

Abstract
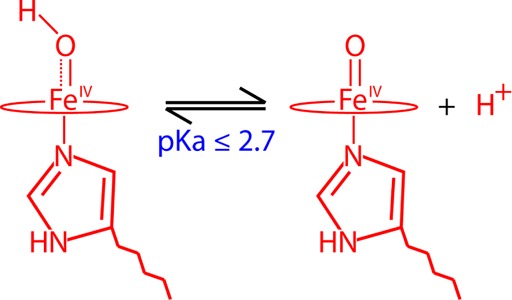
To provide insight into the iron(IV)hydroxide pKa of histidine ligated heme proteins, we have probed the active site of myoglobin compound II over the pH range of 3.9–9.5, using EXAFS, Mössbauer, and resonance Raman spectroscopies. We find no indication of ferryl protonation over this pH range, allowing us to set an upper limit of 2.7 on the iron(IV)hydroxide pKa in myoglobin. Together with the recent determination of an iron(IV)hydroxide pKa ∼ 12 in the thiolate-ligated heme enzyme cytochrome P450, this result provides insight into Nature’s ability to tune catalytic function through its choice of axial ligand.
Introduction
A fundamental understanding of protein structure–function relationships is key to harnessing the catalytic power of enzymes for chemical applications. For some time now, we have been interested in the factors that govern C–H bond activation in heme proteins.1,2 Cytochrome P450s are a class of thiolate ligated heme proteins that utilize dioxygen and the formal equivalents of molecular hydrogen (2H+ + 2e-) to functionalize a broad range of biologically active molecules.3 These enzymes have been shown to function through a high-valent ferryl (or iron(IV)oxo) radical species called compound I.4 Compound I abstracts hydrogen from substrate to form an iron(IV)hydroxide species (compound II) and a substrate radical, which rapidly recombine to give hydroxylated product.4
An enigmatic aspect of P450 catalysis has been the enzyme’s use of an electron-rich thiolate ligated heme to catalyze the oxidation of inert hydrocarbons. We have shown that strong donation from the axial thiolate results in basic ferryl species.1 UV–vis and 57Fe Mössbauer experiments place P450s compound II (or iron(IV)hydroxide) pKa at ∼12.5 This elevated pKa may provide an answer to the question posed by Groves more than a decade ago: “How can a protein create an iron intermediate reactive enough to hydroxylate even as inert a substrate as cyclohexane and not oxidize the relatively fragile protein superstructure?”6
We have argued that the elevated pKa afforded by thiolate ligation shifts the relative free energy for the productive and nonproductive pathways to a regime where the rate constant for productive C–H bond activation dominates that for nonproductive oxidations of the protein superstructure.5 One can obtain insight into the role of thiolate ligation in cytochrome P450 by considering a system in which the compound II pKa can be modulated/tuned, while keeping the driving force for productive substrate oxidation constant. Given this constraint, one can use Marcus theory to derive eq 1, which allows for a comparison of rate constants for nonproductive oxidations by compound I, knp, as a function of compound II pKas.5
| 1 |
Eq 1 shows that the rate constant for nonproductive oxidations varies dramatically with changes in the iron(IV)hydroxide pKa. We have argued that, all things being equal, thiolate ligation provides (relative to histidine ligation) for a > 10,000-fold reduction in the rate constant for nonproductive oxidations of the protein framework, explaining Nature’s use of thiolate ligation in P450.5 Through suppression of the nonproductive pathway—minimizing peroxidase like chemistry—thiolate ligation biases P450 toward C–H bond activation.
The use of eq 1 outlined above requires knowledge of the iron(IV)hydroxide pKa in a histidine ligated heme protein. To obtain the factor of 10,000, we have made use of resonance Raman data, reported by Terner and co-workers for horseradish peroxidase compound II (HRP-II). They observed an iron-oxo stretch in HRP-II at pD 4, suggesting an iron(IV)hydroxide pKa ≤ 3.6.7−9
We note, however, that the existence of iron(IV)hydroxide species in peroxidases and globins has been a subject of debate. A number of crystallographic investigations of compound II species have reported long iron–oxygen bonds, which have been interpreted in terms of a protonated ferryl (i.e., an iron(IV)hydroxide) moiety.10−14 These crystallographic assignments have generally been at odds with spectroscopic measurements that indicate the presence of authentic iron(IV)oxo species.7,15−22 This discrepancy has been explained in terms of photoreduction in the synchrotron beam. It has been argued that the long iron–oxygen bond distances obtained from crystallography may more accurately represent ferric and ferrous forms of the proteins.15,23 Recent crystallographic experiments, designed to minimize and study the effects of photoreduction on these systems, have produced shorter iron–oxygen bond distances that are more in line with the values obtained from spectroscopy.13,24
In spite of these results, reports of iron(IV)hydroxide species in histidine ligated heme proteins have continued to surface.13 Eq 1 reveals the importance of determining the pKa of these systems. This thermodynamic parameter can provide critical insight into Nature’s ability to tune the function of heme proteins through the choice of axial ligation.5 We have shown that the best way to address this problem is through the use of multiple spectroscopies.1,4,5,15,23,25−27 In hopes of providing insight into the existence of iron(IV)hydroxide species in histidine ligated heme proteins, we present an exhaustive spectroscopic characterization of myoglobin compound II (Mb-II). Mb-II represents a particularly convenient system with which to examine the problem. Mb-II has long served as a model for intermediates in the catalytic cycles of heme peroxidase. It can be prepared in the high purities (without contamination from other oxidation states) and the high yields required for spectroscopic measurements. Importantly, Mb-II can be obtained over a wide pH range using the pH jump method.
Myoglobin Compound II and the Status of Ferryl Protonation
Mb-II has been at the center of the debate concerning the existence of iron(IV)hydroxide species in histidine ligated heme proteins. The controversy stems from a series of conflicting X-ray crystallographic and spectroscopic reports.15,23 High-resolution crystal structures of Mb-II, horseradish peroxidase compound II (HRP-II), and cytochrome c peroxidase compound I (CCP-I), which is best described as a ferryl porphyrin (i.e., compound II) intermediate with a distant uncoupled protein-based radical, have indicated long (1.84–1.92 Å) Fe–O bonds between the heme iron and distal oxygen ligand.10−14 These distances are too long to be associated with a traditional iron(IV)oxo species.15,23 To make sense of these distances, crystallographers have invoked ferryl protonation. They have assigned these species as iron(IV)hydroxide complexes. This assignment, however, is at odds with the data obtained from spectroscopic measurements on these intermediates. Extended X-ray absorption fine structure (EXAFS) and resonance Raman spectroscopies provide Fe–O bond distances (∼1.65 Å)1,16−18 and Fe–O vibrational frequencies (∼790 cm–1)7,15,19−22,28 that are consistent with the values obtained from synthetic ferryl porphyrins.22
In hopes of clarifying the ferryl protonation status of histidine ligated heme proteins, Green examined the applicability of Badger’s rule (an empirical formula relating bond distance and vibrational frequency) to the available experimental data.23 Good correlation was found between EXAFS determined bond distances and reported Fe–O vibrational frequencies, while the crystallographically determined bond distances deviated substantially from the values predicted by Badger’s rule. Through comparisons with a plot of Fe–O distance versus iron oxidation state, it was concluded that the long Fe–O bonds obtained from crystallography were the hallmarks of ferric and ferrous complexes. That is, they were a direct result of radiation damage.23
The research groups of Poulos, Schlichting, and Andersson have provided additional insight into the discrepancies between the Fe–O bond distances obtained from X-ray crystallography and spectroscopy.24,29,30 These groups systematically studied the photoreduction that occurs during the collection of an X-ray crystallographic data set. Specifically, they examined how prolonged exposure of metalloprotein crystals to synchrotron radiation results in reduction of the redox-active metal center. They studied the effects on structure and coordination of the active site. Reduction of the metal center was monitored by microspectrophotometry, and changes in the single crystal visible absorption spectra were recorded as a function of radiation dosage.24,29,30
Poulos and co-workers examined photoreduction in crystals of CCP-I.24 They found that <30% of the ferryl intermediate remained after a dose of 1 MGy, only ∼3% of the theoretical dosage limit for protein crystals.31,32 By piecing together data sets collected for 19 crystals, Poulos and co-workers obtained the Fe–O bond distance as a function of radiation dosage. They found that the Fe–O distance varied linearly with dosage, extrapolating to 1.72 ± 0.02 Å at zero exposure and increasing by 0.25 Å/MGy up to 1.90 Å at the highest doses.24
Schlichting and co-workers examined photoreduction in crystals of ferric Mb.29 Substantial changes in the visible absorption spectrum occurred at radiation doses as small as 0.21 MGy, only 1.5 s after data collection had begun. By 2 MGy of exposure, just 15 s into data collection, the cryoreduced state was the major component of the crystals.29
Schlichting and co-workers did not examine Mb-II, but it is clear that crystals of this system are also prone to photoreduction. A Badger’s rule analysis of the long Fe–O bond (1.86 Å at pH 5.2) obtained from the Mb-II crystal structure indicates that the heme center is in a ferric or ferrous state.23 Consistent with this result, Hersleth and Andersson reported X-ray dosage-dependent changes in the UV/visible spectrum of Mb-II crystals. Here again, X-ray doses as small as 0.3 MGy resulted in significant changes in the Q-band spectra. In light of the available data, Hersleth and Andersson reassigned the long crystallographically determined Fe–O bond to a “radiation induced state” of Mb-II,30 consistent with quantum refinement studies that found an Fe(III)–OH state provided the best description for the atypically long Fe–O bond in the Mb-II crystal structure.33
It is clear that X-ray crystallography is not the method of choice for the accurate determination of metal–ligand bond distances in high-valent metalloproteins. Instead, one must turn to spectroscopy to determine these metrics. Mb-II has been examined by nuclear resonance vibrational (pH 8), Mössbauer (pH 7), EXAFS (pH not given), and resonance Raman spectroscopies (pH 5.0).15,17,34,35 The measurements indicate that Mb-II possesses an authentic iron(IV)oxo center, with an iron(IV)hydroxide pKa ≤ 5.
Interestingly, spectroscopic and kinetic measurements indicate that there is a protonation event in Mb-II near pH 5. Changes have been observed in the MCD, UV–vis, and resonance Raman spectra of Mb-II near this pH.15,36,37 Additionally, Mb-II shows an increase in peroxidase activity as the pH decreases below 5.38 These changes have traditionally been assigned to protonation of the proximal or distal histidine and/or a conformational change associated with this protonation. However, Wilson and co-workers have argued that these changes are consistent with protonation of the ferryl moiety, suggesting that Mb-II has an iron(IV)hydroxide pKa ∼ 5.37 As of yet, there is no evidence to support this assignment.
In what follows we report a detailed characterization of horse heart Mb-II over the pH range of 3.9–9.5, using X-ray absorption, Mössbauer, and resonance Raman spectroscopies. The measurements have allowed us to set an upper limit of 2.7 on the pKa of the iron(IV)hydroxide state in Mb.
Results and Discussion
X-ray Absorption Spectroscopy of Mb-II
Mb-II at pH 3.9 was the lowest pH sample (prepared via the rapid freeze-quench pH-jump method, 9.5 → 3.9) that we could prepare before significant protein degradation was observed (by Mössbauer spectroscopy). To gain insight on the ferryl pKa in histidine ligated systems, this intermediate was examined via extended X-ray absorption fine structure (EXAFS) spectroscopy. For comparison, samples of the well-characterized alkaline Mb (MbIII–OH, pH 10.9) were also examined. Sample composition was determined by Mössbauer spectroscopy before XAS data collection. These measurements revealed that Mb-II was prepared in >90% yield.
Figure 1 shows the Fe K-edge absorption spectra for Mb-II and MbIII–OH. The Mb-II edge lies at a higher energy than the MbIII–OH edge, consistent with the increased binding energy of the 1s electrons in the Fe(IV) state. No significant change in the absorption edge was observed between first and second scans, indicating that photoreduction was not a concern (Supporting Information). Mb-II (pH 3.9) exhibits a more intense 1s → 3d pre-edge transition than MbIII–OH. This transition is formally forbidden, but gains intensity through 4p-mixing as the iron center deviates from octahedral symmetry. The intensity of the pre-edge feature is known to track inversely with the Fe–O bond length.39,40
Figure 1.
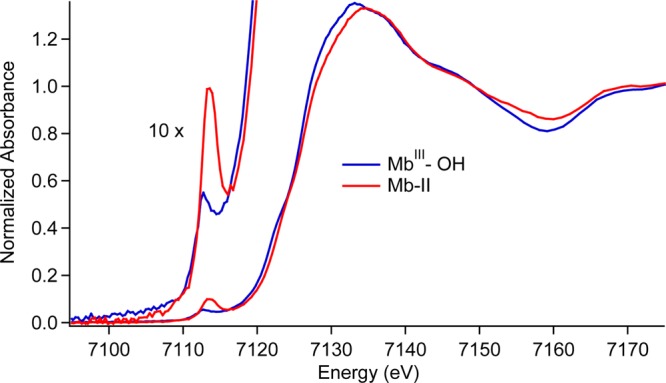
Fe K-edge X-ray absorption edges for MbIII–OH and Mb-II.
EXAFS data sets were constructed from first and second scans only (Figure 2). Analysis of the data yielded an Fe–O bond distance of 1.86 Å for MbIII–OH (Table 1), consistent with a ferric hydroxide state. The Fe–O bond distance in Mb-II is significantly shorter, 1.66 Å at pH 3.9, in good agreement with results from a previous EXAFS investigation of Mb-II (1.69 Å at unknown pH)17 and with Fe–O distances reported for other iron(IV)oxo species.15 Our EXAFS determined distances are also in good agreement with the results of DFT calculations (1.85 and 1.65 Å, respectively) and Badger’s rule predictions utilizing previously reported Fe–O stretching frequencies (νFe(III)–OH = 556 cm–1 → 1.83 Å and νFe(IV)–O = 804 cm–1 → 1.65 Å).15,41,42
Figure 2.
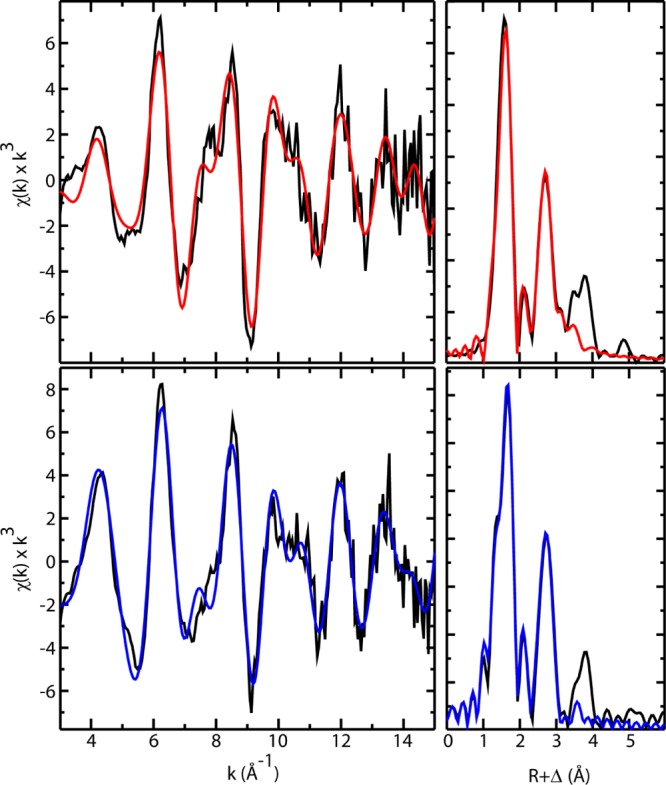
Fe K-edge EXAFS data (left) and Fourier transforms (right) of Mb-II (top) and MbIII–OH (bottom). Black lines show experimental data, and colored lines show the best fits. The fits shown were obtained over the region k = 3–15 Å–1. All EXAFS samples (∼3 mM) were analyzed by Mössbauer spectroscopy prior to data collection.
Table 1. EXAFS fitting results for Mb-II and MbIII–OHa.
| Fe–N |
Fe–O |
||||||
|---|---|---|---|---|---|---|---|
| N | R | σ2 | N | R | σ2 | E0 | error |
| Mb-II | |||||||
| 4 | 2.012 | 0.0027 | 1 | 1.651 | 0.0045 | –13.1 | 0.390 |
| 4 | 2.005 | 0.0026 | 0 | –16.8 | 0.437 | ||
| 5 | 2.011 | 0.0038 | 1 | 1.658 | 0.0047 | -13.7 | 0.377 |
| 5 | 2.006 | 0.0036 | 0 | –16.6 | 0.444 | ||
| MbIII–OH | |||||||
| 4 | 2.016 | 0.0008 | 1 | 1.873 | –0.0002 | –12.6 | 0.309 |
| 4 | 2.015 | 0.0025 | 0 | –8.5 | 0.367 | ||
| 5 | 2.011 | 0.0021 | 1 | 1.858 | 0.0005 | -12.9 | 0.326 |
| 5 | 2.013 | 0.0036 | 0 | –8.8 | 0.353 | ||
Raw data were fit over the region k = 3–15 Å–1. Coordination number N, interatomic distance R (Å), mean square deviation in R (the Debye–Waller factor), σ2 (Å2), and the threshold energy shift E0 (eV). The fit error F is defined as [∑k6(χexptl – χcalc)2/ ∑k6 χexptl2]1/2. Best fits are shown in boldface. Alternative fits include the scattering contribution of the nitrogen (N = 5) from the axial histidine ligand. Coordination numbers, N, were constrained during the fits.
Mössbauer Spectroscopy of Mb-II
Mössbauer samples of Mb-II were prepared at pH 9.5, 4.7, and 3.9 in order to track any changes that may be indicative of ferryl protonation, Figure 3. All three samples contain one major species (≥90%), a quadrupole doublet with ΔEq = 1.47 mm/s (pH 9.5), 1.53 mm/s (pH 4.7), and 1.58 mm/s (pH 3.9). These values are in agreement with previous Mössbauer characterizations of Mb-II at pH 7 (ΔEq ∼ 1.5)34 and similar to the results obtained for synthetic ferryl (FeIV=O) porphyrins.43 The slightly larger quadrupole splittings at lower pH could result from protonation of the proximal or distal histidine and/or hydrogen bonding to the ferryl oxygen. The change in the Mb-II ΔEq as a function of pH is small in comparison to changes that have been observed following ferryl protonation. P450s have been shown to exhibit a ∼ 55% increase in the ΔEq upon ferryl protonation (ΔEq = 1.30 mm/s for FeIV=O and 2.02 mm/s for FeIV–OH).5 If Mb-II was protonated at low pH, the sensitivity of Mössbauer spectroscopy allows for a contribution as small as ∼5% (for integer spin systems) to be seen. Because there is no indication of a protonated species at pH 3.9, we can set an upper limit on the ferryl pKa of Mb at 2.7.
Figure 3.

Mössbauer data of Mb samples. Black is experimental and red is the fit. (A) Mb-II pH 9.5, δ = 0.09 mm/s, ΔEq = 1.47 mm/s. (B) Mb-II pH 4.7, δ = 0.08 mm/s, ΔEq = 1.53 mm/s. (C) Mb-II pH 3.9, δ = 0.07 mm/s, ΔEq = 1.58 mm/s. (D) Ferric Mb pH 7. (E) MbIII–OH pH 10.9. All samples were ∼3 mM. (C) and (E) are the spectra of the samples used for EXAFS.
Resonance Raman Spectroscopy of Mb-II
Resonance Raman investigations of Mb-II have been reported, but they have not found evidence of an iron(IV)hydroxide state. Terner and co-workers examined compound II in sperm whale myoglobin.19 Using 406.7 nm excitation, they observed an Fe–O vibrational mode at 797 cm–1 (at pH 8.6), corresponding to an Fe–O bond distance of 1.65 Å,23 which shifted to 771 cm–1 with 18O substitution. This 26 cm–1 shift is significantly less than the 35 cm–1 shift predicted for an Fe–O diatomic harmonic oscillator, indicating that the Fe–O stretching mode is coupled to other out-of-plane motions.19 Behan and Green examined horse heart Mb-II, using 431 nm excitation.15 They reported an Fe–O vibrational mode at 804 cm–1 at pH 8 that shifted to 790 cm–1 at pH 5. With 18O substitution, these vibrations shifted to 769 and 754 cm–1, respectively. The pH-dependent 14 cm–1 shift in Fe–O stretching frequency has been attributed to hydrogen bonding with a distal histidine residue.15
In support of their crystallographic work, Hersleth and co-workers performed resonance Raman experiments on horse heart Mb-II.12,44 They observed an 18O sensitive Fe–O stretch at 687 cm–1 (pH 7.2), corresponding to an Fe–O bond distance of 1.72 Å,23 a difference of 0.2 Å from their reported crystallographic value. The 687 cm–1 Fe–O stretching frequency is approaching the value expected for an iron(IV)hydroxide complex, but the data are far from conclusive. The 687 cm–1 vibrational mode did not shift in D2O. This is in contrast to chloroperoxidase compound II and the alkaline forms of hemoglobin and myoglobin, where the Fe–OH stretching frequency down shifts ∼13 cm–1 in deuterated buffer.26,41,42 We note, however, that the absence of a deuterium-dependent downshift in the Fe–O(H) stretching frequency does not rule out the presence of a hydroxide ligand. The Fe(III)–OH stretching frequency of alkaline HRP upshifts 4 cm–1 in D2O, while the Fe(III)–OH stretch of Scapharca inaequivalvis hemoglobin has been reported to be independent of deuterium substitution.45 In cases were the Fe–O shift is inconclusive, the application of Badger’s rule can provide considerable insight into the oxygen ligand’s protonation state.
In the current study, we utilize a 501.7 nm laser line that almost exclusively enhances the Fe–O mode. The use of laser excitation red of the Soret band has previously been reported to increase the enhancement of heme iron-oxo stretches as well as decrease the risk for photochemical damage with respect to more blue laser lines.46 As with X-rays, laser irradiation can lead to sample decomposition, and this proved to be particularly problematic in early efforts to characterize enzymatic compound I intermediates. To circumvent these issues, researchers have turned to continuous-flow systems, spinning cells, low-power excitation, data collection at cryogenic temperatures, and, as noted above, alternative excitation frequencies.22 Importantly, we found the photolability of Mb-II at 501.7 nm to be insignificant.
Resonance Raman measurements were performed over the pH range of 4.4–9.5 to determine the Fe–O stretching frequencies, Figure 4. Studies below pH 4.4 were limited by sample fluorescence. Isotopic labeling experiments (18O and 2H) were paired with diatomic harmonic oscillator predictions to identify features indicative of H-bonding and/or a ferryl protonation event. One could argue that the observation of an iron(IV)oxo stretch, particularly at low pH, does not rule out the existence of an iron(IV)hydroxide species, as the sample may be near the iron(IV)hydroxide pKa and the FeIV–OH vibration may not be enhanced. It is for this reason that we have employed multiple spectroscopies in our investigation of Mb-II. Importantly, our Mössbauer measurements on Mb-II indicate the presence of a single species (>90%) with no significant change in metal–ligand coordination from pH 9.5 to pH 3.9. Additionally, our EXAFS measurements indicate an Fe–O distance of 1.65 Å at pH 3.9. Thus, in this case, the observation of an iron(IV)oxo stretch does rule out the existence of an iron(IV)hydroxide state.
Figure 4.
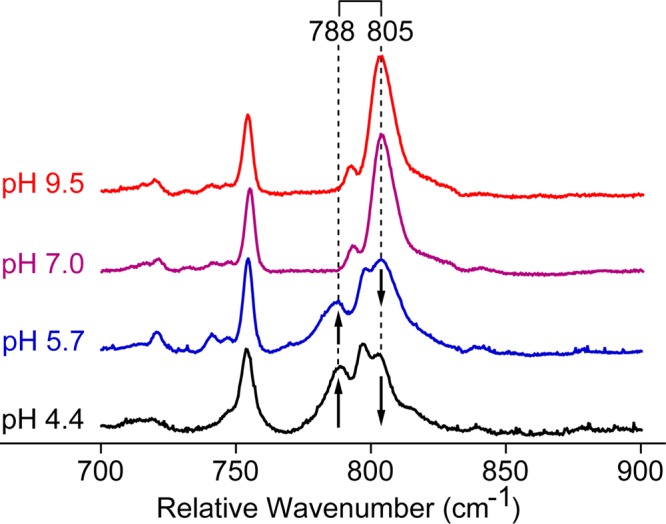
Low-frequency resonance Raman of 16O Mb-II at various pHs. The spectra were normalized by scaling to the intensity of the ∼755 cm–1 peak. Data were collected using the 501.7 nm argon-ion laser line.
Using 501.7 nm excitation, an Fe–O stretching frequency was identified at 805 cm–1 (corresponding to an Fe–O bond distance of 1.65 Å)23 at pH 7 and 9.5 (Figure 4). This resonance shifts 36 cm–1 to 769 cm–1 upon 18O isotopic substitution, in good agreement with the predictions of an Fe–O diatomic harmonic oscillator, Figure 5. At pH 5.7, the 805 cm–1 Fe–O stretch partially shifts to 788 cm–1 (corresponding to a distance of 1.66 Å).23 This shift coincides with changes previously reported in the MCD, UV–vis, and resonance Raman spectra.15,36,37 Spectra obtained at pH 4.4 are similar, but indicate that a larger percentage of Mb-II lies in this alternative, low pH (acidic) conformation.
Figure 5.
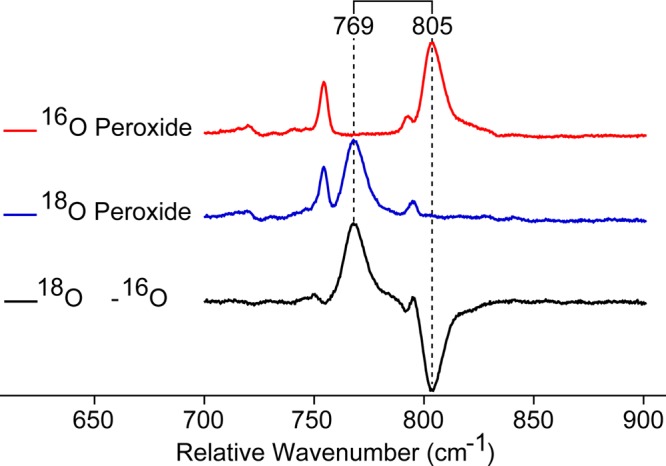
Low-frequency resonance Raman of 16O/18O Mb-II at pH 9.5 (100 mM borate buffer). Results at pH 7 were virtually identical. The peak at ∼755 cm–1 was used to scale the spectra. Data were collected using the 501.7 nm argon-ion laser line.
Interestingly, the Fe–O stretch in the acidic form of Mb-II shifts from 788 to 760 cm–1 upon 18O substitution, a difference of only 28 cm–1, and not the full 36 cm–1 observed for the high pH form and expected for an Fe–O diatomic oscillator. As a result, the Fe–O vibrational modes in the 18O isotopically labeled low-pH samples are not resolvable. They appear as one broad peak in the 18O – 16O difference spectra. The 28 cm–1 shift in the Fe–O stretching frequency is similar to that observed by Terner in sperm whale Mb.19 It indicates that there is coupling between the Fe–O stretch and additional out of plane vibrational modes in the acidic form of horse heart Mb-II.19
The pH-dependent shift in Fe–O stretching frequency is likely due to hydrogen-bonding interactions with the ferryl oxygen. This type of phenomenon has been observed in HRP-II, where the FeIV=O stretch shifts from 790 cm–1 at high pH to 778 cm–1 as the pH is titrated below the pKa of the distal histidine.7,20,22 Crystal structures of HRP-I and HRP-II show that the distal histidine does not interact directly with the ferryl oxygen but organizes an intervening water molecule for hydrogen bonding.10,22 In Mb-II, the pH-dependent shift in the Fe–O stretching frequency is slightly larger (17 cm–1) than HRP-II, which may be due to the closer proximity of the distal histidine in Mb, allowing for a direct interaction with the ferryl oxygen.10,12 It is important to note that the pH-dependent 17 cm–1 shift in Mb-II corresponds to a change of 0.009 Å in Fe–O bond length, which is inconsistent with protonation of the ferryl moiety.23
To examine if the pH-dependent shift in the Mb-II Fe–O stretching frequency can be attributed to H-bonding to the ferryl oxygen, isotopically labeled (18O/16O) samples were also prepared in deuterated buffer, Figure 6. Previous experiments have shown that H-bonding to the ferryl oxygen is weakened by deuterium substitution. In these cases, a 2–3 cm–1 upshift in the Fe–O stretching frequency is observed.7,20−22 Experiments on the high pH form of Mb-II show that the 805 cm–1 stretch appears at the same energy in deuterated buffer, indicating a lack of hydrogen bonding. Similar studies on the acidic form of Mb-II reveal that the Fe–16O stretch at 788 cm–1 shifts to 790 cm–1 in D2O, while the Fe–18O stretch at 760 cm–1 is now clearly seen at 762 cm–1 upon 2H substitution. These upshifts in stretching frequency suggest H-bonding between the distal histidine and ferryl oxygen.
Figure 6.
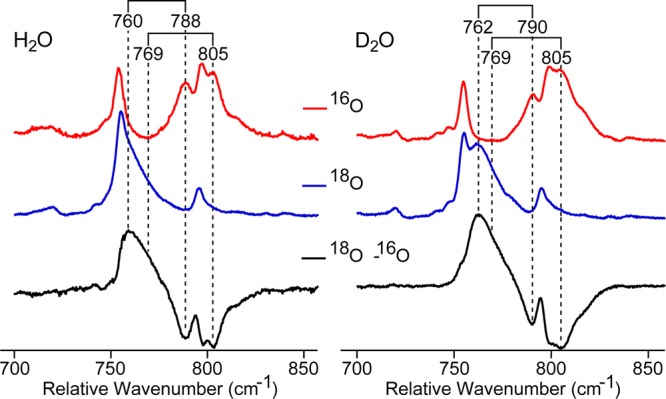
Low-frequency resonance Raman of 16O/18O Mb-II at pH 4.4 (left) and pD 4.8 (right) in acetate buffer. The resonances near ∼720 cm–1 were used to scale the spectra. Data were collected using the 501.7 nm argon-ion laser line.
Conclusion
This analysis has utilized three different spectroscopic techniques (EXAFS, Mössbauer, and resonance Raman spectroscopies) to conclusively show that Mb-II exists as an iron(IV)oxo complex at pH ≥ 3.9. Additionally, the sensitivity of our Mössbauer measurements has allowed us to place an upper limit, pKa ≤ 2.7, on the iron(IV)hydroxide pKa in Mb-II. A similar value for the histidine-ligated HRP-II (pKa ≤ 3.6) is suggested by the work of Sitter et al.7 Thus, it appears that a proximal histidine ligand is not donating enough to stabilize an iron(IV)hydroxide state. These results stand in contrast to those obtained for thiolate ligated systems. We have recently shown that the P450s CYP158 and CYP119 have elevated iron(IV)hydroxide pKas ∼12.0. We have argued that this difference in iron(IV)hydroxide pKa may be used to tune catalytic function in heme enzymes.5
Materials and Methods
Preparation of 57 Mb for Mössbauer
57Fe Heme Synthesis
57Heme synthesis was adapted from the metalloporphyrin synthesis of Adler et al.47 10 mg of 57Fe metal was anaerobically dissolved (60 °C) in 1 M HCl to form ferrous chloride. The HCl was evaporated to dryness. The white, powdered, FeCl2 was combined with 25 mg protoporphyrin IX in a flask containing 50 mL of deaerated DMF. After refluxing the materials for 15 min, the mixture was cooled on ice and exposed to air. The mixture was then diluted 10× with diethyl ether. Excess ferric salts were separated from the ether layer with 100 mM HCl containing 100 mM NaCl. Excess protoporphyrin IX was separated out with 1 M HCl. The remaining ether phase was then washed to neutrality with H2O and evaporated to dryness. 57Heme was stored in a −80 °C freezer until use.
Generation of Apo Mb
Apo Mb was generated using the Teale method.48,49 Lyophilized Mb from equine heart (Sigma) was dissolved into solution (water) and acidified to pH 2. 56Heme was separated from the apo protein by extraction with methyl–ethyl ketone. Several rounds of dialysis, first in H2O and then in potassium phosphate buffer (50 mM, pH 7), were carried out to remove excess methyl-ethyl ketone.
Reconstitution
A slight excess of 57heme was dissolved in 100 mM NaOH. The solution was then mixed with tris-HCl buffer to a final pH ∼ 8.5 and added to the apo Mb (pH 7) solution. The mixture was allowed to stir at 4 °C for 30 min. Excess heme was removed by anion exchange chromatography (Whatman DE-52 resin). Fractions with Rz > 5 were pooled for use.
Freeze-Quenched Samples
Freeze-quench methods were used to generate the ferryl intermediates in Mb. A four syringe ram freeze-quench apparatus from Update Instruments (Madison, WI) was used for all freeze-quench experiments. Aqueous reaction mixtures were sprayed into liquid ethane (89 K). Liquid ethane was subsequently removed under vacuum in an isopentane bath (∼120 K), and samples for resonance Raman, Mössbauer, and EXAFS were packed under liquid nitrogen.
Preparation of Mb-II at Low pH
Mb-II (4.5 mM) was generated in borate buffer (20 mM, pH 9.5, 4 °C) prior to freeze-quenching by mixing with ∼2.5 equiv H2O2 (in water). Under these conditions Mb-II is stable at ≥90% yield for at least 2 h. The solution of Mb-II was then loaded into the freeze-quench syringe (4 °C) and reacted against a high-strength buffer near the desired pH (potassium phosphate 5.5–7 and acetate 3.9–5.5) in a 2:1 mixture (v/v). The reaction was quenched into a liquid ethane bath ∼2 ms after mixing. Samples were packed for spectroscopic analysis at a final protein concentration of 3 mM. Portions of the quenched samples were set aside to confirm the final pH of the solution. For resonance Raman labeling experiments 18H2O2 (2% soln in water, 90% 18O, Icon Isotopes), D2O (99.9% D, Cambridge Isotope Laboratories, Inc.), phosphoric acid (99% D3, MP Biomedicals, LLC), and NaOD (30% [w/w] in D2O, Alfa Aesar) were used.
Preparation of Aqua (MbIII–OH2) and Alkaline Ferric Mb (MbIII–OH)
Solutions of 4 mM MbIII–OH2 (100 mM potassium phosphate buffer, pH 7) and MbIII–OH (100 mM borate buffer, pH 10.9) were pipetted into Mössbauer cups and frozen into liquid ethane. Ferric Mb samples were never hand-quenched into liquid nitrogen because of the formation of hemochrome during the (slower) liquid nitrogen freezing process.50 For XAS samples, the same stock of protein used for Mössbauer samples was loaded into a freeze-quench syringe and quenched into a liquid ethane bath ∼2 ms after mixing. Samples were packed into XAS cups under liquid nitrogen at 77K.
Mössbauer Spectroscopy
A spectrometer from WEB Research (Edina, MN) was used to collect data in constant acceleration mode with a transmission geometry. Spectra were recorded with a 53 mT magnetic field applied parallel to the γ-beam. All measurements were recorded at 4.2 K using a Janis SVT400 cryostat. Isomer shifts were calibrated relative to the centroid of the spectrum of a metallic foil of α-Fe at room temperature. Data analysis was performed using the program WMOSS from WEB research.
X-ray Absorption Spectroscopy (XAS)
XAS data were collected in fluorescence mode at ∼10 K with a 30-element germanium detector (SSRL, BL7-3) using a Si(220) Φ = 90° double monochromator with a 9.5 keV cutoff for harmonic rejection. To minimize the effects of photoreduction, samples were moved in the beam so that unexposed portions of the sample were examined every 2 scans (exposure time was ∼37 min per scan). XAS were obtained by averaging 16 total scans (8 first scans and 8 second scans) for Mb-II and 18 total scans (10 first scans and 8 second scans) for MbIII–OH. The effects of photoreduction were monitored via the analysis of data obtained during the second acquisition scan. Background removal was performed with AUTOBK as found in the ATHENA package. Curve fitting was performed with EXAFSPAK (available at http://www-ssrl.slac.stanford.edu/exafspak.html) using ab initio phases and amplitudes generated with FEFF version 7.0. Data sets were fit over the range k = 3–15 Å–1. Coordination numbers, N, were constrained during the fits. Fits included first- and second-shell atoms and one multiple scattering component. In all cases, the second shell was composed of α- and meso-carbons and the Fe–Cα–N–Fe multiple scattering paths (n = 8, 4, and 16, respectively). All distances, R, and Debye–Waller factors, σ2, were treated as adjustable parameters, and all threshold energy shifts, E0, were linked but allowed to vary. The passive electron reduction factor, So, was held at 0.9. Edge energies were calibrated using α-Fe metal foil (7111.3 eV). Edge positions were obtained from the first derivative of the data using EXAFSPAK (1.0 eV smoothing, third-order polynomial).
Resonance Raman Spectroscopy
Resonance Raman spectra were recorded on a triVista 555 triple monochromator (900/900/2400 gr/mm) equipped with a CCD camera (1340 × 100 pixels). A 501.7 nm line of an argon-ion laser was used for excitation. The power was <5 mW at the sample. Samples were held in a Janis STVP-100 cryostat with a custom holder at 77 K using a ∼135° back-scattering arrangement. Raw spectra were analyzed using the program Igor Pro for background subtraction. No smoothing procedures were performed on the raw data.
Acknowledgments
This work was supported by NIH (R01-GM101390). We thank M. Latimer and E. Nelson for onsite assistance at the synchrotron. Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under contract no. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393).
Supporting Information Available
XAS edges (first and second scans) for MbIII–OH and Mb-II are included to show that neither species undergoes a significant amount of photoreduction (<10%) during data collection. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
† MassBiologics of the University of Massachusetts Medical School, 460 Walk Hill St., Boston, MA 02126.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Green M. T.; Dawson J. H.; Gray H. B. Science 2004, 304, 1653. [DOI] [PubMed] [Google Scholar]
- Green M. T. Curr. Opin. Chem. Biol. 2009, 13, 84. [DOI] [PubMed] [Google Scholar]
- de Montellano P. R. O.Cytochrome P450: Structure, Mechanism, and Biochemistry; 3rd ed.; Kluwer Academic/Plenum Publishers: Dordrecht, The Netherlands, 2005. [Google Scholar]
- Rittle J.; Green M. T. Science 2010, 330, 933. [DOI] [PubMed] [Google Scholar]
- Yosca T. H.; Rittle J.; Krest C. M.; Onderko E. L.; Silakov A.; Calixto J. C.; Behan R. K.; Green M. T. Science 2013, 342, 825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groves J. T. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitter A. J.; Reczek C. M.; Terner J. J. Biol. Chem. 1985, 260, 7515. [PubMed] [Google Scholar]
- The drop in pKa relative to P450 is consistent with the observations of Sono et al., who reported that sulfur ligands bound trans to P450s axial thiolate have enhanced basicity relative to analogous myoglobin adducts.
- Sono M.; Andersson L. A.; Dawson J. H. J. Biol. Chem. 1982, 257, 8308. [PubMed] [Google Scholar]
- Berglund G. I.; Carlsson G. H.; Smith A. T.; Szoke H.; Henriksen A.; Hajdu J. Nature 2002, 417, 463. [DOI] [PubMed] [Google Scholar]
- Bonagura C. A.; Bhaskar B.; Shimizu H.; Li H. Y.; Sundaramoorthy M.; McRee D. E.; Goodin D. B.; Poulos T. L. Biochemistry 2003, 42, 5600. [DOI] [PubMed] [Google Scholar]
- Hersleth H. P.; Uchida T.; Rohr A. K.; Teschner T.; Schuenemann V.; Kitagawa T.; Trautwein A. X.; Gorbitz C. H.; Andersson K. K. J. Biol. Chem. 2007, 282, 23372. [DOI] [PubMed] [Google Scholar]
- Gumiero A.; Metcalfe C. L.; Pearson A. R.; Raven E. L.; Moody P. C. E. J. Biol. Chem. 2011, 286, 1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cytochrome c peroxidase compound I (CCP-I) is not a typical compound I species. It does not possess a porphyrin-based radical. CCP-I is best described as a ferryl porphyrin—that is, a compound II—species with a distant uncoupled protein-based radical. CCP-I is not green in color. It is red like compound II intermediates.22
- Behan R. K.; Green M. T. J. Inorg. Biochem. 2006, 100, 448. [DOI] [PubMed] [Google Scholar]
- Penner-Hahn J. E.; Eble K. S.; McMurry T. J.; Renner M.; Balch A. L.; Groves J. T.; Dawson J. H.; Hodgson K. O. J. Am. Chem. Soc. 1986, 108, 7819. [DOI] [PubMed] [Google Scholar]
- Chance M.; Powers L.; Kumar C.; Chance B. Biochemistry 1986, 25, 1259. [DOI] [PubMed] [Google Scholar]
- Chance M.; Powers L.; Poulos T.; Chance B. Biochemistry 1986, 25, 1266. [DOI] [PubMed] [Google Scholar]
- Sitter A. J.; Reczek C. M.; Terner J. Biochim. Biophys. Acta 1985, 828, 229. [DOI] [PubMed] [Google Scholar]
- Hashimoto S.; Tatsuno Y.; Kitagawa T. Proc. Natl. Acad. Sci. U. S. A. 1986, 83, 2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto S.; Teraoka J.; Inuubushi T.; Yonetani T.; Kitagawa T. J. Biol. Chem. 1986, 261, 11110. [PubMed] [Google Scholar]
- Terner J.; Palaniappan V.; Gold A.; Weiss R.; Fitzgerald M. M.; Sullivan A. M.; Hosten C. M. J. Inorg. Biochem. 2006, 100, 480. [DOI] [PubMed] [Google Scholar]
- Green M. T. J. Am. Chem. Soc. 2006, 128, 1902. [DOI] [PubMed] [Google Scholar]
- Meharenna Y. T.; Doukov T.; Li H. Y.; Soltis S. M.; Poulos T. L. Biochemistry 2010, 49, 2984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone K. L.; Behan R. K.; Green M. T. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 16563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone K. L.; Behan R. K.; Green M. T. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 12307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behan R. K.; Hoffart L. M.; Stone K. L.; Krebs C.; Green M. T. J. Am. Chem. Soc. 2006, 128, 11471. [DOI] [PubMed] [Google Scholar]
- Reczek C. M.; Sitter A. J.; Terner J. J. Mol. Struct. 1989, 214, 27. [Google Scholar]
- Beitlich T.; Kuhnel K.; Schulze-Briese C.; Shoeman R. L.; Schlichting I. J. Synchrotron Radiat. 2007, 14, 11. [DOI] [PubMed] [Google Scholar]
- Hersleth H. P.; Andersson K. K. Biochim. Biophys. Acta, Proteins Proteomics 2011, 1814, 785. [DOI] [PubMed] [Google Scholar]
- Henderson R. Proc. R. Soc. London, Ser. B 1990, 241, 6. [Google Scholar]
- Owen R. L.; Rudino-Pinera E.; Garman E. F. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 4912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson K.; Hersleth H. P.; Rod T. H.; Andersson K. K.; Ryde U. Biophys. J. 2004, 87, 3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda Y.; Morita Y.; Yoshida C. J. Biochem. (Tokyo, Jpn.) 1971, 70, 509. [DOI] [PubMed] [Google Scholar]
- Zeng W.; Barabanschikov A.; Zhang Y.; Zhao J.; Sturhahn W.; Alp E. E.; Sage J. T. J. Am. Chem. Soc. 2008, 130, 1816. [DOI] [PubMed] [Google Scholar]
- Foote N.; Gadsby P. M.; Greenwood C.; Thomson A. J. Biochem. J. 1989, 261, 515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silaghi-Dumitrescu R.; Reeder B. J.; Nicholls P.; Cooper C. E.; Wilson M. T. Biochem. J. 2007, 403, 391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsen C. U.; Skovgaard I. M.; Skibsted L. H. J. Agric. Food Chem. 2003, 51, 5815. [DOI] [PubMed] [Google Scholar]
- Penner-Hahn J. E.; Hodgson K. O.. Iron Porphyrins; VCH: New York, 1989; Vol. III. [Google Scholar]
- Rohde J. U.; Torelli S.; Shan X. P.; Lim M. H.; Klinker E. J.; Kaizer J.; Chen K.; Nam W. W.; Que L. J. Am. Chem. Soc. 2004, 126, 16750. [DOI] [PubMed] [Google Scholar]
- Feis A.; Marzocchi M. P.; Paoli M.; Smulevich G. Biochemistry 1994, 33, 4577. [DOI] [PubMed] [Google Scholar]
- Behan R. K. Ph.D. Dissertation, Pennsylvania State University, 2008. [Google Scholar]
- Simonneaux G.; Scholz W. F.; Reed C. A.; Lang G. Biochim. Biophys. Acta 1982, 716, 1. [Google Scholar]
- Hersleth H. P.; Varnier A.; Harbitz E.; Rohr A. K.; Schmidt P. P.; Sorlie M.; Cederkvist F. H.; Marchal S.; Gorren A. C. F.; Mayer B.; Uchida T.; Schunemann V.; Kitagawa T.; Trautwein A. X.; Shimizu T.; Lange R.; Gorbitz C. H.; Andersson K. K. Inorg. Chim. Acta 2008, 361, 831. [Google Scholar]
- Das T. K.; Boffi A.; Chiancone E.; Rousseau D. L. J. Biol. Chem. 1999, 274, 2916. [DOI] [PubMed] [Google Scholar]
- Ikemura K.; Mukai M.; Shimada H.; Tsukihara T.; Yamaguchi S.; Shinzawa-Itoh K.; Yoshikawa S.; Ogura T. J. Am. Chem. Soc. 2008, 130, 14384. [DOI] [PubMed] [Google Scholar]
- Adler A. D.; Longo F. R.; Kampas F.; Kim J. J. Inorg. Nucl. Chem. 1970, 32, 2443. [Google Scholar]
- Teale F. W. Biochim. Biophys. Acta 1959, 35, 543. [DOI] [PubMed] [Google Scholar]
- Wagner G.; Perez M.; Toscano W. A.; Gunsalus I. J. Biol. Chem. 1981, 256, 6262. [PubMed] [Google Scholar]
- Bizzarri A. R.; Iakovleva O. A.; Parak F. Chem. Phys. 1995, 191, 185. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.