

Abstract
We have determined the structure of P2, the self-priming RdRp from cystovirus φ12 in two crystal forms (A, B) at resolutions of 1.7 Å and 2.1 Å. Form A contains Mg2+ bound at a site that deviates from the canonical non-catalytic position seen in form B. These structures provide insight into the temperature sensitivity of a ts-mutant. However, the tunnel through which template ssRNA accesses the active site is partially occluded by a flexible loop; this feature, along with sub-optimal positioning of other structural elements that prevent the formation of a stable initiation complex, indicate an inactive conformation in crystallo.
Keywords: bacteriophage, cystovirus, RNA-directed RNA polymerase, de novo initiation, transcription, replication
Introduction
In the double-stranded RNA viruses of the cystovirus family (φ6- φ14, φ2954), P2, an RNA-directed polymerase (RdRp), is the critical component of a four-protein polymerase complex (PX) that includes in addition to P2, the major capsid protein, P1, a packaging NTPase, P4, and an accessory protein, P71. P2 performs the dual tasks of transcription and replication de novo without the use of a primer (i. e. P2 is a self-priming RdRp), and plays a critical role in the cystoviral life cycle. The structure of the P2 protein from the cystovirus φ6 was solved about a decade ago2. Since then multiple crystal structures of φ6 P2 complexed with single-stranded DNA (ssDNA)2, single-stranded RNA (ssRNA), catalytic (Mg2+), inhibitory (Ca2+), non-catalytic (Mn2+) metal ions, and multiple substrate NTPs (see Salgado et. al. and Wright et. al. and additional references therein)3,4 have provided valuable insight into its metal-ion requirement, in addition to its substrate and template specificity. However, the availability of structural data on P2 proteins from additional cystoviruses is essential, to ascertain in structural terms, the functional variations of the RdRps across the cystovirus family5,6. Towards this goal, we have focused on the RdRp (P2) from the φ12 cystovirus. This choice is motivated by apparent differences in template utilization5,6 between the φ6 and φ12 RdRps together with their low sequence similarity (∼18% sequence identity)[Supporting Information Fig. S1].
Here, we present the high-resolution structure of φ12 P2 in two different crystal forms at resolutions of 1.7 Å and 2.1 Å. Our work here expands the structural data on the φ12 PX that includes the previously determined structures of P47 and P78. In fact all of the available structures of cystovirus PX proteins, except that of φ6 P2, come from φ12. Only the P1 protein from φ12 (or from any other cystovirus) now lacks high-resolution structural information. Therefore, our present results constitute a major step towards obtaining structural insight into the functional differences between the cystoviral RdRps on one hand, while providing a common platform in which to survey the structure, dynamics and interactions between the constituent proteins of the PX from a common cystoviral species, on the other.
Materials and Methods
Protein expression and purification
The plasmid pPG24 containing the φ12 P2 expression fragment (a kind gift from Dr. Paul Gottlieb, CCNY) was transformed into BL21 (DE3) cells and grown at 37 °C in LB containing ampicillin at a concentration of 150 μg/mL. When the OD600 reached 0.8, isopropyl-β-D-thiogalactopyranoside (IPTG) was added to a final concentration of 1 mM to induce protein overexpression and protein expression was allowed to proceed overnight at 17 °C. Cells were harvested after centrifugation at 5,000 rpm for 25 min at 4 °C, re-suspended in a buffer containing 50 mM Tris, 100 mM NaCl and 4 mM DTT at pH 9.0, and lysed by sonication. The lysate was centrifuged for 25 min at 16,500 rpm at 4 °C, the resulting supernatant was loaded onto a High-Trap Q column (GE Healthcare Biosciences) and eluted using a linear 0.1–1 M NaCl gradient of a buffer containing 50 mM Tris and 4 mM DTT at pH 9.0. Fractions containing P2 were pooled, concentrated, filtered, and then injected into a gel-filtration column (Superdex-200 10/300, GE Healthcare Biosciences) where P2 eluted as a monomer. Purified φ12 P2 was dialyzed in a buffer containing 50 mM Tris, 100 mM NaCl, and 2 mM DTT at pH 8.0, and concentrated using a Centricon-30K micro-concentrator (Amicon Inc) for crystallization trials.
To enable the determination of crystallographic phases a selenomethionine (Se-Met) substituted version of φ12 P2 (19 native methionines in a total of 659 amino acids) was prepared. Se-Met labeling of φ12 P2 was achieved without the use of an auxotropic strain using a protocol similar to that proposed by Van Duyne et. al.9. 1 mL of an overnight LB culture of pPG24 transformed E. coli BL21 (DE3) cells was used to inoculate 1 L of M9 medium and grown overnight at 37 °C. 10 mL of the overnight M9 growth was then used to inoculate 1 L of M9 medium, followed by growth at 17 °C. When the OD600 reached 0.8, 1.0 g each of lysine, threonine and phenylalanine; 0.5 g each of leucine, isoleucine and phenylalanine; and 0.5 g L(+) selenomethionine were added as solids and mixed thoroughly. The culture was induced after 15 minutes of growth using IPTG to a final concentration of 1 mM. The subsequent purification steps were identical to those described above for the native protein.
Protein crystallization
In order to determine the optimal crystallization conditions for φ12 P2, crystallization trials involved multiple screens, including the Nucleix, Mb Class, Classics, PEGs, JCSG+ and JCSG I- IV Suites (Qiagen), as well as the Additive Screen from Hampton Research. Crystallization experiments were assembled with a Mosquito drop setter (TTP Labtech). Native P2 was screened at concentrations that ranged from 6 to 20 mg/mL with a drop size of 300 nL using 96-well crystallization trays (Axygen Biosciences) at room temperature. Crystals grew after 14-30 days under a number of different conditions. Further optimization of growth conditions produced large crystals in hanging drops with 4.3 M NaCl, 0.1 M HEPES and 30 mM tri-glycine. Crystals of Se-Met substituted P2 were also obtained under the same crystallization conditions. These crystals were harvested for data collection. In sitting-drop trays, native crystals that grew in 4.0 M sodium formate and 0.2 M magnesium formate with 20% w/v PEG 3350 at 20 °C were also harvested for data collection. For cryo-protection, individual crystals were transferred to a drop containing mother liquor and 10% ethylene glycol, then flash-cooled in liquid nitrogen prior to data collection.
Data collection and structure determination
X-ray diffraction data for native and Se-Met substituted φ12 P2 crystals were collected at the National Synchrotron Light Source (NSLS) beamline X4C at 100 K. Data were processed and scaled using the HKL2000 suite of programs10. Data collection and refinement statistics are provided in Table I. The φ12 P2 structure was solved by Se-Met SAD using PHENIX11. The initial auto-built structure was manually rebuilt in Coot12 and refined with REFMAC13. This structure was then used as a molecular replacement search model to solve the structures of the crystals that grew in the presence of sodium formate (form A) and magnesium formate (form B), which were also built and refined using Coot and REFMAC. Waters were automatically built using ARP/wARP and then manually edited14. Bound metal ions in the form A and form B structures were identified based on coordination of waters and protein atoms. The metal ion in form A was assigned to be Mg2+ (rather than Mn2+) based on the inspection of anomalous difference Fourier maps and the absence of a peak at the metal ion position expected for Mn2+ at the wavelength of data collection (see [Supporting Information Fig. S2]). The bound metal ions in the form B structure were assigned to be Mg2+ by analogy with form A, as well as due to the presence of 0.2 M magnesium formate in the crystallization solution.
Table I. X-Ray Data Collection and Refinement Statistics.
| Data Collection | ||
|---|---|---|
| Space group | P21212 (form A) | P1 (form B) |
| Cell dimensions | ||
| a, b, c (Å) | 134.8, 72.3, 96.8 Å | 87.9, 94.5, 96.5 Å |
| α, β, γ (°) | 90.0, 90.0, 90.0 ° | 75.2, 63.1, 83.8 Å |
| Wavelength (Å) | 0.979 | 0.979 |
| Resolution (Å) | 1.70 (1.73-1.70 Å) | 2.10 (2.14-2.10 Å) |
| Rsym | 0.069 (0.411) | 0.048 (0.202) |
| I/σI (Last shell) | 39.5 (6.1) | 17.8 (2.9) |
| Completeness (last shell) | 99.9% (100%) | 92.6% (87.1%) |
| Redundancy (last shell) | 14.3 (11.7) | 1.9 (1.7) |
| Refinement | ||
| Resolution (Å) | 50-1.70 Å | 50-2.1 Å |
| Number of unique reflections | 105,419 | 144,887 |
| Rwork/Rfree | 0.130/0.167 | 0.217/0.278 |
| Number of atoms | ||
| Protein | 5306 | 20945 |
| Ligand/Ion | 5 Mg2+, 1 formate | 4 Mg2+ |
| Water | 1165 | 1654 |
| B factors (Å2) | ||
| All | 16.51 | 14.77 |
| Main-chain | 18.24 | 17.53 |
| Side-chain | 21.36 | 18.46 |
| Waters | 43.17 | 32.46 |
| RMS deviations | ||
| Bond lengths (Å) | 0.017 | 0.008 |
| Bond angles (°) | 1.533 | 1.100 |
| Ramachandran Statistics | ||
| Most favored (%) | 91.2 | 90.9 |
| Additionally allowed (%) | 8.8 | 9.1 |
| Generously allowed (%) | 0 | 0 |
| Disallowed (%) | 0 | 0 |
| PDB ID | 4GZK | 4IEG |
The coordinates and structure factors for the structures presented here have been deposited in the Protein Data Bank with codes 4GZK (form A) and 4IEG (form B). The original Se-Met structure that was used as the model for molecular refinement (refined to 2.35 Å) was found to be essentially identical to the form A structure (identical space-group and unit cell dimensions) in spite of being obtained under different crystallization conditions. This structure is therefore not discussed further.
Results and Discussion
Structure of φ12 P2
φ12 P2 crystallized in two different forms both of which yielded high-resolution structures (Table I). The structure from the form A crystals (P21212) was refined to 1.7 Å and represents the highest resolution obtained for a full-length viral polymerase till date. The final model consisted of a single P2 molecule in the asymmetric unit; no electron density was seen for the first nine residues. The structure generated by the form B crystals (P1) was refined to 2.1 Å and contained 4 molecules in the asymmetric unit. In the final model, the following residues could not be modeled - chain A: 1-9, 68; chain B: 1-10, 25-34 and 66-70; chain C: 1-9; and chain D: 66-72. Overall, the structures from both crystal forms were very similar, and only the higher resolution structure (form A) is discussed in detail below unless explicitly specified.
The 34 helices and 13 β-strands (forming 1 mixed and 4 antiparallel β-sheets) of φ12 P2 are arranged in a classic RNA polymerase fold with fingers, thumb and palm domains [Fig. 1]2,15. In addition, the structure contains a largely α-helical C-terminal domain, the outward motion of which has been suggested to facilitate the egress of the elongating dsRNA2. The template tunnel of φ12 P2 leading to the catalytic site, is encircled by loops (“fingertips”) that connect the thumb and fingers domains. This narrow channel is capable of accommodating ssRNA but not dsRNA2. Also seen are the conserved RdRp sequence motifs A-E in the palm domain and F in the fingers domain [Supporting Information Figs. S1, S3]2,15. Motif C contains the polymerase catalytic triad 468GDD470 (SDD in φ6 P2).
Fig 1.
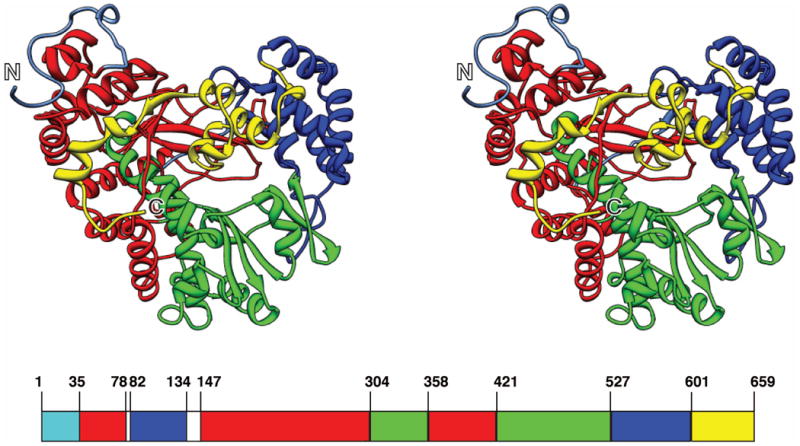
The structure of φ12 P2 (in cross-eyed stereo view) displaying the structural domains – fingers (35-78, 147-303, 358-420; red), thumb (82-134, 527-600; blue), palm (304-357, 421-526; green) and the C-terminal domain (601-659; yellow). The N- and C-termini are labeled.
Similarity to φ6 P2
The overall structure of φ12 P2 is quite similar to that of φ6 P2 (RMSD 1.22 Å over 338 atom pairs with the largest variability seen in the palm domain) [Supporting Information Fig. S4] despite low sequence identity (18%). Of the conserved RdRp sequence motifs, the largest deviation was seen in motif D [Supporting Information Fig. S5]. Motif D has been noted to be the most dynamic of the RdRp sequence motifs and carries a lysine (K499 in φ12, K487 in φ6; these have approximately the same orientation) that is known to the play the role of general acid by protonating the pyrophosphate leaving-group15.
Metal-ion binding site
The form B structure shows a Mg2+ ion bound at a site that is displaced by about 7.5 Å from the position occupied that would be occupied by the two Mg2+ ions required for catalysis [Fig. 2(A)]2. This Mg2+ ion is coordinated by the Oε1 atom of E503, the Oδ1 atom of D470, the main-chain O atom of V507 [Fig. 2(B)] and three water molecules (not shown). This mode of coordination is nearly identical to that seen in the NTP-free structure of φ6 P2 (PDB ID: 1HI8) (the corresponding φ6 residues are E491, D454 and A495)[Fig. 2(B)]. This site, which we refer to as the canonical non-catalytic site has also been shown to co-ordinate a Mn2+ ion in φ6 P22. However, the form A structure which also contains a non-catalytic Mg2+ion [also see Supporting Material Fig. S2], reveals a unique binding site that displaced by about 2.4 Å from the canonical non-catalytic site. This displacement results from the rotation of the sidechains of D470 and E503 (the Oε2 atom of E503 co-ordinates Mg2+ in contrast to the Oε1 atom in the form B structure) [Fig. 2(B)]. V507 is no longer within the co-ordination sphere of the Mg2+ ion and its main-chain O is now replaced by that of G348. It has been recently noted that Mg2+ (and by proxy, Mn2+) occupies multiple positions [Fig. 2(B)] around its canonical non-catalytic site in φ6 P2 in the presence of nucleic acids oligos and substrate NTPs. It was hypothesized that this shuttling of the non-catalytic metal ion between several positions helps guide substrate NTPs to the catalytic site to facilitate RNA synthesis4. While the metal-ion position seen in the form A structure differs from the positions noted in φ6 P2, it could constitute yet another stop in the Mn2+ shuttle. Alternatively, it could play another distinct, yet undetermined role in φ12 P2 that could lie on or off the catalytic pathway. In spite of the absence of NTPs or metal ions at the catalytic site of φ12 P2, the catalytic aspartates (D349 in motif A, D469 and D470 in motif C for φ12 P2) appear to be in proper orientation to enable catalysis, except for D470 in the form A structure (discussed above) [Fig. 2(A)].
Fig. 2.
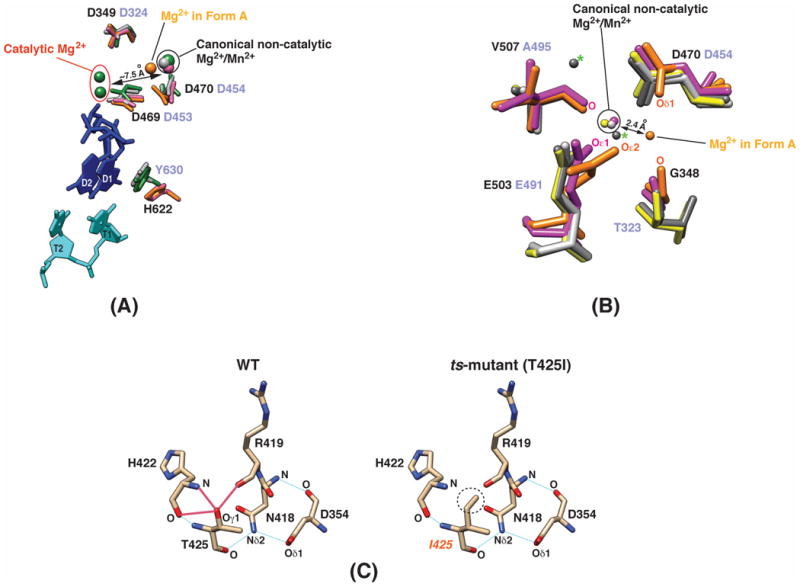
(A) Comparison between key catalytic elements of the P2 proteins from the φ12 and φ6 cystoviruses. Data is shown for φ12 P2 in form A (orange); φ12 P2 in form B (magenta); φ6 P2 (green, PDB ID: 1HI0) in complex with a single-stranded DNA template (5′-TTTCC-3′; cyan; T1, T2 are the template bases from the 3′-end), Mg2+ and Mn2+ ions (both in green), and GTP (blue; D1, D2 are the two GTP molecules that will form the first two bases of the daughter chain); also shown is φ6 P2 + Mg2+ (light grey, PDB ID: 1HI8). The φ12 P2 residues are labeled in black and the corresponding residues of φ6 P2 are labeled in light purple. The average orientation of the catalytic aspartates is similar (except D470 of φ12 P2 in form A, leading to a displacement of the Mg2+ ion from the canonical non-catalytic site). However, the conserved priming loop aromatic residue in φ12 P2 (H622) is not in a position that allows for optimal stacking with GTP (D1) as in the case of the corresponding residue in φ6 P2 (Y630). (B) Location of the binding site for the non-catalytic Mg2+ ions in φ12 and φ6 P2. The canonical non-catalytic metal-ion binding site seen in the form B (magenta) structure is quite similar to that seen in the structures of φ6 P2 (PDB ID: 4A8M, yellow; PDB ID: 1HI8, light grey). The Mg2+ position seen in the form A structure (orange) is unique and is displaced by 2.4 Å from the canonical non-catalytic site. The so-called “upward” and “downward” sites (indicated by the green ‘*’) are occupied by Mg2+ ions in the crystal structure of φ6 P2 (PDB ID: 4A8F, dark grey) in complex with a single-stranded DNA template, ATP and Mg2+. (C) Hydrogen-bonding patterns in the region surrounding the mutation site in wild-type φ12 P2 and the ts-mutant. Three potential hydrogen-bonding interactions involving the Oγ1 atom (left, indicated by the red lines) of T425 are lost upon mutating it to isoleucine (circled on the right panel, as in the ts-mutant).
The φ12 P2 conformation does not favor RNA binding
While φ12 P2 binds ssRNA in solution (data not shown), we were not able to obtain a structure in the presence of ssRNA oligos. The φ12 P2 structures provide some insight into the possible reasons for this. Inspection of the form A structure reveals a narrower template tunnel entrance compared with φ6 [Supporting Material Fig. S6]. A segment between N66 and Q73 forms a flap that covers the template entry tunnel and the sidechains of R68, Q69 and Q73 point towards the mouth of the template tunnel reducing its diameter [Supporting Information Fig. S6]. This corresponding segment in the case of φ6 P2 (I22-K29) forms a helix [Supporting Information Fig. S6] and moves away from the template entry tunnel creating a somewhat larger entry portal. However, this flap in φ12 P2 is highly dynamic and is fully observed only in the form A structure. In addition, the conserved aromatic residue (H622) in the so-called priming loop of φ12 P2 appears to be displaced by almost 6 Å relative to the position seen for the corresponding residue in φ6 P2 (Y630) [Fig. 2(A)]. In its current position, the H622 would be unable to optimally stack with the aromatic ring of the substrate NTP molecule thus preventing the formation of a stable initiation complex with NTPs and template ssRNA. A region (P617-Y621) near the priming residue (H622) forms a helical segment (a loop in the case of φ6 P2) and is also displaced by up to 6 Å, protruding into the template tunnel. Based on the crystal structures of the φ6 P2 initiation complex (with GTP and ssDNA)2, this segment in its current position would clash with the template bases (the clashes would largely involve the N618 sidechain and the last DNA base – T1 in Fig. 2(A)). Additionally, in the structures of φ6 P2 in complex with ssDNA2 or ssRNA3 oligos in the absence of NTPs, the last template base (T1) appears to occupy the same spatial position as H622 in φ12 P2, making this structure poorly suited to bind nucleic acids. This structural alteration does not appear to be an artifact of crystallization; the form A structure and all four independent molecules in the form B structure show nearly identical conformations (<0.2 Å RMSD) in this area, despite the presence of crystal contacts nearby for two of the form B molecules.
Structural explanation for the behavior of a ts-mutant
Yang et. al. noted that a specific mutation (T425I on motif B) on the palm domain of φ12 P2 enhances its temperature sensitivity6. This ts-mutant is active over a very small temperature range with maximum activity around 15 °C. In contrast, the wild-type enzyme shows a broader temperature profile with maximum activity around 30 °C. Inspection of the φ12 P2 structure provides some clues into the origin of this temperature sensitivity. The Oγ1 atom of T425 is in hydrogen-bonding distance with the main-chain oxygens of R419 and H422, in addition to the main-chain nitrogen of H422 [Fig. 2(C)]. These interactions are lost in the T425I mutant, thus increasing the probability of thermally allowed conformational transitions and hence temperature sensitivity. Interestingly, it was noted that the temperature sensitivity of the T425I mutant was largely restricted to the initiation stage6. This again, is not surprising, given that in de novo initiating RdRps, a very precise set of initiation nucleotides and a very precise geometry is required to form an optimal initiation complex. Thus, increased mobility and loss of this precise geometry may affect formation of the correct initiation complex. As shown in Fig. 2(C), loss of hydrogen-bonding with the R419 backbone in the ts-mutant may affect its local geometry, affecting in turn the orientation of the N418 backbone. The latter residue is hydrogen-bonded through its mainchain nitrogen to the mainchain oxygen of D354, and through its sidechain Nδ2 to the sidechain Oδ1 of D354 [Fig. 2(C)]. The highly conserved D354 that lies in the middle of motif A [Supporting Information Fig. S1] has been implicated in NTP selectivity15.
Conclusions
In conclusion, we have presented the structure of the RNA-directed RNA polymerase from the φ12 cystovirus in two distinct crystal forms. One of these structures, that represents the highest resolution achieved for a viral RdRp to date, and reveals a unique binding site for the non-catalytic Mg2+ ion. While several catalytic elements in the φ12 P2 structures are in positions favorable for catalysis, other elements occupy spatial orientations that are not conducive to the formation of a stable initiation complex. Additionally, the structures provide insight into the documented temperature sensitivity of a specific mutant. While attempts to crystallize the complexes of φ12 P2 with NTPs and ssRNA are currently underway, the present structure provides a crucial first step in determining the functional variability across the cystovirus family and extends the high-resolution structural data on the φ12 polymerase complex.
Supplementary Material
Acknowledgments
This work is supported by NSF grant MCB 083141; structural biology facilities at CCNY are partially supported by NIH grant 8G12MD007603. Dr. David Jeruzalmi (CCNY) is thanked for a critical reading of this manuscript. The authors also thank Dr. Leonard Mindich (PHRI) for useful discussions.
References
- 1.Mindich L. Packaging in dsRNA viruses. Adv Exp Med Biol. 2012;726:601–608. doi: 10.1007/978-1-4614-0980-9_26. [DOI] [PubMed] [Google Scholar]
- 2.Butcher SJ, Grimes JM, Makeyev EV, Bamford DH, Stuart DI. A mechanism for initiating RNA-dependent RNA polymerization. Nature. 2001;410:235–240. doi: 10.1038/35065653. [DOI] [PubMed] [Google Scholar]
- 3.Salgado PS, Makeyev EV, Butcher SJ, Bamford DH, Stuart DI, Grimes JM. The structural basis for RNA specificity and Ca2+ inhibition of an RNA-dependent RNA polymerase. Structure. 2004;12:307–316. doi: 10.1016/j.str.2004.01.012. [DOI] [PubMed] [Google Scholar]
- 4.Wright S, Poranen MM, Bamford DH, Stuart DI, Grimes JM. Noncatalytic ions direct the RNA-dependent RNA polymerase of bacterial double-stranded RNA virus φ6 from de novo initiation to elongation. J Virol. 2012;86:2837–2849. doi: 10.1128/JVI.05168-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang H, Makeyev EV, Bamford DH. Comparison of polymerase subunits from double-stranded RNA bacteriophages. J Virol. 2001;75:11088–11095. doi: 10.1128/JVI.75.22.11088-11095.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang H, Gottlieb P, Wei H, Bamford DH, Makeyev EV. Temperature requirements for initiation of RNA-dependent RNA polymerization. Virology. 2003;314:706–715. doi: 10.1016/s0042-6822(03)00460-4. [DOI] [PubMed] [Google Scholar]
- 7.Mancini EJ, Kainov DE, Grimes JM, Tuma R, Bamford DH, Stuart DI. Atomic snapshots of an RNA packaging motor reveal conformational changes linking ATP hydrolysis to RNA translocation. Cell. 2004;118:743–755. doi: 10.1016/j.cell.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 8.Eryilmaz E, Benach J, Su M, Seetharaman J, Dutta K, Wei H, Gottlieb P, Hunt JF, Ghose R. Structure and dynamics of the P7 protein from the bacteriophage φ12. J Mol Biol. 2008;382:402–422. doi: 10.1016/j.jmb.2008.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Van Duyne GD, Standaert RF, Karplus PA, Schreiber SL, Clardy J. Atomic structures of the human immunophilin FKBP-12 complexes with FK506 and rapamycin. J Mol Biol. 1993;229:105–124. doi: 10.1006/jmbi.1993.1012. [DOI] [PubMed] [Google Scholar]
- 10.Otwinowski Z, Minor W. Processing of X-ray Diffraction data collected in oscillation mode. Meth Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 11.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 14.Langer G, Cohen SX, Lamzin VS, Perrakis A. Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nature Protoc. 2008;3:1171–1179. doi: 10.1038/nprot.2008.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cameron CE, Moustafa IM, Arnold JJ. Dynamics: the missing link between structure and function of the viral RNA-dependent RNA polymerase? Curr Opin Struct Biol. 2009;19:768–774. doi: 10.1016/j.sbi.2009.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.