

Abstract
Recent studies have shown that long-term persistence of human cytomegalovirus (HCMV) in mononuclear cells of myeloid lineage is dependent on the UL138 open reading frame, which promotes latent infection. Although T-cell recognition of protein antigens from all stages of lytic HCMV infection is well established, it is not clear whether proteins expressed during latent HCMV infection can also be recognized. This study conducted an analysis of T-cell response towards proteins associated with HCMV latency. Ex vivo analysis of T cells from healthy virus carriers revealed a dominant CD8+ T-cell response to the latency-associated pUL138 protein, which recognized a non-canonical 13 aa epitope in association with HLA-B*3501. These pUL138-specific T cells displayed a range of memory phenotypes that were in general less differentiated than that previously described in T cells specific for HCMV lytic antigens. Antigen-presentation assays revealed that endogenous pUL138 could be presented efficiently by HCMV-infected cells. However, T-cell recognition of pUL138 was dependent on newly synthesized protein, with little presentation from stable, long-lived protein. These data demonstrate that T cells targeting latency-associated protein products exist, although HCMV may limit the presentation of latent proteins, thereby restricting T-cell recognition of latently infected cells.
Introduction
Human cytomegalovirus (HCMV) is a ubiquitous betaherpesvirus. It is an important cause of complications in immunosuppressed individuals, in whom it can replicate unconstrained, leading to HCMV disease that can involve the lungs, gastrointestinal tract, liver and retina (Crough & Khanna, 2009; Gandhi & Khanna, 2004). In transplantation, HCMV has also been implicated in the pathogenesis of graft-versus-host disease and solid-organ allograft rejection (Boeckh et al., 2003; Broers et al., 2000; Crough & Khanna, 2009; Pouteil-Noble et al., 1993). As with all herpesviruses, HCMV establishes lifelong latency after primary infection, and it is the subsequent virus reactivation that accounts for much of the morbidity. Conceptually, persistence can take the form of low-level productive infection or true latent infection. In latent infection, the viral genome is maintained in the absence of production of infectious virions, and this is now thought to be the major form of latency and source of reactivation in HCMV (Sinclair, 2008). The primary sites of HCMV latency are the mononuclear cells of myeloid lineage, from CD34+ haematopoietic stem cells through to peripheral blood monocytes (Mendelson et al., 1996; Reeves et al., 2005; Taylor-Wiedeman et al., 1991). The frequency of infected cells in naturally infected individuals has been quantified and has been estimated at 1 in 104–105, with each infected cell carrying between two and 13 HCMV genome equivalents (Slobedman & Mocarski, 1999), probably in the form of circular episomes (Bolovan-Fritts et al., 1999).
The patterns of viral gene expression in latent and lytic infections are markedly different. Many genes that are critical in lytic infection, for example IE1, are expressed only transiently and weakly in in vitro experimental models of latency (Cheung et al., 2006; Goodrum et al., 2004). Expression of other lytic genes, such as IE2, is not detectable (Cheung et al., 2006; Goodrum et al., 2004). These in vitro findings have been supported by ex vivo analysis of mononuclear cells in natural latency whereby expression of IE1/IE2 and structural genes could not be detected despite detectable levels of HCMV DNA (Mendelson et al., 1996; Taylor-Wiedeman et al., 1994). A number of latency-associated transcripts have now been described; most were initially identified by an in vitro latency infection model, and some have subsequently been confirmed on ex vivo analysis of peripheral blood and bone marrow mononuclear cells. These transcripts include alternative transcripts from the IE1/IE2 region (Kondo et al., 1994, 1996), an antisense transcript from UL81–82 (Bego et al., 2005) known as UL81–82ast or LUNA, an alternatively spliced transcript from UL111.5A encoding LAcmvIL-10 (Jenkins et al., 2004), UL138 (Goodrum et al., 2007; Petrucelli et al., 2009) and a number of less well-characterized transcripts (Cheung et al., 2006; Goodrum et al., 2002, 2004). The mechanistic roles of these transcripts are not fully understood. Their expression is not restricted to latent infection; most are also expressed at some point during productive infection (Jenkins et al., 2008; Lunetta & Wiedeman, 2000). Whilst UL138 has been demonstrated to be an important component of latent infection, it is likely that other viral transcripts also contribute to latent infection (Goodrum et al., 2007; Petrucelli et al., 2009). Indeed, studies from one of our laboratories (F. G.) using UL138-null viruses have indicated a potential role for other viral factors in establishing and maintaining latent infection (Petrucelli et al., 2009).
It is now firmly established that T-cell immunity is central to controlling HCMV infection. CD4+ and CD8+ T-cell numbers and function correlate with the risks of HCMV reactivation and disease (Crough & Khanna, 2009; Gandhi & Khanna, 2004). The T-cell response to HCMV is broadly specific: more than half of the >200 open reading frames in HCMV can be recognized by T cells (Sylwester et al., 2005). These include products from all three phases of lytic infection (immediate-early, early and late) and all categories of structural and non-structural proteins (Elkington et al., 2003; Sylwester et al., 2005). However, latently infected cells do not express most of these dominant antigens and have been presumed not to be subject to T-cell immune surveillance. Here, we conducted an in-depth analysis of T-cell responses to latency-associated proteins and demonstrated that, similar to lytic proteins, latency-associated proteins can also be recognized by T cells. Hence, latently infected cells could also potentially come under T-cell control.
Results
In vitro and ex vivo characterization of T-cell responses towards HCMV-encoded latency-associated transcripts
In the first set of experiments, we sought to determine whether T cells specific for proteins encoded by HCMV latency-associated transcripts were present in healthy virus carriers. We focused on two recently identified transcripts: UL138 (Goodrum et al., 2007; Petrucelli et al., 2009) and LUNA (Bego et al., 2005). Both transcripts have been found in latently infected cells ex vivo and shown to encode protein products (Bego et al., 2005; Goodrum et al., 2007; Petrucelli et al., 2009). The amino acid sequences of LUNA and pUL138 are well conserved across HCMV strains. LUNA sequence alignment from seven HCMV strains showed only minor polymorphisms in five of the 133 aa residues: each variation was represented by a single strain alone, and none was present in more than one strain (Bego et al., 2005). The sequence used in our overlapping peptide set was from laboratory strain AD169, which differs from the clinical strain Merlin in only one residue (Bego et al., 2005). pUL138 is polymorphic at two residues: strain Merlin has alanine and asparagine at aa 112 and 124, respectively, whereas strains Toledo and Fix have valine and serine at these positions. The UL138 gene from the clinical isolate used in this study and cloned into recombinant adenovirus encoded alanine at aa 112 and serine at aa 124.
Peripheral blood was obtained from 22 healthy HCMV-seropositive individuals representing a broad spectrum of human leukocyte antigen (HLA) alleles: HLA-A1, -A2, -A3, -A11, -A23, -A24, -A26, -A31 and -A32; HLA-B7, -B8, -B13, -B14, -B18, -B27, -B35, -B37, -B40(60), -B41, -B44, -B46 and -B62; HLA-Cw1, -Cw2, -Cw3, -Cw4, -Cw5, -Cw7 and -Cw17; HLA-DRB1*01, -DRB1*03, -DRB1*04, -DRB1*07, -DRB1*08, -DRB1*09, -DRB1*1101, -DRB1*1401 and -DRB1*15; and HLA-DQB1*02, -DQB1*03, -DQB1*05 and -DQB1*06. Because ex vivo identification of low-frequency T cells could be both masked and masqueraded by background cytokine secretion, we used a two-step method to increase stringency. We stimulated peripheral blood mononuclear cells (PBMCs) with overlapping peptide pools or recombinant adenovirus encoding LUNA or pUL138 and cultured the cells for 10 days to expand any antigen-specific T cells. Next, the presence of LUNA- or pUL138-specific T cells was determined by restimulation with overlapping peptide pools in a 6 h intracellular cytokine-secretion assay. The first stimulation was performed with overlapping peptides in 18 cases and recombinant adenovirus in 12 cases; eight individuals were screened using both methods and the results from the two were comparable. pUL138-specific CD8+ T cells could be expanded from four individuals (Fig. 1a); representative data from a non-responder are also shown. The CD8+ T-cell response was directed towards an epitope located on the N terminus of pUL138 (MDDLPLNVGLPIIGVMLVLI). We were unable to detect any pUL138-specific CD4+ or LUNA-specific CD8+ or CD4+ T-cell responses.
Fig. 1.

CD8+ T-cell recognition of HCMV-encoded latency determinant pUL138. (a) PBMCs from healthy virus carriers were stimulated with Ad5f35.UL138 and expanded for 10 days, and T-cell specificity was assessed in a 6 h intracellular cytokine-secretion assay. The results show the CD8+ T-cell responses to the pUL138 20mer peptide MDDLPLNVGLPIIGVMLVLI in four responders (N01, N02, N06 and N11) and a representative non-responder (N13). (b) Epitope minimization. pUL138-specific T-cell lines expanded from two donors were tested against peptides of different lengths. The minimum epitope was a 13mer non-canonical sequence, LPLNVGLPIIGVM. (c) Stabilization of surface HLA-B35 molecule on T2.B*3501 cells by LPLNVGLPIIGVM peptide. Results from three independent experiments are shown (mean±sem); the P value was calculated using a paired, two-tailed Student’s t-test.
All four individuals with a CD8+ T-cell response to pUL138 were HLA-B*3501+. To map precisely the minimum epitope sequence, we expanded polyclonal T-cell lines from two of these individuals and tested the response to progressively shortened peptides in an intracellular cytokine-secretion assay (Fig. 1b). This mapped the minimum epitope to a non-canonical 13 aa sequence: LPLNVGLPIIGVM (referred to as LPL; Fig. 1b). This 13mer peptide epitope effectively stabilized surface HLA-B35 expression in the transporter associated with antigen processing (TAP)-deficient T2.B*3501 cell line (Fig. 1c), which further confirmed its HLA restriction. This epitope is probably restricted to the HLA-B*3501 subtype: a T-cell response was detected in four out of five HLA-B*3501+ HCMV-seropositive individuals, but in none of four HCMV-seropositive individuals with other HLA-B35 subtypes. Of note, the T-cell response against this epitope was not detected in either of two tested HCMV-seronegative HLA-B*3501+ individuals (data not shown).
To characterize further the T-cell response to pUL138, we stained fresh PBMCs from five HLA-B*3501+ HCMV-seropositive individuals with custom-synthesized HLA-B*3501–LPL major histocompatibility complex (MHC)–peptide tetramer, with co-staining for T-cell surface memory markers. Ex vivo analysis with the MHC–peptide tetramer showed a precursor frequency in fresh PBMCs ranging from 0.02 to 0.55 % of CD8+ T cells (Fig. 2a). Phenotypic analysis revealed a heterogeneous population, ranging from predominantly early (CD27+CD57−) through to late (CD27−CD57+) memory phenotype (Fig. 2b). However, re-expression of CD45RA, a feature of late CD8+ T-cell differentiation commonly found in HCMV-specific T cells (Appay et al., 2002; Hislop et al., 2007), was observed in only one out of five donors. Within the five individuals with a pUL138 response, we could detect an ex vivo HLA-B*3501-restricted IPSINVHHY (pp65) population in four, and an HLA-A23-restricted AYAQKIFKIL (IE1) and HLA-B8-restricted ELRRKMMYM (IE1) population in one individual each. We compared the ex vivo memory phenotype of T cells of different specificities within these individuals and found that pUL138-specific T cells were more likely to express the early phenotype marker CD27 and a trend towards lower expression of the late phenotype marker CD57 or the re-expression of CD45RA (Fig. 2c). Similar to HCMV-specific T cells targeting immediate-early or late antigens, pUL138-specific T cells could also have strong ex vivo functionality and secrete multiple cytokines [i.e. gamma interferon (IFN-γ), tumour necrosis factor alpha (TNF-α) and macrophage inflammatory protein 1β (MIP-1β)] following stimulation with pUL138 peptide (Fig. 2d).
Fig. 2.

Ex vivo characterization of pUL138-specific T cells from healthy virus carriers. (a, b) Fresh PBMCs were stained with anti-CD8, anti-CD27 and anti-CD57 antibody and HLA-B*3501–LPL tetramers. Flow cytometry dot plots gated on CD8+ T cells are shown. (a) Tetramer staining from three representative HLA-B35+ HCMV-seropositive donors and two HLA-B35+ HCMV-seronegative controls. (b) Expression of CD27, CD57, CD45RA and CD45RO in pUL138 and lytic antigen-specific T cells from two representative donors, N02 (i) and N11 (ii). (c) Expression of T-cell memory markers from five HLA-B35+ HCMV-seropositive individuals. T cells were gated by tetramer staining and had the following specificities: pUL138 (n = 5), pp65 (n = 4) and IE1 (n = 2). Results are shown as means±sem; the P value was calculated using an unpaired, two-tailed Student’s t-test. (d) Flow cytometry dot plots from a 6 h intracellular cytokine-secretion assay performed on PMBCs ex vivo from donor N06.
Endogenous processing and presentation of pUL138 in virus-infected cells
We sought to determine whether endogenously expressed pUL138 could be presented by the relevant cell types. Because the frequency of latently infected cells ex vivo is estimated at 1 in 10−4–10−5, in vitro infection was necessary. We found that peripheral blood monocytes infected with recombinant adenovirus encoding pUL138 could process the LPL epitope endogenously and efficiently activated antigen-specific T cells (Fig. 3a). However, there was only very low-level activation of pUL138-specific T cells in response to monocytes infected with HCMV strain Toledo. Whilst poor pUL138 T-cell recognition may be due to inefficient HCMV infection of monocytes in vitro (Kitajima et al., 2001; Scott et al., 1989), which has been reported at 0.01–15 % depending on viral strain (Lathey & Spector, 1991; Rice et al., 1984), the monocytes were relatively more efficient in presenting HLA class I epitopes from pp65 and pp50 at multiple time points (Fig. 3b, c). In vitro HCMV infection of monocytes is usually abortive, and the observed low-level T-cell activation could be in response to monocyte presentation of exogenous, rather than endogenous, antigens. In order to confirm endogenous presentation following HCMV infection, we also studied primary fibroblasts, which are highly permissive to HCMV infection in vitro. As shown in Fig. 3(a), fibroblasts infected with either recombinant adenovirus or HCMV strain Toledo could efficiently present LPL epitope, leading to activation of pUL138-specific T cells.
Fig. 3.

Endogenous processing and presentation of pUL138. (a) HLA-B35+ monocytes or primary fibroblasts were infected with Ad5f35.UL138 or HCMV strain Toledo. At 16 h p.i., these cells were used to stimulate pUL138-specific T-cell lines in intracellular cytokine-secretion assays. Results are shown as representative flow cytometry dot plots from one of three independent experiments. (b) Presentation of lytic HCMV antigens by monocytes (HLA-A1+ and -B35+) 16 h after infection with HCMV strain Toledo. Flow cytometry dot plots were gated on HLA-A*0101–VTE (pp50) or HLA-B*3501–IPS (pp65) pentamer-positive T-cell populations. (c) Time course of presentation of pUL138, pp50 and pp65 CD8+ T-cell epitopes by monocytes (HLA-A1+ and -B35+) after infection with HCMV strain Toledo. Results are shown as T-cell responses to HCMV-infected monocytes after subtracting the responses to non-infected monocytes. Results from two independent experiments are shown (mean±sem).
Kinetics of pUL138 presentation in virus-infected cells
We next studied the time course of pUL138 expression and T-cell recognition in HCMV infection. Primary fibroblasts (HLA-A1+ and -B35+) were infected with HCMV strain Toledo. At multiple time points between 6 and 84 h post-infection (p.i.), the HCMV-infected fibroblasts were used as antigen-presenting cells in intracellular cytokine-secretion assays. The response of CD8+ T cells specific for pUL138 was determined by co-staining with allophycocyanin (APC)-conjugated HLA-B*3501–LPL tetramer and fluorescein isothiocyanate (FITC)-conjugated antibody to IFN-γ. As a comparator, the response of T cells specific for an HLA-A1-restricted epitope from pp50, an antigen expressed during early–late infection, was determined concurrently by co-staining with phycoerythrin (PE)-conjugated HLA-A*0101–VTE pentamer. In parallel, pUL138 expression was determined by immunoblot analysis of a HCMV-infected MRC5 primary lung fibroblast cell line. As shown in Fig. 4(a), pUL138 expression reached a maximum at around 12 h, remained at high levels up to 24 h p.i. and then progressively dropped to low but detectable levels at 84 h. Coincident with the protein expression pattern, T-cell recognition of the pUL138 epitope also showed a steady increase from 6 h p.i., reaching a maximum at 24 h. At 30 h, T-cell recognition dropped significantly and remained at low to undetectable levels for the duration of the experiment, until the final time point at 84 h (Fig. 4b). In contrast, T-cell recognition of pp50 showed a biphasic pattern with high levels of presentation at 6–24 h and 72–84 h. The parallel fall in pUL138 and pp50 recognition at 30 h was suggestive of a common mechanism, such as immune evasion, although other factors, as discussed below, could also be involved.
Fig. 4.
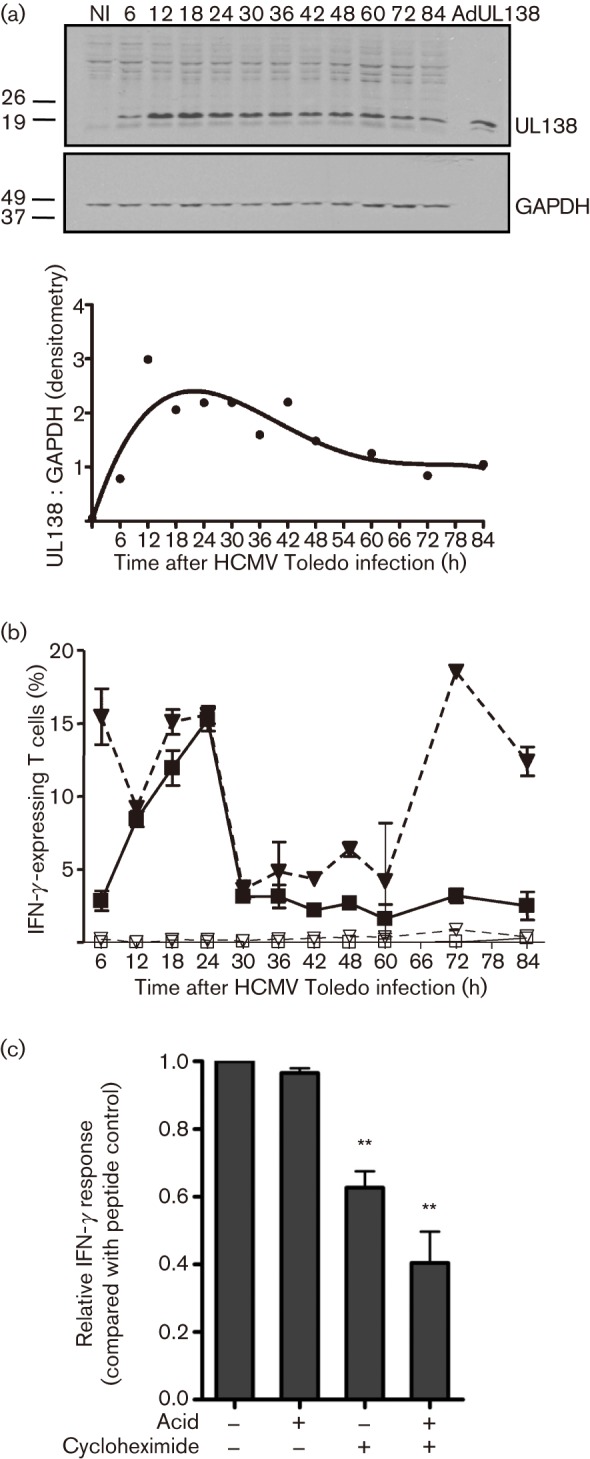
Kinetics of pUL138 expression and antigen presentation. (a) Immunoblot of primary lung fibroblasts (MRC5) infected with HCMV strain Toledo. The graph shows the intensity of pUL138 bands measured by densitometry and normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) expression. (b) IFN-γ response of the pUL138-specific T-cell line to HCMV-infected primary fibroblasts (HLA-A1+ and -B35+). The response to pp50, an early–late antigen, was used for comparison. The response was assessed simultaneously by co-staining with IFN-γ (FITC), HLA-B*3501–LPL tetramer (APC) and HLA-A*0101–VTE pentamer (PE). ▪, pUL138-specific T-cell response to HCMV-infected fibroblasts; □, pUL138-specific T-cell response to uninfected fibroblasts; ▾, pp50-specific T-cell response to HCMV-infected fibroblasts; ▿, pp50-specific T-cell response to uninfected fibroblasts. Results are shown as means±sd (n = 2). (c) Effect of MHC–peptide stripping (acid) and cycloheximide treatment on pUL138 antigen presentation and T-cell recognition. T-cell response was normalized against peptide-pulsed controls. Results are shown as means±sem from three independent experiments. **P<0.05 compared with untreated controls; the P value was calculated using a paired, two-tailed Student’s t-test.
We hypothesized that the rapid reduction in pUL138 recognition, despite persistence of pUL138 at moderate levels, was in part because the epitope was predominantly derived from newly synthesized rather than long-lived, stable protein. We used an intracellular cytokine-secretion assay in which pUL138-specific T cells were stimulated with HLA-B*3501+ HCMV-infected fibroblasts that had been treated with citrate buffer to strip off MHC–peptide complexes and/or cycloheximide to block fresh protein synthesis. The presentation of pUL138 by MHC–peptide complexes was determined by intracellular cytokine secretion in pUL138-specific T cells. As shown in Fig. 4(c), pUL138-specific T cells showed strong intracellular IFN-γ expression after stimulation with untreated HLA-B*3501+ HCMV-infected fibroblasts. Treatment with either citrate buffer or cycloheximide alone had some effect on the stimulating capability of the fibroblasts; however, treatment with citrate buffer followed by cycloheximide significantly reduced the T-cell response. These observations indicated that endogenously processed pUL138 epitope was primarily derived from newly synthesized rather than long-lived, stable protein. This would provide an additional explanation for the steep reduction in pUL138 T-cell recognition after 24 h p.i., which was significantly more pronounced than the fall in pUL138 protein antigen. These experiments do not, however, rule out a contribution from other factors, particularly the expression of HCMV immune-evasion genes.
Discussion
Although the role of virus-specific T cells in controlling the replicative phase of HCMV infection has been firmly established, their potential role in controlling latent HCMV infection has not yet been documented. Here we have shown for the first time that CD8+ T cells can efficiently recognize the HCMV latency-associated determinant pUL138. The ex vivo frequency and functionality of this T-cell response were comparable to responses towards structural and immediate-early antigens expressed only in lytic infection. However, the HCMV latent antigen-specific T cells, at least in some donors, had an earlier memory phenotype (CD27+/CD57−/CD45RA−/+) compared with that reported for HCMV lytic-specific T cells (CD27−/CD57+/CD45RA+) (Appay et al., 2002). This observation, whilst preliminary, is reminiscent of a similar phenomenon seen in the Epstein–Barr virus (EBV) latent antigen-specific T-cell response (Hislop et al., 2001, 2002). In vitro differentiation of CD34+ cells and monocytes from HCMV-seropositive donors into dendritic cells is associated with activation of the HCMV major immediate-early promoter, transcription of virus immediate-early genes and production of infectious virions (Reeves et al., 2005). It is possible that recurrent reactivation in professional antigen-presenting cells contributes to the terminal differentiation and memory inflation seen in HCMV-specific T cells directed towards immediate-early and structural proteins (Appay et al., 2002; Vescovini et al., 2007). It will be interesting to determine whether the expression of an early memory phenotype by T cells targeting latent antigens is related to antigen expression in immature precursor cells.
In EBV infection, latent antigen-specific T cells play a crucial role in controlling EBV-associated malignancies that do not express lytic antigens; they are also thought to be more important than the lytic-specific response in controlling virus reactivation (Hislop et al., 2007). The precise in vivo role of HCMV-specific T cells directed towards latency-associated determinants such as pUL138 remains to be fully determined. As the pUL138 determinant is expressed during both latent and replicative phases of infection, T cells specific to this protein may play an important role in controlling HCMV infection, regardless of the phase of infection. Whilst we have not investigated T-cell recognition in a latent infection model, latently infected cells should have the required antigen-processing machinery and should be able to present the T-cell epitope. One can postulate that targeting latently infected cells may help to control the size of the viral reservoir or limit virus reactivation. On the other hand, continuous antigen stimulation and T-cell activation may have detrimental indirect immune effects and promote immune senescence (Hadrup et al., 2006; Waller et al., 2008). The differential presentation of pUL138 compared with structural antigens may have a role in shaping the phenotype of the immune response towards latency-associated antigens. If this turns out to be a general phenomenon, this may have significant implications on our understanding of latent HCMV infection in healthy virus carriers.
Antigen presentation in HCMV-infected cells followed interesting kinetics that indicated that CD8+ T-cell epitopes from pUL138 are preferentially presented in the early phase of viral infection. As the viral infection progressed, pUL138 presentation and T-cell recognition fell steeply, even though pUL138 remained readily detectable in virus-infected cells. Further analysis revealed that the pUL138 epitope was derived predominantly from newly synthesized protein rather than from long-lived, stable protein. These observations are consistent with recent findings in other viral proteins whereby CD8+ T-cell epitopes have been found to be derived primarily from rapidly degrading polypeptides rather than long-lived protein (Tellam et al., 2004, 2007). The ongoing identification and characterization of latency-associated transcripts is likely to be followed by the discovery of other latency-associated T-cell epitopes. As this response is studied across a broader range of epitopes, its general applicability and functional significance will become clearer. It may be that the T-cell response against latency-associated antigens plays an important role in controlling HCMV infection as has been documented for EBV latent infection and this may, in future, provide new approaches to HCMV immunotherapy.
Methods
Blood samples and HCMV stocks.
Peripheral blood samples were obtained from healthy individuals after informed consent. Stocks of HCMV strain Toledo were produced in the human fetal lung fibroblast cell line MRC5 and harvested by repeated freeze–thaw cycles.
Recombinant adenovirus.
Recombinant adenovirus encoding UL138 or LUNA, referred to as Ad5f35.UL138 and Ad5f35.LUNA, respectively, were constructed using an Adeno-X Expression System (Clontech). UL138 was amplified by PCR from plasma of a solid-organ transplant recipient with HCMV reactivation; LUNA was amplified from HCMV strain AD169. Primers were (restriction sites underlined: forward XbaI, reverse KpnI): UL138 forward 5′-GCTCTAGAATGGACGATCTGCCGCTGAACG-3′, UL138 reverse 5′-CGGGGTACCTCACGTGTATTCTTGATGATAATG-3′, LUNA forward 5′-CTAGTCTAGAATGACGAGCGTGAGAGCCCCG-3′, and LUNA reverse 5′-GGGGTACCTCATTTGGAACACCGACAGCCG-3′. The PCR products were ligated into a pShuttle2 cassette, amplified in E. coli, and the inserts excised with Pl-SceI and I-CeuI and ligated into the Ad5f35 vector (Yotnda et al., 2001). Purified plasmids were PacI-digested and transfected into HEK293 cells. Recombinant adenovirus was harvested by repeated freeze–thaw cycles.
Peptides.
Peptides (20mers overlapping by 10 aa) spanning the 168 aa pUL138 protein (strain Merlin; GenBank accession no. NC_006273) and 133 aa LUNA protein (antisense of UL82, strain AD169, GenBank accession no. NC_001347), were synthesized by Mimotopes. Stocks were resuspended at 2 mg ml−1 in 10 % DMSO and used at concentrations of 1–5 µg ml−1.
In vitro culture and expansion of antigen-specific T cells.
PBMCs (5×105) were pulsed for 1 h with pools of five to six overlapping 20mers at 5 µg ml−1 per peptide, washed and added to 5×105 autologous responder PBMCs. Alternatively, 1×106 PBMCs were infected with Ad5f35.UL138 or Ad5f35.LUNA at an m.o.i. of 10 for 90 min, washed and added to 2×106 autologous responder PBMCs. Cultures were supplemented on days 3, 7 and 10 with recombinant human interleukin-2 (IL-2) at 10–20 U ml−1 and 30 % supernatant from the MLA144 T-cell line (a gibbon lymphoma cell line, which constitutively expresses IL-2; ATCC). On day 10, cultures were screened for antigen-specific T cells by a peptide-stimulated 6 h intracellular cytokine-secretion assay. To establish antigen-specific T-cell lines, the cultures were expanded by weekly restimulation with a peptide-pulsed irradiated (8000 rad) autologous EBV-transformed lymphoblastoid cell line (Moss et al., 1988).
Intracellular cytokine-secretion assay.
T cells were stimulated with peptides or virus-infected cells for 4–6 h in the presence of brefeldin A (1 µg ml−1; BD Biosciences). Cells were stained with cell surface antibodies (anti-CD3, -CD4, and/or -CD8), fixed and permeabilized, and then stained with IFN-γ antibody (BD Pharmingen). Flow cytometry was performed on a FACSCanto II (BD Biosciences) using BD FACSDiva software and the results were analysed with FlowJo software (Tree Star). For ex vivo functionality, PBMCs were stimulated with peptides in a 6 h intracellular cytokine-secretion assay and stained with the following antibodies: anti-CD8 conjugated to peridinin-chlorophyll-protein complex (PerCP)–Cy5.5 (Ebioscience), anti-IFN-γ conjugated to fluorescein isothiocyanate (FITC), anti-TNF-α conjugated to APC, and anti-MIP-1β conjugated to PE (all from BD Pharmingen).
MHC–peptide tetramer analysis.
HLA-B*3501–LPLNVGLPIIGVM tetramer conjugated to APC was synthesized by the NIH Tetramer Core Facility at Emory University, Atlanta, GA, USA. PBMCs or T-cell lines were stained with the tetramer for 20 min at 4 °C, followed by cell surface antibodies (FITC-conjugated anti-CD57or anti-CD45RA, PE-conjugated anti-CD27 or anti-CD45RO, PerCP–Cy5.5-conjugated anti-CD8, and/or Alexa Fluor 700-conjugated anti-CD4; BD Biosciences or EBioscience) for 15 min at 4 °C, washed and analysed by flow cytometry. For intracellular cytokine secretion, cells were stained with HLA-B*3501–LPLNVGLPIIGVM tetramer, HLA-A*0101–VTEHDTLLY (pp50) pentamer or HLA-B*3501–IPSINVHHY (pp65) pentamer (ProImmune) followed by surface antibody staining and intracellular cytokine staining as described above.
Antigen-presentation assays.
Monocytes were isolated by CD14 immunomagnetic selection according to the manufacturer’s instructions (Miltenyi Biotec) and infected with Ad5f35.UL138 or HCMV strain Toledo at an m.o.i. of 10. Primary human fibroblasts were infected with Ad5f35.UL138 at an m.o.i. of 5 or HCMV strain Toledo at m.o.i. of 1. Cells were exposed to virus for 90 min, washed and, unless otherwise indicated, used as stimulators in intracellular cytokine-secretion assays 16 h after infection. The responder to simulator ratios were 4 : 1 for fibroblasts and 1 : 10 for monocytes.
To study the kinetics of endogenous presentation of pUL138, HLA-B35+ fibroblasts were infected with HCMV strain Toledo and harvested at multiple time points between 6 and 84 h for use as antigen-presenting cells in intracellular cytokine-secretion assays with an LPL-specific T-cell line as the responder.
To study the relative contribution of newly synthesized versus stable protein in antigen presentation, HCMV-infected fibroblasts were treated with citric acid to strip MHC–peptide from the cell surface, cycloheximide to block protein synthesis, or both, prior to use as antigen-presenting cells. To strip MHC–peptide from the cell surface, HCMV-infected fibroblasts were treated with citrate phosphate buffer (0.131 M citric acid, 0.066 M Na2HPO, pH 3) for 2 min on ice, neutralized by 100-fold dilution in RPMI 1640 with 10 % fetal calf serum, and washed twice. The cells were further incubated at 37 °C in the presence or absence of 50 µM cycloheximide for 5 h and then used as stimulators in intracellular cytokine-secretion assays. To control for viability, equal numbers of fibroblasts were pulsed with peptide, washed at least four times and used as positive-control stimulators. The IFN-γ response was adjusted by calculating: (response to non-peptide-pulsed fibroblasts)/(response to peptide-pulsed fibroblasts). Experiments were performed in duplicate wells and three independent experiments were performed.
MHC stabilization assay.
T2.B*3501 cells are a TAP-deficient T2 cell line stably transfected with HLA-B*3501 (Takiguchi et al., 1994). Cells (2×105) were incubated with 100 µg peptide ml−1 for 1 h at 37 °C with 5 % CO2, at 26 °C for 14–16 h and then at 37 °C for 2 h. Cells were washed and stained with anti-HLA-Bw6 (SFR8-B6; ATCC HB-152) followed by FITC-conjugated goat anti-mouse antibody, and analysed by flow cytometry.
Immunoblot.
MRC5 primary lung fibroblasts were infected with HCMV strain Toledo at an m.o.i. of 2. Protein lysates from multiple time points (12 µg per lane) and positive controls from Ad5f35.UL138-infected fibroblasts (0.2 µg per lane) were separated by SDS-PAGE, transferred to nitrocellulose membrane and blocked overnight at 4 °C in Tris-buffered saline containing 0.1 % Tween 20, 5 % skimmed milk and 2.5 mg BSA ml−1. To detect pUL138, the membrane was probed with polyclonal rabbit anti-pUL138 antibody at a 1 : 1000 dilution (Petrucelli et al., 2009), followed by polyclonal sheep anti-rabbit horseradish peroxidase (HRP)-conjugated antibody. Protein bands were detected using enhanced chemiluminescence substrate (Western Lighting Plus ECL; PerkinElmer). Membranes were stripped and reprobed with monoclonal mouse anti-GAPDH (Abcam) followed by polyclonal sheep anti-mouse HRP-conjugated antibody. Protein levels were compared by densitometric analysis using Quantity One software (Bio-Rad Laboratories).
Acknowledgements
S.-K. T. is supported by a Clinical Fellowship from the Leukaemia Foundation of Australia. R. K. is supported by a Principal Research Fellowship from National Health and Medical Research Council (NH&MRC) of Australia. This study was supported by a Program Grant from the NH&MRC. The authors gratefully acknowledge Dr John Altman and the NIH Tetramer Core Facility at Emory University for production of the tetramer. Author contributions: S.-K. T. designed and performed the experiments, analysed the data and wrote the manuscript; F. G. provided reagents, analysed the data and critically reviewed the manuscript; R. K. designed the study and wrote the manuscript with S.-K. T.
References
- Appay V., Dunbar P. R., Callan M., Klenerman P., Gillespie G. M., Papagno L., Ogg G. S., King A., Lechner F. & other authors (2002). Memory CD8+ T cells vary in differentiation phenotype in different persistent virus infections. Nat Med 8, 379–385 [DOI] [PubMed] [Google Scholar]
- Bego M., Maciejewski J., Khaiboullina S., Pari G., St Jeor S. (2005). Characterization of an antisense transcript spanning the UL81–82 locus of human cytomegalovirus. J Virol 79, 11022–11034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeckh M., Nichols W. G., Papanicolaou G., Rubin R., Wingard J. R., Zaia J. (2003). Cytomegalovirus in hematopoietic stem cell transplant recipients: current status, known challenges, and future strategies. Biol Blood Marrow Transplant 9, 543–558 [DOI] [PubMed] [Google Scholar]
- Bolovan-Fritts C. A., Mocarski E. S., Wiedeman J. A. (1999). Peripheral blood CD14+ cells from healthy subjects carry a circular conformation of latent cytomegalovirus genome. Blood 93, 394–398 [PubMed] [Google Scholar]
- Broers A. E., van Der Holt R., van Esser J. W., Gratama J. W., Henzen-Logmans S., Kuenen-Boumeester V., Lowenberg B., Cornelissen J. J. (2000). Increased transplant-related morbidity and mortality in CMV-seropositive patients despite highly effective prevention of CMV disease after allogeneic T-cell-depleted stem cell transplantation. Blood 95, 2240–2245 [PubMed] [Google Scholar]
- Cheung A. K., Abendroth A., Cunningham A. L., Slobedman B. (2006). Viral gene expression during the establishment of human cytomegalovirus latent infection in myeloid progenitor cells. Blood 108, 3691–3699 [DOI] [PubMed] [Google Scholar]
- Crough T., Khanna R. (2009). Immunobiology of human cytomegalovirus: from bench to bedside. Clin Microbiol Rev 22, 76–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkington R., Walker S., Crough T., Menzies M., Tellam J., Bharadwaj M., Khanna R. (2003). Ex vivo profiling of CD8+-T-cell responses to human cytomegalovirus reveals broad and multispecific reactivities in healthy virus carriers. J Virol 77, 5226–5240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi M. K., Khanna R. (2004). Human cytomegalovirus: clinical aspects, immune regulation, and emerging treatments. Lancet Infect Dis 4, 725–738 [DOI] [PubMed] [Google Scholar]
- Goodrum F. D., Jordan C. T., High K., Shenk T. (2002). Human cytomegalovirus gene expression during infection of primary hematopoietic progenitor cells: a model for latency. Proc Natl Acad Sci U S A 99, 16255–16260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodrum F., Jordan C. T., Terhune S. S., High K., Shenk T. (2004). Differential outcomes of human cytomegalovirus infection in primitive hematopoietic cell subpopulations. Blood 104, 687–695 [DOI] [PubMed] [Google Scholar]
- Goodrum F., Reeves M., Sinclair J., High K., Shenk T. (2007). Human cytomegalovirus sequences expressed in latently infected individuals promote a latent infection in vitro. Blood 110, 937–945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadrup S. R., Strindhall J., Kollgaard T., Seremet T., Johansson B., Pawelec G., thor Straten P., Wikby A. (2006). Longitudinal studies of clonally expanded CD8 T cells reveal a repertoire shrinkage predicting mortality and an increased number of dysfunctional cytomegalovirus-specific T cells in the very elderly. J Immunol 176, 2645–2653 [DOI] [PubMed] [Google Scholar]
- Hislop A. D., Gudgeon N. H., Callan M. F., Fazou C., Hasegawa H., Salmon M., Rickinson A. B. (2001). EBV-specific CD8+ T cell memory: relationships between epitope specificity, cell phenotype, and immediate effector function. J Immunol 167, 2019–2029 [DOI] [PubMed] [Google Scholar]
- Hislop A. D., Annels N. E., Gudgeon N. H., Leese A. M., Rickinson A. B. (2002). Epitope-specific evolution of human CD8+ T cell responses from primary to persistent phases of Epstein–Barr virus infection. J Exp Med 195, 893–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hislop A. D., Taylor G. S., Sauce D., Rickinson A. B. (2007). Cellular responses to viral infection in humans: lessons from Epstein–Barr virus. Annu Rev Immunol 25, 587–617 [DOI] [PubMed] [Google Scholar]
- Jenkins C., Abendroth A., Slobedman B. (2004). A novel viral transcript with homology to human interleukin-10 is expressed during latent human cytomegalovirus infection. J Virol 78, 1440–1447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins C., Garcia W., Abendroth A., Slobedman B. (2008). Expression of a human cytomegalovirus latency-associated homolog of interleukin-10 during the productive phase of infection. Virology 370, 285–294 [DOI] [PubMed] [Google Scholar]
- Kitajima H., Okubo Y., Honda J., Yonemitsu J., Yoshida N., Fumimori T., Oizumi K. (2001). Interleukin-4 is needed for the infection of monocytes by human cytomegalovirus. Intervirology 44, 264–270 [DOI] [PubMed] [Google Scholar]
- Kondo K., Kaneshima H., Mocarski E. S. (1994). Human cytomegalovirus latent infection of granulocyte–macrophage progenitors. Proc Natl Acad Sci U S A 91, 11879–11883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo K., Xu J., Mocarski E. S. (1996). Human cytomegalovirus latent gene expression in granulocyte–macrophage progenitors in culture and in seropositive individuals. Proc Natl Acad Sci U S A 93, 11137–11142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lathey J. L., Spector S. A. (1991). Unrestricted replication of human cytomegalovirus in hydrocortisone-treated macrophages. J Virol 65, 6371–6375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunetta J. M., Wiedeman J. A. (2000). Latency-associated sense transcripts are expressed during in vitro human cytomegalovirus productive infection. Virology 278, 467–476 [DOI] [PubMed] [Google Scholar]
- Mendelson M., Monard S., Sissons P., Sinclair J. (1996). Detection of endogenous human cytomegalovirus in CD34+ bone marrow progenitors. J Gen Virol 77, 3099–3102 [DOI] [PubMed] [Google Scholar]
- Moss D. J., Misko I. S., Burrows S. R., Burman K., McCarthy R., Sculley T. B. (1988). Cytotoxic T-cell clones discriminate between A- and B-type Epstein–Barr virus transformants. Nature 331, 719–721 [DOI] [PubMed] [Google Scholar]
- Petrucelli A., Rak M., Grainger L., Goodrum F. (2009). Characterization of a novel Golgi apparatus-localized latency determinant encoded by human cytomegalovirus. J Virol 83, 5615–5629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pouteil-Noble C., Ecochard R., Landrivon G., Donia-Maged A., Tardy J. C., Bosshard S., Colon S., Betuel H., Aymard M., Touraine J. L. (1993). Cytomegalovirus infection – an etiological factor for rejection? A prospective study in 242 renal transplant patients. Transplantation 55, 851–857 [DOI] [PubMed] [Google Scholar]
- Reeves M. B., MacAry P. A., Lehner P. J., Sissons J. G., Sinclair J. H. (2005). Latency, chromatin remodeling, and reactivation of human cytomegalovirus in the dendritic cells of healthy carriers. Proc Natl Acad Sci U S A 102, 4140–4145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice G. P., Schrier R. D., Oldstone M. B. (1984). Cytomegalovirus infects human lymphocytes and monocytes: virus expression is restricted to immediate-early gene products. Proc Natl Acad Sci U S A 81, 6134–6138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott D. M., Rodgers B. C., Freeke C., Buiter J., Sissons J. G. (1989). Human cytomegalovirus and monocytes: limited infection and negligible immunosuppression in normal mononuclear cells infected in vitro with mycoplasma-free virus strains. J Gen Virol 70, 685–694 [DOI] [PubMed] [Google Scholar]
- Sinclair J. (2008). Human cytomegalovirus: latency and reactivation in the myeloid lineage. J Clin Virol 41, 180–185 [DOI] [PubMed] [Google Scholar]
- Slobedman B., Mocarski E. S. (1999). Quantitative analysis of latent human cytomegalovirus. J Virol 73, 4806–4812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sylwester A. W., Mitchell B. L., Edgar J. B., Taormina C., Pelte C., Ruchti F., Sleath P. R., Grabstein K. H., Hosken N. A. & other authors (2005). Broadly targeted human cytomegalovirus-specific CD4+ and CD8+ T cells dominate the memory compartments of exposed subjects. J Exp Med 202, 673–685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takiguchi M., Kawaguchi G., Sekimata M., Hiraiwa M., Kariyone A., Takamiya Y. (1994). The role of the conserved residue in pocket A and the polymorphic residue in pocket E of HLA-B*3501 in presentation of human minor histocompatibility peptides to T cells. Int Immunol 6, 1345–1352 [DOI] [PubMed] [Google Scholar]
- Taylor-Wiedeman J., Sissons J. G., Borysiewicz L. K., Sinclair J. H. (1991). Monocytes are a major site of persistence of human cytomegalovirus in peripheral blood mononuclear cells. J Gen Virol 72, 2059–2064 [DOI] [PubMed] [Google Scholar]
- Taylor-Wiedeman J., Sissons P., Sinclair J. (1994). Induction of endogenous human cytomegalovirus gene expression after differentiation of monocytes from healthy carriers. J Virol 68, 1597–1604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tellam J., Connolly G., Green K. J., Miles J. J., Moss D. J., Burrows S. R., Khanna R. (2004). Endogenous presentation of CD8+ T cell epitopes from Epstein–Barr virus-encoded nuclear antigen 1. J Exp Med 199, 1421–1431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tellam J., Fogg M. H., Rist M., Connolly G., Tscharke D., Webb N., Heslop L., Wang F., Khanna R. (2007). Influence of translation efficiency of homologous viral proteins on the endogenous presentation of CD8+ T cell epitopes. J Exp Med 204, 525–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vescovini R., Biasini C., Fagnoni F. F., Telera A. R., Zanlari L., Pedrazzoni M., Bucci L., Monti D., Medici M. C. & other authors (2007). Massive load of functional effector CD4+ and CD8+ T cells against cytomegalovirus in very old subjects. J Immunol 179, 4283–4291 [DOI] [PubMed] [Google Scholar]
- Waller E. C., Day E., Sissons J. G., Wills M. R. (2008). Dynamics of T cell memory in human cytomegalovirus infection. Med Microbiol Immunol 197, 83–96 [DOI] [PubMed] [Google Scholar]
- Yotnda P., Onishi H., Heslop H. E., Shayakhmetov D., Lieber A., Brenner M., Davis A. (2001). Efficient infection of primitive hematopoietic stem cells by modified adenovirus. Gene Ther 8, 930–937 [DOI] [PubMed] [Google Scholar]