

Abstract
The mechanism of autophagy relies on complex cell signaling and regulatory processes. Each cell contains many proteins that lack a rigid 3-dimensional structure under physiological conditions. These dynamic proteins, called intrinsically disordered proteins (IDPs) and protein regions (IDPRs), are predominantly involved in cell signaling and regulation. Yet, very little is known about their presence among proteins of the core autophagy machinery. In this work, we characterized the autophagy protein Atg3 from yeast and human along with 2 variants to show that Atg3 is an IDPRs-containing protein and that disorder/order predicted for these proteins from their amino acid sequence corresponds to their experimental characteristics. Based on this consensus, we applied the same prediction methods to all known Atg proteins from Saccharomyces cerevisiae. The data presented here provide an insight into the structural dynamics of each Atg protein. They also show that intrinsic disorder at various levels has to be taken into consideration for about half of the Atg proteins. This work should become a useful tool that will facilitate and encourage exploration of protein intrinsic disorder in autophagy.
Keywords: Atg3, autophagy, intrinsically disordered protein, stress, vacuole, yeast
Introduction
A number of studies published within the past 15 years show that not all proteins fall into the category of globular structures that operate on the principle of “lock-and-key” and that have a well-defined 3-dimensional structure in solution. A rapidly growing group of biologically-active proteins possess a structure that is entirely or substantially flexible under physiological conditions.1-6 These polypeptides, called intrinsically disordered proteins or intrinsically disordered protein regions, exist in aqueous solution as a dynamic ensemble of interconverting structures with multiple free-energy minima.7 IDPs are divided into several subclasses, ranging from completely unstructured proteins (that resemble the unfolded state of a globular protein) through pre-molten globule-like and molten globule-like proteins, retaining some elements of secondary/tertiary structure, up to hybrid proteins consisting of IDPRs along with ordered domains. The current view of IDPs and IDPRs recognizes that the structural organization of proteins can be affected by intrinsic disorder on many different levels and that, rather than classes and subclasses, it is better to consider the structural heterogeneity of IDPs/IDPRs as a continuum. Many IDPs and IDPRs undergo disorder-to-order transition upon binding to a biological partner (such as a protein, nucleic acid, or a cofactor), but some retain their disorder in the biologically active state.8-11
The conformational plasticity of IDPs and IDPRs confers unique biological properties that provide many advantages over the function of globular proteins.10,12-14 For example, this structural flexibility increases the speed of interaction and overcomes steric restrictions, which enables larger interaction surfaces. It also allows for many-to-one signaling, where many unstructured regions can bind to a single structured partner, or one-to-many signaling, where one disordered region can bind to many structurally diverse partners. Flexibility of IDPs and IDPRs also brings high accessibility for posttranslational modifications, and allows for a cascade-type of binding, where the first binding partner induces folding and thus creates a binding site for the second partner. Importantly, disorder decouples binding affinity and specificity, which allows for the formation of specific complexes with low binding strength. Conversely, some IDPs/IDPRs can form very stable complexes through their intertwining with other proteins.15 IDPRs in hybrid proteins are located primarily at the termini. So far, more than 24 functions have been described for these unstructured regions.11,16
IDPs are involved predominantly in cell processes that require prompt responses, such as cell signaling or pathway regulation.8,17 The nature and unique properties of IDPs and IDPRs associate them with pathological conditions that lead to various human diseases. To date, IDPs and IDPRs have been implicated in cancer, amyloidosis, neurodegenerative and cardiovascular diseases, and diabetes.12,18-20
One cell process that involves multiple aspects of signaling and a complex regulatory network mediated through a wide range of protein-protein interactions is autophagy,21 which is a mechanism through which cells degrade and recycle cytoplasmic components to maintain homeostasis. Autophagy includes several types of self-eating, which differ in substrates and mechanisms of sequestration.22 The most robust of these is macroautophagy that involves the sequestration of parts of the cytoplasm within a double-membrane vesicle, an autophagosome, that, once completed, fuses with the vacuole/lysosome in order to degrade its cargo and release the resulting macromolecules back into the cytoplasm.23 Macroautophagy (hereafter autophagy) operates through a network of core autophagy-related (Atg) proteins, many of which have conserved homologs from yeast to mammals. The Atg proteins interconnect with numerous cytoplasmic proteins involved in other cell pathways, which makes autophagy a highly complex cellular process.24 Defects in autophagy are associated with numerous pathophysiologies including aging, cancer, neurodegenerative diseases, cardiac and liver disease, macular degeneration, immune deficiency, and metabolic disorders such as diabetes.25-27
Autophagy must be tightly regulated—either too little or too much can lead to cellular dysfunction—and therefore we propose that it is subject to precise and rapid modulation, a characteristic that could be well suited to the use of IDPs. Accordingly, we decided to examine the presence of intrinsic disorder in autophagy proteins. The crystal structure of yeast Atg3 is available28-30 and the protein is predicted to contain a disordered domain of approximately 70 amino acids; these residues are indispensable for autophagic function.30 We analyzed this protein along with 2 mutated variants that represent opposite extremes in terms of protein plasticity, and also compared Atg3 from yeast to its human homolog, ATG3. We show that Atg3 contains IDPRs with dynamic properties such as those that are described for many other IDPRs. The experimental results presented here are consistent with multiple computational predictions of protein disorder from the protein amino acid sequence. To obtain insights into the molecular dynamics of autophagy proteins, we used prediction methods that have been demonstrated to be consistent with empirical data, and applied them to the amino acid sequences of all of the known Atg proteins in S. cerevisiae. The results presented here show that intrinsic disorder, along with its advantages and potential weaknesses, should be taken into consideration when contemplating the function of many autophagy proteins.
Results
Atg3 is a dynamic protein lacking a rigid structure in both solution and in a bound state
IDPs/IDPRs exhibit characteristics that clearly distinguish them from globular proteins, including a difficulty in protein crystallization. Since each IDP/IDPR represents a highly dynamic ensemble of interconverting structures, capturing its solution structure is very difficult. Many disordered regions are usually missing in the crystal structures of IDPs1,31-34 due to noncoherent X-ray scattering. Atg3 is the ubiquitin-carrier-like (E2-like) protein that mediates conjugation of Atg8 to phosphatidylethanolamine (PE), an essential step for autophagy.24 S. cerevisiae Atg3 was crystallized in several studies. The first showed the crystal structure of the protein in solution at 2.5-Å resolution.30 The core region of Atg3 is similar to canonical E2 enzymes, but it has a unique trans-architecture that involves dynamic protein conformational changes that are essential for function. The region encoding amino acids 84–161 is unstructured and crucial for the function of Atg3 in autophagy.30 As expected, most of this region is missing in the Atg3 crystal structure, as are several other protein domains (Fig. 1A). Other studies28,29 show the crystal structure of yeast Atg3 in a complex with its biological partner, Atg7 (Fig. 1B). Even in the presence of the structured Atg7 protein, the predicted disordered region remains in a mostly disordered state and the majority of this part of the protein is still missing in the crystal structure. Together, the missing domains account for more than one-third of the Atg3 protein. Moreover, comparison of the position of the small α−helical domain between I129 and K142 in both Atg3 crystal structures reveals significant conformational change of Atg3 that involves extensive repositioning of the disordered domain upon its binding to Atg7. Together, these data indicate that Atg3 is a rapidly moving dynamic protein that lacks rigid structure not only in solution, but also in the context of a physiological complex.

Figure 1. Models of yeast Atg3. (A) The crystal structure of Atg3 in solution (PDB ID: 2DYT30). The missing protein regions are listed on the right side of the panel, and the amino acid residues at the border with the missing regions are denoted directly in the ribbon model. (B) The crystal structure of Atg3 in a complex with Atg7 (Atg7 not shown) (PDP ID: 4GSL28). The missing regions and border residues are shown as in (A). (C) Hypothetical structural model of Atg3 in solution. The extended disordered conformation of Atg3 is depicted schematically, and includes disordered domains that are not visible in the published crystal structure.30 Structural models were created using DeepView/Swiss-PDBViewer v.4.0.4 available at http://spdbv.vital-it.ch/.
Atg3 adopts an extended disordered conformation in solution
The compactness of a globular protein can generally be determined by its hydrodynamic radius (Rh). A globular protein can unfold from its native state to an unfolded state through 2 distinct conformations, the molten globule and the pre-molten globule. An increasing amount of disorder in each of these conformations is accompanied by an increase in hydrodynamic radius (which essentially corresponds to the protein’s volume). The hydrodynamic volume of a protein in the molten globule, pre-molten globule, and the unfolded state increases approximately 1.5-, 3-, and 12-fold, respectively, compared with the volume in the native state.2 Furthermore, the log(Rh) vs. log(Mw) dependencies for the native state, the molten globule, the pre-molten globule, and the unfolded state are clearly different.35,36 Thus, the hydrodynamic radius of a protein is a very good indicator as to whether a protein is in the compact state, or in the extended disordered conformation, especially if the crystal structure of a protein is unavailable or is incomplete. IDPs adopt a far less compact conformation than globular proteins of comparable molecular mass in their native state.2,5,37 To examine the compactness of Atg3, we determined its hydrodynamic radius along with the hydrodynamic radii of 4 standard globular proteins, comparing the log(Rh) vs. log(Mw) dependency. Atg3 exhibited a significantly displaced dependency of its hydrodynamic radius on protein molecular mass from the linear dependency observed for standard globular proteins (Fig. 2). The experimental hydrodynamic radius of Atg3 exceeds the calculated hydrodynamic radius of a native-coil-like IDP (perhaps due to its highly negative charge and a possible prolate ellipsoid shape), and is similar to the calculated hydrodynamic radius of a globular protein of comparable molecular mass that was unfolded by treatment with guanidinium chloride (GdmCl; Table 1). This indicates that Atg3 adopts a largely disordered conformation in solution. An oligomeric form of Atg3 in solution can be excluded, because the experimentally determined molecular mass of Atg3 (data not shown) was comparable to the predicted mass based on the amino acid sequence (Table 1).
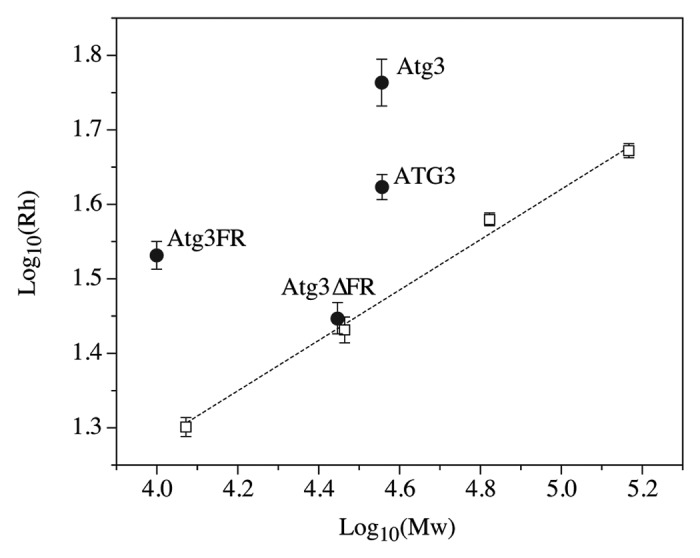
Figure 2. Dependence of log(Rh) on log(Mw) for disordered and ordered proteins. Black circles, Atg3/ATG3 proteins; white squares, globular standard proteins (carbonic anhydrase, cytochrome c, bovine serum albumin, and alcohol dehydrogenase). The hydrodynamic radii (Rh) were determined by dynamic light scattering and molecular masses were calculated from the corresponding amino acid sequences.
Table 1. Molecular mass and hydrodynamic radii of the Atg3 proteins.
| Protein | Molecular massa (kDa) | Calculated Rhb | Experimental Rhc | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Folded | MG | PMG | Unfolded by urea | Unfolded by GdmCl (Å) | Native coil | Native PMG | |||
| Atg3 | 36 | 27 | 29 | 38 | 53 | 56 | 50 | 40 | 58 |
| Atg3ΔFR | 28 | 24 | 27 | 34 | 47 | 49 | 44 | 36 | 28 |
| Atg3FR | 10 | 17 | 19 | 23 | 27 | 28 | 26 | 24 | 34 |
| ATG3 | 36 | 27 | 29 | 38 | 53 | 56 | 50 | 40 | 42 |
a The molecular masses were determined from the amino acid sequence. bThe calculated values, using equations 2–8 in reference 36, are the hydrodynamic radii of a corresponding coil-like IDP, PMG-like IDP, or various folding states of a globular protein of comparable molecular mass. MG, molten globule; PMG, pre-molten globule; Rh, hydrodynamic radius. cHydrodynamic radii were experimentally determined by dynamic light scattering.
To extend the analysis of Atg3 conformation in solution, we utilized 2 circular dichroism (CD) measurements, the near-UV CD spectrum and protein melting curve. The near-UV CD spectrum in general reflects the environment of aromatic amino acid residues (Tyr, Phe, Trp) and is very diminished when these residues are highly mobile and exposed to a solvent by protein unfolding.38 The near-UV CD spectrum of Atg3 was very flat (Fig. S1), suggesting that much of Atg3 is not in a rigid structure. The protein melting curve, measured as an increase in ellipticity at 222 nm, reflects the heat-induced loss of α-helices, and for pre-molten globules is noncooperative and occurs over a wide range of temperatures.39 The Atg3 melting curve was noncooperative and gradually increased over the range of 50 °C (Fig. S2), indicating that Atg3 lacks a tightly-packed hydrophobic core.38,39
To further verify this result, we also analyzed 2 purified mutated variants of Atg3, Atg3FR and Atg3∆FR (Fig. S3). Atg3FR is the recombinant flexible domain of Atg3 encoding amino acids 82–165, and Atg3∆FR is the recombinant protein where the region encoding amino acids 86–159 was deleted. As expected, the log(Rh) vs. log(Mw) dependency for Atg3FR was significantly displaced from the linear dependency log(Rh) vs. log(Mw) of standard globular proteins, and its experimental hydrodynamic radius exceeds the calculated hydrodynamic radius of both a native-coil-like IDP and a GdmCl-unfolded globular protein with comparable molecular mass (Table 1). This suggests that Atg3FR adopts a conformation even more disordered than that of the full-length Atg3. In contrast, the Atg3∆FR mutant lacking the major disordered region adopts the overall compactness near to the compactness of a comparable globular protein in the molten globule state (Fig. 2; Table 1).
We also compared the hydrodynamic properties of Atg3 with the properties of its human homolog, ATG3 (see Fig. S3 for the sequence alignment). We found that ATG3 was present in solution as a largely disordered protein compared with the globular standard, but its overall conformation was more compact than that of Atg3 from yeast; the Rh of ATG3 was comparable to the Rh of a native pre-molten globule in aqueous solution (Fig. 2; Table 1).
Atg3 is a largely disordered protein with some secondary structure elements
IDPs can exist as a structural ensemble either at the secondary or at the tertiary level.40,41 The above-presented hydrodynamic data suggest that Atg3 in solution adopts a largely disordered conformation at the tertiary structure level. To see whether it is a random-coil-like protein with no secondary structure, or rather if it behaves as a slightly ordered protein with limited secondary structure elements, we examined the far-UV CD spectra of Atg3. The α-helical species in a protein are generally manifested by a negative CD signal at 208 nm and a deeper minimum at 222 nm, while random coils and β-sheet structures exhibit pronounced negative ellipticity around 200 nm and 218 nm, respectively.5,38 We determined the far-UV CD spectrum of Atg3 at 25 °C (Fig. 3A). The far-UV CD spectrum of Atg3 lacked the pronounced minimum at approximately 200 nm that would be expected for a protein composed entirely or largely of random coils, and therefore Atg3 cannot be considered to display the properties of such a protein at the secondary structure level. In comparison, the far-UV CD spectrum of Atg3FR (Fig. 3B) did resemble the spectrum of unordered structures with a negative minimum at approximately 200 nm,38,39 indicating that the secondary structure is missing in Atg3FR. The far-UV CD spectrum of Atg3∆FR (Fig. 3C) revealed a more pronounced negative minimum at 222 nm compared with that of Atg3, which would indicate a higher content of α-helical elements in Atg3∆FR than in Atg3.
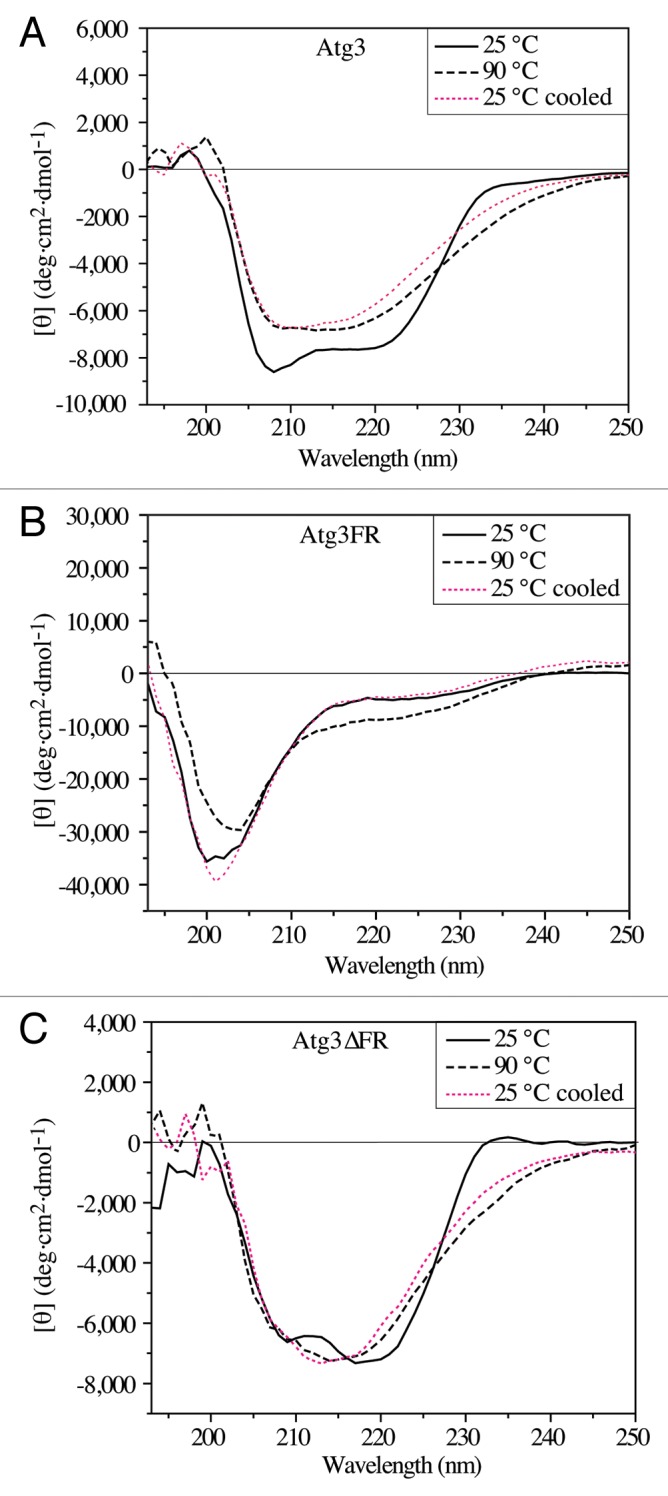
Figure 3. Far-UV CD spectra (protein solution minus buffer) of the yeast Atg3 proteins in 50 mM potassium buffer, pH 8.0. The spectra for Atg3 (A), Atg3FR (B), and Atg3∆FR (C) were measured at 25 °C, at 90 °C, and at 25 °C after heating to 90 °C (25 °C cooled). Each spectrum is the average of 15 scans. Experimental conditions were as follows: time constant, 1s; bandwidth, 1 nm; path length, 1 mm; sample volume, 300 μl.
In agreement with these findings, heating of Atg3 and, in particular, Atg3∆FR induced some irreversible changes to the protein secondary structure, whereas the same treatment induced reversible folding of the random-coil-like Atg3FR (Fig. 3). We note that the attempts to deconvolve the far-UV CD spectra using DICHROWEB42 failed to provide reliable results (data not shown), as the values for the normalized root means square deviation were higher than what is required for reliable data (in general less than 0.1), and also the experimental input differed from the spectra calculated by various algorithms. One of the reasons for this discrepancy could be that reference databases applied by the algorithms were derived from globular proteins, and disordered structures are not well represented in these databases.43
Atg3 exhibits aberrant migration during SDS-PAGE
IDPs/IDPRs typically display aberrant migration during SDS-PAGE5 due to poor SDS binding that reflects their unusual amino acid composition, which consists of a high ratio of charged to hydrophobic residues.1,2,4,5 We found that purified Atg3 similarly displayed an anomalous migration pattern following SDS-PAGE (Fig. 4A; Table 2). The predicted molecular mass determined from the amino acid sequence of Atg3 is 36 kDa, but the protein exhibited a 10-kDa shift, and migrated as a 46-kDa polypeptide. We also examined the migration patterns of the Atg3 variants and human ATG3. Atg3FR, which behaved as an extended random-coil-like protein (Fig. 2; Fig. 3), has a predicted molecular mass of 10 kDa, but migrated at the position of a polypeptide twice as large (Fig. 4A; Table 2). In contrast, Atg3∆FR migrated on the gel at its predicted molecular mass of 28 kDa, which agrees with the above-mentioned results showing that it behaved more like a globular protein. ATG3 exhibited a less dramatic migration shift following SDS-PAGE than its yeast counterpart, and ran on the gel as a 1.17-times larger protein than would correspond to its predicted molecular mass (Fig. 4A; Table 2), again in agreement with its hydrodynamic properties.

Figure 4. Properties of the Atg3/ATG3 proteins resulting from their amino acid sequence. (A) Migration of the Atg3/ATG3 proteins on a Coomassie Blue-stained gel following SDS-PAGE. The left lane in each panel shows the band corresponding to the purified protein, and the right lane shows protein standard molecular mass markers. The size in kDa is indicated on both sides of each panel. (B) Compositional profiling of Atg3 shows the fractional difference in amino acid composition of Atg3 and a set of intrinsically disordered proteins in the DisProt database relative to a reference set of ordered, globular proteins in the PDB.
Table 2. Migration patterns of the Atg proteins on SDS-PAGE.
| Protein | m calculateda (kDa) | m SDS-PAGEb (kDa) | Times higher on SDS-PAGE than calculated |
|---|---|---|---|
| Atg3 | 36 | 46 | 1.28 |
| Atg3ΔFR | 28 | 28 | 1 |
| Atg3FR | 10 | 20 | 2 |
| ATG3 | 36 | 42 | 1.17 |
Consistent with the migration on SDS-PAGE, amino acid compositional profiling showed that Atg3 is highly enriched in negatively charged amino acid residues, and also in Gln and Ser, compared with globular proteins in the Protein Data Bank (PDB) database (Fig. 4B). All of these residues are flexible, and disorder promoting.1 The enrichment in negatively charged Asp is even higher than what is observed in IDPs from the DisProt database.44 Among rigid, order-promoting residues,1 Atg3 is depleted in Cys, Val, Leu, and His, as are IDPs from DisProt compared with globular proteins, but Atg3 contains more Trp, Ile, Tyr, and Met than the proteins included in DisProt.
Bioinformatics analyses classify Atg3 as a hybrid protein with IDPRs
In the past decade, many bioinformatics tools were developed to predict protein disorder from amino acid sequence.4,45-47 These predictors show that many proteins have at least 1 long disordered region of ≥ 40 consecutive disordered residues and that IDPs/IDPRs possess a unique combination of charge and hydrophobicity. Here we applied several predictors of protein intrinsic disorder on all 4 Atg3/ATG3 proteins/domains to show that the predicted results were very consistent with the experimental data.
First, we used 3 PONDR (predictors of natural disordered regions) predictors, PONDR-VSL2, PONDR-VL3, and PONDR-FIT.46,48,49 These programs provide a score for each residue in the linear amino acid sequence, indicating its likelihood of being disordered or structured (see Materials and Methods for a more detailed description). Based on all 3 PONDRs (Fig. 5A), Atg3 is predicted to contain some structured domains along with a few short and 1 long unstructured region of more than 70 consecutive disordered residues (with a score above the cutoff of 0.5), which qualifies Atg3 as a hybrid protein possessing IDPRs. Human ATG3 is also predicted to be a hybrid protein with IDPRs, but one that is slightly more structured than yeast Atg3. The PONDR predictors also show that Atg3FR and Atg3∆FR represent 2 opposite extremes in terms of protein disorder; Atg3FR is an unfolded protein fragment, while Atg3∆FR resembles an ordered protein.

Figure 5. Intrinsic disorder predictions. (A) PONDR scores from PONDR-FIT, PONDR-VSL2, and PONDR-VL3 predictors plotted as a function of residue position in the amino acid sequence of yeast Atg3, Atg3FR, Atg3∆FR, and human ATG3. (B) ∆CH-∆CDF analysis of the Atg3 variants. The coordinates of each protein represent a distance of the corresponding protein from the boundary in the CH plot and the CDF plot. Q1 contains proteins predicted to be disordered based on the CH plot, but ordered based on the CDF plot; the Q2 quadrant includes proteins predicted to be ordered by both CH and CDF plots; the Q3 quadrant contains proteins predicted to be ordered by the CH plot, but disordered by the CDF plot; the Q4 quadrant includes proteins predicted to be disordered by both the CH and CDF plots. The ∆CDF parameter was calculated from PONDR-VL3.
We also applied the ∆CH-∆CDF method (see Materials and Methods for further details) to the amino acid sequences of the Atg3/ATG3 proteins (Fig. 5B). This analysis shows that Atg3 and ATG3 are at the boundary of the Q3 and Q4 quadrants, a location which includes proteins predicted to adopt a hybrid conformation possessing approximately equal amounts of ordered and disordered regions. In this plot, Atg3 exhibits a lower ΔCDF and a higher ΔCH value than ATG3, indicating a larger extent of disorder, in agreement with experimental data. In comparison, Atg3FR and Atg3∆FR occupy 2 opposite quadrants, Q4 and Q2, respectively; Atg3FR is positioned as an extended random-coil-like IDP and Atg3∆FR as an ordered protein predicted by both the CH and CDF methods, again in agreement with our experimental data.
Insight into the molecular dynamics of autophagy proteins
The consensus between the experimental and predicted data shown here for the Atg3 proteins confirms what has been found for many other disordered proteins,4,45,50 which is that protein propensity for disorder is encoded in its amino acid sequence and can be reliably predicted. To gain insight into the molecular dynamics of autophagy proteins, we applied predictors of protein disorder (PONDR, CH and CDF) on the amino acid sequences of all of the Atg proteins in S. cerevisiae. The ∆CH-∆CDF plots generated using PONDR-VSL2 and PONDR-VL3 showed that at least 50% of the Atg proteins from yeast occupy the Q1, Q3, or Q4 quadrants, indicating that they are disordered based on the CH, CDF, or both CH and CDF methods (Fig. 6). Of these, Atg16, Atg29, and Atg31 in Q4 are the most disordered yeast Atg proteins (putative pre-molten globules) that can undergo significant conformational changes and/or disorder-to-order transitions upon posttranslational modification and/or binding to a physiological partner. In comparison, Atg3, Vps30/Atg6, Atg8, Atg9, Atg12, Atg13, Atg19, Atg20, Atg21, Atg23, Snx4/Atg24, Atg32, Atg33, Atg34, Atg36, and Atg38 are molten globule-like or hybrid (mixed) proteins with approximately equal amounts of ordered and disordered regions. They are less dynamic in solution than the proteins in the Q4 quadrant, which suggests more feasible capturing of their intact solution structure by crystallography. At the same time, flexible domains of the proteins in Q3 can be expected to play an important functional role in autophagy. The remaining Atg proteins from yeast, specifically Atg1, Atg2, Atg4, Atg5, Atg7, Atg10, Atg11, Atg14, Atg15, Atg17, Atg18, Atg22, Atg26, and Atg27, are proteins that are mostly structured in solution, and are likely to operate on the “lock-and-key” principle. IDPRs, if any, present in their structure represent only a small portion of their overall conformation. The far right location of Atg5, Atg7, Atg10, and Atg22 in the Q2 quadrant indicates that these proteins gain the most rigid structure in solution, and thus are most suitable for crystallographic studies.
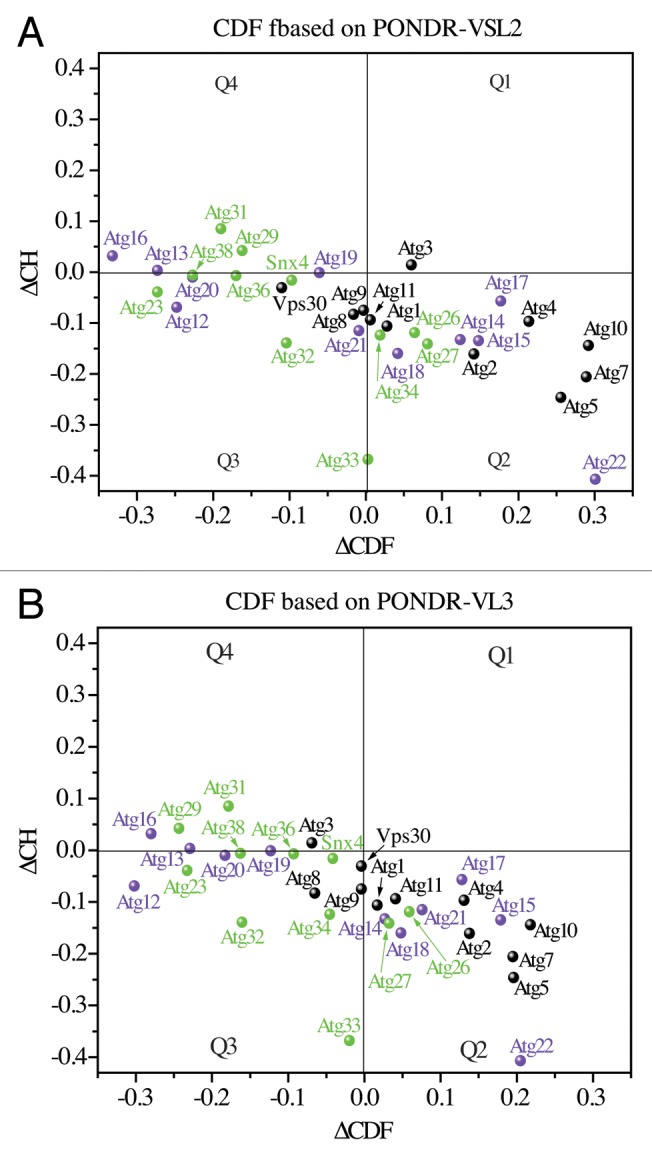
Figure 6. ∆CH-∆CDF analysis of Atg proteins from yeast. The ∆CH-∆CDF plots, where the ∆CDF parameter was calculated based on PONDR-VSL2 (A) and PONDR-VL3 (B), show the distribution of the Atg proteins from yeast in the Q1, Q2, Q3, and Q4 quadrants. For description of quadrants, see the legend for Figure 5. For better clarity, Atg1, Atg2, Atg3, Atg4, Atg5, Vps30/Atg6, Atg7, Atg8, Atg9, Atg10, and Atg11 are shown in black, Atg12, Atg13, Atg14, Atg15, Atg16, Atg17, Atg18, Atg19, Atg20, Atg21, and Atg22 are labeled in purple, and Atg23, Snx4/Atg24, Atg26, Atg27, Atg29, Atg31, Atg32, Atg33, Atg34, Atg36, and Atg38 are marked in green.
Distribution of the Atg proteins from yeast in the Q1, Q2, Q3, and Q4 quadrants based on the actual ∆CH and ∆CDF values (Tables S1 and S2) is consistent with their PONDR-FIT scores (Figs. S4–S9); namely, amino acid residues of the structured Atg proteins in the Q2 quadrant have mostly low PONDR-FIT scores, whereas the residues of the highly disordered Atg proteins in the Q4 quadrant exhibit mostly high PONDR-FIT scores (often above the 0.5 threshold), and the residues of the hybrid Atg proteins in the Q3 quadrant (or at its border) equally alternate between high and low PONDR-FIT scores. From these PONDR data, we found that 24 Atg proteins from yeast are predicted to have IDPRs of 30 or more disordered (with a score of ~0.5 or higher) residues (Table S3). This result shows the widespread intrinsic disorder among autophagy proteins. It should be noted, however, that the number of these IDPRs in itself is not a good indication of the extent of intrinsic disorder for a particular protein in solution. All amino acid residues in the sequence and their order/disorder scores have to be taken jointly into account (as is the case in the ΔCH-ΔCDF plot) to reveal the overall dynamics and behavior of a protein.
At this point, we emphasize that Figure 6, Tables S1 and S2, and Figures S4–S9 together create a tool that allows for a quick assessment of the structure and dynamics of any of the currently known yeast Atg proteins, and this information has many practical applications. For example, this tool can be applied in a quick search for the disorder propensity of a domain harboring a mutation, in a rapid evaluation of the accessibility of a protein domain to a posttranslational modification, or in the assessment of the suitability of application of an experimental technique on a particular Atg protein.
Discussion
The protein-protein interactions involved in the signaling and regulation of autophagy are complex.21,24 The number of studies on autophagy and the disease-related consequences of its malfunction have increased exponentially within the past 15 years.51 In parallel, during this same time, IDPs/IDPRs were shown to be highly abundant in cells, and to play a crucial role in cellular signaling and regulation owing to their unique set of characteristics and capabilities.3,4,8 The failure of various IDPs/IDPRs to be engaged in these functions is associated with numerous human diseases.12,18,20 Yet, very little is known about the connection of autophagy proteins with protein intrinsic disorder.
To investigate this connection, we used the characterization of yeast Atg3 as a source of experimental data. Atg3 appeared to be a promising candidate for intrinsic disorder, because it has missing domains in its crystal structures,28-30 and possesses a flexible domain that is essential for its function.30 The consensus of experimental data with prediction results based strictly on the amino acid sequence that are presented here revealed that free Atg3 in solution is likely a hybrid (mixed) protein possessing ordered domains along with functionally important intrinsically disordered regions. The presence of the ordered domains is supported by the observations that the far-UV CD spectrum of Atg3 shows some secondary structure elements, PONDR-FIT predicts ordered regions (residues 15–90 and 165–238; Fig. 5A) in Atg3 that coincide with its crystallized domains (residues 21–83 and 165–238; Fig. 1A), the spot corresponding to Atg3 in the ΔCH-ΔCDF plot is close to the boundary between the Q3 and Q4 quadrants (Fig. 6), and Atg3ΔFR is able to fold to a near-structured protein conformation (Fig. 2; Table 1). In support of intrinsic disorder, Atg3 exhibits anomalous migration on SDS-PAGE (Fig. 4) and a hydrodynamic radius that far exceeds that of an ordered protein with comparable molecular mass (Fig. 2), both in contrast to Atg3ΔFR. Furthermore, the near-UV CD spectrum and the melting curve of Atg3 indicate the absence of a hydrophobic core (Figs. S1 and S2), and PONDR predictors show that nearly half of the Atg3 protein has the capacity to be disordered (with a score ≥ 0.5) in solution in the absence of a binding partner (Fig. 5A), including the longest α-helix seen in the Atg3 crystal (residues 239–265). Finally, several domains including the functionally important flexible domain (residues 90–165) of Atg3 are missing in its crystal (Fig. 1A). All these results together are consistent with the view that Atg3 may freely float in solution as a structure with a small ordered head (without a tightly packed hydrophobic core) and a long disordered tail. In the crystal, some protein regions can undergo disorder-to-order transition induced by crystal contacts or sulfate. To better illustrate this result, Figure 1C shows a model that schematically depicts a possible dynamic solution structure of Atg3 that cannot be seen in full from the crystal structure.
A sulfate ion was suggested to mimic the native ligand of Atg3, the phosphate group of PE.30 Ligand binding is reported to induce folding in disordered proteins,4 and Atg3 retains significant disorder in a physiological complex with Atg7.28,29 Thus, it is reasonable to assume that disorder-to-order transition and other functions of disorder can be engaged when Atg3 interacts with Atg8 and mediates its conjugation to PE. A disordered N terminus of Atg3 (Fig. 1; Fig. 5A) plays an essential role in Atg8–PE conjugation.52 Moreover, our protein disorder predictions (Fig. 6; Tables S1 and S2) indicate that Atg8 is also affected by intrinsic disorder, albeit to a lesser degree than Atg3, while Atg7 is their very structured partner (Fig. 6; Tables S1 and S2), in agreement with previous studies.28,29,53-55 Clearly, elucidation of precise order-disorder interplay within the Atg7-Atg3-Atg8–PE complex will not be a trivial task for future research.
The results with yeast Atg3 are strengthened here by characterization of 2 of its mutants (Atg3FR and Atg3∆FR) and of its human homolog (ATG3). The earlier study by Yamada et al.30 shows, using 1H-15N HSQC spectra, that the recombinant yeast Atg3 domain similar to Atg3FR used here is a random coil in solution. The CD spectra of Atg3FR along with other experimental and computational data presented here are in agreement with this result. Atg3FR can be categorized as an unfolded extended IDP in solution.3,39 In contrast, Atg3∆FR exhibits experimental and computational characteristics of a protein that gains mostly compact and ordered structure in solution. Together, Atg3FR and Atg3∆FR represent the opposites that occupy 2 extreme positions on the disorder-order scale. Interestingly, both experimental and computational characterization of human ATG3 revealed that it is a hybrid protein with a slightly lower propensity for disorder than the yeast protein. Amino acid sequence alignment of Atg3 from various organisms (Fig. S10) reveals that the loss of flexibility of the Atg3 protein during evolution may have occurred partially due to the loss of the yeast disordered domain between V239 and S280, and perhaps through insertions and changes in the yeast disordered domain between G100 and K125. As expected, the disordered regions of Atg3 show very little conservation, in contrast to the structured domains (Fig. S10; Fig. 5A). Why the dynamic structure of Atg3 lost some plasticity during evolution remains an open intriguing question for mammalian autophagy. It is interesting particularly because the extent of intrinsic disorder tends to increase, rather than decrease, with increasing complexity of the organism.10,12
The consensus between experimental features of IDPs and the prediction of their disorder from amino acid sequence was demonstrated for proteins of various cellular pathways.4,13,45,50 Identification of Atg3 as a protein with IDPRs in this study extended the validity of this consensus into autophagy. Elucidation of the mechanism of autophagy is currently coming to the point where more detailed information on the structure of each Atg protein is in demand. Some Atg proteins or their rigid domains have been crystallized,28-30,34,53-55 but a lot of structural information is still needed, and data on disorder in Atg proteins are very limited.56,57 The intrinsic disorder prediction presented here yields the first systematic insight into the molecular dynamics of proteins of the core autophagy machinery, and reveals that intrinsic disorder is very important in autophagy, as it affects at least 50% of the Atg proteins in yeast (Fig. 6). The wide-range layout of these proteins in the Q3 and Q4 quadrants of the ∆CH-∆CDF plot indicates that they are disordered to various degrees.
The highest concentration and level of disorder is present among Atg proteins involved in autophagy induction (Atg13, Atg29, and Atg31). One plausible explanation could be the feasibility of fast regulation of the autophagy induction complex owing to the high plasticity of its components that are proposed to form stable constitutive interactions in both growing and starvation conditions;56 efficient regulation is one of the typical features of IDPs.3,8,57 A high plasticity of the intrinsically disordered domains of Atg29 (residues 1–11) and Atg31 (residues 1–50 and 180–196) (Fig. S8) is clearly important for formation of the Atg17-Atg31-Atg29 complex. These domains must undergo disorder-to-order transition and form, based on the crystal structure of the Atg17-Atg31-Atg29 complex,34 either β-sheets involved in the binding of Atg29 and Atg31 or α-helices involved in the binding of Atg31 to Atg17. Thus, mutations that would prevent these transitions may reduce the capability to recognize binding partners and lead to protein aggregation. This would likely interfere with the proper assembly of the Atg17-Atg31-Atg29 complex and efficient autophagy. In agreement with these possibilities, attempts to purify Atg29 alone failed due to the formation of soluble aggregates.56
Localization of Atg16 in the Q4 quadrant indicates that the ubiquitin-like system also requires a significant amount of protein plasticity in order to facilitate Atg8 lipidation during phagophore formation. It also opens several puzzling questions, such as what role, if any, does the intrinsic disorder of Atg16 play in Atg8 lipidation and in the functional conformation of the Atg12–Atg5-Atg16 complex. The proteins located in or at the border of the Q3 quadrant (Vps30/Atg6, Atg8, Atg9, Atg12, Atg13, Atg19, Atg20, Atg21, Atg23, Snx4/Atg24, Atg32, Atg33, Atg34, Atg36, and Atg38) are also intriguing. Finding the functions of their disordered domains in autophagy promises new and exciting discoveries.
In summary, IDPs have unique structural attributes and capabilities.3,7,12,40 Finding that many Atg proteins in yeast possess these attributes suggests that intrinsic disorder is very relevant to autophagy. This outcome has important implications for elucidation of the mechanism of autophagy. First, unstructured domains of disordered autophagy proteins require detailed analysis, because flexible domains in IDPs are important contributors to their biological function.10-14 Second, the structure-function paradigm and function based on the principle of lock-and-key, largely valid for globular rigid proteins, fundamentally differ from the characteristics and abilities of IDPs/IDPRs.7,12,19 Therefore, Atg proteins that qualify as IDPs or carry IDPRs require assumptions and the formulation of hypotheses different from those derived for globular proteins. Finally, the presence of intrinsic disorder in proteins affects the suitability of experimental techniques applied to their analysis. While some methods used on compact rigid proteins are very informative and revealing, application of the same techniques on IDPs/IDPRs can become cumbersome, especially if it requires removal of important flexible domains. We hope that the work presented here will be a useful tool that helps to navigate autophagy research toward a direction where the role of disorder still waits to be discovered.
Materials and Methods
Protein expression and purification
The Atg3 expression constructs were generated by standard molecular techniques, and coding regions were verified by automated sequencing. Constructs were designed such that tag removal by either thrombin (Sigma, T4648) or TEV protease leaves 2 extra residues (sequence: Gly-Ser) on the N-termini of proteins. S. cerevisiae Atg3, Atg3FR and Atg3∆FR were expressed as GST-fusion proteins from pGEX-4T1 (GE Healthcare Life Sciences, 28-9545-49), and full-length human ATG3 was expressed from a version of pGEX-4T1 modified to contain a TEV site. S. cerevisiae Atg3FR has 5 extra vector-derived residues (sequence: Arg-Pro-His-Arg-Asp) on its C terminus (Fig. S3). Protein expression was performed in BL21 CodonPlus (λDE3) RIL cells (Agilent Technologies/Stratagene, 230245) growing in LB medium induced with 0.6 mM IPTG at 16 °C overnight and harvested by centrifugation. After glutathione sepharose (GE Healthcare Life Sciences, 17-5132-01) affinity chromatography, S. cerevisiae Atg3FR was cleaved with thrombin (1:100 ratio at 4 °C overnight) and purified by anion exchange chromatography before glutathione sepharose passback to remove cleaved GST. After thrombin cleavage, S. cerevisiae Atg3 and Atg3∆FR were purified by anion exchange and size exclusion chromatography (Superdex S200 10/300, GE Healthcare Life Sciences, 17-5175-01). Human ATG3 was purified by glutathione sepharose affinity chromatography and cleaved from the column with TEV protease (1:100 ratio at 4 °C overnight). Proteins were concentrated by centrifugal ultrafiltration, then frozen in liquid nitrogen, and stored at −80 °C.
Dynamic light scattering
Dynamic light scattering was used to determine hydrodynamic radii of purified proteins. Proteins were loaded on a size-exclusion chromatography column for multi-angle light scattering (Wyatt Technology Corporation, WTC-030S5). The column was attached to an ÄKTA purifier liquid chromatography system (GE Healthcare) and in-line DAWN HELEOS MALS and Optilab T-rEX differential refractive index detector. The system was equilibrated and chromatography performed at room temperature in 50 mM potassium phosphate buffer, pH 8.0, 100 mM NaCl. Data were analyzed by the ASTRA 6 software package (Wyatt Technology Corporation).
Circular dichroism
The Atg3/ATG3 proteins that were used for CD spectra measurement were dialyzed twice in 50 mM potassium phosphate buffer, pH 8.0. The CD spectra were obtained using an AVIV 62 DS CD spectrometer. The raw signal was used to calculate the mean residue ellipticity that takes into account the protein concentration.38 Experimental conditions for CD measurements are shown in the figure legends.
SDS-PAGE
Samples were analyzed by SDS-PAGE as described previously,58 except that the 10% acrylamide gel did not contain urea and the sample buffer contained 120 mM DTT. Protein standards were the PageRulerTM Plus Prestained Protein Ladder (Fermentas Life Sciences, SM1812).
Intrinsic disorder predictions
Amino acid compositional profiler was used as described previously.45 Disorder of Atg proteins was predicted using PONDR-VSL2,49 PONDR-VL3,48 and PONDR-FIT46 predictors. PONDR-VSL2 applies artificial neural networks that predict both short and long disordered regions from the amino acid sequence, and also employs secondary structure prediction. PONDR-VL3 has an advantage in accuracy in predicting longer disordered regions.48,49 PONDR-FIT is the most recent meta-predictor that combines 6 existing predictors, and yields the best accuracy in PONDR family predictors.46 In this work we use results from 3 different PONDR predictors since this is an accepted practice in the field due to the fact that each predictor has its own advantages and limitations and can be used for finding some specific disorder-related features. In essence, this is similar to the application of a multiparametric approach in the experimental characterization of protein structure.
The peculiarities of the PONDR score distribution within a protein sequence can be used for the calculation of the so-called cumulative distribution function (CDF). The CDF is essentially a graphed representation of the percent of the entire protein that corresponds to each particular disorder score (from low to high propensity for disorder) from PONDR. The CDF algorithm was developed for PONDR-VSL2 and PONDR-VL3, but not for PONDR-FIT. Training and testing of each CDF algorithm on protein data sets generated a boundary that separates ordered and disordered proteins.47 Disordered proteins occupy a lower portion of the CDF plot, below the boundary, while ordered proteins have a CDF curve in an upper part of the CDF plot. We present data from the CDFs based on PONDR-VSL2 and PONDR-VL3, because they both have a good accuracy,47 but generate slightly different results. The separating boundary in a CDF plot is represented by a collection of 7 discrete points. The average of vertical distances from the protein’s CDF curve to these 7 boundary points determines the ∆CDF parameter for a particular protein.59
Another analysis of amino acid sequence used in this work, which is independent of CDF, is the calculation of the mean net charge and mean hydrophobicity for a particular protein. This calculation determines the position of a protein in a charge-hydrophobicity (CH) plot. Disordered proteins occupy different areas in this plot than ordered proteins, and the separating border line is described by a linear equation.3 The perpendicular distance from the protein’s position to the boundary in the CH plot determines the ∆CH parameter for a particular protein. A combination of the independent parameters ∆CDF and ∆CH together generates the ∆CH-∆CDF plot, where ∆CH is the function of ∆CDF.57,59,60 This method separates all proteins into 4 groups, and each group is located in 1 of 4 quadrants (Q1-Q4), based on the propensity for disorder. Q1 contains proteins predicted to be disordered based on the CH plot, but ordered based on the CDF plot. Very few proteins are predicted to be in this quadrant; the proteins that are assigned to Q1 are referred to as rare proteins and their common structural features are still under investigation. The Q2 quadrant includes proteins predicted to be ordered by both CH and CDF plots. Proteins in Q2 are likely to gain a stable well-folded structure in solution. The Q3 quadrant contains proteins predicted to be ordered by the CH plot, but disordered by the CDF plot. Proteins located in Q3 are native molten globules or hybrid proteins that consist of both disordered and ordered regions. The Q4 quadrant includes proteins predicted to be disordered by both the CH and CDF plots. These proteins have a substantial amount of extended disorder, and adopt conformations of pre-molten globules or random coils.
Supplementary Material
Disclosure of Potential Conflicts of Interests
No potential conflicts of interests were disclosed.
Acknowledgments
We thank Dr DR Southworth (University of Michigan) for assistance with dynamic light scattering, Drs Y Qiu, AM Taherbhoy, SE Kaiser, and BA Schulman (St. Jude Children’s Research Hospital) for providing protein samples, and Dr CK Yip (University of British Columbia) for helpful comments. This work was supported by NIH grant R01GM053396 to DJK, and through the Protein Folding Diseases FastForward Initiative, University of Michigan.
Glossary
Abbreviations:
- Atg
autophagy-related
- CD
circular dichrosism
- CDF
cumulative distribution function
- CH
charge hydrophobicity
- DisProt
Database of Protein Disorder
- FR
flexible region
- GdmCl
guanidinium chloride
- IDP
intrinsically disordered proteins
- IDPR
intrinsically disordered protein region
- PDB
Protein Data Bank
- PE
phosphatidylethanolamine
- PONDR
predictors of natural disordered regions
- UV
ultraviolet
References
- 1.Dunker AK, Lawson JD, Brown CJ, Williams RM, Romero P, Oh JS, Oldfield CJ, Campen AM, Ratliff CM, Hipps KW, et al. Intrinsically disordered protein. J Mol Graph Model. 2001;19:26–59. doi: 10.1016/S1093-3263(00)00138-8. [DOI] [PubMed] [Google Scholar]
- 2.Uversky VN. Natively unfolded proteins: a point where biology waits for physics. Protein Sci. 2002;11:739–56. doi: 10.1110/ps.4210102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Uversky VN. A decade and a half of protein intrinsic disorder: biology still waits for physics. Protein Sci. 2013;22:693–724. doi: 10.1002/pro.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Uversky VN, Gillespie JR, Fink AL. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins. 2000;41:415–27. doi: 10.1002/1097-0134(20001115)41:3<415::AID-PROT130>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 5.Weinreb PH, Zhen W, Poon AW, Conway KA, Lansbury PT., Jr. NACP, a protein implicated in Alzheimer’s disease and learning, is natively unfolded. Biochemistry. 1996;35:13709–15. doi: 10.1021/bi961799n. [DOI] [PubMed] [Google Scholar]
- 6.Wright PE, Dyson HJ. Intrinsically unstructured proteins: re-assessing the protein structure-function paradigm. J Mol Biol. 1999;293:321–31. doi: 10.1006/jmbi.1999.3110. [DOI] [PubMed] [Google Scholar]
- 7.Kragelj J, Ozenne V, Blackledge M, Jensen MR. Conformational propensities of intrinsically disordered proteins from NMR chemical shifts. Chemphyschem. 2013;14:3034–45. doi: 10.1002/cphc.201300387. [DOI] [PubMed] [Google Scholar]
- 8.Ferreon ACM, Ferreon JC, Wright PE, Deniz AA. Modulation of allostery by protein intrinsic disorder. Nature. 2013;498:390–4. doi: 10.1038/nature12294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Janin J, Sternberg MJ. Protein flexibility, not disorder, is intrinsic to molecular recognition. F1000 Biol Rep. 2013;5:2. doi: 10.3410/B5-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tompa P. The interplay between structure and function in intrinsically unstructured proteins. FEBS Lett. 2005;579:3346–54. doi: 10.1016/j.febslet.2005.03.072. [DOI] [PubMed] [Google Scholar]
- 11.Uversky VN. The most important thing is the tail: multitudinous functionalities of intrinsically disordered protein termini. FEBS Lett. 2013;587:1891–901. doi: 10.1016/j.febslet.2013.04.042. [DOI] [PubMed] [Google Scholar]
- 12.Cumberworth A, Lamour G, Babu MM, Gsponer J. Promiscuity as a functional trait: intrinsically disordered regions as central players of interactomes. Biochem J. 2013;454:361–9. doi: 10.1042/BJ20130545. [DOI] [PubMed] [Google Scholar]
- 13.Dunker AK, Brown CJ, Lawson JD, Iakoucheva LM, Obradović Z. Intrinsic disorder and protein function. Biochemistry. 2002;41:6573–82. doi: 10.1021/bi012159+. [DOI] [PubMed] [Google Scholar]
- 14.Mao AH, Lyle N, Pappu RV. Describing sequence-ensemble relationships for intrinsically disordered proteins. Biochem J. 2013;449:307–18. doi: 10.1042/BJ20121346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Uversky VN. Multitude of binding modes attainable by intrinsically disordered proteins: a portrait gallery of disorder-based complexes. Chem Soc Rev. 2011;40:1623–34. doi: 10.1039/c0cs00057d. [DOI] [PubMed] [Google Scholar]
- 16.Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- 17.Iakoucheva LM, Brown CJ, Lawson JD, Obradović Z, Dunker AK. Intrinsic disorder in cell-signaling and cancer-associated proteins. J Mol Biol. 2002;323:573–84. doi: 10.1016/S0022-2836(02)00969-5. [DOI] [PubMed] [Google Scholar]
- 18.Babu MM, van der Lee R, de Groot NS, Gsponer J. Intrinsically disordered proteins: regulation and disease. Curr Opin Struct Biol. 2011;21:432–40. doi: 10.1016/j.sbi.2011.03.011. [DOI] [PubMed] [Google Scholar]
- 19.Deleersnijder A, Gerard M, Debyser Z, Baekelandt V. The remarkable conformational plasticity of alpha-synuclein: blessing or curse? Trends Mol Med. 2013;19:368–77. doi: 10.1016/j.molmed.2013.04.002. [DOI] [PubMed] [Google Scholar]
- 20.Uversky VN, Oldfield CJ, Dunker AK. Intrinsically disordered proteins in human diseases: introducing the D2 concept. Annu Rev Biophys. 2008;37:215–46. doi: 10.1146/annurev.biophys.37.032807.125924. [DOI] [PubMed] [Google Scholar]
- 21.He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;43:67–93. doi: 10.1146/annurev-genet-102808-114910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jin M, Liu X, Klionsky DJ. SnapShot: Selective autophagy. Cell. 2013;152:368–U400, e2. doi: 10.1016/j.cell.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klionsky DJ, Codogno P. The mechanism and physiological function of macroautophagy. J Innate Immun. 2013;5:427–33. doi: 10.1159/000351979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol. 2010;22:124–31. doi: 10.1016/j.ceb.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Czaja MJ, Ding WX, Donohue TM, Jr., Friedman SL, Kim JS, Komatsu M, Lemasters JJ, Lemoine A, Lin JD, Ou JHJ, et al. Functions of autophagy in normal and diseased liver. Autophagy. 2013;9:1131–58. doi: 10.4161/auto.25063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lieberman AP, Puertollano R, Raben N, Slaugenhaupt S, Walkley SU, Ballabio A. Autophagy in lysosomal storage disorders. Autophagy. 2012;8:719–30. doi: 10.4161/auto.19469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–75. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kaiser SE, Mao K, Taherbhoy AM, Yu S, Olszewski JL, Duda DM, Kurinov I, Deng A, Fenn TD, Klionsky DJ, et al. Noncanonical E2 recruitment by the autophagy E1 revealed by Atg7-Atg3 and Atg7-Atg10 structures. Nat Struct Mol Biol. 2012;19:1242–9. doi: 10.1038/nsmb.2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Taherbhoy AM, Tait SW, Kaiser SE, Williams AH, Deng A, Nourse A, Hammel M, Kurinov I, Rock CO, Green DR, et al. Atg8 transfer from Atg7 to Atg3: a distinctive E1-E2 architecture and mechanism in the autophagy pathway. Mol Cell. 2011;44:451–61. doi: 10.1016/j.molcel.2011.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yamada Y, Suzuki NN, Hanada T, Ichimura Y, Kumeta H, Fujioka Y, Ohsumi Y, Inagaki F. The crystal structure of Atg3, an autophagy-related ubiquitin carrier protein (E2) enzyme that mediates Atg8 lipidation. J Biol Chem. 2007;282:8036–43. doi: 10.1074/jbc.M611473200. [DOI] [PubMed] [Google Scholar]
- 31.Fujioka Y, Noda NN, Nakatogawa H, Ohsumi Y, Inagaki F. Dimeric coiled-coil structure of Saccharomyces cerevisiae Atg16 and its functional significance in autophagy. J Biol Chem. 2010;285:1508–15. doi: 10.1074/jbc.M109.053520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Le Gall T, Romero PR, Cortese MS, Uversky VN, Dunker AK. Intrinsic disorder in the protein data bank. J Biomol Struct Dyn. 2007;24:325–42. doi: 10.1080/07391102.2007.10507123. [DOI] [PubMed] [Google Scholar]
- 33.Muchmore SW, Sattler M, Liang H, Meadows RPH, Harlan JE, Yoon HS, Nettesheim D, Chang BS, Thompson CB, Wong SL, et al. X-ray and NMR structure of human Bcl-xL, an inhibitor of programmed cell death. Nature. 1996;381:335–41. doi: 10.1038/381335a0. [DOI] [PubMed] [Google Scholar]
- 34.Ragusa MJ, Stanley RE, Hurley JH. Architecture of the Atg17 complex as a scaffold for autophagosome biogenesis. Cell. 2012;151:1501–12. doi: 10.1016/j.cell.2012.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Uversky VN. Use of fast protein size-exclusion liquid chromatography to study the unfolding of proteins which denature through the molten globule. Biochemistry. 1993;32:13288–98. doi: 10.1021/bi00211a042. [DOI] [PubMed] [Google Scholar]
- 36.Uversky VN. What does it mean to be natively unfolded? Eur J Biochem. 2002;269:2–12. doi: 10.1046/j.0014-2956.2001.02649.x. [DOI] [PubMed] [Google Scholar]
- 37.Gast K, Fiedler C. Dynamic and static light scattering of intrinsically disordered proteins. Methods Mol Biol. 2012;896:137–61. doi: 10.1007/978-1-4614-3704-8_9. [DOI] [PubMed] [Google Scholar]
- 38.Kelly SM, Jess TJ, Price NC. How to study proteins by circular dichroism. Biochim Biophys Acta. 2005;1751:119–39. doi: 10.1016/j.bbapap.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 39.Uversky VN. Intrinsically disordered proteins and their environment: effects of strong denaturants, temperature, pH, counter ions, membranes, binding partners, osmolytes, and macromolecular crowding. Protein J. 2009;28:305–25. doi: 10.1007/s10930-009-9201-4. [DOI] [PubMed] [Google Scholar]
- 40.Borcherds W, Kashtanov S, Wu H, Daughdrill GW. Structural divergence is more extensive than sequence divergence for a family of intrinsically disordered proteins. Proteins. 2013;81:1686–98. doi: 10.1002/prot.24303. [DOI] [PubMed] [Google Scholar]
- 41.Uversky VN, Dunker AK. Understanding protein non-folding. Biochim Biophys Acta. 2010;1804:1231–64. doi: 10.1016/j.bbapap.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Whitmore L, Wallace BA. DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res. 2004;32:W668–73. doi: 10.1093/nar/gkh371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Whitmore L, Wallace BA. Protein secondary structure analyses from circular dichroism spectroscopy: methods and reference databases. Biopolymers. 2008;89:392–400. doi: 10.1002/bip.20853. [DOI] [PubMed] [Google Scholar]
- 44.Sickmeier M, Hamilton JA, LeGall T, Vacic V, Cortese MS, Tantos A, Szabo B, Tompa P, Chen J, Uversky VN, et al. DisProt: the database of disordered proteins. Nucleic Acids Res. 2007;35:D786–93. doi: 10.1093/nar/gkl893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vacic V, Uversky VN, Dunker AK, Lonardi S. Composition Profiler: a tool for discovery and visualization of amino acid composition differences. BMC Bioinformatics. 2007;8:211–8. doi: 10.1186/1471-2105-8-211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xue B, Dunbrack RL, Williams RW, Dunker AK, Uversky VN. PONDR-FIT: a meta-predictor of intrinsically disordered amino acids. Biochim Biophys Acta. 2010;1804:996–1010. doi: 10.1016/j.bbapap.2010.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xue B, Oldfield CJ, Dunker AK, Uversky VN. CDF it all: consensus prediction of intrinsically disordered proteins based on various cumulative distribution functions. FEBS Lett. 2009;583:1469–74. doi: 10.1016/j.febslet.2009.03.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Obradovic Z, Peng K, Vucetic S, Radivojac P, Brown CJ, Dunker AK. Predicting intrinsic disorder from amino acid sequence. Proteins. 2003;53:566–72. doi: 10.1002/prot.10532. [DOI] [PubMed] [Google Scholar]
- 49.Peng K, Vucetic S, Radivojac P, Brown CJ, Dunker AK, Obradovic Z. Optimizing long intrinsic disorder predictors with protein evolutionary information. J Bioinform Comput Biol. 2005;3:35–60. doi: 10.1142/S0219720005000886. [DOI] [PubMed] [Google Scholar]
- 50.Dasgupta I, Sanglas L, Enghild JJ, Lindberg I. The neuroendocrine protein 7B2 is intrinsically disordered. Biochemistry. 2012;51:7456–64. doi: 10.1021/bi300871k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol. 2007;8:931–7. doi: 10.1038/nrm2245. [DOI] [PubMed] [Google Scholar]
- 52.Hanada T, Satomi Y, Takao T, Ohsumi Y. The amino-terminal region of Atg3 is essential for association with phosphatidylethanolamine in Atg8 lipidation. FEBS Lett. 2009;583:1078–83. doi: 10.1016/j.febslet.2009.03.009. [DOI] [PubMed] [Google Scholar]
- 53.Hong SB, Kim BW, Lee KE, Kim SW, Jeon H, Kim J, Song HK. Insights into noncanonical E1 enzyme activation from the structure of autophagic E1 Atg7 with Atg8. Nat Struct Mol Biol. 2011;18:1323–30. doi: 10.1038/nsmb.2165. [DOI] [PubMed] [Google Scholar]
- 54.Noda NN, Satoo K, Fujioka Y, Kumeta H, Ogura K, Nakatogawa H, Ohsumi Y, Inagaki F. Structural basis of Atg8 activation by a homodimeric E1, Atg7. Mol Cell. 2011;44:462–75. doi: 10.1016/j.molcel.2011.08.035. [DOI] [PubMed] [Google Scholar]
- 55.Schwarten M, Stoldt M, Mohrlüder J, Willbold D. Solution structure of Atg8 reveals conformational polymorphism of the N-terminal domain. Biochem Biophys Res Commun. 2010;395:426–31. doi: 10.1016/j.bbrc.2010.04.043. [DOI] [PubMed] [Google Scholar]
- 56.Mao K, Chew LH, Inoue-Aono Y, Cheong H, Nair U, Popelka H, Yip CK, Klionsky DJ. Atg29 phosphorylation regulates coordination of the Atg17-Atg31-Atg29 complex with the Atg11 scaffold during autophagy initiation. Proc Natl Acad Sci U S A. 2013;110:E2875–84. doi: 10.1073/pnas.1300064110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Peng Z, Xue B, Kurgan L, Uversky VN. Resilience of death: intrinsic disorder in proteins involved in the programmed cell death. Cell Death Differ. 2013;20:1257–67. doi: 10.1038/cdd.2013.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Betts SD, Ross JR, Hall KU, Pichersky E, Yocum CF. Functional reconstitution of photosystem II with recombinant manganese-stabilizing proteins containing mutations that remove the disulfide bridge. Biochim Biophys Acta. 1996;1274:135–42. doi: 10.1016/0005-2728(96)00023-0. [DOI] [PubMed] [Google Scholar]
- 59.Huang F, Oldfield C, Meng J, Hsu WL, Xue B, Uversky VN, Romero P, Dunker AK. Subclassifying disordered proteins by the CH-CDF plot method. Pacific Symposium on Biocomputing Pacific Symposium on Biocomputing 2012:128-39 [PubMed] [Google Scholar]
- 60.Mohan A, Sullivan WJ, Jr., Radivojac P, Dunker AK, Uversky VN. Intrinsic disorder in pathogenic and non-pathogenic microbes: discovering and analyzing the unfoldomes of early-branching eukaryotes. Mol Biosyst. 2008;4:328–40. doi: 10.1039/b719168e. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.