

Abstract
The sensing of accurate homologous recognition and pairing between discreet chromosomal regions and/or entire chromosomes entering meiosis is an essential step in ensuring correct alignment for recombination. A component of this is the recognition of heterology, which is required to prevent recombination at ectopic sites and between non-homologous chromosomes. It has been observed that a number of diverged organisms add an additional layer to this process: regions or chromosomes without a homologous counterpart are targeted for silencing during meiotic prophase I. This phenomenon was originally described in filamentous fungi, but has since been observed in nematodes and mammals. In this review we will generally group these phenomena under the title of meiotic silencing, and describe what is known about the process in the organisms in which it is observed. We will additionally propose that the functions of meiotic silencing originate in genome defense, and discuss its potential contributions to genome evolution and speciation.
Keywords: chromosome homology, meiosis, meiotic silencing, prophase 1, recombination, trans-sensing
Introduction
The hallmark of sexual reproduction is meiosis, a complex yet highly conserved process that allows for controlled genetic exchange between organisms. Mediated through the process of meiotic recombination, this genetic exchange involves alignment of homologous chromosomes that itself requires extensive searching and satisfaction of stringent homology criteria before the onset of recombination is allowed. Concomitant with this process is the establishment of a unique chromatin architecture mediated by regulated modifications of nucleosomal histones, and specialized non-histone proteins (Ivanovska & Orr-Weaver 2006). The extensive alignment of entire chromosomes, initiated early in meiosis by poorly understood homology trans-sensing mechanisms, allows genomes from two distinct organisms to be compared for significant differences prior to recombination and passage on to the next generation. These differences may be as dramatic as chromosome-wide polymorphisms between species, or as minor as novel and recent transposon insertions. The former may trigger checkpoints and other alarms that lead to hybrid sterility; the latter may be used to recognize and trigger an immune response to genomic invaders such as viruses and transposable elements (TE).
Sexual reproduction is good for organisms, but also beneficial to the propagation of TE. The elements themselves are mutagenic, and thus the benefits of sex must outweigh this potential harm to the species. TE may not always be detrimental, however, and probably contribute to aspects of genetic diversity and/or chromosome stability (Pardue & DeBaryshe 2003). The genomic landscape is thus shaped to a great extent by the ongoing battles waged between genetic programs contributing ‘constructively’ to the regulated expression and stable inheritance of a species’ DNA, and invasive, parasitic genetic elements such as transposons. It is also clear that different species have dealt with this invasive consequence of sexual reproduction with varying degrees of success. Some organisms, such as Neurospora, exhibit a very rigorous genome defense and hence have relatively small genomes with low repetitive content. Others, such as plants, have multiple genome defense mechanisms in place but exhibit little success in countering TE spreading. As a result, plants genomes can be enormous, with contributions from TE and TE remnants as high as 80% of their sequence content – although this has not been noticeably detrimental, and may even have been beneficial, to plant representation in the biosphere (Wessler 1988, 1998, 2001, Wessler et al. 1995, Feschotte et al. 2002).
Complex genomes have evolved sophisticated ways to sense the presence, and control the behavior, of repeated DNA sequences. At risk is their chromosomal integrity and, with it, the very existence of the species. Control is especially critical in meiosis since there is both an opportunity for new TE to exchange between genomes, and a risk that pre-existing elements may create ectopic homology and cause heterologous chromosome alignment and exchange. A lack of control of either can have catastrophic consequences for the organism (e.g. Bourc’his & Bestor 2004). It is therefore not surprising to find that molecular mechanisms used by many types of cells to recognize and suppress viruses, retrotransposons, and insertion sequences have been either adapted from, or adapted to, meiotic events. In this review we will attempt to summarize those meiotic defense mechanisms that have been observed in highly diverged organisms. As each of these meiotic processes ultimately lead to strong post-transcriptional or transcriptional repression of genes, genetic regions, or chromosomes, we will use the general term meiotic silencing to encompass them all. As we will discuss, in some experimental organisms (Neurospora crassa, Caenorhabditis elegans) these processes share obvious mechanistic overlap involving RNA interference (RNAi) pathways, whereas mammals may have adapted other mechanisms toward the same end. Still other organisms, such as flies and plants, do not obviously exhibit meiotic silencing as defined within this review. The evolutionary consequences of meiotic silencing, and its potential roles and impacts on genome organization will be discussed.
Meiotic silencing in Neurospora
A single cytoplasm is shared by many nuclei in filamentous fungi, so their genomes are at greater risk of expansive invasion by TE than those of plants and animals. Genomes like that of N. crassa have developed a number of complex defenses to preserve their integrity (Galagan et al. 2003, Borkovich et al. 2004). At least four distinct but potentially interrelated mechanisms are known: DNA methylation, quelling, repeat induced point mutation (RIP), and meiotic silencing (Figure 1). During haploid vegetative growth, AT-rich elements that invade the genome may be detected and densely methylated (Romano & Macino 1992, Singer et al. 1995, Miao et al. 2000, Freitag et al. 2001). Multi-copy elements can also activate quelling, a post-transcriptional silencing mechanism (Cogoni & Macino 1999b, 2000, Cogoni 2001, Pickford et al. 2002). Once activated, quelling produces diffusible signals (i.e. small interfering RNA or siRNA) that block the propagation of the repeated element by RNAi-mediated degradation of its transcript (Catalanotto et al. 2002). If nuclei containing repeated DNA elements enter the sexual phase of the life cycle, the sequences are subjected to repeat-induced point-mutation (RIP) (Selker 1990, 1997, Freitag et al. 2002). In this process, G:C-to-A:T transition mutations are introduced into the duplicated sequences, presumably by unrepaired deamination of methylated cytosines. Many of the remaining, non-mutated cytosine bases are kept methylated by DIM-2, a DNA methyltransferase responsible for all known cytosine methylation in Neurospora (Kouzminova & Selker 2001). Additionally, these sequences can be associated with trimethylated histone-H3, lysine-9 (H3K9me3) heterochromatin and transcriptionally repressed (Rountree & Selker 1997, Tamaru et al. 2003). Remaining elements are scrutinized during meiosis by meiotic trans-sensing (or trans-vection) (Aramayo & Metzenberg 1996), a process that checks the identity and the presence of ectopic or novel sequences in the genomes with which they are about to exchange genetic material. Such elements are then silenced post-transcriptionally by meiotic silencing. Given this extensive ‘genome paranoia’, it is not surprising that duplicated elements of any sort, including paralogous genes, are rare in Neurospora (Galagan et al. 2003).
Figure 1.
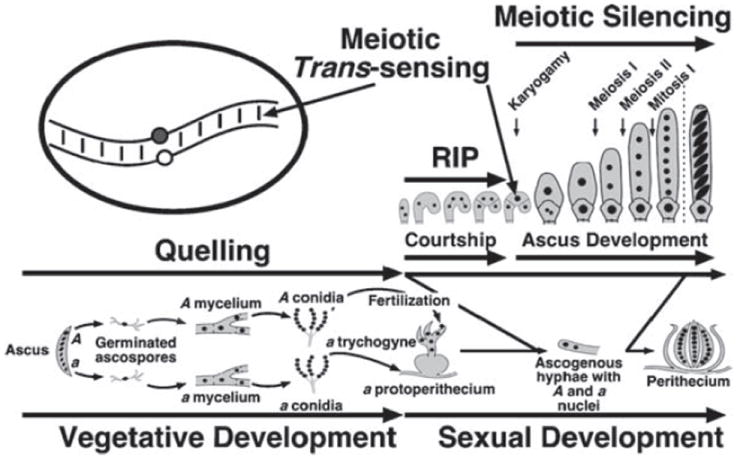
Life cycle of Neurospora crassa. Neurospora has two mating types, A and a. During vegetative development, strains of either mating type form asexual spores (conidia). Sexual spores (ascospores) are formed when strains of opposite mating type mate and undergo sex. Mating occurs when, in response to nitrogen starvation, a strain (either A or a) forms a protoperithecium (female element) that is fertilized by a male element of the opposite mating type to initiate perithecium development. After fertilization the male- and the female-derived nuclei coexist in a heterokaryotic tissue and divide mitotically until they are sorted into a dikaryotic tissue (dikaryotic cells contain only one nucleus of each mating type). The nuclei then pair and undergo a series of synchronous mitoses until the tip of the hyphal cell in which they reside bends to form a hook-shaped cell called a crozier. One crozier will originate one ascus, containing eight ascospores. In the figure the time and places in the life cycle where quelling, RIP, meiotic trans-sensing and meiotic silencing occur are indicated. Homologs pair inside the only diploid nucleus present in the zygote, immediately after karyogamy. The oval representing a diploid nucleus shows only one pair of chromosomes in the process of ‘sensing’.
Meiosis in Neurospora occurs inside an ascusmother cell called the zygote, a cell that initially contains the two haploid nuclei of opposite mating type (Figure 1). The zygote develops inside the perithecium – the complex, multicellular sexual reproductive apparatus (Raju 1992). The nuclei inside the zygote undergo karyogamy, meiosis, post-meiotic mitosis, and are then compartmentalized, resulting in an ascus cell that contains eight haploid spores arrayed in an order that reflects their lineage (Raju 1980, 1992). One perithecium may contain up to 200 developing asci. Meiotic segregation and recombination can be studied in Neurospora by analyzing individual asci (tetrads) or random spores ejected from numerous asci (Perkins 1966, 1988, Davis & de Serres 1970). The zygote is the only known diploid cell of the Neurospora life cycle, and thus the only known cell in Neurospora in which trans-sensing and meiotic silencing occurs.
Pairing between homologous chromosomes is essential for meiotic recombination and hence proper chromosome segregation, and this occurs in the Neurospora zygote. Homolog pairing requires poorly understood thresholds of assessing genetic differences among chromosomes in order to promote homologous recognition and to prevent heterologous pairing. In Neurospora, successful pairing of chromosomes is subjected to a another level of analysis mediated by trans-sensing, which identifies the unsuccessful pairing of discrete DNA regions due to the presence of a heterologous region, or the presence of a deletion on the opposite chromosome (i.e. the lack of a homologous region). This failure to ‘sense’ a homologous region triggers an RNAi-mediated silencing mechanism that has been termed MSUD (meiotic silencing of unpaired DNA) (Shiu et al. 2001). This meiotic trans-sensing process was discovered during studies of the Ascospore maturation-1 (Asm-1) gene (Aramayo & Metzenberg 1996). ASM-1 (the product of asm-1+) is an abundant nuclear protein essential for the formation of aerial hyphae, the development of protoperithecia (the haploid female sexual structures), and the maturation of the ascospores (the haploid sexual spores) (Aramayo & Metzenberg 1996, Aramayo et al. 1996). Recessive, loss-of-function mutations in Asm-1 are spore-autonomous: spores inheriting the mutant allele fail to develop, whereas spores with the asm-1+ allele mature normally. A deletion allele of Asm-1 (Asm-1Δ), however, was ascus dominant: all spores within the ascus failed to develop, including the ones carrying the asm-1+ allele. Surprisingly, heterozygous strains (Asm-1Δ; Asm-1+) into which an additional, ectopically integrated normal copy was introduced were still sterile, showing that the dominance of Asm-1Δ was not due to haploinsufficiency (Aramayo & Metzenberg 1996). Further experiments proved that pairing of the Asm-1 gene, regardless of where it occurs, is essential for its normal expression and ascus development, and it was postulated that paired alleles could ‘sense’ (i.e. trans-sense or undergo meiotic trans-vection) the presence or absence of their partners in homologous chromosomes (Aramayo & Metzenberg 1996). Furthermore, failure to sense an intact and appositionally localized copy on the homologous chromosome caused silencing of copies positioned elsewhere in the genome, regardless of their own pairing status (Shiu et al. 2001, Lee et al. 2004). Meiotic trans-sensing and meiotic silencing are two highly interrelated but different mechanisms that, together, scan and control the integrity of the genomes that participate in meiosis.
What induces meiotic silencing?
Chromosomes appear to ‘sense’ the presence of homology and/or identify small regions of nonidentity within their homologs in early meiotic stages. If the regional identities are equivalent above a certain threshold, development proceeds normally. If not, meiotic trans-sensing machinery is activated and targets homologous sequences in the genome, regardless of their own pairing status (Figure 2). This means that genes present at either their own canonical chromosomal positions (Figure 3a), or at the same ectopic chromosomal position (Figure 3b), do not trigger silencing. Some of the quantitative and qualitative aspects of the sensing threshold have been addressed experimentally and three types of silencing (i.e. cis-, trans-, and cis-trans) have been described (Figure 3c to 3f) (Lee et al. 2004). Quantitatively, the following observations have been made: (1) If given one small and one large loop of unpaired DNA, both carrying the same identical length of DNA homologous to a set of paired reporter genes, the large loop will silence more efficiently than the smaller one. (2) Given two loops of identical size, but one carrying a greater proportion DNA homologous to a set of paired reporter genes, this loop will silence more efficiently than the one carrying less homologous DNA. (3) The silencing signal produced by an unpaired loop is confined to the unpaired region and does not ‘spread’ to neighboring regions (e.g. paired reporter genes can be located next to a region of unpaired DNA without being significantly perturbed). (4) The canonical promoter of a gene need not be present in the loop of unpaired DNA for a gene to be silenced. (5) Meiotic silencing does not affect the ability of a promoter to direct transcription at a later developmental time (Kutil et al. 2003, Lee et al. 2003, 2004, Pratt et al. 2004).
Figure 2.
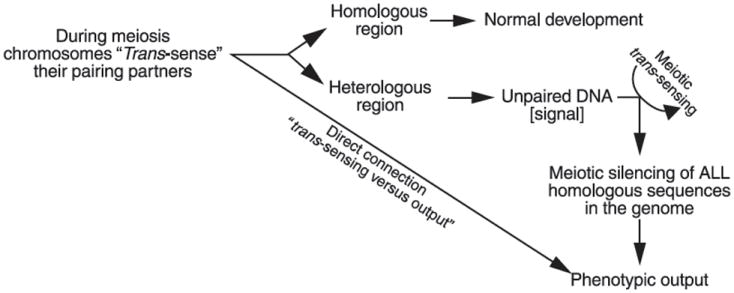
A model for meiotic trans-sensing; see text for details.
Figure 3.
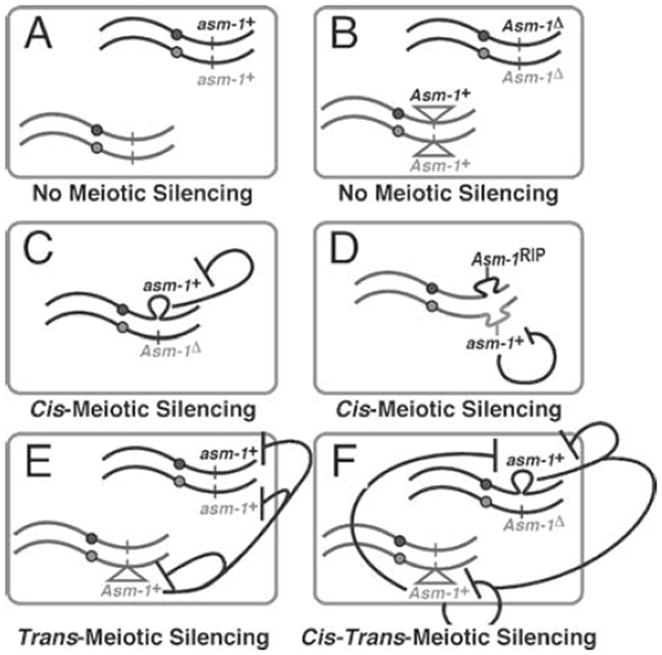
How can meiotic silencing be induced? Heterologies and insertions/deletions located on both homologs do not induce silencing (A and B). In general, only significant lack of homology on an opposite chromosomal region induces silencing and results in the induction of either: cis-silencing or trans-silencing. Cis-silencing can be induced in two different ways: by the presence of deletions (C) or by the presence of heterologous regions (D). In contrast, trans-silencing occurs only when a fragment of the gene is inserted elsewhere in the genome (E). The lack of homology at that particular ectopic chromosomal position then results in the silencing, in-trans, of paired functional copies of the gene. In contrast to cis-silencing, the fragment present in-trans does not need to be functional but, if it were, then its unpairing would, in addition, result in its own cis-silencing. It is therefore possible to have both cis-, and trans-silencing occurring at the same time (E and F).
Significant sequence deviation between homologous alleles, as opposed to deletions or insertions, is also sensed and can trigger silencing. The degree of identity that is required to escape trans-sensing and avoid triggering meiotic silencing suggests that the Neurospora system has very stringent standards for defining homology (Pratt et al. 2004). This has been assessed by crossing strains carrying wild-type alleles of meiotic reporter genes with strains carrying varying degrees of mutation (generated by RIP) in the homologous locus. The mutated alleles, if sufficiently different, would confer a dominant phenotype by triggering meiotic silencing and repressing the wild-type gene. Amazingly, as little as 6% sequence divergence (or 94% identity) could trigger low detectable silencing, yet a slight increase of identity to 97% (or 3% divergence) prevented significant silencing (Pratt et al. 2004). Furthermore, the methylation status of the RIP-mutated alleles shifted the sequence identity threshold, such that methylated mutant alleles were detected as having worse identity to the wild-type allele than unmethylated ones (Pratt et al. 2004). This suggests that either the 5-methylcytosines contribute to heterology as a ‘fifth base’, or that their effects on chromatin structure (by recruitment of methylated DNA-binding proteins, etc.) impact on homology recognition. These contributions are not mutually exclusive, and thus both may contribute to identification and pairing.
At what level is the known meiotic machinery involved at this level of homology sensing? Recent data suggest that the stable long-distance pairing (i.e. alignment), short-distance pairing (i.e. synapsis), recombination and segregation steps of chromosome pairing in meiosis are all dispensable for meiotic silencing. Mutations in spo-11, ski-8, Msh-4, mei-1, Mei-2 and mei-3, which cause severe defects in, or eliminate synapsis and/or recombination, do not abrogate gene-specific silencing despite global asynapsis (R. J. Pratt & R. Aramayo, unpublished). spo-11, and likely ski-8, mutants fail to stably align their chromosomes (Tesse et al. 2003, Bowring et al. 2006). This suggests that trans-sensing and the triggering of meiotic silencing may occur during an earlier, unstable chromosome alignment stage of meiosis upstream of mechanisms guiding synapsis. Very little is understood about these early events in chromosome recognition; but it is conceivable that the mechanisms involved in this trans-sensing use features of homology recognition mechanisms present in all organisms. Alternatively, trans-sensing could occur through a novel parallel independent pathway, perhaps through a mechanism similar to programmed DNA elimination in ciliates (discussed below).
RNAi and meiotic silencing
A role for RNAi has been strongly inferred by the identification of genes required for Neurospora meiotic silencing. Shiu et al. showed that mutations in Suppressor of ascus dominance-1 (Sad-1), encoding a putative RNA-dependent RNA polymerase (RdRP), suppress the ascus dominance of Asm-1Δ and other reporter genes (Shiu et al. 2001). RdRP have been identified as essential components of RNAi in several organisms, and a separate RdRP, QDE-1, has been shown to be essential for quelling the vegetative RNAi silencing process (Cogoni & Macino 1999a). Further screens for meiotic silencing factors identified two other additional RNAi-related genes: one coding for an Argonaute-like protein, Suppressor of meiotic silencing-2 (Sms-2) (Lee et al. 2003), and the other coding for a Dicer-like protein, Suppressor of meiotic silencing-3 (Sms-3) (Lee et al. 2007). Several other Sms mutants have also been identified (R. J. Pratt, D. W. Lee & R. Aramayo, unpublished). In addition, the post-transcriptional gene silencing (PTGS) nature of meiotic silencing was confirmed using transgene reporters. Only regions containing homology to the reporter transcript result in silencing when unpaired (Lee et al. 2004). Based on these observations a model for RNAi involvement in meiotic silencing was postulated (Figure 4; Lee et al. 2003).
Figure 4.
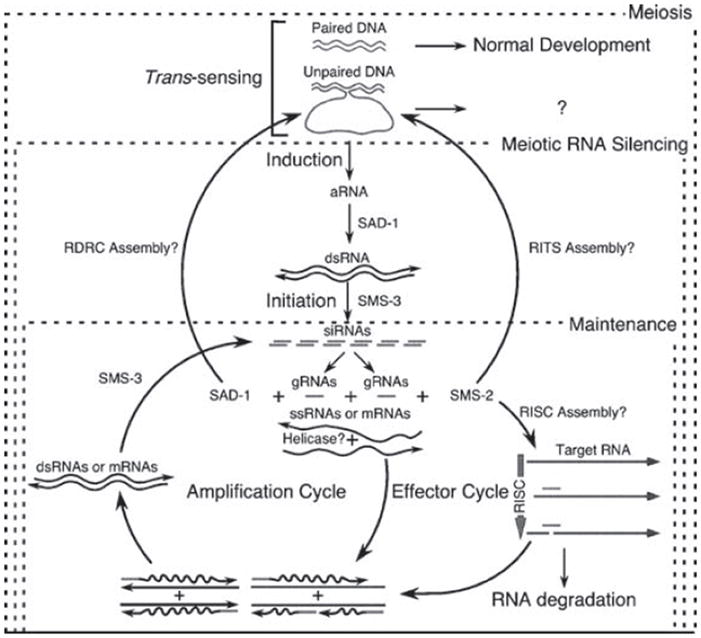
A model for meiotic silencing. Paired DNA does not induce silencing. Unpaired DNA does. The degree of unpairing determines the strength of the induction step, which presumably involves the synthesis of aberrant RNA (aRNA) and its conversion to double-stranded RNA (dsRNA) by the SAD-1 RNA-dependent RNA polymerase (RdRP). The presence of dsRNA triggers the initiation of the meiotic RNA silencing process, which is composed of: The conversion of the dsRNA trigger into siRNAs via the SMS-3 Dicer (initiation step). The use of guide RNA (gRNA), as primers and single-stranded RNA (ssRNA), as template by SAD-1 RdRP to generate dsRNA (amplification cycle). The incorporation of the gRNA generated by both the initiation step and the amplification cycles into the RNA-inducing silencing complex – RISC, to direct the endonucleolytic cleavage of mRNA or ssRNA (effector cycle). It is possible that SAD-1 and SMS-2 maintain the silencing by participating in complexes related to the RNA-directed RNA polymerase (RDRC), and the RNA-induced transcriptional silencing (RITS) complexes detected in S. pombe, respectively.
The Sad-1, Sms-2, and Sms-3 orthologs in the fission yeast Schizosaccharomyces pombe (Rdp1, Ago1, and Dcr1, respectively) constitute the main RNA interference (RNAi) pathway in this organism (Volpe et al. 2002). The RNAi pathway in S. pombe is essential for normal chromosome biology, including heterochromatin formation (e.g. histone H3K9 methylation) and normal centromere and telomere functions (Martienssen et al. 2005). In contrast, Neurospora strains carrying loss-of-function mutations in any of the components of the meiotic silencing pathway (i.e. Sad-1, Sms-2, or Sms-3) do not have a discernible vegetative phenotype. In addition, whereas DNA methylation in Neurospora requires a histone H3K9-specific methyltransferase, this activity is independent of RNAi (Freitag et al. 2004), and there is no evidence that transcriptional repression accompanies meiotic silencing (Lee et al. 2004). However, all of the genes required for meiotic silencing in Neurospora are also required for fertility (Shiu et al. 2001; Lee et al. 2003, 2007), indicating that components of the meiotic RNA silencing pathway are required for the completion of sexual development and/or meiosis and that these processes are intimately co-dependent.
A model for meiotic RNA silencing
Once identified, regions of non-homology are predicted to produce some form of aberrant RNA, which is converted into dsRNA by the action of the RdRP, SAD-1 (Figure 4). The presence of dsRNA triggers the initiation step of the pathway, which could involve the conversion of the dsRNA trigger into siRNA, although the presence of small RNA with homology to the target sequence have not as yet been observed. This step is predicted to be executed by the SMS-3/DCL-1 Dicer, because it is required for meiotic silencing (Galagan et al. 2003, Lee et al. 2003, Borkovich et al. 2004, Catalanotto et al. 2004). The maintenance of silencing probably involves amplification, also through SAD-1, and degradation of the target mRNA via an RNA induced silencing complex (RISC), of which the Argonaute-like protein SMS-2 is predicted to be an essential component, as in other conserved RNAi pathways (Lee et al. 2003). The lack of detectable siRNA-like molecules may be due to their rapid utilization and turnover, which may also contribute to the lack of silencing ‘memory’ once sporulation is complete and homolog interactions are over (R. J. Pratt & R. Aramayo, unpublished). Interestingly, the SAD-2 protein is also required for meiotic silencing, but its role appears to be to anchor or recruit the SAD-1 RdRP to the cytoplasmic face of the nuclear periphery (Shiu et al. 2006). This indicates that SAD-1’s RdRP activity is required outside of the nucleus, perhaps to ‘screen’ RNA as they transit nuclear pores as a sort of quality check. In any case, RdRP activity is likely a downstream effector of meiotic silencing, and not a component of the trans-sensing mechanism that must necessarily be nuclear. An ‘RNA quality’ monitoring system, however, implies that deficient pairing triggers production of aberrant transcripts to alert the system, and hence that proficient recognition and pairing suppresses this transcription. Determining any role(s) for aberrant transcription in meiotic silencing awaits further analyses.
Meiotic silencing in C. elegans
The nematode C. elegans is a hermaphroditic species in which sex determination is governed by the ratio of X chromosomes to autosomes (X:A). In wild-type populations grown under laboratory conditions, over 99% of the animals are XX hermaphrodites produced as self-progeny by self-fertilization, and thus have an X:A of 2X:2A. This self-fertilization is made possible because XX animals produce both sperm and oocytes. In the larval hermaphrodite ovaries, the first germ cells to progress through meiosis enter spermatogenesis. Sperm are produced until the last larval molt; at this point the germline sex identity switches, and cells progressing through meiosis undergo oogenesis for the rest of the life of the animal (reviewed in Seydoux & Schedl 2001).
On rare occasion non-disjunction of the X chromosome occurs, yielding an embryo with sex chromosome genotype XO and thus an X:A ratio of 1X:2A. This ratio triggers development into true males that produce only sperm and are somatically equipped for mating with hermaphrodites – and hence capable of cross-fertilization. Therefore, the X chromosome can encounter male meiosis either fully synapsed with its homolog (XX larval spermatogenesis), or completely devoid of a partner (XO spermatogenesis). The natural occurrence of spermatogenesis in which the X chromosome has or lacks a partner during meiosis has made it an excellent organism for studying sex-specific determinants of meiotic silencing, as discussed below. Before this discussion, however, it is important to introduce what is known about epigenetic regulation of the endogenous target of meiotic silencing in this organism: the X chromosome.
The C. elegans X chromosome
Goldstein first reported that the univalent X chromosome of XO males forms a ‘heterochromatic X-body’ in pachytene nuclei during adult male meiosis (Goldstein 1982, Goldstein & Slaton 1982). This structure was highly reminiscent of the XY body or sex body in mammalian male meiosis, which is a consequence of meiotic sex chromosome inactivation (MSCI), and will be discussed later in this review. Subsequent studies have shown that meiotic chromatin is substantially different on the X chromosome(s) between the two sexes, and both X states are dramatically different from autosomal chromatin in both sexes. In both adult germlines the X chromatin is highly depleted of nucleosomal histone modifications that normally correlate with gene activity. For example, acetylation of histone H3 at lysines 9 and 14, acetylation of histone H4 at lysines 8, 12, and 16 and methylation of H3 at lysine 4 all correlate with transcriptional activity. All of these marks are readily detected on the autosomes at all stages of meiosis in both sexes; none is detected on the X chromosome in pre-meiotic and early meiotic stages in male and hermaphrodite adult germ cells (Table 1; Reuben & Lin 2002, Kelly et al. 2002, Fong et al. 2002).
Table 1.
X chromatin and structural differences in XX and XO meiosis.
| X chromosome histone modifications | Pre-meiosis | Lep/Zyg | Pachy | Diplo | Oocyte | Spermatid | |
|---|---|---|---|---|---|---|---|
| XX oogenesis | H3K4me2 | − | − | −/+ | + | + | n.a. |
| H3Acetyl | − | − | −/+ | + | + | n.a. | |
| H3K9me2 | − | − | − | − | − | n.a. | |
| H3K27me2,3 | + | + | + | + | + | n.a. | |
| XX spermatogenesis | H3K4me2 | − | − | − | − | n.a. | − |
| H3Acetyl | − | − | − | − | n.a. | − | |
| H3K9me2 | − | − | − | − | n.a. | − | |
| H3K27me2,3 | + | + | + | + | n.a. | − | |
| X Body | − | − | + | + | n.a. | n.d. | |
| XO spermatogenesis | H3K4me2 | − | − | − | − | n.a. | − |
| H3Acetyl | − | − | − | − | n.a. | − | |
| H3K9me2 | − | − | + | + | n.a. | − | |
| H3K27me2,3 | + | + | + | + | n.a. | − | |
| X body | − | − | + | + | n.a. | n.d. |
n.a.=Not applicable, n.d.=not detected. Bold symbols indicate significant differences between XX and XO spermatogenesis and/or oogenesis. X body indicates premature condensation of the X chromosome(s).
The C. elegans polycomb group MES proteins play a role in germline X inactivation, since mutations in the mes genes result in the aberrant accumulation of ‘active’ histone modifications on the X chromosomes in hermaphrodite germ cells (Fong et al. 2002). mes mutants exhibit severe sterility, and the phenotypic severity rises with X chromosome dosage, indicating that the sterility is linked to X misregulation. Males are therefore less affected than hermaphrodites, suggesting that the Polycomb system is not a major component of the meiotic silencing pathway and the male-specific X chromatin assembly that will be discussed next (Fong et al. 2002).
A germline sex-specific difference in X inactivation is observed in late pachynema. In male meiosis, ‘active’ epigenetic marks remain depleted from the X chromosome through all stages, including the meiotic divisions; i.e. there is no evidence of post-meiotic activation of the X in male spermatogenesis. In contrast, as oocytes progress through late pachynema and enter diplonema, the X chromosomes begin to rapidly accumulate ‘active’ histone modifications. This correlates with late-pachynema/early diplonema transcriptional activation of X-linked oocyte-enriched genes (Kelly et al. 2002). This activation, however, is a transient process since RNA polymerase II is depleted from all chromosomes as they enter diakinesis shortly thereafter (Kelly et al. 2002, Fong et al. 2002, Bean et al. 2004). However, although transcription is shut down, the oocyte chromatin retains active histone marks through the meiotic divisions, and the maternal genome is transmitted to the embryo with an epigenetic memory of transcriptional activity. Importantly, the fully synapsed XX pair in larval hermaphrodites also does not activate during spermatogenesis (Kelly et al. 2002). Indeed, formation of the hypercondensed ‘X body’ occurs in both XX and XO spermatogenesis. Therefore, while X inactivation occurs in both oogenic and spermatogenic germ lines in C. elegans, it is reversed in oogenesis and maintained in spermatogenesis. Furthermore, as spermatogenesis progresses all the chromosomes lose all assayed histone modifications and hypercondense as the histones are probably replaced, presumably by recently identified protamine-like proteins (Chu et al. 2006). In short, the epigenetic regulation of X chromosomes going though spermatogenesis and oogenesis differ significantly.
Synapsis-dependent heterochromatin assembly
One epigenetic process is unique to XO spermatogenesis and is not observed in XX animals. During XO male spermatogenesis, the X chromosome uniquely and transiently accumulates high levels of a histone modification predominantly associated with heterochromatin, histone H3 dimethylated on lysine 9 (H3K9me2). The appearance of this epitope occurs in early pachytene and remains associated with the X chromosome until diplotene, when it progressively disappears as the autosomes begin to condense before the meiotic divisions. The H3K9me2 enrichment precedes ‘X body’ formation, but it is not required for it (discussed below). Interestingly, the enrichment of H3K9me2 is limited to the X chromosome in XO meiosis; it does not occur in either hermaphrodite XX spermatogenesis or oogenesis (Kelly et al. 2002, Reuben & Lin 2002, Bean et al. 2004). Conversely, enrichment is observed on the single X in XO oogenesis in sex reversal mutants mutants and this can repress X-linked oocyte-specific genes (Bean et al. 2004). H3K9me2 targeting is thus strictly dependent on the absence of a homolog for the X chromosome as it enters pachynema. Prevention of H3K9me2 targeting requires pairing and synapsis of the homologs, as a mutation in which X chromosome pairing is specifically defective causes both asynapsed X chromosomes to be enriched in H3K9me2 during XX oogenesis (Bean et al. 2004). Surprisingly, the mechanism has no particular preference for the X chromosome – any unsynapsed chromosome or chromosomal fragment is sufficient to activate this deposition. Defective synapsis of all chromosomes, due to mutation of a synaptonemal complex (SC) component, causes widespread decoration of H3K9me2, and autosome-derived free duplications also attract the modification (Bean et al. 2004). These results indicate that the X chromosome in male meiosis is actually a derived target for a mechanism that identifies and modifies the chromatin of any DNA entering the pachytene stage of meiosis with defective synapsis.
RNAi and C. elegans meiotic silencing
The restriction of the C. elegans H3K9me2 enrichment mechanism to unsynapsed chromatin in meiosis (hereafter referred to as meiotic silencing for simplicity) makes a very appealing comparison with the fungal and ciliate meiotic silencing mechanisms (described below). In both fungi and ciliates a requirement for factors linked to RNAi has been demonstrated – is there any evidence for this in C elegans?
Several genes have been identified that play essential roles in H3K9me2 targeting to unsynapsed chromatin. One, RHA-1, is a putative RNA helicase (Walstrom et al. 2005). Another, EGO-1, is a putative RdRP that is required for normal meiosis and fertility, and RNAi in germ cells (Smardon et al. 2000, Maine et al. 2005, Vought et al. 2005). As discussed earlier, the SAD-1 putative RdRP is essential for meiotic silencing in Neurospora (Shiu et al. 2001, Shiu & Metzenberg 2002, Lee et al. 2003; Pratt et al. 2004), suggesting a strong mechanistic link between meiotic silencing in worms and the fungal process(es). RdRP play essential roles in RNAi-mediated transcriptional gene silencing and are required for the formation of H3K9me2-enriched heterochromatin in fission yeast (Volpe et al. 2002, Martienssen et al. 2005). However, although in Neurospora one newly identified Sms mutant suggests a connection (D. W. Lee & R. Aramayo, unpublished), there is currently no conclusive evidence that histone modifications play a role in fungal meiotic silencing. The C. elegans mechanism may represent a subsequent (or parallel) adaptation of the transcriptional silencing mechanisms found in S. pombe.
Preliminary studies have identified two additional factors, an Argonaute family member and a protein with a helicase motif, that are required for meiotic silencing in C. elegans, and each is also required for RNAi in germ cells (X. She & E. Maine, personal communication, Duchaine et al. 2006, Yigit et al. 2006). C. elegans meiotic silencing is therefore intricately linked with RNAi pathways, yet perhaps in a unique manner. Unlike Neurospora, the Dicer endonuclease is not required for meiotic silencing in C. elegans (Maine et al. 2005). It has been proposed that unsynapsed regions may be targets for aberrant transcription, which then recruits the RNA silencing machinery to those sites, similar to mechanisms described in S. pombe (Maine et al. 2005, Irvine et al. 2006).
Another factor required for worm meiotic silencing is him-17, as H3K9 methylation of the male X is defective in mutants (Reddy & Villeneuve 2004). HIM-17 contains multiple THAP domains, a C2CH Zinc finger motif that mediates sequence-specific DNA binding (Reddy & Villeneuve 2004, Clouaire et al. 2005). Meiotic double-strand breaks (DSB) also do not form in him-17 mutants, although synapsis can occur. The lack of DSB is not linked to the meiotic silencing defect in him-17 animals since H3K9me2 targeting is not disrupted by defects in SPO-11, the meiotic endonuclease required for DSBs (Reddy & Villeneuve 2004). HIM-17 thus seems to act upstream of both meiotic silencing and DSB formation, and may be a component of a system that recognizes or creates chromatin structure that is appropriate for both processes. Interestingly, him-17 interacts genetically with Lin35/Rb, which has established roles in chromatin organization (Reddy & Villeneuve 2004). The signals that detect asynapsed chromatin and/or prevent meiotic silencing of normally synapsed chromatin in C. elegans are poorly understood.
A connection with imprinted X inactivation?
A consequence of meiotic silencing is an X chromosome that is largely inactive in male meiosis (discussed below). Interestingly, the X chromosome that ‘experiences’ spermatogenesis appears to be uniquely inactive in the early XX embryo (Bean et al. 2004). An epigenetic imprint is observed, characterized by an initial absence of several histone H3-specific modifications, which is strikingly limited to the sperm-derived X chromosome in XX embryos. It is clear that the ‘paternal’ X (Xp) is inherited in this state, as the imprint is visible in early stages of sperm chromatin decondensation before it forms the paternal pronucleus. This period coincides with active histone replacement and/or insertion into the paternal chromatin in the absence of transcription and DNA replication (W. G. Kelly, unpublished). The Xp is thus either uniquely refractory to H3-specific histone-modifying enzymes and/or refractory to replication-and transcription-dependent histone replacement, or the Xp is enriched in an H3-like molecule that is difficult to modify and/or remove. Importantly, there is no similar disparity in histone H4 modifications between the Xp and all other chromosomes.
The Xp accumulates H3 modifications after a number of rounds of cell division, suggesting a replication-coupled replacement or dilution of the imprint. It is important to note that delayed activation is also seen in self-progeny of hermaphrodites, albeit with faster kinetics than cross progeny sired by XO males (Bean et al. 2004). This indicates that meiotic silencing of the X chromosome in sperm meiosis is not required for imprint establishment. Likewise, a period of meiotic X inactivation is not required per se since, as discussed, the X is also inactive throughout most of meiosis during oogenesis. The major differences between the Xp and Xm meiosis are: (a) the Xm reactivates prior to fertilization (i.e. is not maintained as inactive); and (b) the Xp forms a condensed ‘X body’ during spermatogenesis (even when fully synapsed, as in XX larval spermatogenesis). Whether X reactivation in oogenesis prevents imprinted X inactivation, or X body formation in spermatogenesis is required, remains to be shown. However, it is intriguing to note that the X ‘imprint’ is limited to histone H3 modifications, which may suggest that X body formation is the result of spermspecific incorporation of a histone H3-like molecule that either lacks modification sites or is somehow refractory to acetylation and methylation. Gradual substitution of the unknown molecule with S-phase histone H3 (and its subsequent modification) during DNA replication could account for the disappearance of the imprint during embryogenesis. The conserved H3.3 variant histones are unlikely to play a major role since (a) they do not lack residues that can be modified, and (b) H3.3::GFP fusion proteins are absent from the X during spermatogenesis and do not appear enriched on the Xp in the embryo (Ooi et al. 2006).
The haploid male X chromosome is therefore a significant endogenous target of meiotic silencing in C. elegans, and (as discussed below) this appears to have had an impact on the genetic content of the X to make it generally transcriptionally inactive in all stages of spermatogenesis. This overall inactivity may have made it an enriched substrate for an epigenetic imprint that slows its reactivation in the embryo. As we will see, there are similarities between worms and some mammals in this regard.
Meiotic silencing in Drosophila
Lifschytz and Lindsley proposed, almost four decades ago, that X chromosome inactivation during male meiosis is a common theme and requirement for male fertility in many organisms, including Drosophila (Lifschytz & Lindsley 1972). Their conclusion for Drosophila was drawn from interpretation of the dominant male sterility caused by X:autosomal translocations, the cause of which is still not understood in flies. Importantly, the sterile phenotypes are all post-meiotic and it is not until this stage that the X appears to be transcriptionally inactive (Rastelli & Kuroda 1998). A complicating factor is that many classical fly spermatogenesis genes with post-meiotic functions are actually transcribed earlier in spermatocytes and translated after the meiotic divisions. However, some male fertility factors are Y-linked in Drosophila species and sequences are highly expressed from Y lampbrush loops in spermatocytes (e.g. Vogt et al. 1982). Indeed, recent microarray experiments showed no overall difference in X-linked gene expression in male versus female gonads, and it was concluded that the X chromosome is dosagecompensated upwards in male gonads to match female and autosome expression (Gupta et al. 2006). It is therefore unclear whether meiotic silencing such as exists in C. elegans and mammals (discussed below) also occurs in Drosophila.
Interestingly, there is no obvious RdRP ortholog in the Drosophila genome, an activity essential for meiotic silencing in Neurospora and worms. However, RNAi-related processes are certainly present in fly germ cells and are essential for normal male meiosis, due at least in part to a role in regulating the repetitive Stellate locus and retrotransposons (Aravin et al. 2001). RdRP are proposed to be required for amplification of RNAi ‘triggers’ for a systemic response (Yigit et al. 2006); such amplification via RdRP activity may be required to recruit silencing complexes to sites of asynapsis, but not to high-copy elements. Hence, the silencing of high-copy elements in Drosophila could occur via an RNAi-mediated mechanism despite the apparent absence of an RdRP. Male meiosis is also unique in flies because it is achiasmatic; a synaptonemal complex does not form and recombination does not occur, although homologs are paired by other mechanisms (McKee 2004). It would seem hazardous to maintain a mechanism for silencing unsynapsed chromatin in cells where all chromosomes would be targeted.
Meiotic silencing in mammals
Mammalian genomes also appear to lack recognizable RdRP yet meiotic silencing has recently been described in mice (Baarends et al. 2005, Turner et al. 2005). The degeneration of the sex-delimited X partner, the Y chromosome, is not as complete in mammals as has presumably occurred in C. elegans. However, the sequence homology between the X and Y chromosomes is limited to small regions termed the pseudo-autosomal regions, or PAR (reviewed in Handel 2004). Similar to the unsynapsed X chromosome in C. elegans, the poorly synapsed XY pair forms a heterochromatic XY or sex body during male pachynema, and uniquely attracts or is highly enriched in a number of proteins and protein modifications (Handel 2004). The X chromosome is also transcriptionally inactivated in male meiosis in a process named meiotic sex chromosome inactivation, or MSCI. MSCI is initiated in prophase I and although it was originally thought to be reversed post-meiotically, evidence now strongly suggests that very few X-linked loci are activated in round spermatids and later stages (Namekawa et al. 2006, Turner et al. 2006).
Unique chromatin architecture accompanies MSCI. As in C. elegans, the sex body becomes enriched in H3K9me2 and is lacking in histone acetylation (Khalil et al. 2004). Interestingly, the chromatin is also enriched in H3K4me2, a mark that is found in active and/or poised chromatin (Khalil et al. 2004). Another nucleosomal histone, H2A, is highly targeted for ubiquitylation in sex body chromatin by Rad 18 (van der Laan et al. 2004, Baarends et al. 2005, de Vries et al. 2005). Furthermore, variant forms of H2A, macroH2A1.2 and the phosphorylated from of H2AX, γH2AX, are enriched on the sex body (Handel 2004). γH2AX is a conserved mark of DSB breaks in DNA, a normal event in meiotic recombination. However, whereas γH2AX disappears from the autosomes as meiotic recombination proceeds, it persists on the sex body through pachynema (Mahadevaiah et al. 2001). The ATR kinase is required for γH2AX accumulation on the sex body, and it is directed there by BRCA1; lesions in H2AX or Brca1 abrogate sex body formation and MSCI (Fernandez-Capetillo et al. 2003, 2004a,b, Fernandez-Capetillo & Nussenzweig 2004, Turner et al. 2004, Cuadrado et al. 2006, Kruhlak et al. 2006). Disruption of the synaptonemal complex protein Sycp1also disrupts sex body formation and MSCI, but it is likely that this is due to ATR mislocalization (de Vries et al. 2005).
Recent evidence suggests that histone replacement accompanies MSCI, resulting in a massive loss of the canonical S-phase histones H3.1 and H3.2, and enrichment for the H3.3 replacement histone variant specifically in sex body chromatin (van der Heijden et al. 2007). The appearance of γH2AX on the sex body precedes H3.1/H3.2 replacement, and there is no replacement in Sycp1–/– spermatocytes, indicating that the ATR/γH2AX system is upstream of, and may guide, the histone replacement mechanism. The replacement of H3.1/H3.2 also correlates with the loss of histone modifications observed during sex body formation, and thus may be the cause of this loss. It has been proposed that this allows erasure of the epigenetic marks that are added pre-pachynema in advance of post-meiotic regulation (van der Heijden et al. 2007). Another replacement histone variant, H2A.Z accumulates in X and Y chromatin post-meiotically, but is depleted from the sex body during pachynema/diplonema (Greaves et al. 2006). Interestingly, H2A.Z is also depleted from sex chromatin during meiosis in C. elegans, albeit in both sexes (W. Kelly, unpublished).
It has long been thought that sex body formation is a protective mechanism to ensure progression of XY nuclei though meiosis without triggering a ‘pachytene checkpoint’ that senses synaptic defects (Handel 2004). Recent data, however, suggest that MSCI and sex body formation are consequences of meiotic silencing. For each modification listed above, as in H3K9me2 targeting in C. elegans, the assembly of unique sex body chromatin during pachynema is neither germline sex–nor sex chromosome-specific, and the process has been named meiotic silencing of unsynapsed chromatin (MSUC; Schimenti 2005). Therefore, and also as in C. elegans, X chromosome inactivity during male meiosis (MSCI) is a consequence of it being a (probably evolved) target of a meiotic silencing mechanism (MSUC). Similarly, unsynapsed autosomal segments can attract all of the chromatin modifications found on the XY body, and are likewise inactivated in male meiosis (Turner et al. 2004, 2005, Baarends et al. 2005). The histone H3 replacement mechanisms described above can also occur on asynapsed autosomes, and are thus a component of MSUC (van der Heijden et al. 2007). The X chromosome in XO oogenesis is similarly inactivated (Baarends et al. 2005, Turner et al. 2005). Conversely, forcing YY pairing in male meiosis prevents inactivation of the Y, which causes arrest and subsequent loss of these spermatocytes, presumably because of inappropriate Y transcription (Turner et al. 2006). MSUC thus appears related to meiotic silencing in C. elegans. However, although perhaps the objective is the same in both organisms – identifying and silencing unsynapsed chromatin – the effector mechanisms involved have little apparent overlap. The C. elegans homolog of BRCA1, for example, is not required for male X silencing and H2AX is not found in C. elegans (W. Kelly, unpublished).
The observed loss of YY spermatocytes that escape MSUC has led to the suggestion that escape from meiotic silencing, and consequential ectopic activation of sex-linked genes, causes pachytene arrest of XYY cells and others with aberrant synapsis, rather than a pachytene checkpoint (Roeder & Bailis 2000, Turner et al. 2006). A corollary to this is that defects in homolog synapsis may lead to repression of essential loci, which may also trigger pachytene arrest in organisms with meiotic silencing (Turner et al. 2005). Some consequences of asynapsis that have been ascribed to a meiotic checkpoint function in mammals may therefore be due to degeneration triggered by meiotic silencing prior to progression to any checkpoint (P. Burgoyne, personal communication).
RNAi roles in meiotic silencing in mammals
All of the known factors required for meiotic silencing in mice also play roles in DNA damage recognition and repair pathways, suggesting that this is an adaptive mechanism unrelated to the RNAi-related mechanisms in other organisms. Is there any evidence for an underlying connection to RNAi? Recent data showed that the mouse Maelstrom (MAEL) protein is localized to both the XY body and structures called chromatoid bodies during male meiosis, and this has been proposed to represent such a link (Costa et al. 2006). Within the germ cell nuage, MAEL interacts with MILLI and MIWI, two Argonaute-related proteins that are both essential for spermatogenesis and are implicated in germline-specific RNAi pathways (Aravin et al. 2006 and references therein). The functional relevance of MAEL to either meiotic silencing or RNAi, however, remains to be determined.
More intriguingly, transgenes passed as hemizygous through mouse meiosis have been shown to induce an epigenetically repressed status on the opposing homologous allele that is heritable. Amazingly, the repressed allele is further able to induce a similar state when passaged in a heterozygous state opposite another allele with identical (and normal) sequence, albeit with decreasing efficiency (Rassoulzadegan et al. 2002, 2006, Herman et al. 2003). This is highly reminiscent of the paramutation phenomenon in plants (Chandler et al. 2000), which was recently shown to require a putative RdRP (Alleman et al. 2006). Although paramutation in plants is not necessarily linked to meiotic events, the mouse phenomena are clearly initiated by meiotic heterozygosity. Importantly, small siRNA-like molecules have been observed in one case, suggesting a link with RNAi mechanisms (Rassoulzadegan et al. 2006). This suggests that meiotic silencing-style mechanisms related to epigenetic events can and do operate in mammalian meiosis. Further, it also indicates that non-homology can be recognized with DNA elements as small as transgenes, which is similar to the limits observed in Neurospora.
Meiotic silencing and imprinting
As discussed earlier, the X chromosome in C. elegans that is a substrate for meiotic silencing, the Xp, arrives into the embryo in an apparently ‘pre-inactivated’ state. In mammals the Xp is preferentially inactivated in early embryos in a specific process called imprinted X inactivation (reviewed in Heard & Disteche 2006). In placental (eutherian) mammals, this is maintained only in the extraembryonic tissues, whereas in marsupials and monotremes the Xp remains inactive in most female tissues (Huynh & Lee 2005). The imprint for Xp inactivity is presumably established during spermatogenesis in both worms and mammals. However, other than their consequences and their shared X chromosome target, the meiotic silencing mechanisms in worms and mice have precious little overlap. Furthermore, there are no significant similarities in the respective mechanisms that inactivate the X in the early embryo. In the case of worms, the Xp arrives inactive like all chromosomes, but is less readily activated than the others during zygotic gene activation. In mice, robust transcription of the Xist non-coding RNA is required for X inactivation and a subsequent series of complicated and highly regulated events (Heard & Disteche 2006). There is evidence that the Xp arrives pre-inactivated in mice, but this is controversial and the opposite conclusion has also been reached (Okamoto et al. 2004, 2005, Huynh & Lee 2005). On the surface this debate may seem minor, since all agree that the Xp is preferentially inactivated in very early pre-implantation embryos. However, preinactivation has been proposed to provide continuity with the X inactivity during spermatogenesis, and is thus a potential paradigm for understanding this and other imprinting mechanisms. Does pairing and/or meiotic silencing play any role in imprinted X inactivation?
In mice the Xist locus is essential for imprinted inactivation, and Xist transgenes at ectopic (autosomal) sites have been tested for their ability to induce hallmarks of imprinted inactivation in a parent of origin-dependent manner. Such transgenes show paternal imprinting and most, if not all, of its hallmarks at these ectopic loci – yet this regulation is independent of pairing status during meiosis in the paternal germ line (Okamoto et al. 2005). Furthermore, the establishment of the imprint is independent of the hallmarks of MSCI, as none of the characteristic modifications associated with the sex body (γH2AX, etc.) was detected on the transgene during meiosis.
Note, however, the conundrum associated with mammalian X inactivation: the near global inactivation of the X chromosome relies on activation of the X-linked Xist locus. Therefore, imprinted X inactivation in mouse is a combination of a paternal imprint on the Xist locus that prevents its repression, and a maternal imprint that prevents its expression in the early embryo. Furthermore, parental Xist regulation in the embryo is opposite to the chromosome as a whole in adult meiosis: inactive in males and active in females. This suggests that the Xist ‘promoter’ has evolved a specific, imprinted regulation that may be unique to this locus in eutherian mammals. Indeed, imprinted inactivation of the Xp is maintained through all developmental stages in marsupial mammals, and this occurs in the absence of an Xist locus (Duret et al. 2006, J. McCarrey, personal communication). Therefore, the Xp in non-eutherian mammals may indeed have an underlying inherent propensity for inactivity that is independent of Xist regulation – as exists in C. elegans. Is there any evidence for this?
Recent microarray studies have shown that in worms, flies, and mammals the single X chromosome in males has an overall transcriptional activity that approaches two times that of any autosome (Nguyen & Disteche 2006, Gupta et al. 2006). This was originally hypothesized by Charlesworth to be the primary goal of dosage compensation: rather than a system to equalize X gene dosage between the sexes, the goal may be primarily to balance haplo-X gene expression with diplo-autosome expression in males, and secondarily to suppress this hyperactivation in diplo-X females (Charlesworth 1991). Since males normally inherit their X chromosome solely from the oocyte, this may indicate that the hyperactivity is an epigenetic consequence of transcriptional activity in oogenesis, as a hallmark of oogenesis is high transcription of maternal products. Conversely, there may be a similar but opposite epigenetic memory of the Xp’s inactivity in spermatogenesis. This could be weakened to different extents in eutherian mammals, with differing reliances on the Xist system and its separate and specific imprinted regulation of the Xist locus. Underlying imprinted repression of some loci may contribute to why XpO mice are smaller and grow slower than XmO mice (Thornhill & Burgoyne 1993). Weakening or loss of inherited inactivity as a consequence of Xist regulation may be why Xp inactivation is only stabilized in the earliest somatic tissues in eutherians (perhaps as a consequence of early differentiation), but is more persistent in the developing marsupial (Huynh & Lee 2005). Preliminary analyses suggest that the marsupial X chromosome is also largely inactivated in male meiosis (J. McCarrey, personal communication), lending support to the idea that paternal X meiotic inactivation, and hence meiotic silencing, could be connected to Xist-independent imprinted X inactivation in noneutherian mammals.
A role for meiotic silencing in X chromosome biology
In worms and mammals the genes required for spermatogenesis are poorly represented on the X chromosome. This bias is particularly dramatic in C. elegans, as genes required for germline function in both sexes are also depleted from the X, and even genes with enriched expression in oogenesis are significantly under-represented (Reinke et al. 2004). Indeed, there is evidence that essential loci have not escaped pressures against X-linkage. A number of examples of ‘housekeeping’ genes (e.g. a ribosomal protein, a poly-A binding protein, etc.) with autosome/X-linked pairs have been studied in worms, and in each case the autosomal copy is required for fertility, whereas the X-linked copy has somatic functions. Mutations in the former therefore cause fertility defects, whereas mutations in the latter yield growth or developmental defects (Maciejowski 2005; S. L’Hernault, personal communication). Analyses of glycolytic and TCA cycle genes indicate a similar trend: genes that are X-linked show a very strong bias for having an autosomal paralog (W. Kelly, unpublished).
The situation in mammals is similar, albeit apparently less restrictive. Although earlier studies indicated enrichment for sperm-functioning genes on the X, it was shown that this enrichment was restricted to early-acting genes, and that X-linked late- and post-meiotic genes are deficient (Khil et al. 2004, Wang et al. 2005). Retrotransposition appears to be one mode of movement of sperm-functioning genes from the X. A bioinformatics study of retroposed genes and pseudogenes in the mouse and human genomes found a striking enrichment of retroposed gene trafficking in both the X-to-autosome and autosomes-to-X directions (Emerson et al. 2004). Importantly, the movement of genes from the X to autosomes (in mice) seems heavily biased towards genes with late spermatogenic expression or function.
It seems obvious that there would be a strong selection to remove genes required for spermatogenesis from the X chromosome in species exhibiting MSCI. It then follows that, since MSCI is a consequence of meiotic silencing, meiotic silencing must play a significant role in shaping the genetic profile of the X chromosome. However, it has long been proposed that the genetic content of the X is under the selective force of sexual antagonism, in which genes beneficial only to one sex or and/or antagonistic to the other will undergo stronger selection if linked to a sex chromosome (reviewed in Vallender & Lahn 2004, Vicoso & Charlesworth 2006). It has been proposed that the loss of sperm genes from the X because of sexual antagonism creates a chromosome that by default is inactivated; i.e. MSCI is a consequence of the loss of large blocks of genes from the X due to antagonistic pressure. This model, termed the sexual antagonism X inactivation (SAXI) model, was proposed prior to the discovery of meiotic silencing in worms and mice, and hence may be incomplete (Wu & Xu 2003). It seems likely that progressive degeneration of a male-limited X homolog (Y chromosome), and a resulting enrichment for targets of meiotic silencing on the X in male meiosis, also play important roles in gene loss from the X chromosome. Meiotic silencing is an ancient process that predates multicellular organisms; it is therefore highly likely that it is a contributing force for shaping sex chromosomes in organisms that retain it.
Evolutionary conservation and function of meiotic silencing
The ancient origins of meiotic silencing in all of its current manifestations are likely grounded in RNAi-mediated genome defense mechanisms. The role of RNAi in genome defense has been identified in many organisms, including the ones discussed in this review. It is likely that the recognition of linear sequence heterology in meiosis, crucial for productive recombination, is also (and perhaps originally) a component of genome defense. Sexual reproduction is beneficial to, and may have been driven by, transposable elements (Hickey 1993). The meiotic processes that we observe today may be partly a combined product of escalating warfare between genomes and invasive elements, with homolog recognition originating as a friend-or-foe assessment. Such trans-sensing and its connection to the meiotic recombination machinery is very poorly understood, but could be either related to RNAi processes, or a separate process that has adapted RNAi mechanisms as the effector process in some organisms.
Although meiotic silencing is evident in highly diverse organisms, it also appears to be absent in many, including yeast and flies, and at least some plants. It is difficult to see a pattern that explains these differences, but perhaps a connection between meiotic events and RNAi components may be instructive to explore. Fission yeast, for example, seems to have all of the RNAi components required, albeit a minimal set, and has no meiotic silencing (A. Klar & R. Aramayo, unpublished). However, recombination in this organism occurs without synapsis. Conversely, budding yeast have synapsis, but no RNAi components and no meiotic silencing (Aravind et al. 2000, Anantharaman et al. 2002). In Drosophila, synapsis is absent only in males, but there is no evidence for meiotic silencing in either sex, and an RdRP seems to be missing from the fly RNAi toolkit. Similarly, there are no obvious RNAi-related RdRP encoded in mammalian genomes, yet mammals have both synapsis and meiotic silencing. However, meiotic silencing in mammals, as defined by MSCI and MSUC, appears to be largely an adaptation of DNA repair pathways and, apparently, mechanistically quite distinct from RNAi. It remains to be determined, however, if the widespread antisense transcription observed in the mammalian transcriptome replaces the roles played by RdRP in other organisms (Katayama et al. 2005). Finally, there are flowering plants, which have multiple isoforms of all RNAi components and full synaptonemal complex formation – but no clear demonstration as yet for meiotic silencing (V. Chandler, personal communication). A related phenomenon in plants, paramutation, is an inter-allelic, trans-sensing epigenetic process that requires an RdRP, mop-1 (Chandler et al. 2000, Chandler & Stam 2004). Paramutation may have originated from meiotic silencing, but adapted later to operate in mitotic tissues.
The absence of either an RdRP or a synaptonemal complex may select against meiotic RNAi silencing, or select for adaptation of a non-RNAi-related mechanism. As mentioned for flies, the loss of meiotic synapsis could predispose for a further loss of meiotic silencing, if unsynapsed chromatin can be targeted for repression by this process. Similarly, loss of synapsis in fission yeast could have caused the loss of a mechanistic link between RNAi and meiotic silencing. The loss of an RdRP, on the other hand, would not cripple RNAi-related mechanisms, but might decrease their sensitivity to endogenous substrates such as unpaired DNA.
Irrespective of the effector response in meiotic silencing, all sexually reproducing organisms must utilize some aspects of trans-sensing to detect homology between chromosomes while also creating a barrier to heterologous pairing. A number of transvective processes have been identified in diverse species – operating either within or outside of meiosis. In many cases these processes have either been determined, or are predicted to have a mechanistic link to RNAi (Wu & Morris 1999, Herman et al. 2003; Alleman et al. 2006, Rassoulzadegan et al. 2006). Genomic DNA elimination in Tetrahymena is a dramatic example of a trans-sensing process that involves RNAi (for a more informative review of thei complex process, see Mochizuki & Gorovsky 2004). These ciliates have two nuclei that share one cytoplasm: a transcriptionally inactive, diploid micronucleus and a transcriptionally active, polyploid macronucleus. The macronuclear DNA is a fragmented and amplified version of the micronucleus in which all repetitive DNA elements, including transposons, have been deleted (reviewed in Mochizuki & Gorovsky 2004, Matzke & Birchler 2005). After conjugation, meiosis, and genome mixing, the old macronucleus is destroyed and a new macronucleus arises from a new, genetically mixed micronucleus – but any sequence not present in the old macronucleus is eliminated during new macronuclear maturation. The recognition process – a trans-sensing process between two separate nuclei – involves production of small, scanning RNA (scRNA) from micronuclear transcription that are proposed to ‘scan’ the old macronuclear genome. Those with homology (e.g. encoding essential loci) are degraded, whereas those with no homology (e.g. repetitive elements) are then used to guide elimination of homologous sequences in the new macronucleus. This results in a fragmented genome in which all repetitive elements have been eliminated, and the remaining gene-encoding sequences are amplified and transcribed during vegetative growth. The elimination targeting mechanism is highly related to RNAi-mediated transcriptional silencing in plants and S. pombe, but ciliates have clearly found a more permanent way to silence the targets – delete them! It is interesting to consider that related genome scanning mechanisms may be at work in meiotic trans-sensing.
From a genome protection perspective it makes sense that organisms have developed a molecular mechanism like meiotic silencing to check the integrity of the genomes participating in meiosis. Meiotic silencing allows prevention of the expression of novel insertions (e.g. transposons) without necessarily affecting the regulation of adjacent genes. From an evolutionary perspective, however, meiotic silencing could also participate in speciation. Genomes evolving significant differences in genes required for germ cell/spore viability or meiotic progression could trigger trans-sensing alerts and meiotic silencing, leading to reproductive isolation from each other. In this context, Shiu et al. (2001), showed that mutations in Sad-1-RdRP significantly increased interbreeding efficiencies among various Neurospora species, indicating that a component of the species barrier in this organism is meiotic silencing (Shiu et al. 2001). Genomes could also accumulate a series of mutations or duplications in regions that house genes that are not required for meiosis without consequences to fertility. Heterochromatic regions of genomes, for example, may have become transposon ‘traps’ partly because they are regions in which transposons can invade without creating a meiotic silencing consequence. Heterozygous (novel) insertions would be recognized by trans-sensing and repressed to prevent further spread, but unless the genomic neighborhood housed meiotic loci that would be affected by any potential ‘spreading’ of the repression there would be no consequence to fertility. Duplications of genes involved in development, considered a major source of evolutionary complexity, would also be allowed.
The study of meiotic trans-sensing and meiotic silencing is therefore not only important from a mechanistic perspective (i.e. to understand how chromosomes pair and how homologous regions are detected), but it is also important from an evolutionary point of view. Meiotic silencing may be another reproductive fitness test in the assessment of genomes that are brought together by sexual reproduction, and its potential role in genome evolution merits consideration.
Acknowledgments
We apologize for the exclusion, due to limited space considerations, of information pertinent to organisms other than those included here. We also wish to thank the following colleagues for helpful discussions and/or the sharing of data unpublished at the time of this writing: Yun Tao, John Lucchesi, Eleanor Maine, Xingyu She, Bruce McKee, John McCarrey, Paul Burgoyne, Vicki Chandler, Brian Oliver, Edith Heard, Steve L’Hernault, Jeff Chen and Amar Klar. This work was supported by US Public Health Service Grants GM58770 to R.A. and GM63102 to W.K.
Footnotes
Note added in proof
Recent results from the Villeneuve lab indicate that CHK-2, a kinase required for efficient homolog pairing in worm meiosis, is also required for H3K9me2 accumulation on the male X. CHK-2 is related to the cds1/chk2 DNA repair checkpoint kinases, which may provide a conserved mechanistic link between worm and mammalian meiotic silencing (Bessler JB et al. (2007) Genetics epub 10.1534/genetics.107.070987).
References
- Alleman M, Sidorenko L, McGinnis K, et al. An RNA-dependent RNA polymerase is required for paramutation in maize. Nature. 2006;442:295–298. doi: 10.1038/nature04884. [DOI] [PubMed] [Google Scholar]
- Anantharaman V, Koonin EV, Aravind L. Comparative genomics and evolution of proteins involved in RNA metabolism. Nucl Acids Res. 2002;30:1427–1464. doi: 10.1093/nar/30.7.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aramayo R, Metzenberg RL. Meiotic transvection in fungi. Cell. 1996;86:103–113. doi: 10.1016/s0092-8674(00)80081-1. [DOI] [PubMed] [Google Scholar]
- Aramayo R, Peleg Y, Addison R, Metzenberg R. Asm-1+, a Neurospora crassa gene related to transcriptional regulators of fungal development. Genetics. 1996;144:991–1003. doi: 10.1093/genetics/144.3.991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravin A, Gaidatzis D, Pfeffer S, et al. A novel class of small RNAs bind to MILI protein in mouse testes. Nature. 2006;442:203–207. doi: 10.1038/nature04916. [DOI] [PubMed] [Google Scholar]
- Aravin AA, Naumova NM, Tulin AV, et al. Double-stranded RNA-mediated silencing of genomic tandem repeats and transposable elements in the D. melanogaster germline. Curr Biol. 2001;11:1017–1027. doi: 10.1016/s0960-9822(01)00299-8. [DOI] [PubMed] [Google Scholar]
- Aravind L, Watanabe H, Lipman DJ, Koonin EV. Lineage-specific loss and divergence of functionally linked genes in eukaryotes. Proc Natl Acad Sci USA. 2000;97:11319–11324. doi: 10.1073/pnas.200346997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baarends WM, Wassenaar E, van der Laan R, et al. Silencing of unpaired chromatin and histone H2A ubiquitination in mammalian meiosis. Mol Cell Biol. 2005;25:1041–1053. doi: 10.1128/MCB.25.3.1041-1053.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bean CJ, Schaner CE, Kelly WG. Meiotic pairing and imprinted X chromatin assembly in Caenorhabditis elegans. Nat Genet. 2004;36:100–105. doi: 10.1038/ng1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borkovich KA, Alex LA, Yarden O, et al. Lessons from the genome sequence of Neurospora crassa: tracing the path from genomic blueprint to multicellular organism. Microbiol Mol Biol Rev. 2004;68:1–108. doi: 10.1128/MMBR.68.1.1-108.2004. table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourc’his D, Bestor TH. Meiotic catastrophe and retrotransposon reactivation in male germ cells lacking Dnmt3L. Nature. 2004;431:96–99. doi: 10.1038/nature02886. [DOI] [PubMed] [Google Scholar]
- Bowring FJ, Yeadon PJ, Stainer RG, Catcheside DE. Chromosome pairing and meiotic recombination in Neurospora crassa spo11 mutants. Curr Genet. 2006;50:115–123. doi: 10.1007/s00294-006-0066-1. [DOI] [PubMed] [Google Scholar]
- Catalanotto C, Azzalin G, Macino G, Cogoni C. Involvement of small RNAs and role of the qde genes in the gene silencing pathway in Neurospora. Genes Dev. 2002;16:790–795. doi: 10.1101/gad.222402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catalanotto C, Pallotta M, ReFalo P, et al. Redundancy of the two dicer genes in transgene-induced posttranscriptional gene silencing in Neurospora crassa. Mol Cell Biol. 2004;24:2536–2545. doi: 10.1128/MCB.24.6.2536-2545.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler VL, Stam M. Chromatin conversations: mechanisms and implications of paramutation. Nat Rev Genet. 2004;5:532–544. doi: 10.1038/nrg1378. [DOI] [PubMed] [Google Scholar]
- Chandler VL, Eggleston WB, Dorweiler JE. Paramutation in maize. Plant Mol Biol. 2000;43:121–145. doi: 10.1023/a:1006499808317. [DOI] [PubMed] [Google Scholar]
- Charlesworth B. The evolution of sex chromosomes. Science. 2001;251:1030–1033. doi: 10.1126/science.1998119. [DOI] [PubMed] [Google Scholar]
- Chu DS, Liu H, Nix P, et al. Sperm chromatin proteomics identifies evolutionarily conserved fertility factors. Nature. 2006;443:101–105. doi: 10.1038/nature05050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clouaire T, Roussigne M, Ecochard V, et al. The THAP domain of THAP1 is a large C2CH module with zinc-dependent sequence-specific DNA-binding activity. Proc Natl Acad Sci USA. 2005;102:6907–6912. doi: 10.1073/pnas.0406882102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cogoni C. Homology-dependent gene silencing mechanisms in fungi. Annu Rev Microbiol. 2001;55:381–406. doi: 10.1146/annurev.micro.55.1.381. [DOI] [PubMed] [Google Scholar]
- Cogoni C, Macino G. Gene silencing in Neurospora crassa requires a protein homologous to RNA-dependent RNA polymerase. Nature. 1999a;399:166–169. doi: 10.1038/20215. [DOI] [PubMed] [Google Scholar]
- Cogoni C, Macino G. Homology-dependent gene silencing in plants and fungi: a number of variations on the same theme. Curr Opin Microbiol. 1999b;2:657–662. doi: 10.1016/s1369-5274(99)00041-7. [DOI] [PubMed] [Google Scholar]
- Cogoni C, Macino G. Post-transcriptional gene silencing across kingdoms. Curr Opin Genet Dev. 2000;10:638–643. doi: 10.1016/s0959-437x(00)00134-9. [DOI] [PubMed] [Google Scholar]
- Costa Y, Speed RM, Gautier P, et al. Mouse Maelstrom: the link between meiotic silencing of unsynapsed chromatin and microRNA pathway? Hum Mol Genet. 2006;15:2324–2334. doi: 10.1093/hmg/ddl158. [DOI] [PubMed] [Google Scholar]
- Cuadrado M, Martinez-Pastor B, Murga M, et al. ATM regulates ATR chromatin loading in response to DNA double-strand breaks. J Exp Med. 2006;203:297–303. doi: 10.1084/jem.20051923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RH, de Serres FJ. Genetic and microbiological research techniques for Neurospora crassa. In: Colowick SP, Kaplan NO, editors. Metabolism of Amino Acids and Amines. New York: Academic Press; 1970. pp. 79–143. [Google Scholar]
- de Vries FA, de Boer E, van den Bosch M, et al. Mouse Sycp1 functions in synaptonemal complex assembly, meiotic recombination, and XY body formation. Genes Dev. 2005;19:1376–1389. doi: 10.1101/gad.329705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchaine TF, Wohlschlegel JA, Kennedy S, et al. Functional proteomics reveals the biochemical niche of C. elegans DCR-1 in multiple small-RNA-mediated pathways. Cell. 2006;124:343–354. doi: 10.1016/j.cell.2005.11.036. [DOI] [PubMed] [Google Scholar]
- Duret L, Chureau C, Samain S, Weissenbach J, Avner P. The Xist RNA gene evolved in eutherians by pseudogenization of a protein-coding gene. Science. 2006;16:1653–1655. doi: 10.1126/science.1126316. [DOI] [PubMed] [Google Scholar]
- Emerson JJ, Kaessmann H, Betran E, Long M. Extensive gene traffic on the mammalian X chromosome. Science. 2004;303:537–540. doi: 10.1126/science.1090042. [DOI] [PubMed] [Google Scholar]
- Fernandez-Capetillo O, Allis CD, Nussenzweig A. Phosphorylation of histone H2B at DNA double-strand breaks. J Exp Med. 2004a;199:1671–1677. doi: 10.1084/jem.20032247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Capetillo O, Lee A, Nussenzweig M, Nussenzweig A. H2AX: the histone guardian of the genome. DNA Repair. 2004b;3:959–967. doi: 10.1016/j.dnarep.2004.03.024. [DOI] [PubMed] [Google Scholar]
- Fernandez-Capetillo O, Mahadevaiah SK, Celeste A, et al. H2AX is required for chromatin remodeling and inactivation of sex chromosomes in male mouse meiosis. Dev Cell. 2003;4:497–508. doi: 10.1016/s1534-5807(03)00093-5. [DOI] [PubMed] [Google Scholar]
- Fernandez-Capetillo O, Nussenzweig A. Linking histone deacetylation with the repair of DNA breaks. Proc Natl Acad Sci USA. 2004;101:1427–1428. doi: 10.1073/pnas.0307342101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feschotte C, Jiang N, Wessler SR. Plant transposable elements: where genetics meets genomics. Nat Rev Genet. 2002;3:329–341. doi: 10.1038/nrg793. [DOI] [PubMed] [Google Scholar]
- Fong Y, Bender L, Wang W, Strome S. Regulation of the different chromatin states of autosomes and X chromosomes in the germ line of C. elegans. Science. 2002;296:2235–2238. doi: 10.1126/science.1070790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freitag M, Ciuffetti LM, Selker EU. Expression and visualization of green fluorescent protein (GFP) in Neurospora crassa. Fungal Gene Newsl. 2001;48 [Google Scholar]
- Freitag M, Lee DW, Kothe GO, et al. DNA methylation is independent of RNA interference in Neurospora. Science. 2004;304:1939. doi: 10.1126/science.1099709. [DOI] [PubMed] [Google Scholar]
- Freitag M, Williams RL, Kothe GO, Selker EU. A cytosine methyltransferase homologue is essential for repeat-induced point mutation in Neurospora crassa. Proc Natl Acad Sci USA. 2002;99:8802–8807. doi: 10.1073/pnas.132212899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galagan JE, Calvo SE, Borkovich KA, et al. The genome sequence of the filamentous fungus Neurospora crassa. Nature. 2003;422:859–868. doi: 10.1038/nature01554. [DOI] [PubMed] [Google Scholar]
- Goldstein P. The synaptonemal complexes of Caenorhabditis elegans: pachytene karyotype analysis of male and hermaphrodite wild-type and him mutants. Chromosoma. 1982;86:577–593. doi: 10.1007/BF00330128. [DOI] [PubMed] [Google Scholar]
- Goldstein P, Slaton DE. The synaptonemal complexes of Caenorhabditis elegans: comparison of wild-type and mutant strains and pachytene karyotype analysis of wild-type. Chromosoma. 1982;84:585–597. doi: 10.1007/BF00292857. [DOI] [PubMed] [Google Scholar]
- Greaves IK, Rangasamy D, Devoy M, Marshall Graves JA, Tremethick DJ. The X and Y chromosomes assemble into H2A.Z-containing [corrected] facultative heterochromatin [corrected] following meiosis. Mol Cell Biol. 2006;26:5394–5405. doi: 10.1128/MCB.00519-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta V, Parisi M, Sturgill D, et al. Global analysis of X-chromosome dosage compensation. J Biol. 2006;5:3. doi: 10.1186/jbiol30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handel MA. The XY body: a specialized meiotic chromatin domain. Exp Cell Res. 2004;296:57–63. doi: 10.1016/j.yexcr.2004.03.008. [DOI] [PubMed] [Google Scholar]
- Heard E, Disteche CM. Dosage compensation in mammals: fine-tuning the expression of the X chromosome. Genes Dev. 2006;20:1848–1867. doi: 10.1101/gad.1422906. [DOI] [PubMed] [Google Scholar]
- Herman H, Lu M, Anggraini M, et al. Trans allele methylation and paramutation-like effects in mice. Nat Genet. 2003;34:199–202. doi: 10.1038/ng1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickey DA. Molecular symbionts and the evolution of sex. J Hered. 1993;84:410–414. doi: 10.1093/oxfordjournals.jhered.a111363. [DOI] [PubMed] [Google Scholar]
- Huynh KD, Lee JT. X-chromosome inactivation: a hypothesis linking ontogeny and phylogeny. Nat Rev Genet. 2005;6:410–418. doi: 10.1038/nrg1604. [DOI] [PubMed] [Google Scholar]
- Irvine DV, Zaratiegui M, Tolia NH, et al. Argonaute slicing is required for heterochromatic silencing and spreading. Science. 2006;313:1134–1137. doi: 10.1126/science.1128813. [DOI] [PubMed] [Google Scholar]
- Ivanovska I, Orr-Weaver TL. Histone modifications and the chromatin scaffold for meiotic chromosome architecture. Cell Cycle. 2006;5:2064–2071. doi: 10.4161/cc.5.18.3253. [DOI] [PubMed] [Google Scholar]
- Katayama S, Tomaru Y, Kasukawa T, et al. Antisense transcription in the mammalian transcriptome. Science. 2005;309:1564–1566. doi: 10.1126/science.1112009. [DOI] [PubMed] [Google Scholar]
- Kelly WG, Schaner CE, Dernburg AF, et al. X-chromosome silencing in the germline of C. elegans. Development. 2002;129:479–492. doi: 10.1242/dev.129.2.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalil AM, Boyar FZ, Driscoll DJ. Dynamic histone modifications mark sex chromosome inactivation and reactivation during mammalian spermatogenesis. Proc Natl Acad Sci USA. 2004;101:16583–16587. doi: 10.1073/pnas.0406325101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khil PP, Smirnova NA, Romanienko PJ, Camerini-Otero RD. The mouse X chromosome is enriched for sex-biased genes not subject to selection by meiotic sex chromosome inactivation. Nat Genet. 2004;36:642–646. doi: 10.1038/ng1368. [DOI] [PubMed] [Google Scholar]
- Kouzminova E, Selker EU. dim-2 encodes a DNA methyltransferase responsible for all known cytosine methylation in Neurospora. EMBO J. 2001;20:4309–4323. doi: 10.1093/emboj/20.15.4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruhlak MJ, Celeste A, Dellaire G, et al. Changes in chromatin structure and mobility in living cells at sites of DNA double-strand breaks. J Cell Biol. 2006;172:823–834. doi: 10.1083/jcb.200510015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutil BL, Seong KY, Aramayo R. Unpaired genes do not silence their paired neighbors. Curr Genet. 2003;43:425–432. doi: 10.1007/s00294-003-0412-5. [DOI] [PubMed] [Google Scholar]
- Lee DW, McLaughlin M, Pratt RJ, Aramayo R. SMS-3, a Dicer-like protein is required for development and meiotic silencing, in Neurospora. 2007 (Submitted) [Google Scholar]
- Lee DW, Pratt RJ, McLaughlin M, Aramayo R. An Argonaute-like protein is required for meiotic silencing. Genetics. 2003;164:821–828. doi: 10.1093/genetics/164.2.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DW, Seong K-Y, Pratt RJ, Baker K, Aramayo R. Properties of unpaired DNA required for efficient silencing in Neurospora crassa. Genetics. 2004;167:131–150. doi: 10.1534/genetics.167.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lifschytz E, Lindsley DL. The role of X-chromosome inactivation during spermatogenesis (Drosophila-allocycly-chromosome evolution-male sterility-dosage compensation) Proc Natl Acad Sci USA. 1972;69:182–186. doi: 10.1073/pnas.69.1.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maciejowski J, Ahn JH, Cipriani PG, et al. Autosomal genes of autosomal X-linked duplicated gene pairs and germline proliferation in Caenorhabditis elegans. Genetics. 2005;169:1997–2011. doi: 10.1534/genetics.104.040121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahadevaiah SK, Turner JM, Baudat F, et al. Recombinational DNA double-strand breaks in mice precede synapsis. Nat Genet. 2001;27:271–276. doi: 10.1038/85830. [DOI] [PubMed] [Google Scholar]
- Maine EM, Hauth J, Ratliff T, et al. EGO-1, a putative RNA-dependent RNA polymerase, is required for heterochromatin assembly on unpaired dna during C. elegans meiosis. Curr Biol. 2005;15:1972–1978. doi: 10.1016/j.cub.2005.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martienssen RA, Zaratiegui M, Goto DB. RNA interference and heterochromatin in the fission yeast Schizosaccharomyces pombe. Trends Genet. 2005;21:450–456. doi: 10.1016/j.tig.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Matzke MA, Birchler JA. RNAi-mediated pathways in the nucleus. Nat Rev Genet. 2005;6:24–35. doi: 10.1038/nrg1500. [DOI] [PubMed] [Google Scholar]
- McKee BD. Homologous pairing and chromosome dynamics in meiosis and mitosis. Biochim Biophys Acta. 2004;1677:165–180. doi: 10.1016/j.bbaexp.2003.11.017. [DOI] [PubMed] [Google Scholar]
- Miao VP, Freitag M, Selker EU. Short TpA-rich segments of the zeta-eta region induce DNA methylation in Neurospora crassa. J Mol Biol. 2000;300:249–273. doi: 10.1006/jmbi.2000.3864. [DOI] [PubMed] [Google Scholar]
- Mochizuki K, Gorovsky MA. Small RNAs in genome rearrangement in Tetrahymena. Curr Opin Genet Dev. 2004;14:181–187. doi: 10.1016/j.gde.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Namekawa SH, Park PJ, Zhang LF, et al. Postmeiotic sex chromatin in the male germline of mice. Curr Biol. 2006;16:660–667. doi: 10.1016/j.cub.2006.01.066. [DOI] [PubMed] [Google Scholar]
- Nguyen DK, Disteche CM. Dosage compensation of the active X chromosome in mammals. Nat Genet. 2006;38:47–53. doi: 10.1038/ng1705. [DOI] [PubMed] [Google Scholar]
- Okamoto I, Arnaud D, Le Baccon P, et al. Evidence for de novo imprinted X-chromosome inactivation independent of meiotic inactivation in mice. Nature. 2005;438:369–373. doi: 10.1038/nature04155. [DOI] [PubMed] [Google Scholar]
- Okamoto I, Otte AP, Allis CD, Reinberg D, Heard E. Epigenetic dynamics of imprinted X inactivation during early mouse development. Science. 2004;303:644–649. doi: 10.1126/science.1092727. [DOI] [PubMed] [Google Scholar]
- Ooi SL, Priess JR, Henikoff S. Histone H3.3 variant dynamics in the germline of Caenorhabditis elegans. PLoS Genet. 2006;2:e97. doi: 10.1371/journal.pgen.0020097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardue ML, DeBaryshe PG. Retrotransposons provide an evolutionarily robust non-telomerase mechanism to maintain telomeres. Annu Rev Genet. 2003;37:485–511. doi: 10.1146/annurev.genet.38.072902.093115. [DOI] [PubMed] [Google Scholar]
- Perkins DD. Details for collection of asci as unordered groups of eight projected ascospores. Neurospora Newsl. 1966;9:11. [Google Scholar]
- Perkins DD. Comments on Metzenberg’s procedure for isolating unordered Neurospora asci. Fungal Genet Newsl. 1988;35:29. [Google Scholar]
- Pickford AS, Catalanotto C, Cogoni C, Macino G. Quelling in Neurospora crassa. Adv Genet. 2002;46:277–303. doi: 10.1016/s0065-2660(02)46010-5. [DOI] [PubMed] [Google Scholar]
- Pratt RJ, Lee DW, Aramayo R. DNA methylation affects meiotic trans-sensing, not meiotic silencing, in Neurospora. Genetics. 2004;168:1925–1935. doi: 10.1534/genetics.104.031526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raju NB. Meiosis and ascospore genesis in Neurospora. Eur J Cell Biol. 1980;23:208–223. [PubMed] [Google Scholar]
- Raju NB. Genetic control of the sexual cycle in Neurospora. Mycol Res. 1992;96:241–262. [Google Scholar]
- Rassoulzadegan M, Grandjean V, Gounon P, et al. RNA-mediated non-Mendelian inheritance of an epigenetic change in the mouse. Nature. 2006;441:469–474. doi: 10.1038/nature04674. [DOI] [PubMed] [Google Scholar]
- Rassoulzadegan M, Magliano M, Cuzin F. Transvection effects involving DNA methylation during meiosis in the mouse. EMBO J. 2002;21:440–450. doi: 10.1093/emboj/21.3.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rastelli L, Kuroda MI. An analysis of maleless and histone H4 acetylation in Drosophila melanogaster spermatogenesis. Mech Dev. 1998;71:107–117. doi: 10.1016/s0925-4773(98)00009-4. [DOI] [PubMed] [Google Scholar]
- Reddy KC, Villeneuve AM. C. elegans HIM-17 links chromatin modification and competence for initiation of meiotic recombination. Cell. 2004;118:439–452. doi: 10.1016/j.cell.2004.07.026. [DOI] [PubMed] [Google Scholar]
- Reinke V, Gil IS, Ward S, Kazmer K. Genome-wide germline-enriched and sex-biased expression profiles in Caenorhabditis elegans. Development. 2004;131:311–323. doi: 10.1242/dev.00914. [DOI] [PubMed] [Google Scholar]
- Reuben M, Lin R. Germline X chromosomes exhibit contrasting patterns of histone H3 methylation in Caenorhabditis elegans. Dev Biol. 2002;245:71–82. doi: 10.1006/dbio.2002.0634. [DOI] [PubMed] [Google Scholar]
- Roeder GS, Bailis JM. The pachytene checkpoint. Trends Genet. 2000;16:395–403. doi: 10.1016/s0168-9525(00)02080-1. [DOI] [PubMed] [Google Scholar]
- Romano N, Macino G. Quelling: transient inactivation of gene expression in Neurospora crassa by transformation with homologous sequences. Mol Microbiol. 1992;6:3343–3353. doi: 10.1111/j.1365-2958.1992.tb02202.x. [DOI] [PubMed] [Google Scholar]
- Rountree MR, Selker EU. DNA methylation inhibits elongation but not initiation of transcription in Neurospora crassa. Genes Dev. 1997;11:2383–2395. doi: 10.1101/gad.11.18.2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schimenti J. Synapsis or silence. Nat Genet. 2005;37:11–13. doi: 10.1038/ng0105-11. [DOI] [PubMed] [Google Scholar]
- Selker EU. Premeiotic instability of repeated sequences in Neurospora crassa. Annu Rev Genet. 1990;24:579–613. doi: 10.1146/annurev.ge.24.120190.003051. [DOI] [PubMed] [Google Scholar]
- Selker EU. Epigenetic phenomena in filamentous fungi: useful paradigms or repeat-induced confusion? Trends Genet. 1997;13:296–301. doi: 10.1016/s0168-9525(97)01201-8. [DOI] [PubMed] [Google Scholar]
- Seydoux G, Schedl T. The germline in C. elegans: origins, proliferation, and silencing. Int Rev Cytol. 2001;203:139–185. doi: 10.1016/s0074-7696(01)03006-6. [DOI] [PubMed] [Google Scholar]
- Shiu PK, Metzenberg RL. Meiotic silencing by unpaired DNA: properties, regulation and suppression. Genetics. 2002;161:1483–1495. doi: 10.1093/genetics/161.4.1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiu PK, Raju BN, Zickler D, Metzenberg R. Meiotic silencing by unpaired DNA. Cell. 2001;107:905–916. doi: 10.1016/s0092-8674(01)00609-2. [DOI] [PubMed] [Google Scholar]
- Shiu PK, Zickler D, Raju NB, Ruprich-Robert G, Metzenberg RL. SAD-2 is required for meiotic silencing by unpaired DNA and perinuclear localization of SAD-1 RNA-directed RNA polymerase. Proc Natl Acad Sci USA. 2006;103:2243–2248. doi: 10.1073/pnas.0508896103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer MJ, Marcotte BA, Selker EU. DNA methylation associated with repeat-induced point mutation in Neurospora crassa. Mol Cell Biol. 1995;15:5586–5597. doi: 10.1128/mcb.15.10.5586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smardon A, Spoerke JM, Stacey SC, et al. EGO-1 is related to RNA-directed RNA polymerase and functions in germ-line development and RNA interference in C. elegans. Curr Biol. 2000;10:169–178. doi: 10.1016/s0960-9822(00)00323-7. [DOI] [PubMed] [Google Scholar]
- Tamaru H, Zhang X, McMillen D, et al. Trimethylated lysine 9 of histone H3 is a mark for DNA methylation in Neurospora crassa. Nat Genet. 2003;34:75–79. doi: 10.1038/ng1143. [DOI] [PubMed] [Google Scholar]
- Tesse S, Storlazzi A, Kleckner N, Gargano S, Zickler D. Localization and roles of Ski8p protein in Sordaria meiosis and delineation of three mechanistically distinct steps of meiotic homolog juxtaposition. Proc Natl Acad Sci USA. 2003;100:12865–12870. doi: 10.1073/pnas.2034282100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornhill AR, Burgoyne PS. A paternally imprinted X chromosome retards the development of the early mouse embryo. Development. 1993;118:171–174. doi: 10.1242/dev.118.1.171. [DOI] [PubMed] [Google Scholar]
- Turner JM, Aprelikova O, Xu X, et al. BRCA1, histone H2AX phosphorylation, and male meiotic sex chromosome inactivation. Curr Biol. 2004;14:2135–2142. doi: 10.1016/j.cub.2004.11.032. [DOI] [PubMed] [Google Scholar]
- Turner JM, Mahadevaiah SK, Ellis PJ, Mitchell MJ, Burgoyne PS. Pachytene asynapsis drives meiotic sex chromosome inactivation and leads to substantial postmeiotic repression in spermatids. Dev Cell. 2006;10:521–529. doi: 10.1016/j.devcel.2006.02.009. [DOI] [PubMed] [Google Scholar]
- Turner JM, Mahadevaiah SK, Fernandez-Capetillo O, et al. Silencing of unsynapsed meiotic chromosomes in the mouse. Nat Genet. 2005;37:41–47. doi: 10.1038/ng1484. [DOI] [PubMed] [Google Scholar]
- Vallender EJ, Lahn BT. How mammalian sex chromosomes acquired their peculiar gene content. Bioessays. 2004;26:159–169. doi: 10.1002/bies.10393. [DOI] [PubMed] [Google Scholar]
- van der Heijden GW, Derijck AA, Posfai E, et al. Chromosome-wide nucleosome replacement and H3.3 incorporation during mammalian meiotic sex chromosome inactivation. Nat Genet. 2007 doi: 10.1038/ng1949. [DOI] [PubMed] [Google Scholar]
- van der Laan R, Uringa EJ, Wassenaar E, et al. Ubiquitin ligase Rad18Sc localizes to the XY body and to other chromosomal regions that are unpaired and transcriptionally silenced during male meiotic prophase. J Cell Sci. 2004;117:5023–5033. doi: 10.1242/jcs.01368. [DOI] [PubMed] [Google Scholar]
- Vicoso B, Charlesworth B. Evolution on the X chromosome: unusual patterns and processes. Nat Rev Genet. 2006;7:645–653. doi: 10.1038/nrg1914. [DOI] [PubMed] [Google Scholar]
- Vogt P, Hennig W, Siegmund I. Identification of cloned Y chromosomal DNA sequences from a lampbrush loop of Drosophila hydei. Proc Natl Acad Sci USA. 1982;79:5132–5136. doi: 10.1073/pnas.79.17.5132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpe TA, Kidner C, Hall IM, et al. Regulation of heterochromatic silencing and histone H3 lysine-9 methylation by RNAi. Science. 2002;297:1833–1837. doi: 10.1126/science.1074973. [DOI] [PubMed] [Google Scholar]
- Vought VE, Ohmachi M, Lee MH, Maine EM. EGO-1, a putative RNA-directed RNA polymerase, promotes germline proliferation in parallel with GLP-1/notch signaling and regulates the spatial organization of nuclear pore complexes and germline P granules in Caenorhabditis elegans. Genetics. 2005;170:1121–1132. doi: 10.1534/genetics.105.042135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walstrom KM, Schmidt D, Bean CJ, Kelly WG. RNA helicase A is important for germline transcriptional control, proliferation, and meiosis in C. elegans. Mech Dev. 2005;122:707–720. doi: 10.1016/j.mod.2004.12.002. [DOI] [PubMed] [Google Scholar]
- Wang PJ, Page DC, McCarrey JR. Differential expression of sex-linked and autosomal germ-cell-specific genes during spermatogenesis in the mouse. Hum Mol Genet. 2005;14:2911–2918. doi: 10.1093/hmg/ddi322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessler SR. Phenotypic diversity mediated by the maize transposable elements Ac and Spm. Science. 1988;242:399–405. doi: 10.1126/science.2845581. [DOI] [PubMed] [Google Scholar]
- Wessler SR. Transposable elements and the evolution of gene expression. Symp Soc Exp Biol. 1998;51:115–122. [PubMed] [Google Scholar]
- Wessler SR. Plant transposable elements. A hard act to follow. Plant Physiol. 2001;125:149–151. doi: 10.1104/pp.125.1.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessler SR, Bureau TE, White SE. LTR-retrotransposons and MITEs: important players in the evolution of plant genomes. Curr Opin Genet Dev. 1995;5:814–821. doi: 10.1016/0959-437x(95)80016-x. [DOI] [PubMed] [Google Scholar]
- Wu CI, Xu EY. Sexual antagonism and X inactivation–the SAXI hypothesis. Trends Genet. 2003;19:243–247. doi: 10.1016/s0168-9525(03)00058-1. [DOI] [PubMed] [Google Scholar]
- Wu CT, Morris JR. Transvection and other homology effects. Curr Opin Genet Dev. 1999;9:237–246. doi: 10.1016/S0959-437X(99)80035-5. [DOI] [PubMed] [Google Scholar]
- Yigit E, Batista PJ, Bei Y, et al. Analysis of the C. elegans Argonaute family reveals that distinct Argonautes act sequentially during RNAi. Cell. 2006;127:747–757. doi: 10.1016/j.cell.2006.09.033. [DOI] [PubMed] [Google Scholar]