern can be a very efficient fingerprint for a protein fold.
iv) Minimization of the fingerprint size embedded in the beta-strands
In the above studies, the OCRS fingerprint composed of just 12% of the conserve amino acids in the beta-strands regions could be used to detect the homologous proteins of the IgV-set domain quite efficiently, whereas the OCR residues in the loop regions were not essential for detecting the structural fold. The next question was whether further non-essential residues were included in the identified OCRS fingerprint and whether their elimination could improve the efficiency of the OCRS fingerprint further. To examine this possibility, an attempt was made to reduce the number of conserved residues from the OCRS, which represents the protein signature, by eliminating the conserved positions individually, and investigating the efficiency of the reduced fingerprints. Generally, a further reduction in the sequence pattern length resulted in an increase in the fold scan sensitivity, but at the same time, the occurrence of false positive hits was increased by multiple folds, resulting in an overall decrease in the fold scan efficiency (Supplementary Table S2 online). On the other hand, two exceptions were observed, where an elimination of the hydrophobic conserved positions, i.e. either F837or V877 in 1g6vK, improved the fold scan efficiency compared to the efficiency of OCRS. For example, OCRS without the conserved F837 residue, which is designated as OCRMIN in Table 1, showed 95% fold detection efficiency despite the detection of some false positives. Further elimination of both the hydrophobic conserved positions, together, decreased the fold detection efficiency significantly. Overall, the sequence pattern length could be reduced by only 1 position with an increase in the fold detection efficiency, and the number of false positives increased as more conserved positions in OCRS were eliminated. These results provide two insights. First, the OCRS fingerprint for the IgV-set domain proteins may be composed of almost the minimal critical residues, and are very close to OCRMIN, which determine the similar structural fold quite efficiently. Second, further elimination of the non-essential residues can enhance the fold detection efficiency further similar to the above studies.
Benchmarking the OCR-based approach on Dataset
The above results confirm that the OCR based approach can be a simple way of identifying the efficient fingerprint to detect protein homologs. Here, this study examined whether the OCR-based approach could be also used to identify such efficient fingerprints for other proteins with a range of folds and sequence similarities. Similar to the model study, two OCR-based fingerprints, i.e. OCR and OCRS, were generated for the various target folds, and their fold detection efficiencies were compared with the fingerprints created by the MSA, SBA and SSS-based approaches. This study also examined if the OCRS fingerprint was close to the minimal fingerprint to detect the structural fold.
i) Selection of protein folds and generation of fingerprints
The datasets consist of three different fold classes of proteins in the Structural Classification of Proteins (SCOP) database, i.e. all-beta, α + β, and α/β. Each fold class contained 4 structural folds, where the members in each fold were structurally homologous with a range of sequence identities. Each fold class had 2 representative structural folds at low sequence identity and 2 representative structural folds at high sequence identity. Each protein fold consisted of the protein members of single or multiple protein families, and 10 representative protein structures with the most sequence diversity were selected. Table 2 lists the structural and sequence properties of the selected protein folds. The conserved sequence patterns for the target folds in Table 2 were generated using MSA, SBA, SSS and OCR-based approaches (Supplementary Figure S1 online).
Table 2. Target dataset consists of 12 protein fold with structurally similar sequence dissimilar protein sequences.
| β-Strands | α-Helix | ||||||
|---|---|---|---|---|---|---|---|
| Fold Class and Fold Type | Sequence Length | #Strand | #res | #Helix | #res | #Loop res | Min SEQ ID |
| All beta Proteins | |||||||
| GFP-like protein | 212 ~ 238 | 11 | 125 | 5 | 24 | 89 | 10 |
| Cupredoxin-like proteins | 102 ~ 108 | 6 | 37 | 1 | 3 | 65 | 15 |
| Acid Proteases | 99 ~ 113 | 5 | 45 | 1 | 4 | 50 | 27 |
| Ribosomal Protein L14 | 122 ~ 138 | 5 | 35 | 2 | 10 | 76 | 35 |
| Alpha and beta (α + β) Proteins | |||||||
| β-Grasp (Ubiquitin-like) protein | 72 ~ 79 | 4 | 22 | 2 | 14 | 39 | 10 |
| Nucleoside Triphosphate Hydrolase | 165 ~ 180 | 6 | 46 | 6 | 49 | 84 | 16 |
| RNAase A-like proteins | 101 ~ 133 | 7 | 41 | 3 | 25 | 58 | 28 |
| Cysteine Proteinases | 201 ~ 218 | 6 | 36 | 6 | 55 | 129 | 33 |
| Alpha and beta (α/β) Proteins | |||||||
| 50 S Ribosomal Protein L25 | 94 ~ 98 | 6 | 43 | 3 | 20 | 31 | 16 |
| 50 S Ribosomal Protein L6P | 164 ~ 191 | 13 | 70 | 3 | 36 | 71 | 23 |
| Difydrofolate reductase-like proteins | 159 ~ 186 | 10 | 49 | 4 | 34 | 76 | 27 |
| Pyruvate kinase N-terminal domain | 97 ~ 101 | 8 | 35 | 2 | 7 | 80 | 30 |
Here, table lists the 12 protein folds with structurally similar sequence dissimilar protein sequences which are used as target dataset. Fold class and title is listed in first column, second column shows the sequence length of representative structures of each fold. For each fold, secondary structure elements information, i.e. total number of residues involved in strand, helix and loop, are listed. Here, Min SEQ ID indicates the minimum sequence identity among the sequences representing the particular fold.
ii) Homology detection
Homology detection was performed against the PDB using the generated fingerprints and their fold detection efficiencies were compared. Table 3 lists the percentage of the sequence residues involved in the generated fingerprints as well as the detection efficiencies of the respective fingerprints for the target folds. As shown in the results, the general trend of the fold detection efficiency was similar to the result of the model protein study using the IgV-set domain proteins. The detection efficiencies of the OCR fingerprints generally showed improved detection efficiency compared to the MSA, SBA, and SSS-based fingerprints for most of the target folds. The use of the OCRS fingerprint enhanced the detection efficiency further. For example, in the cases of the cysteine proteinases and pyruvate kinase N-terminal domain-like protein, a dramatic change in efficiency was observed, where the fold detection efficiency of OCRS fingerprints increased from 61% and 48% to 86% and 94%, respectively, compared to the efficiencies of the OCR fingerprints. The sizes of the respective OCRS fingerprints ranged from 6% to 17% of the total residue numbers of the target protein folds. The maximum efficiency of the OCR-based fingerprints, either OCR or OCRS, was in the range of 84%–100%, whereas the MSA, SBA and SSS-based fingerprints showed relatively low and very different detection efficiencies depending on the target folds.
Table 3. Database Scan results using various fingerprints for Target Dataset.
| MSA | SBA | SSS | OCR | OCRS | OCRMIN | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Fold Class and Fold Type | #Protein | %res | EFF | %res | EFF | %res | EFF | %res | EFF | %res | EFF | %res | EFF |
| All beta Proteins | |||||||||||||
| GFP-like protein | 273 | 23 | 33 | 18 | 44 | 19 | 98 | 10 | 99 | 7 | 100 | 7 | 100 |
| Cupredoxin-like proteins | 120 | 22 | 0 | 26 | 24 | 20 | 35 | 10 | 91 | 6 | 17 | 9 | 99 |
| Acid Proteases | 578 | 60 | 76 | 59 | 67 | 55 | 78 | 46 | 80 | 13 | 84 | 11 | 89 |
| Ribosomal Protein L14 | 291 | 66 | 64 | 64 | 70 | 44 | 73 | 41 | 83 | 8 | 87 | 7 | 90 |
| Alpha and beta (α + β) Proteins | |||||||||||||
| β-Grasp (Ubiquitin-like) protein | 400 | 45 | 58 | 45 | 62 | 31 | 70 | 27 | 75 | 12 | 90 | 12 | 90 |
| Nucleoside Triphosphate Hydrolase | 530 | 33 | 34 | 26 | 47 | 18 | 57 | 14 | 91 | 10 | 93 | 9 | 94 |
| RNAase A-like proteins | 319 | 45 | 83 | 42 | 81 | 33 | 88 | 31 | 93 | 17 | 98 | 15 | 98 |
| Cysteine Proteinases | 198 | 39 | 33 | 39 | 34 | 37 | 40 | 33 | 61 | 6 | 86 | 6 | 90 |
| Alpha and beta (α/β) Proteins | |||||||||||||
| 50 S Ribosomal Protein L25 | 111 | 52 | 86 | 47 | 88 | 26 | 88 | 14 | 97 | 6 | 35 | 14 | 97 |
| 50 S Ribosomal Protein L6P | 317 | 47 | 57 | 45 | 62 | 36 | 74 | 28 | 81 | 12 | 89 | 12 | 98 |
| Difydrofolate reductase-like proteins | 272 | 38 | 56 | 38 | 57 | 33 | 67 | 25 | 67 | 13 | 91 | 12 | 96 |
| Pyruvate kinase N-terminal domain | 65 | 57 | 48 | 54 | 60 | 49 | 14 | 44 | 48 | 17 | 94 | 16 | 100 |
Here, table lists the percentage of the sequence residues involved in the generated fingerprints as well as the fold detection efficiencies of the respective fingerprints for the target dataset. Here, #Protein, %res and EFF indicates the total number of structural homologs the PDB, percentage of the conserved positions used to generate each fingerprints, and the fold detection efficiency, respectively.
In two exceptional cases, the fold detection efficiency of OCR was higher than the OCRS. In the cases of the Cupredoxin-like proteins and 50 S Ribosomal Protein L25-like proteins, the fold detection efficiency of the OCRS fingerprints decreased significantly from 91% and 97% to 17% and 35%, respectively, compared to their OCR fingerprints. In these cases, the high number of false positives was detected in the database scan (Supplementary Table S3 online). The OCR in the loop region of two protein folds was presumed to include some critical residues for homology detection, and the omission of the critical residues in the OCRS fingerprints may result in a substantial decrease in specificity.
iii) Minimization of the beta-strands embedded OCR fingerprint size
These results suggest that the size of the OCRS fingerprints are only 5–15% of the total residue numbers of the target protein folds. Interestingly, the fingerprint sizes of the protein folds with low or high similarity were not so different. An attempt was made to identify the fingerprints with lower numbers by reducing the OCRS fingerprints and examining their detection efficiencies. The OCRS fingerprints for the target folds β-Grasp (ubiquitin-like) and Ribosomal protein L25 presented the minimum size sequence pattern, for which any further conserved positions could not be eliminated without sacrificing the fold detection efficiency. For the other target folds, the sequence pattern length could be reduced at a maximum by only 1–2 residues. These results suggest that the identified OCRS fingerprints for the target folds are close to the minimum critical residues needed to detect the target folds efficiently, like the Immunoglobulin V-set domain case. On the other hand, the use of the minimized OCRS, i.e. OCRMIN, led to further enhancement of the detection efficiency. Their detection efficiencies were at approximately 90% to 100% for most of the target folds (Table 3).
Overall, the fold detection study for the target dataset confirmed the following three important outcomes of the model study. First, the OCR-based approach showed very high fold detection efficiency for the target folds. The fold detection efficiency of the MSA, SBA and SSS methods were relatively low and the efficiency of these methods differed from fold to fold. In contrast, the fingerprints obtained from the OCR based approach, i.e. OCR fingerprint, OCRS fingerprint and OCRMIN fingerprint, showed significantly improved efficiency and more than 90% fold detection efficiency at the maximum. Second, reducing the fingerprint size using the OCR-based approach proved to be efficient in eliminating the non-essential residues while retaining the critical conserved residues. Third, the OCRS fingerprint was almost the minimal fingerprint to detect the structure fold.
Properties of the OCR-based fingerprints embedded in beta-strands
To determine if there were any common features of the identified OCR-based fingerprints above, the residues comprising the OCRS fingerprints was characterized at various aspects. No specific features were found for the target dataset common in the aspects of the side chain properties and their positional properties. The identified residues showed irregular patterns in terms of their polar and non-polar properties, and they were distributed unevenly from the core to surface regions (data not shown).
On the other hand, an analysis of the distribution of the minimum conserved positions stated the clustering of the conserved positions across the entire sequence length. The sequence patterns were a cluster-like pattern where the conserved residues were grouped into several blocks separated by irregular gaps. For example, as shown in Figure 3, the distribution of the overlapped-conserved residues for the Immunoglobulin V-set domain showed five different clusters. Each cluster consisted of 2–3 amino acids and the distance between the clusters was varied. Figure S2 shows the clusters of the other target folds. The fingerprint for each target fold contains 3–5 conserved residue clusters. Most of the conserved residue clusters contained 3–5 identified positions but the cluster size might be 12 residues long, as found in the RNAase A-like fold. The general length of the irregular gaps was 10–20 amino acids, but it could be more than 40 residues, as in the case of the GFP-like protein.
Figure 3. Distribution of OCRS across the protein sequence.

Conserved positions in OCRS fingerprints of Immunoglobulin V-set domain are plotted across the entire protein sequence length for easy visualization. The figure shows conserved positions are not distributed equally but as the multiple conserved blocks.
Comparison of fold detection efficiency with traditional methods
The OCRS fingerprints in the above results were proven to be extremely effective to detect the homologous structures. Benchmarking of the fold detection efficiency of the OCR-based approach, to check the practical importance of the method, was performed along the traditional methods such as PSI-BLAST, HMMER, HHpred and FASTA search and the results were listed in Table 4. Fold detection efficiency of the PSI-BLAST were in the range of 42% to 92%, which varied depending on the fold type. HMMER showed an improvement in fold detection efficiency with the detection of over 65% protein homologs for each fold in dataset, except in the case of β-Grasp (Ubiquitin-like) fold where it showed just 39% of fold detection efficiency. HHpred and FASTA search showed a significant increase in fold detection efficiency with the detection of over 75% of sequence homologs for each fold. In some cases, HHpred and FASTA search showed better fold detection efficiency than the OCR-based approach. The results showed that the fold detection efficiency of the fingerprints obtained using the OCR-based approach is either competitive or better than the traditional approaches.
Table 4. Database Scan results using various homology detection methods for Target Dataset.
| OCRS | PSI-BLAST | HMMER | HHpred | FASTA search | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Fold Class and Title | #Fold | #Hits | EFF | #Hits | EFF | #Hits | EFF | #Hits | EFF | #Hits | EFF |
| All beta Proteins | |||||||||||
| GFP-like protein | 273 | 273 | 100 | 216 | 79 | 228 | 84 | 263 | 96 | 248 | 91 |
| Cupredoxin-like proteins | 120 | 117* | 98 | 110 | 92 | 108 | 90 | 106 | 88 | 103 | 86 |
| Acid Proteases | 578 | 485 | 84 | 480 | 83 | 462 | 80 | 472 | 82 | 514 | 89 |
| Ribosomal Protein L14 | 291 | 260 | 87 | 123 | 42 | 210 | 72 | 234 | 80 | 221 | 76 |
| Alpha and beta (α + β) Proteins | |||||||||||
| β-Grasp (Ubiquitin-like) protein | 400 | 363 | 90 | 265 | 66 | 156 | 39 | 327 | 82 | 304 | 76 |
| Nucleoside Triphosphate Hydrolase | 530 | 492 | 93 | 370 | 70 | 369 | 70 | 508 | 96 | 514 | 97 |
| RNAase A-like proteins | 319 | 312 | 98 | 251 | 79 | 289 | 91 | 310 | 97 | 299 | 94 |
| Cysteine Proteinases | 198 | 170 | 86 | 132 | 66 | 144 | 73 | 164 | 83 | 172 | 87 |
| Alpha and beta (α/β) Proteins | |||||||||||
| 50 S Ribosomal Protein L25 | 111 | 111* | 100 | 74 | 66 | 74 | 67 | 107 | 96 | 100 | 90 |
| 50 S Ribosomal Protein L6P | 317 | 282 | 89 | 155 | 49 | 212 | 67 | 264 | 82 | 285 | 90 |
| Difydrofolate reductase-like proteins | 272 | 248 | 91 | 216 | 79 | 238 | 88 | 237 | 87 | 242 | 89 |
| Pyruvate kinase N-terminal domain | 65 | 61 | 94 | 46 | 71 | 48 | 74 | 63 | 97 | 60 | 93 |
Here, table lists the total number of identified protein homolog during the fold scan against the PDB and the fold detection efficiencies of each method. Here #hits indicates structural fold detected by each method and EFF indicates the fold detection efficiency. Fold detection efficiency is calculated as the ratio of total true positive hits to the total number of structural folds in the PDB.
*Fold detection results for Cupredoxin-like proteins and 50S Ribosomal Protein L25 are obtained using the OCR-fingerprint.
Discussion
A major concern in the design of ideal-like protein fingerprints is how to improve their sensitivity for homology detection without sacrificing their specificity. This suggests that the non-essential residues that can decrease the sensitivity should be excluded in the design with retaining the critical residues for a protein fold. This study demonstrated that such design was possible by extracting the beta-strand embedded residues that are conserved in terms of sequence, structure and hydrogen bonding pattern from a group of related protein sequences. The OCR-based fingerprints were found to be very efficient in detecting the homologous protein folds of the various classes, such as the beta-strand rich, alpha + beta proteins and alpha/beta proteins regardless their sequence similarities. Our results may provide an important conceptual improvement in the design of ideal fingerprint for a protein fold, which may make a contribution to the understanding of the relation between protein sequences and structures.
In our study, the OCR-based approach was utilized to prepare the fingerprints for the protein folds including beta-strands. In the case of the α-helix rich proteins, the OCR-based approaches could not be applied efficiently to define the critical residues due to the lack of consistent intramolecular interactions such as the hydrogen bonds between the beta-strands. Nevertheless, the importance of eliminating non-essential residues in the fold detection for α-helix rich proteins was also confirmed. The OCRH-fingerprint consisting of the overlapped conserved residues from α-helical region showed higher fold detection efficiency compared to each fingerprint generated respectively by MSA or SBA method. When an attempt was made to reduce the fingerprint size by eliminating the overlapped conserved positions individually, the efficiencies were improved gradually and the minimum fingerprints, OCRMIN, were quite sensitive and specific to identify the structural folds. Supplementary Table S4 and S5 list the α-helix rich target folds description and the fold detection efficiency of the various fingerprints for the folds.
The sizes of the OCRS fingerprints were only 5–15% of the target protein, but the small fingerprints were sufficient to detect the sequences for a given fold regardless of the protein folds and their similarities with perfect specificity. What makes the high specificity of these small size fingerprints? The overlapped conversed residues across the sequence length formed a small subset of clusters with neighboring or consecutive amino acids that resulted in the form of local sequence motif (Figure 3 and Supplementary Figure S2 online). Any disturbance to these small subsets of clusters, while searching for the minimum crucial positions for the target folds, decreased the fold detection specificity significantly (Supplementary Table S2 online). We presume that the high specificity of the OCR-based fingerprints was due to the presence of these clustered sequence motifs in the pattern, despite their small size.
In the Table 4, fold detection efficiency of the OCR-based approach was compared with the traditional methods, demonstrating that the OCR-based approach was quite competitive or even showed higher efficiency compared to other methods. In fact, the OCR-based approach and other traditional methods follow different algorithms in the detection of homologous proteins. Therefore, such direct comparison may not be perfectly legitimate to evaluate the performance of the methods. However, such comparison provides the insight that OCR-based approach can be very useful to detect protein homology.
In our study, OCR-based sequence patterns could detect all or most of the known structure homologs of a protein from protein structure database. In particular, database scan using the OCR-based patterns was confirmed to be also efficient in the detection of remote homologous proteins. For example, OCR-based pattern developed using the 10 representative GFP-like sequences successfully identified the domain G2 of Nidogen-1 (PDB ID: 1GL4 and 1H4U) as a homolog in our study (Supplementary Table S10 online). In fact, it is not easy to identify such relationship due to the low sequence similarity between the proteins. Fold detection using the protein sequence of avGFP or other GFP variant by the traditional approaches such as PSI-BLAST, HMMER, HHpred and FASTA search was unable to identify Domain G2 of nidogen-1 as structural homolog (Supplementary Table S10, S11, S12, S13 and S14 online). The relationship could be identified only after the structure of mouse nidogen globular fragment 2 was solved using X-ray crystallography26. Further, to check the possibility that novel homologous proteins can be identified using the OCR-fingerprints, we attempted to perform the fold scan against the larger database such as NCBI non-redundant (nr) protein sequence database. We expected that fold detection against the sequence database will provide more sequence hits which might not be well studied due to the lack of any structural or functional annotation. Identification of such remote homologous proteins was quite successful. For instance, several sequences with no significant sequence similarity were identified using the OCR-based pattern for Cupredoxin-like proteins. The accession numbers of the identified sequences were WP_010687666, WP_019121393, WP_021320206, WP_004263537, WP_008217106, WP_019379850, etc. The identified sequences share around 15 ~ 24% of the sequence similarity with the representative Cupredoxin-like protein (Supplementary Figure S15 online). Tertiary structures of the identified sequences were modeled successfully, which showed that the sequences are homologous to the Cupredoxin-like proteins (More details about these results will be presented elsewhere). The identified sequences have been also annotated as Cupredoxin-like protein in NCBI sequence database while we were preparing this report, which also confirmed our results. Although we focused on demonstrating the characterization and efficiency of OCR-based approach in this report, these results implicate that the OCR-based approach can be an efficient tool in the search of novel homologous proteins for a specific target fold. We also expect that OCR-based approach/fingerprints can be combined with other efficient algorithms or database such as PROSITE, which may generate much more efficient sequence patterns to characterize protein sequences and structures.
Methods
Selection of protein folds
In the present study, evolutionary-related protein folds were derived from the Structural Classification of Proteins (SCOP) database27. Three β-strand rich protein fold classes, i.e. all-beta, alpha + beta (α + β) and alpha/beta (α/β), were used. The protein folds in each class and protein structures of a particular fold were selected according to the following criteria:
Protein structures are shown to be more conserved than the sequences during the evolutionary mechanism. Protein sequences representing a particular protein fold within a superfamily can either be highly similar (sequence homologs) or dissimilar (remote homologs) in nature. Therefore, in the dataset, two structural folds consist of the homologous proteins with high sequence identity (around 30% or more) and two structural folds consist of the homologous proteins with low sequence identity (20% or less), were selected to identify the conserved sequence patterns.
For each structural fold, 10 representative protein sequences within a superfamily were selected in a way that no sequences have >90% sequence identity to each other. The sequence pattern generated from such sequences will be a fingerprint for a wider range of sequences for a fold.
Structurally similar but sequence dissimilar protein family members or members missing one or two α-helices or β-strands represents the cases of evolutionary pressure, where structure is fully or mostly intact regardless of the sequence change, were included in this study.
Protein structural folds with different sizes, i.e. sequence length from 80 to 260 amino acids, were selected.
Low resolution protein structures, i.e. below 2.5Å, were eliminated from the selection.
Alignment of the sequences and mining of the conserved sequence pattern
Three sequence alignment methods were used: multiple sequence alignment (MSA) by ClustalW21, structure based alignment (SBA) by Dali server22, and SSS-based alignment. These alignment methods were performed for each fold using the ten representative protein sequences and/or structures. In the present study, the amino acid properties, such as hydrophobicity and hydrophilicity were used as the criteria to consider the conservedness of a position in the alignment to maximize the number of conserved positions in the alignments. A conserved position in this study was defined as the presence of either only hydrophobic or only hydrophilic residues at a particular position of the alignment. The amino acid residues V, I, L, M, F, W, C, A and Y are interchangeable at the hydrophobic conserved positions whereas residues Q, N, E, D, R, K, H, T, S, G, and P are interchangeable at the hydrophilic conserved positions.
Multiple sequence alignment was performed by ClustalW web server for the 10 representative protein sequences using the default parameters. Multiple structure alignment was performed using the DALI server. It performs a database search using an input query structure against the database of known structures (PDB) and returns the list of structural neighbors28. Now, protein structures, which correspond to the 10 representative protein sequences used for MSA, were selected and automated structural alignment option were used to perform the multiple structure alignment. Further, the conserved positions in both the alignments were redefined based on hydrophobicity and hydrophilicity criterion. In the case of SSS, the alignment was performed separately for each strand and loop rather than the entire sequence. The alignment in the strand was performed using the inter-strand hydrogen (H)-bonds. The alignment of the residues in the loop region was performed manually using the physical properties of the amino acids. From the resulting alignment, conserved residue positions were identified and the conserved sequence patterns were obtained from each sequence alignment method.
Overlapped Conserved Residues (OCR) and homologous fold detection
To identify the critical conserved residues at three aspects, i.e. sequence, structure, and intramolecular interaction, simultaneously, the above three independent alignment methods for each of the target fold were performed, and the common positions were extracted from the identified conserved positions, which are called the Overlapped Conserved Residues (OCRs). The OCR was used to generate an OCR-fingerprint. Similarly, the OCRS fingerprint was obtained utilizing the overlapped residues embedded in the strand region. The syntax of the OCR-fingerprint was similar to the PROSITE patterns. Therefore, they could be used directly for fold detection against the structure database.
The standalone version of the EXPASY ScanProsite tool was used for fold detection using various sequence patterns as an input29. Over 78000 protein sequence from the PDB was downloaded and used as the input for the ScanProsite tool. Fold detection using the specific sequence patterns against the structure database was performed. The step by step process to obtain OCR-based fingerprint is detailed in Supplementary Information (Supplementary Text and Supplementary Table S6, S7, S8 and S9 online). The search picked up structural hits, which are classified into ‘True Positives, TP', ‘False Negatives, FN' and ‘False Positives, FP' proteins. Identified structural hits (proteins) which are the members of the same superfamily as the representative proteins used to generate the pattern for the fold, are defined as ‘true positives' hits, whereas members of the superfamily, which are not identified by the sequence pattern in fold detection are defined as ‘false negatives'. Further, the identified hits which do not belong to the superfamily in consideration are defined as ‘false positives'.
The effectiveness of an OCR-based pattern is determined in the terms of “sensitivity” and “specificity”. A fingerprint is defined as highly specific if it detects only ‘true positives' hits and no or minimum ‘false positives' hits. “Specificity” is calculated as the ratio of ‘true positives' hits to the total of ‘true positives' and ‘false positives'.
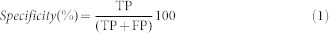 |
A sequence pattern is highly sensitive if it detects all or most of the structure homologs. “Sensitivity” is calculated as the ratio of ‘true positives' hits to the total number of structure homologs in PDB.
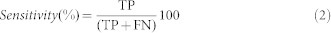 |
A sequence pattern is highly efficient if it detects all or most of the homologous proteins, ‘true positives' and no or minimum ‘false positive'. “Efficiency” is calculated as the ratio of ‘true positives' hits to the total number of hits.
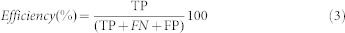 |
If, FP is ‘zero' or ‘low';
 |
Fold detection efficiency using the OCR-fingerprints were identified and compared with the efficiency of the three independent alignment methods.
Benchmarking of OCR-based approach against the target dataset
Fold detection efficiency of the OCR-based approached was tested against the target datasets, consists 12 protein folds in 3 different structural classes in SCOP, to benchmark the approach. For each fold, fingerprints such as MSA, SBA, SSS, OCR, OCRS and OCRMIN were obtained and fold detection against the PDB was performed. Fold detection efficiency for each fingerprint were listed and compared.
Fold detection efficiency of the OCR-based approach was compared with fold detection efficiencies of the traditional methods such as PSI-BLAST, HMMER, HHpred and FASTA search30,31,32,33. Fold detection using PSI-BLAST and FASTA search were performed using one representative protein sequence for each fold against the Protein Data Bank. HMMER, using the default Significance E-values, were utilized to detect homologous protein sequence against the protein structure database. Similarly, HMM-HMM comparison based homology search tool HHpred was used for homology detection, using one representative protein sequence for each fold, against the manually uploaded PDB sequence database. Fold detection efficiency of OCR-fingerprints with the PSI-BLAST, HMMER, HHpred and FASTA search were listed and compared.
Author Contributions
A.G., S.S., K.S.H. and S.G.L. designed research; A.G. performed research; A.G. and S.G.L. analyzed data; and A.G., K.S.H. and S.G.L. wrote the paper. All authors reviewed the manuscript.
Supplementary Material
Supplementary Information
Acknowledgments
This research was supported by Basic Science Program through the National Research Foundation of Korea (NRF) funded by the Korea government (MSIP) (NRF-2012R1A2A2A01045306).
References
- Geer L. Y., Domrachev M., Lipman D. J. & Bryant S. H. CDART: protein homology by domain architecture. Genome Res. 12, 1619–1623 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punta M. et al. The Pfam protein families database. Nucleic Acids Res. 40, D290–D301 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic I., Doerks T. & Bork P. SMART 7: recent updates to the protein domain annotation resource. Nucleic Acids Res. 40, D302–D305 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haft D. H., Selengut J. D. & White O. The TIGRFAMs database of protein families. Nucleic Acids Res. 31, 371–373 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L., White J. V. & Smith T. F. A homology identification method that combines protein sequence and structure information. Protein Sci. 7, 2499–2510 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Lazikani B., Sheinerman F. B. & Honig B. Combining multiple structure and sequence alignments to improve sequence detection and alignment: Application to the SH2 domains of Janus kinases. Proc. Natl. Acad. Sci. USA 98, 14796–14801 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang C. L. et al. On the role of structural information in remote homology detection and sequence alignment: new methods using hybrid sequence profiles. J. Mol. Biol. 334, 1043–1062 (2003). [DOI] [PubMed] [Google Scholar]
- Sigrist C. J. et al. New and continuing developments at PROSITE. Nucleic Acids Res. 41, D344–347 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigrist C. J. et al. PROSITE: a documented database using patterns and profiles as motif descriptors. Brief. Bioinform. 3, 265–274 (2002). [DOI] [PubMed] [Google Scholar]
- Jonassen I., Collins J. F. & Higgins D. G. Finding flexible patterns in unaligned protein sequences. Protein Sci. 4, 1587–1595 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley P., Kim P. S. & Berger B. TRILOGY: Discovery of sequence-structure patterns across diverse proteins. Proc. Natl. Acad. Sci. USA 99, 8500–8505 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attwood T. K., Beck M. E., Bleasby A. J. & Parry-Smith D. J. PRINTS - A database of protein motif fingerprints. Nucleic Acids Res. 22, 3590–3596 (1994). [PMC free article] [PubMed] [Google Scholar]
- Hunter S. et al. InterPro in 2011: new developments in the family and domain prediction database. Nucleic Acids Res. 40, D306–312 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remmert M., Biegert A., Hauser A. & Söding J. HHblits: Lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat. Methods. 9, 173–175 (2011). [DOI] [PubMed] [Google Scholar]
- Söding J., Biegert A. & Lupas A. N. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 33, W244–W248 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens F. J. Efficient recognition of protein fold at low sequence identity by conservative application of Psi-BLAST: validation. J. Mol. Recogn. 18, 139–149 (2005). [DOI] [PubMed] [Google Scholar]
- Heger A. & Holm L. Sensitive pattern discovery with ‘fuzzy' alignments of distantly related proteins. Bioinformatics 19, 130–137 (2003). [DOI] [PubMed] [Google Scholar]
- Jonassen I., Eidhammer I., Conklin D. & Taylor W. R. Structure motif discovery and mining the PDB. Bioinformatics 18, 362–367 (2002). [DOI] [PubMed] [Google Scholar]
- Friedberg I. & Margalit H. Persistently conserved positions in structurally similar, sequence dissimilar proteins: roles in preserving protein fold and function. Protein Sci. 11, 350–360 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin M. A. et al. Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 (2007). [DOI] [PubMed] [Google Scholar]
- Holm L. & Rosenstrom P. Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–549 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheiner S. Contributions of NH···O and CH···O Hydrogen Bonds to the Stability of β-Sheets in Proteins. J. Phys. Chem. B. 110, 18670–18679 (2006). [DOI] [PubMed] [Google Scholar]
- Kister A. E. & Gelfand I. Finding of residues crucial for supersecondary structure formation. Proc. Natl. Acad. Sci. USA 106, 18996–19000 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. et al. Structure of the Vdelta domain of a human gammadelta T-cell antigen receptor. Nature 391, 502–506 (1998). [DOI] [PubMed] [Google Scholar]
- Hopf M., Göhring W., Ries A., Timpl R. & Hohenester E. Crystal structure and mutational analysis of a perlecan-binding fragment of nidogen-1. Nat. Struct. Biol. 8, 634–640 (2001). [DOI] [PubMed] [Google Scholar]
- Murzin A. G., Brenner S. E., Hubbard T. & Chothia C. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 247, 536–540 (1995). [DOI] [PubMed] [Google Scholar]
- Bernstein F. C. et al. The Protein Data Bank: a computer-based archival file for macromolecular structures. J. Mol. Biol. 112, 535–542 (1977). [DOI] [PubMed] [Google Scholar]
- de Castro E. et al. ScanProsite: detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins. Nucleic Acids Res. 34, W362–365 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul S. F., Madden T. L., Schäffer A. A., Zhang J., Zhang Z., et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn R. D., Clements J. & Eddy S. R. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39, W29–W37 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Söding J., Biegert A. & Lupas A. N. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 33, W244–W248 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski R. A. Enhancing the functional annotation of PDB structures in PDBsum using key figures extracted from the literature. Bioinformatics 23, 1824–1827 (2007). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information