

Abstract
Antifreeze proteins and glycoproteins [AF(G)Ps] have been well-known for more than three decades for their ability to inhibit the growth and recrystallization of ice through binding to specific ice crystal faces, and they show remarkable structural compatibility with specific ice crystal faces. Here, we show that the crystal growth faces of methyl α-d-mannopyranoside (MDM), a representative pyranose sugar, also show noteworthy structural compatibility with the known periodicities of AF(G)Ps. We selected fish AFGPs (AFGP8, AFGP1–5), and a beetle AFP (DAFP1) with increasing antifreeze activity as potential additives for controlling MDM crystal growth. Similar to their effects on ice growth, the AF(G)Ps can inhibit MDM crystal growth and recrystallization, and more significantly, the effectiveness for the AF(G)Ps are well correlated with their antifreeze activity. MDM crystals grown in the presence of AF(G)Ps are smaller and have better defined shapes and are of higher quality as indicated by single crystal X-ray diffraction and polarized microscopy than control crystals, but no new polymorphs of MDM were identified by single crystal X-ray diffraction, solid-state NMR, and attenuated total reflectance infrared spectroscopy. The observed changes in the average sizes of the MDM crystals can be related to the changes in the number of the MDM nuclei in the presence of the AF(G)Ps. The critical free energy change differences of the MDM nucleation in the absence and presence of the additives were calculated. These values are close to those of the ice nucleation in the presence of AF(G)Ps suggesting similar interactions are involved in the molecular recognition of MDM by the AF(G)Ps. To our knowledge this is the first report where AF(G)Ps have been used to control crystal growth of carbohydrates and on AFGPs controlling non-ice-like crystals. Our finding suggests MDM might be a possible alternative to ice for studying the detailed mechanism of AF(G)P–crystal interactions. The relationships between AF(G)Ps and carbohydrate binding proteins are also discussed. The structural compatibility between AF(G)Ps and growing crystal faces demonstrated herein adds to the repertoire of molecular recognition by AF(G)Ps, which may have potential applications in the sugar, food, pharmaceutical, and materials industries.
Introduction
Antifreeze proteins and antifreeze glycoproteins [AF(G)Ps] are defined by their ability to inhibit the growth and recrystallization of ice by binding to specific ice crystal surfaces.1−3 AF(G)Ps have diverse structures. For examples, AFGPs (e.g., AFGP1–5, AFGP8) found in the blood of the Antarctic fish Pagothenia borchgrevinki consist of glycotripeptide repeats (Ala-Ala-Thr), where the disaccharide, β-d-galactosyl(1 → 3)-α-N-acetylgalactosamine, is joined to each threonine through a glycosidic linkage. AFP isoform 1 isolated from the hemolymph of the beetle Dendroides canadensis (DAFP1) is composed of 12- or 13-mer repeats including Thr-Xxx-Thr (where Xxx is any amino acid) without glycosylation (Figure 1). AF(G)Ps also possess different antifreeze activities. The difference between the melting point and the noncolligative freezing point of H2O in the presence of AF(G)Ps is referred to as thermal hysteresis (TH), the value of which is generally used as a measure of antifreeze activity of the various AF(G)Ps. Among the AF(G)Ps mentioned above, at the same protein concentration, DAFP1 has the greatest antifreeze activity (i.e., producing the highest TH), followed by AFGP1–5, and then AFGP8. Despite large differences in their structures and antifreeze activities, all the AF(G)Ps share remarkable capabilities for binding to specific ice crystal faces and modifying the ice crystal habits within the thermal hysteresis gap, representing a paradigmatic case of controlling crystal growth.4,5
Figure 1.
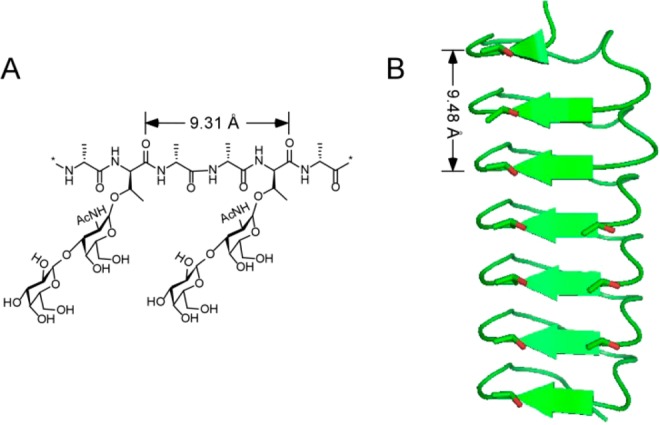
Structures of antifreeze biomolecules. (A) The structure of the repeat unit of antifreeze glycoproteins (AFGPs). (B) A model structure of beetle antifreeze protein isoform 1 from Dendroides canadensis (DAFP1). The distances in the structures indicate the periodicities.
Controlling the shape and size of crystals is central to many practical processes like pharmaceutical, food, materials, and chemical manufacturing since the shape and size can have a great impact on the chemical and physical properties.6−9 For example, milling is commonly used in manufacturing active pharmaceutical ingredients to reduce the particle size and ensure homogeneity, but it costs energy, causes localized heating, makes dusts, and can result in various defects in crystals.9 Thus, the use of additives holds promise for processes requiring control of crystal growth. Though the detailed mechanism of how additives work is not yet fully understood, substantial efforts have been directed toward the identification and design of new additives. However, the molecular recognition phenomena are generally limited to the cases of inorganic salts, carbohydrates, amino acids, and benzene ring containing compounds. Moreover, the known additives, except for a few inorganic salts (e.g., sodium chloride), are all limited to molecules having similar structures to the crystal substances of interest and are only effective at concentrations of 2–20 wt %.8,10,11 In the cases of inorganic salts, the controls of the nucleation and/or crystal growth are based on ionic–ionic interactions. For example, in shell biomineralization, shell proteins control shell crystal growth by ionic interactions using a relatively large amount (usually about 1:10 in molar ratio).12,13
Proteins and/or peptides are known to play an amazing role in both living organisms and the inanimate world to control the growth of minerals and produce new forms of solids with different properties.12−15 It has been reported that AFPs can inhibit the growth of clathrate hydrates, a class of ice-like crystalline solids.16 More recently, the molecular recognition repertoire of AFPs has been shown to include non-ice-like crystals, such as 5-methyluridine, and interestingly, the distances between the hydroxyl oxygen atoms on the putative ice-binding sites of AFPs closely match those between the hydroxyl oxygen atoms on the fast growth faces of ice as well as 5-methyluridine.17 However, the effects of AFPs on clathrate hydrates and nucleoside crystals are not directly related to their antifreeze activities and the required dosages for the control of nucleoside crystal growth are significantly less than those for ice growth control.17,18
The structural compatibility between AF(G)Ps and the fast growth faces of ice prompted us to explore their potential use as agents to control crystal growth of non-ice-like crystalline solids. We chose methyl α-d-mannopyranoside (MDM), a representative compound in the family of pyranose sugars and their derivatives for multiple reasons. MDM has wide industrial applications, such as in the synthesis of resins, plastics, and explosives.19,20 Moreover, MDM tends to crystallize from its melt in large blocks with a high percent of twin defects, an unwelcome crystal habit, which is unsuitable for X-ray diffraction. Thus, far no effective additives for control of size and shape of MDM crystal growth are known. Furthermore, the exposed hydroxyls on the fast growth faces of MDM crystals have a repeat distance which is similar to that of the hydrogen bonding residues present in the AF(G)Ps, making them promising candidates as additives for the control of MDM crystal growth (Figure 2).
Figure 2.
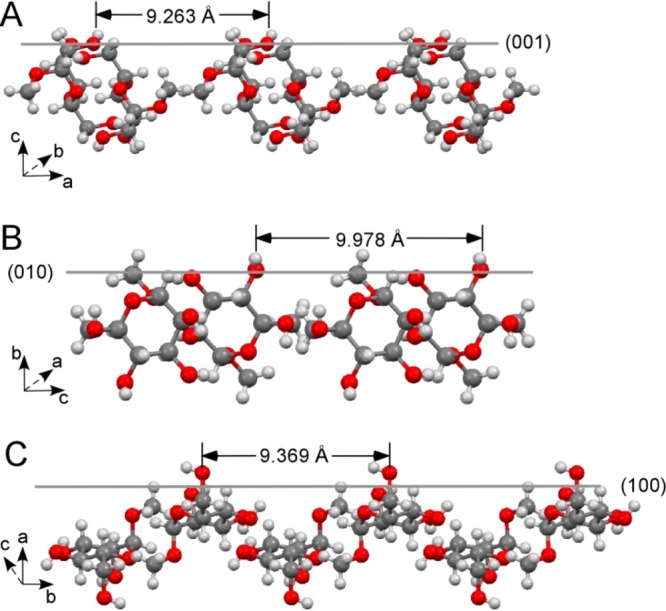
Periodicities of the hydroxyl oxygen atoms on the growing faces of methyl α-d-mannopyranoside (MDM) crystals. (A) The hydroxyl oxygen atoms (O5) show repeat distances of 9.263 Å on the (001) face along a axis. (B) The hydroxyl oxygen atoms (O3) have repeat distances of 9.978 Å on the (010) face along c axis. (C) The hydroxyl oxygen atoms (O2) have repeat distances of 9.369 Å on the face of (100) along b axis.
We made use of three different AF(G)Ps, DAFP1, AFGP1–5, and AFGP8 with descending antifreeze activity, as additives to control MDM crystal growth. We demonstrate for the first time that the AF(G)Ps can inhibit the crystal growth and the recrystallization of MDM and the effectiveness is related to their antifreeze activity. It is also the first report of AF(G)Ps controlling the growth and inhibiting recrystallization of non-ice carbohydrate crystals. Significantly, the effective amounts of AF(G)Ps for MDM recrystallization and crystal growth control can be very small and the additive/MDM molar ratio was shown to be as low as 10–7. The success of this new methodology will have a great impact in further expansion of the molecular recognition repertoire of AF(G)Ps, and the results thus will greatly expand the potential applications of AF(G)Ps in industry.
Experimental Section
Materials
The disaccharide, 2-acetamido-2-deoxy-3-O-(β-d-galactopyranosyl)-d-galactose, also called β-galactosyl (1 → 3)-α-N-acetylgalactosamine, was purchased from Santa Cruz Biotechnology (San Diego, CA). Lens culinaris lectin or agglutinin (LCA) was purchased from Aniara (West Chester, OH 45069). All other chemicals were purchased from Sigma-Aldrich (St. Louis, MO) at ACS grade or better and used without additional purification, unless otherwise indicated. All of the aqueous solutions were prepared using Milli-Q water produced from a Synergy water system (Millipore) with a minimum resistivity of 18 MΩ·cm. All the sample solutions were filtered through 0.1 μm filters before use unless otherwise indicated. National Scientific glass sample vials, 8 mL, were used for crystallization. All glassware and stir bars were cleaned in a KOH/2-propanol bath and rinsed with distilled water. After soaking in 1 M HCl for 24 h and rinsing with distilled water, the glassware and stir bars were further cleaned using RBS35 (Pierce), a surface-active detergent. After rinsing with distilled water and then with deionized water, the glassware and stir bars were air-dried at room temperature.
Synthesis of Methyl α-d-Mannopyranoside (MDM)
DMSO (10 mL) and methanol (500 mL) were added to a reaction vessel containing 50 g of d-mannose. The mixture was refluxed overnight and then stirred at room temperature for 1 day. The solids were filtered off, dissolved in hot ethanol, and then allowed to crystallize as pure MDM.
Solution NMR Spectroscopy
1H NMR spectra were recorded at 298 K on a 500 MHz Bruker NMR spectrometer. Chemical shifts (δ) are reported in parts per million (ppm) relative to residual solvent [D2O (TSP), δ 4.81, D2O (DSS), 4.40]. Multiplicities are given as s (singlet), d (doublet), m (multiplet). Proton-decoupled 13C NMR spectra were recorded at 125.8 MHz (13C). The 13C chemical shifts are reported relative to CDCl3 (δ 77.2 ppm). The 1H and 13C NMR spectra of the pure MDM are shown in Figures S1 and S2 (Supporting Information).
Liquid Chromatography Electrospray Ionization-Mass Spectrometry (LC-ESI-MS)
LC-ESI-MS data were collected on a Waters LCT Premier XE time-of-flight instrument controlled by MassLynx 4.1 software. MDM samples were dissolved in methanol and infused using direct loop injection from a Waters Acquity UPLC into the multimode ionization source. The solvent was 50/50 (v/v) MeOH/MeCN (LC-MS grade, VWR Scientific). The lock mass standard for accurate mass determination was leucine enkephalin (Sigma L9133). LC-MS: the calculated mass [(C7H14O6) + Na]+ was 217.07 and observed at 217.07 m/z with an isotope pattern consistent with C, H, O incorporation (Figure S3, Supporting Information).
Antifreeze Biomolecule Preparation
AFGP1–5 and AFGP8 were isolated from 5% trichloroacetic acid (TCA) supernatants of the blood serum of the Antarctic toothfish Dissostichus mawsoni. The TCA soluble AFGPs were further purified using HR-100 column chromatography (2.5 × 130 cm) with a 25 mM ammonium bicarbonate buffer. The fractions were lyophilized to remove the volatile buffer. The separation of the AFGP1–5 group from the smaller AFGP8 was verified by gel electrophoresis. MALDI-TOF mass spectrometry gave a value of 2670 Da for AFGP8. The larger isoforms of the AFGP1–5 fraction are not resolved with MALDI-TOF MS but their molecular weights are known from previous studies involving ultracentrifugation. The AFGPs were weighed on an Ohaus Voyager Pro analytical precision balance (Parsippany, NJ) and were dissolved in water.
DAFP-1 was expressed and purified as described previously.21 The purified DAFP-1 was characterized using SDS-PAGE gel electrophoresis, MALDI-TOF mass spectrometer, circular dichroism (CD) spectrometry, and differential scanning calorimetry (DSC), respectively, as previously described22 and the identity of DAFP1 was confirmed. The concentration of stock DAFP-1 solution was determined using a Cary 100 Bio UV–vis spectroscopy (Varian) and the extinction coefficient of 5.47 × 103 M–1 cm–1 at 280 nm was used.23
The denatured DAFP-1 was made by reducing all its disulfide bonds by a method previously reported.24 Briefly, DAFP1 (∼1 mM) was incubated in 0.10 M sodium citrate, pH 3.0, and 15.0 mM tris(2-carboxyethyl)phosphine hydrochloride (TCEP) at 60 °C for 30 min. The denatured DAFP1 was purified using ÄKTA Purifier 10 (GE Healthcare) with a Sephacryl S-100 gel filtration column (GE Healthcare).
Thermal Hysteresis Measurements
Freezing and melting points were determined in aqueous AF(G)P samples using a Clifton nanolitre osmometer (Clifton Technical Physics) following the protocol of Cziko et al. 2006.25 The instrument was calibrated with distilled water (0 mOsm) and a 1000 mOsm NaCl standard (Opti-mole, Wescor Inc.). Samples were suspended in heavy immersion oil in each of the 6 wells in the Clifton Nanoliter sample holder. They were cooled until frozen and then slowly warmed until a single ice crystal (approximately 10 μm) slowly melted while observed at 200×. This temperature was taken as the melting point or equilibrium freezing point. Following determination of the melting point, a 10 μm single ice crystal was slowly cooled to 0.05 °C below the melting point held for 1 min and then cooled to approximately 0.2 °C below the melting point and held for 5 min. They were then cooled at 0.074 °C per minute until sudden rapid growth was observed, and this value was taken as the freezing point. Melting and freezing point determinations for each individual were repeated twice per sample well in five separate wells. The thermal hysteresis, the difference between the melting point and the freezing point, represents the antifreeze activity.
Crystallization of MDM from the Aqueous Solution
Methyl α-d-mannopyranoside (MDM) was previously crystallized only from ethanol solutions.26 Here we found that MDM can be crystallized directly from its aqueous solution. Thus, the AF(G)P and control compounds were added directly to the aqueous MDM. Briefly, on day 1, 20 μL of water, AF(G)Ps, 2-acetamido-2-deoxy-3-O-(β-d-galactopyranosyl)-d-galactose (Gal1-β-3GalNAc), and Lens culinaris lectin or agglutinin (LCA) solutions of different concentrations were added separately into each sample vial containing 2 mL of 515 mM MDM. The final MDM concentration was 510 mM in each vial and the resulting additive/MDM molar ratios were 20 × 10–5 (denatured DAFP1/MDM), 3 × 10–7 (DAFP1/MDM), 2.0 × 10–4 (AFGP1–5/MDM), 2.2 × 10–4 (AFGP8/MDM), 8.0 × 10–4 [Gal1-β-3GalNAc/MDM], and 5.4 × 10–4 (LCA/MDM), respectively. The vials (8 mL, National Scientific) were gently swirled after the additions and remained uncovered and allowed to slowly evaporate at room temperature. At least three weights were recorded everyday (i.e., at least every 8 h) until all the liquid in the vials evaporated. The experiments were repeated five times. Sample results are listed in Table S1 (Supporting Information). The average sizes of the final MDM crystals in each vial were determined using weight analysis and the ratios of the average sizes of the final crystals of MDM in the absence and presence of the AF(G)Ps are listed in Table 2. Optical micrographs of the crystals that formed upon evaporation of the liquid were taken with a DS-Fi2 color camera attached to a Nikon SMZ-1000 microscope.
Table 2. Estimated Difference of the Critical Free Energy Change of the MDM Nucleation in the Absence and Presence of the AF(G)Ps (ΔΔG).
| AFGP8 | AFGP1–5 | DAFP1 | |
|---|---|---|---|
| S/S′ ( × 103)a | 0.2 ± 0.0 | 2.2 ± 0.5 | 34.3 ± 0.5 |
| ΔΔG (kJ/mol)b | –13.2 ± 0.0 | –19.0 ± 0.2 | –25.9 ± 0.0 |
S/S′ is the ratio of the average sizes of the final crystals of MDM in the absence and presence of the AF(G)Ps.
ΔΔG was estimated using eq 4, where R = 8.314 J·mol–1·K–1 and T = 298 K.
Effect of Additives on Nascent MDM Crystals
The crystallization of MDM in the absence of additives was reproducible with respect to the sizes and shapes of the crystals. Thus, the same criterion was used in the habit study in the presence of each of the AF(G)Ps and controls. When the seed MDM crystals were first observed under the microscope, DAFP-1, AFGP1–5 and AFGP8 were added to the vials to final concentrations of 1.53, 102, and 112.2 μM, respectively. After incubation with the additives the induced habit changes by AF(G)Ps were able to be terminated by removing the mother liquor from the vials washing the crystals with ice-cold water three times and transferring them into new vials containing the same amount of fresh saturated MDM. Upon completion of crystallization, the crystal habit was the same as those obtained in the absence of the AF(G)P additives and from MDM solutions in the presence of the control compound. Optical micrographs of the crystals were taken as described above.
Crystallization Kinetics
The crystallization kinetics were estimated based on the rate of crystal mass increase in each sample vial. All liquid in a vial was collected and transferred to a closed cap vial and the weight of the vial or the weight of the vial with crystals was determined with an Oham Discovery semi micro analytical balance. The liquid in the capped vial was then quickly transferred back to the vial containing the crystals and the crystallization continued until the next weight measure. Three vials were used for each additive and the controls. The average weight values are reported.
Single Crystal X-ray Diffraction
Crystals without any additives were sent to X-ray crystallography laboratory at UCSD for analysis. A colorless crystal was mounted on a Cryoloop with Paratone-N oil and data were collected at 125 K with a Bruker APEX II CCD using Cu K alpha radiation. Data corrected for absorption with SADABS and structure solved by direct methods. All non-hydrogen atoms were refined anisotropically by full matrix least-squares on F2. Hydrogen atoms of hydroxyl groups were found from Fourier difference maps and were refined with O–H distance of 0.86 (0.01) Å and 1.20 Ueq of parent O atoms. All other hydrogen atoms were placed in calculated positions with appropriate riding parameters. Additional information on data collection parameters are given in Table 1. The crystallographic data of MDM was deposited in the Cambridge Database (CCDC) and the CCDC deposit number is 985875.
Table 1. Crystallographic Data for Methyl α-d-Mannopyranoside (MDM).
| parameter | MDM |
|---|---|
| formula | C7H14O6 |
| formula weight | 194.18 |
| temperature (K) | 125(2) |
| crystal system | orthorhombic |
| space group | P212121 |
| a/Å | 9.2633(2) |
| b/Å | 9.3690(2) |
| c/Å | 9.9779(2) |
| α = β = γ | 90° |
| cell volume/Å3 | 865.96(3) |
| calc density/g cm–3 | 1.489 |
| Z | 4 |
| data/restraints/parameters | 1543/2/131 |
| final R indices for I > 2σ(I) | R1 = 0.0232 |
| wR2 = 0.0685 |
Twin Defect Determination
The ratios of twin defects of the crystal samples were estimated by partitioned extinction under a polarizing microscope (Nikon SMZ-1000 microscope with a DS-Fi2 color camera). At least 5 pieces were cut from one sample and the diffraction patterns were determined at the Beckman Institute of California Institute of Technology using a Bruker APEXII four circle diffractometer with a SMART 1000 CCD detector using Mo radiation from a sealed-tube X-ray generator both equipped with an Oxford Cryosystems crystal cooling system. The twin defects were confirmed by twin-lattice quasi-symmetry (TLQS).
Solid-State NMR Spectroscopy
Approximately 120 mg of solids were gently ground using mortar and pestle and packed in a 4 mm wide ZrO2 rotor with a Kel-F cap. 13C cross polarized magic angle spinning (CP MAS) solid-state NMR spectra were recorded at 298 K at 75.47 MHz (13C) on a 300 MHz Bruker spectrometer using a 4 mm broadband MAS probe with proton broadband decoupler. Spinning frequency of 10 kHz, CP contact time of 1.5 ms, and a 60 s delay were utilized.
MALDI-TOF Mass Spectrometry
MDM crystals were quickly washed with 1:1 (v:v) cold water–ethanol at ∼0 °C in order to remove any potential nonspecific additives bound to solids. Each resulting sample was lyophilized. MALDI-TOF mass spectra were obtained on a PerSeptive Biosystems/Voyager-DE MALDI-TOF mass spectrometer at the Stanford PAN facility. Calibrations of the mass spectrometer against external mass standards were carried out before sample analyses. The matrix, sinapinic acid, was prepared as a saturated solution in 1:1 (v:v) water:acetonitrile with 0.1% trifluoroacetic acid. A Ziptip C18 resin (Millipore) was used to desalt the mass sample. The sample was dissolved in doubly distilled water–TFA at pH 3.00. 0.5 μL of the sample solution was mixed with an equal volume of the matrix solution in a sample plate and air-dried before analysis.
Fourier Transform Infrared (FT-IR) Spectroscopy
FT-IR attenuated total reflectance (ATR) spectra were collected on a Nicolet iS5 FT-IR spectrometer (Thermo Fischer Scientific Inc., Waltham, MA) equipped with an iD5 ATR accessory. The IR frequencies were recorded in cm–1, and the spectra were measured in a spectral range from 4000 to 200 cm–1.
Results and Discussion
Structural Compatibility
The crystals of methyl α-d-mannopyranoside (MDM) were obtained by slow evaporation of their aqueous solutions at room temperature in the air. The resulting MDM crystals appear as a few colorless blocks with overlaps and about 75% having twin defects (Table S1, Supporting Information). The structure of the MDM crystals were determined using single-crystal X-ray diffraction and the crystallographic parameters are listed in Table 1. The crystal structures of MDM obtained in this study are in very good agreement with those published previously27 but with an improved resolution. The growing faces of MDM crystal include (001), (010), and (100) and all these faces have hydroxyl oxygen groups in a periodic manner: on the (010) face, the hydroxyl oxygen atoms (O3) have the repeat distance of 9.263 Å (or 9.978 Å) along a (or c) axis; on the (001) face, the hydroxyl oxygen atoms (O5) have the repeat distances of 9.263 Å (or 9.369 Å) along a (or b) axis; while the hydroxyl oxygen atoms (O2) are on the face of (100) with the repeat distance of 9.369 Å (or 9.978 Å) along b (or c) axis (Figure 2).
The hydroxyl groups in the O-linked disaccharide on each threonine residue in the AFGP repeat unit of Ala-Ala-Thr have been modeled to bind to the prism planes of hexagonal ice presumably through hydrogen bonding.28 As reported by NMR studies, the length of the 3-mer repeat in the polyproline II backbone of AFGPs is 9.31 Å,29 which is about twice the repeat spacing in ice along the a-axis. The periodicity of the AFGP backbone, 9.31 Å, matches the repeat distance between the hydroxyl oxygen atoms, 9.263 or 9.369 Å, on the growing faces of MDM crystal surprisingly well (Figures 1A and 2A,C). Moreover, the average distance between the hydroxyl groups in the conserved threonine residues in adjacent repeat loops of DAFP-1 is 4.74 Å deduced from homology modeling,22 twice which is 9.48 Å and compatible with all the three possible repeat hydroxyl oxygen spacings, 9.263, 9.369, or 9.978 Å, on the growing faces of MDM crystals (Figures 1B and 2). This remarkable similarity in repeat spacings between AF(G)Ps and MDM crystal faces led us to examine the effect of AF(G)Ps on MDM crystal growth.
Effect of AF(G)Ps on MDM Recrystallization Inhibition and Crystal Growth
On the basis of the apparent structural compatibility between MDM and the AF(G)Ps, we selected two Antarctic fish AFGPs AFGP8 and AFGP1–5 with 4 and approximately 15–65 glycotripeptide repeats, respectively, and a beetle AFP from D. canadensis, DAFP1, as potential additives to control the crystal growth of MDM. The crystallization of MDM was carried out using a wide range of additive concentrations, and the AF(G)Ps prevented the growth of large MDM crystals resulting in smaller MDM crystals with improved quality. Although the direct addition of these AF(G)Ps at the various concentrations had little effect on the crystallization induction time of MDM, both DAFP1 and AFGP1–5 remarkably delayed the time for completion of crystallization (refer to Figure 4). The extent of the delay depended on the additive/MDM molar ratio; the greater the molar ratio, the later the completion of crystallization (Table S1, Supporting Information). A certain additive/MDM molar ratio, referred as to the critical ratio, is needed to efficiently inhibit small MDM crystals from growing into large ones (or inhibit MDM recrystallization). Here, the critical ratios of DAFP1/MDM, AFGP1–5/MDM, and AFGP8/MDM were estimated to be 3.0 × 10–7, 2.0 × 10–6, and 2.2 × 10–4, respectively. At ratios higher than the critical ratios, DAFP1 ranks as the most effective followed by, AFGP1–5, and then AFGP8 in inhibiting the growth of large MDM crystals. As indicated previously, in the absence of antifreeze additives, MDM crystals appear as a large overlapping block with twin defects (Figure 3A). In contrast, in the presence of AFGP8 and AFGP1–5, approximately 30 and 60% of the final MDM crystals appeared as smaller triangular shaped blocks, respectively (Figure 3B,C). Remarkably, with DAFP1, all the final MDM crystals appeared as even smaller grains (Figure 3D). No apparent morphology changes in the MDM crystals obtained in the presence of the AF(G)Ps were observed, suggesting that the adsorption of the additives and the kinetics of the adsorption both affect MDM crystal growth.6 Moreover, the twin defect percentage in the MDM crystals dropped from approximately 75% (in the absence of additives) to 40, 15, and less than 5%, in the presence of AFGP8, AFGP1–5, and DAFP1, respectively. The quality of the MDM crystals obtained with the AF(G)Ps are greatly improved and much more suitable for single X-ray crystallography analysis in comparison to those without additives.
Figure 4.
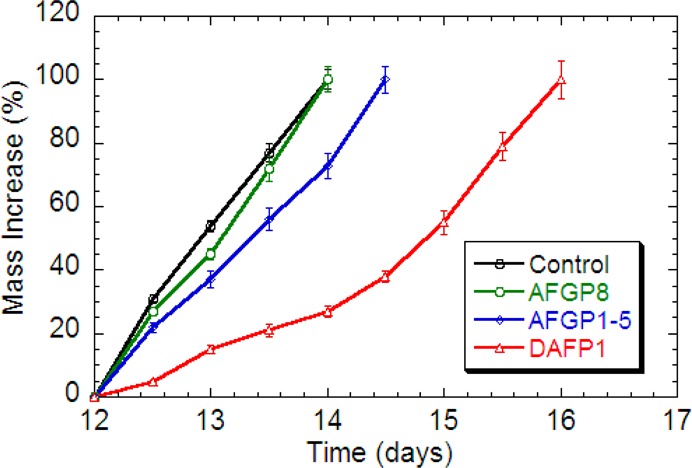
Crystallization kinetics of MDM in the presence of AFGP8 (green), AFGP1–5 (blue), and DAFP1 (red), respectively. The data of the control (black) are the average data of MDM in the absence and presence of the three controls, Gal1-β-3GalNAc, LCA, and denatured DAFP1. The error bars represent the standard deviation from at least three measurements.
Figure 3.
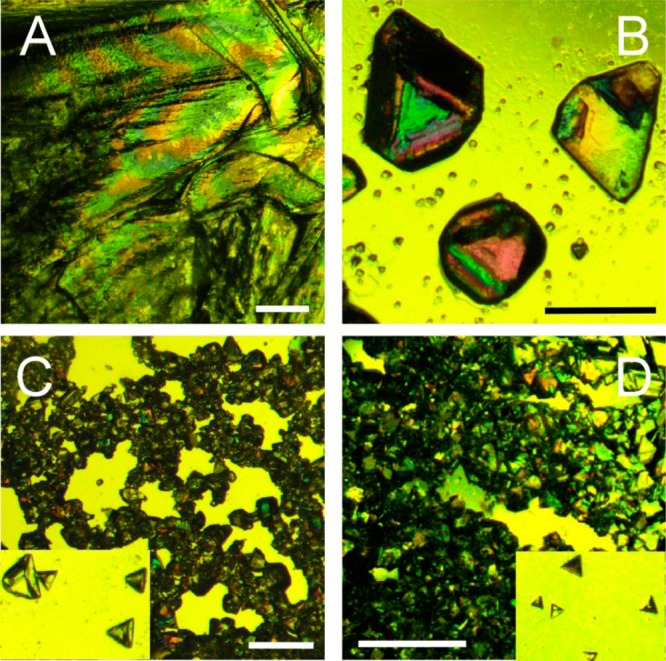
Optical micrographs of the MDM crystals: in the absence of additives (A), in the presence of AFGP8 (B), AFGP1–5 (C) and DAFP1 (D), respectively. The length of the scale bars is 1 mm. The zoom-in views (150%) of the isolated MDM crystals in (C) and (D) are shown in the corresponding insets.
In the absence of additives or in the presence of any of the controls, the crystallization of MDM started during day 12 and was complete by day 14 (Figure 4). With AFGP1–5 and DAFP1 (at the critical ratios of additive/MDM), the crystallization of MDM still started during day 12, but was prolonged, from 14 days to 14.5 and 16 days, respectively (Figure 4). Once the crystals of MDM appear, AFGP8 at its critical ratio can inhibit the further growth of MDM crystals until day 3, but has little effect on prolonging the crystallization process of MDM (Figure 4).
Generally, additives may affect crystal growth by modifying crystal habit and/or affecting nucleation.6,8,17 To understand how AF(G)Ps influence MDM crystal growth, AFGP8, AFGP1–5, DAFP1 were also added to the saturated MDM aqueous solutions in the presence of MDM seed crystals and the additive/MDM ratios were at the critical ratios. MDM crystals continue to grow in all the sample vials, while the final MDM crystals appeared to have better defined shapes with smaller sizes (Figure S4, Supporting Information). Moreover, the crystal quality for X-ray diffraction was also improved compared to those obtained in the absence of the AF(G)Ps (Table S1, Supporting Information). The overall effectiveness of AF(G)Ps as additives in inhibiting the recrystallization and crystal growth of MDM is well correlated with their antifreeze activities (Figure 5). AF(G)Ps are well-known for inhibiting ice recrystallization and modifying ice morphology (e.g., flat-round shaped polycrystalline ice can be modified to hexagonally shaped single crystal ice by AF[G]Ps).2,30,31 The effect of AF(G)Ps on MDM are notably similar to their effect on ice.
Figure 5.
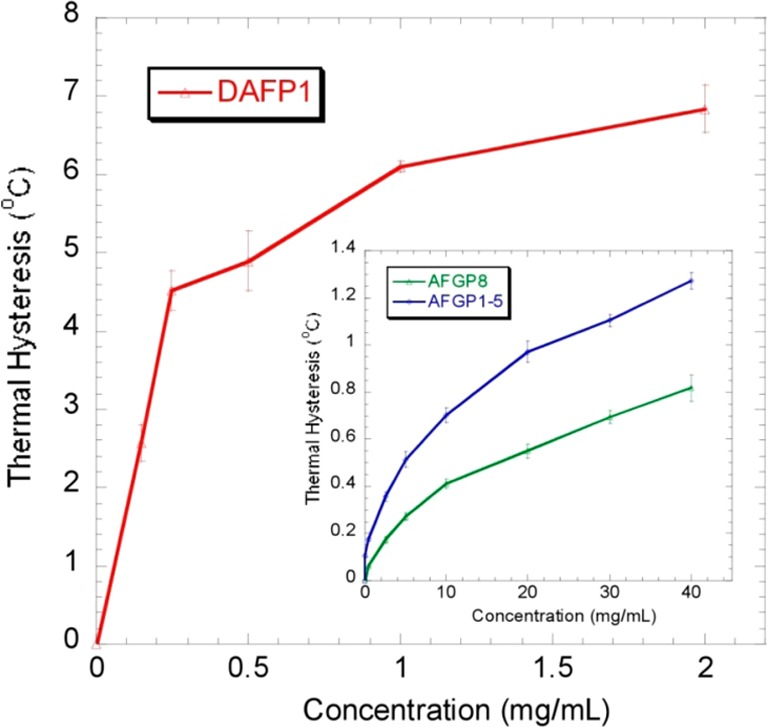
Thermal hysteresis (TH) (or antifreeze) activities of DAFP1, AFGP1–5, and AFGP8 measured using nanoliter. The error bars represent the standard deviation from at least three measurements.
To further demonstrate that the effects of AF(G)Ps on MDM crystal growth are related to the structural compatibility between MDM and the additive, we selected three control compounds and performed the crystallization of MDM in their presence. 2-acetamido-2-deoxy-O-(β-d-galactopyranosyl)-d-galactose (Gal1-β-3GalNAc) is the disaccharide unit attached to threonine residues in the AFGPs but by itself shows no antifreeze activity. Denatured DAFP1 with a disrupted tertiary structure also retains no antifreeze activity. A 46 kDa carbohydrate binding protein, Lens culinaris lectin (LCA), that binds specifically to α-d-mannose and α-d-glucose,32,33 was also used. As expected, none of these compounds inhibited the formation of large MDM crystals or affected the crystallization rate of MDM (Figure S5, Supporting Information). Though LCA is a lectin that binds to some carbohydrates, it cannot recognize MDM and therefore does not affect the crystallization of MDM. The results in the presence of Gal1-β-3GalNAc indicates that the molecular recognition is between the intact AFGPs and the MDM crystal, rather than between the isolated disaccharide and MDM suggesting that the structural compatibility involves the spacings of the disaccharides and their hydroxyl groups as determined by the repeat spacing of the threonines in the peptide backbone. Moreover, the results in the presence of denatured DAFP1 strongly suggest that the structural compatibility between DAFP1 and MDM crystal faces must exist.
Crystal habit is determined by the relative growth rates of specific crystal faces.6 In the absence of additives, the growth rates of (001), (010) and (100) faces of MDM crystals are similar, resulting in the final overlapping block shape of MDM crystals. The altered crystal habit and size of MDM crystals in the presence of AF(G)Ps indicate that AF(G)Ps may selectively adsorb onto one or more of these faces by hydrogen-bonding interactions between the hydroxyl groups in the O-linked disaccharides in AFGPs or in the conserved threonines in DAFP1 and the hydroxyl oxygen atoms in the growing faces of MDM crystal (Figures 1 and 2). The characteristics of useful additives for crystallization include those that have a high selectivity for the fast growing crystal face(s), form a lattice match and bind reversibly.34 Here, the AF(G)Ps are shown to be excellent additives for MDM crystallization that are effective at very low concentrations. In addition to this characteristic, AF(G)Ps have another feature that may account for their superior ability in controlling the crystallization of MDM, which is, they have distinct hydrophilic and hydrophobic faces positioned on opposite sides of the molecule. The hydrophilic face of AF(G)Ps may interact with the MDM lattice sites through hydrogen-bonding interactions, while the hydrophobic faces may be exposed to the bulk solution and play a role in rejecting incoming MDM molecules from joining the crystal lattice until the desorption of AF(G)Ps occurs.
The MDM crystals formed in the presence of the additives as well as with the controls were analyzed using single crystal X-ray diffraction, and no new forms were identified. The hydrogen bonding networks of the resulting MDM crystals were also examined using solid-state NMR spectroscopy and attenuated total reflectance infrared spectroscopy (ATR-IR). The 13C cross-polarization magic angle spinning (CP-MAS) NMR and ATR-IR spectra of the MDM crystals with altered habits obtained in the presence of AF(G)Ps were almost identical to those of the MDM crystals obtained in the absence of AF(G)Ps and with the control compounds. The representative 13C CP-MAS NMR and ATR-IR spectra are shown in Figures 6 and S6 (Supporting Information), respectively. These results further support the absence of any new crystal forms of MDM as well as the absence of amorphous precipitates and indicate that the obtained smaller triangular shaped MDM crystals are just habit modification by the AF(G)Ps.
Figure 6.
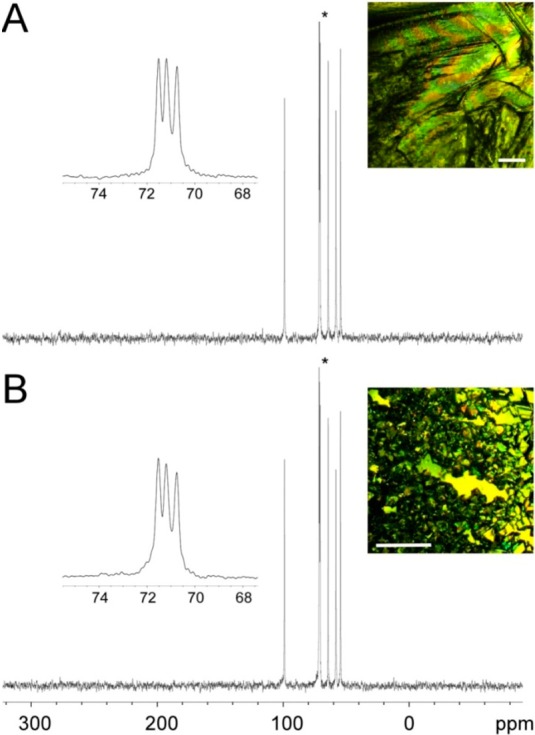
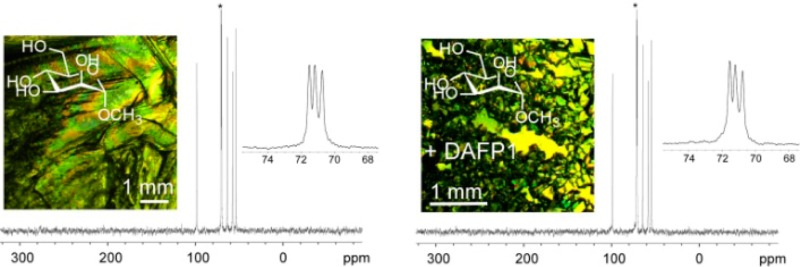
Representative CPMAS 13C NMR spectra of the MDM crystals in the absence and presence of AF(G)Ps. The CPMAS 13C NMR spectra of MDM crystals in the absence of AF(G)Ps (A) and in the presence of DAFP1 (B). The insets show the details of the peak with an asterisk and the habits of the MDM crystals are shown at the right upper corner of each panel (the length of the scale bars is 1 mm).
MALDI-TOF mass spectrometry was utilized to investigate the existence of possible interactions between the additives and MDM crystals. The mass results of AF(G)Ps alone indicated that the molecular weights (MWs) of AFGP8, AFGP1–5, and DAFP1 are 2670, 8968, and 9009 Da. Although the MW of AFGP1–5 gave only the 9009 Da isoform the larger isoforms are most likely also involved in the inhibition process even though they fail to show up in the MALDI spectra. Moreover, the molecular weights of AFGPs determined in this work are close to the reported values using sedimentation equilibrium centrifugation.1 The mass spectra of the resulting MDM crystal samples in the presence of these additives show the peaks of AFGP1–5, AFGP8, and DAFP1, respectively, suggesting adsorption of the AF(G)Ps to MDM crystals (Figure 7). In contrast, no peaks of denatured DAFP1, Gal1-β-3GalNAc, or LCA were observed in the mass spectra of the MDM crystal samples in the presence of these control compounds (data not shown).
Figure 7.
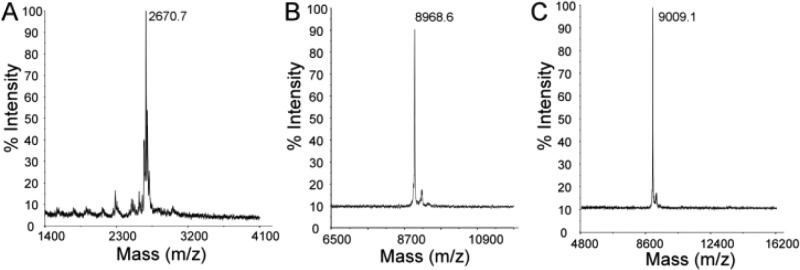
MALDI-TOF mass spectra of the achieved MDM crystals in the presence of AF(G)Ps. The peaks of AF(G)Ps were observed: (A) AFGP8 (m/z = 2670.7), (B) DAFP1 (m/z = 8968.6), and (C) AFGP1–5 (m/z).
It is suggested that the adsorption of AF(G)Ps onto the fast growing faces of MDM crystal delays the growth of MDM crystal or directly inhibits the MDM nuclei at the early stage, while increasing the supersaturation of the MDM solution resulting in the formation of more nuclei of MDM at a later time. As a result, smaller sized MDM crystals grow rather than large overlapping ones. Moreover, the results suggest that the effects of AF(G)Ps on MDM crystallization may be similar to their known effects on ice crystals, though significantly much smaller amounts of AF(G)Ps are needed to control the MDM crystal growth compared to ice.2−4,35,36 Notably, the efficacy of the AF(G)Ps on control of MDM crystallization has been shown to be related to their antifreeze activity, the first demonstration that the efficacy of the control of crystal growth correlates to their antifreeze activity.16−18
Size Changes of MDM by AF(G)Ps: Theoretical Aspects
Crystal sizes are generally determined by the rates of nucleation and crystal growth.6 Since the concentrations of the AF(G)Ps are extremely low in contrast to that of MDM, we can assume that the diffusion of MDM molecules from its bulk solution to nuclei and/or crystal faces remains a constant and the crystallization process is a homogeneous nucleation case. Thus, in the absence of AF(G)Ps, the final crystal sizes of MDM depend only on the rate of nucleation. The number of nuclei formed per unit time per unit volume, J, can be expressed as
| 1 |
where R is the gas constant, T is the experimental temperature, and A is the Arrhenius constant for the system, and ΔG is the critical free energy change for the nucleation of MDM.
The MDM crystallization induction time without any additives is t. The presence of the AF(G)Ps may inhibit the nucleation of MDM until the additives are fully consumed by the nuclei. Such process increases the degree of supersaturation at the time t + Δt, resulting in a different number of nuclei formed per unit time per unit volume, J′, and a different critical free energy change, ΔG′.
| 2 |
The total final masses of MDM crystals should be equal in the absence and presence of additives:
| 3 |
where V is the volume of the MDM solution at the induction time t, V′ is the volume of the MDM solution with the AF(G)Ps at the time t + Δt, and S and S′ are the average sizes of the final crystals of MDM in the absence and presence of additives. According to the experimental observations, we can assume Δt ≪ t and (t +Δt) V′ = t V. In the absence of additives, the crystal growth of MDM starts at t and at t + Δt, the newly formed nuclei may join into the early grown crystals and finally lead to the overlapped block shaped MDM crystals with high percent of twin defects.
Substituting eqs 1 and 2 into eq 3 and rearranging, we obtain
| 4 |
where ΔΔG = ΔG – ΔG′, the critical free energy change differences of the MDM nucleation in the absence and presence of the additives.
By substituting the ratio of the average sizes of the final MDM crystals in the absence and presence of each AF(G)P (S/S′) into eq 4, the ΔΔG can be calculated and the results are consistent with the effectiveness of the additives (Table 2). Moreover, these values are close to those of the ice nucleation in the presence of AF(G)Ps,35 suggesting similar interactions are involved in the molecular recognition of MDM by the AF(G)Ps.
Conclusion
Although the structures and crystal faces of ice and MDM are clearly very distinct: hexagonal ice Ih (the most common and abundant crystal form of ice) belongs to the space group of P63/mmc with a = b = 4.52 Å and c = 7.36 Å;37 MDM crystals belong to the space group of P212121 with a = 9.263 Å, b = 9.369 Å, and c = 9.978 Å, the periodicities in ice Ih and MDM have similarities. In MDM crystals, the periodicity in the direction of a (9.263 Å) is very close to that of b (9.369 Å) and both of these periodicities are approximately double the periodicity in the direction of a (4.52 × 2 = 9.04 Å) in ice Ih. Both ice Ih and MDM have exposed hydroxyl groups on one or more of their crystal faces whose periodicities are approximately n × 4.7 Å (n = 1, 2, 3, ...), matching the periodicities in the AF(G)Ps (Figure 1). The adsorption of AF(G)Ps to MDM crystals and their effect on its crystallization suggest a noteworthy structural compatibility between the AF(G)Ps and the fast growing faces of MDM crystal. Moreover, the theoretical analysis suggested that the molecular recognition of MDM by AF(G)Ps are similar to that of ice by AF(G)Ps. It is possible that the molecular recognition repertoire of AF(G)Ps may involve other crystal compounds, which could be shown by using this methodology we have described. Searches for a structural compatibility between AF(G)Ps and the fast growing crystal faces could result in the expansion of the potential applications of AF(G)Ps in a wide range of industries.
This finding also suggests a new possibility for addressing the details of the mechanisms of the AF(G)Ps adsorption inhibition of ice crystal growth. Physical methods that are available for the mechanistic study of AF(G)P-ice interactions, however, are very limited in ability to resolve these contradictions, largely due to the instability of ice crystals and the difficulties of handling ice crystals with specific crystal faces under well-controlled low-temperatures. MDM crystals are stable at room temperature and the study of AF(G)P-MDM interactions does not require specialized equipment. AF(G)Ps are known for their ability to inhibit ice growth and recrystallization.1 It is still a matter of debate whether AF(G)Ps bind reversibly or irreversibly to ice surfaces.38−42 The theoretical analysis on the size changes of MDM by the AF(G)Ps suggests that the molecular recognitions between AF(G)Ps and MDM and between AF(G)Ps and ice are similar. Thus, MDM might be used as a potential model to study the mechanism of AF(G)P action on ice. Comprising such structurally diverse classes of proteins and ligands, the detailed mechanism of AF(G)P-ligand action is, however, very complex and needs further study.
The discovery of the molecular recognition between AF(G)Ps and MDM crystals, based on their structural compatibility, may be more than a coincidence. In fact, the evolutionary origins of some AFPs are lectins. For examples, type II fish AFPs are homologues of the carbohydrate recognition domain of Ca2+ dependent C-type lectins43 and the origin of type III fish AFP is sialic acid synthase.44 Moreover, winter rye AFPs are homologues of Chitinase and glucanase that bind to polysaccharides,45 and carrot AFP is a homologue of polygalacturonase inhibitor.2 AFGPs have evolved from trypsinogen like protease.46 Though the origin of DAFP1 is currently unknown, DAFP1 has been demonstrated to recognize MDM in this work and certain nucleosides.17 Furthermore, known lectins can usually bind to more than one ligand.47 Thus, it should not come as a surprise if AF(G)Ps can recognize other carbohydrates and/or the origin of DAFP1 might be related to a carbohydrate binding protein.
Efficient control of the crystallization of carbohydrates is crucial in sugar and related food and pharmaceutical industries. Industrial additives for sugar crystallization are usually compounds with similar structures to the target sugar crystals and a concentration of 2–20% (w/w) is required for effects.48,49 The required molar ratio of AF(G)Ps for effects on MDM crystallization can be as low as 10–7. The high efficacy in controlling the size and shape of carbohydrate crystals and the high stability of AF(G)Ps suggests possible applications of the AF(G)Ps as useful additives and/or scaffolds for designing novel additives in sugar and related food and pharmaceutical industries.
Acknowledgments
We thank Mr. Larry Henling at California Institute of Technology for MDM twin defect determination and Dr. Greg Khitrov at the University of California, Los Angeles for LC-MS that was supported by NCRR (S10-RR025631). X.W. acknowledges support by NIH (GM086249).
Supporting Information Available
Figures S1–S6, Table S1, and crystal data (CIF). This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Raymond J. A.; DeVries A. L. Proc. Natl. Acad. Sci. U. S. A. 1977, 74, 2589–2593. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Knight C. A.; DeVries A. L.; Oolman L. D. Nature 1984, 308, 295–296. [DOI] [PubMed] [Google Scholar]
- a Worrall D.; Elias L.; Ashford D.; Smallwood M.; Sidebottom C.; Lillford P.; Telford J.; Holt C.; Bowles D. Science 1998, 282, 115–117. [DOI] [PubMed] [Google Scholar]; b John U. P.; Polotnianka R. M.; Sivakumaran K. A.; Chew O.; Mackin L.; Kuiper M. J.; Talbot J. P.; Nugent G. D.; Mautord J.; Schrauf G. E.; Spangenberg G. C. Plant Cell Environ. 2009, 32, 336–348. [DOI] [PubMed] [Google Scholar]
- Note: Here, recrystallization is defined as the nucleation and growth of stress-free grains, appearing as the formation of larger sized crystals.
- Fletcher G. L.; Hew C. L.; Davies P. L. Annu. Rev. Physiol. 2001, 63, 359–390. [DOI] [PubMed] [Google Scholar]
- Jia Z. C.; Davies P. L. Trends Biochem. Sci. 2002, 27, 101–106. [DOI] [PubMed] [Google Scholar]
- Mullin J. W.Crystallization, 4th ed.; Butterworth-Heinemann: Oxford, U.K., 2001. [Google Scholar]
- Hartel R. W. Annu. Rev. Food Sci. Technol. 2013, 4, 277–292. [DOI] [PubMed] [Google Scholar]
- Sangwal K.Additives and Crystallization Processes: From Fundamentals to Applications; John Wiley & Sons, Ltd.: West Sussex, England, 2007. [Google Scholar]
- Otte A.; Zhang Y.; Carvajal M. T.; Pinal R. CrystEngComm 2012, 14, 2560–2570. [Google Scholar]
- Dowling R.; Davey R. J.; Curtis R. A.; Han G.; Poornachary S. K.; Chow P. S.; Tan R. B. H. Chem. Commun. 2010, 46, 5924–5926. [DOI] [PubMed] [Google Scholar]
- Graham K. R.; Stalder R.; Wieruszewski P. M.; Patel D. G.; Salazar D. H.; Reynolds J. R. ACS Appl. Mater. Interfaces 2012, 5, 63–71. [DOI] [PubMed] [Google Scholar]
- a Belcher A. M.; Wu X. H.; Christensen R. J.; Hansma P. K.; Stucky G. D.; Morse D. E. Nature 1996, 381, 56–58. [Google Scholar]
- Wheeler A. P.; George J. W.; Evans C. A. Science 1981, 212, 1397–1398. [DOI] [PubMed] [Google Scholar]
- Mann S.Biomineralization: Principles and Concepts in Bioinorganic Materials Chemistry; Oxford University Press: Oxford, U.K., 2001. [Google Scholar]
- a Falini G.; Albeck S.; Weiner S.; Addadi L. Science 1996, 271, 67–69. [Google Scholar]; b DeOliveira D. B.; Laursen R. A. J. Am. Chem. Soc. 1997, 119, 10627–10631. [Google Scholar]; c Volkmer D.; Fricke M.; Huber T.; Sewald N. Chem. Commun. 2004, 1872–1873. [DOI] [PubMed] [Google Scholar]
- Zeng H.; Wilson L. D.; Walker V. K.; Ripmeester J. A. J. Am. Chem. Soc. 2006, 128, 2844–2850. [DOI] [PubMed] [Google Scholar]
- Wang S.; Wen X.; Nikolovski P.; Juwita V.; Fnu Arifin J. Chem. Commun. 2012, 11555–11557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S.; Wen X.; Golen J. A.; Arifin J. F.; Rheingold A. L. Chem.—Eur. J. 2013, 19, 16104–16112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derewenda Z.; Yariv J.; Helliwell J. R.; Kalb A. J.; Dodson E. J.; Papiz M. Z.; Wan T.; Campbell J. EMBO J. 1989, 8, 2189–2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duhamel R. C.; Whitehead J. S. Methods Enzymol. 1990, 184, 201–207. [DOI] [PubMed] [Google Scholar]
- Amornwittawat N.; Wang S.; Banatlao J.; Chung M.; Velasco E.; Duman J. G.; Wen X. Biochim. Biophys. Acta 2009, 1794, 341–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S.; Amornwittawat N.; Juwita V.; Kao Y.; Duman J. G.; Pascal T. A.; Goddard W. A.; Wen X. Biochemistry 2009, 48, 9696–9703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace C. N.; Vajdos F.; Fee L.; Grimsley G.; Gray T. Protein Sci. 1995, 4, 2411–2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N.; Chibber B. A. K.; Castellino F. J.; Duman J. G. Biochemistry 1998, 37, 6343–6350. [DOI] [PubMed] [Google Scholar]
- Cziko P. A.; Evans C. W.; Cheng C.-H. C.; DeVries A. L. J. Exp. Biol. 2006, 209, 407–420. [DOI] [PubMed] [Google Scholar]
- Hunt D. J.; Subramanian E. Acta Crystallogr. 1969, B25, 2144–2152. [DOI] [PubMed] [Google Scholar]
- Gatehouse B. M.; Poppleton B. J. Acta Crystallogr. 1970, B26, 1761–1765. [Google Scholar]
- Knight C. A.; Driggers E.; DeVries A. L. Biophys. J. 1993, 64, 252–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao B. N. N.; Bush C. A. Biopolymers 1987, 26, 1227–1244. [DOI] [PubMed] [Google Scholar]
- Knight C. A.; DeVries A. L.; Oolman L. D. Nature 1984, 308, 295–296. [DOI] [PubMed] [Google Scholar]
- Griffith M.; Yaish M. W. Trends Plants Sci. 2004, 9, 399–405. [DOI] [PubMed] [Google Scholar]
- Liener I. E.; Sharon N.. The Lectins: Properties, Functions and Applications in Biology and Medicine; Academic Press: Orlando, FL, 1986. [Google Scholar]
- Foriers A.; Lebrun E.; Van Rapenbusch R.; de Neve R.; Strosberg A. D. J. Biol. Chem. 1981, 256, 5550–5560. [PubMed] [Google Scholar]
- Salvalaglio M.; Vetter T.; Giberti F.; Mazzotti M.; Parrinello M. J. Am. Chem. Soc. 2012, 134, 17221–17233. [DOI] [PubMed] [Google Scholar]
- Wang S.; Amornwittawat N.; Wen X. J. Chem. Thermodyn. 2012, 53, 125–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Raymond J. A.; Wilson P.; DeVries A. L. Proc. Natl. Acad. Sci. U. S. A. 1989, 86, 881–885. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Davies P. L.; Sykes B. D. Curr. Opin. Struct. Biol. 1997, 7, 828–834. [DOI] [PubMed] [Google Scholar]
- Hobbs P. V.Ice Physics; Oxford University Press: Oxford, U.K., 2010. [Google Scholar]
- Meister K.; Ebbinghaus D.; Xu Y.; Duman J. G.; DeVries A.; Gruebele M.; Leitner D. M.; Havenith M. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 1617–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Garnham C. P.; Campbell R. L.; Davies P. L. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 7363–7367. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Sun T.; Lin F.-H.; Campbell R. L.; Allingham J. S.; Davies P. L. Science 2014, 343, 795–798. [DOI] [PubMed] [Google Scholar]
- Pertaya N.; Marshall C. B.; DiPrinzio C. L.; Wilen L.; Thomson E. S.; Wettlaufer J. S.; Davies P. L.; Braslavsky I. Biophys. J. 2007, 92, 3663–3673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zepeda S.; Yokoyama E.; Uda Y.; Katagiri C.; Furukawa Y. Cryst. Growth Des. 2008, 8, 3666–3672. [Google Scholar]
- Knight C. A.; DeVries A. L. Phys. Chem. Chem. Phys. 2009, 11, 5749–5761. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Li Z.; Lin Q.; Kosinski J.; Seetharaman J.; Bujnicki J. M.; Sivaraman J.; Hew C. L. PLoS One 2007, 2, e548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baardsnes J.; Davies P. L. Trends Biochem. Sci. 2001, 26, 468–469. [DOI] [PubMed] [Google Scholar]
- Hon W. C.; Griffith M.; Mlynarz A.; Kwok Y. C.; Yang D. S. C. Plant Physiol. 1995, 109, 879–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng C.-H. C; Chen L. Nature 1999, 401, 443–444. [DOI] [PubMed] [Google Scholar]
- Komath S. S.; Kavitha M.; Swamy M. J. Org. Biomol. Chem. 2006, 4, 973–988. [DOI] [PubMed] [Google Scholar]
- Campana Cue C.; Ruiz Salvador A. R.; Aguilera Morales S.; Falcon Rodriguez F. L.; Perez Gonzalez P. J. Cryst. Growth 2001, 231, 280–289. [Google Scholar]
- Garnier S.; Petit S.; Coquerel G. J. Cryst. Growth 2002, 234, 207–219. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.