

Abstract
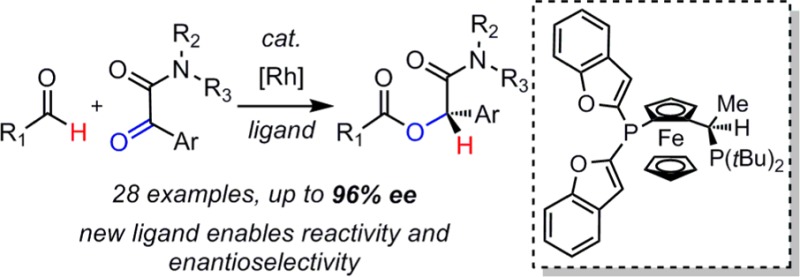
Under Rh(I) catalysis, α-ketoamides undergo intermolecular hydroacylation with aliphatic aldehydes. A newly designed Josiphos ligand enables access to α-acyloxyamides with high atom-economy and enantioselectivity. On the basis of mechanistic and kinetic studies, we propose a pathway in which rhodium plays a dual role in activating the aldehyde for cross-coupling. A stereochemical model is provided to rationalize the sense of enantioinduction observed.
Introduction
The catalytic functionalization of aldehyde C–H bonds represents a mild and atom-economical approach to prepare esters.1 While there has been much progress in developing hydroacylation of aldehydes,2−6 the hydroacylation of ketones is relatively unexplored and warrants attention due to its potential for constructing chiral esters.4 Our laboratory has demonstrated intramolecular ketone hydroacylations, including the first examples of enantioselective lactonizations.7 Due to the propensity of nonchelating aldehydes to engage in decarbonylation,8−10 Tishchenko dimerizations,5 and aldol condensations, intermolecular hydroacylations are considerably more challenging.11−13 Herein, we report the design and development of the first enantioselective intermolecular ketone hydroacylation.
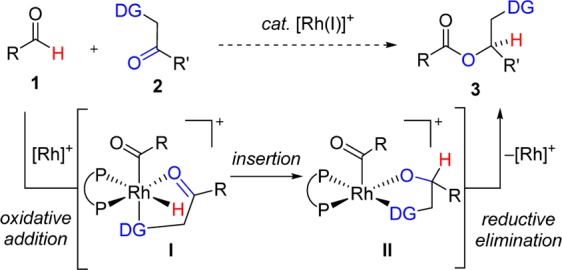
We imagined a novel cross-coupling between a broad range of aldehydes 1 and ketones bearing a directing group (DG) 2 (Scheme 1). On the basis of previous studies, we proposed that this functional group would promote preferential ketone binding to the Rh(I) center and favor chemoselective cross-coupling over aldehyde dimerization or decarbonylation.9d,9e,14 In the presence of a suitable bidentate phosphine ligand, coordination of ketone 2 to Rh, followed by oxidative addition of aldehyde 1, would generate coordinatively saturated Rh(III)-hydride I. Octahedral complex I has a lower propensity to undergo carbonyl deinsertion, a process that deactivates the rhodium catalyst.8 Ketone insertion into acyl-Rh(III)-hydride I would lead to Rh(III)-alkoxide II, followed by reductive elimination to give ester 3 with concomitant regeneration of the Rh(I) catalyst. In comparison to conventional approaches to ester synthesis, this catalytic method obviates the need for prefunctionalization of the acyl component and is amenable to enantioselective synthesis of esters.
Scheme 1. Proposed Rh(I)-Catalyzed Cross-Coupling of Aldehydes and Ketones.
Results and Discussion
Method Development
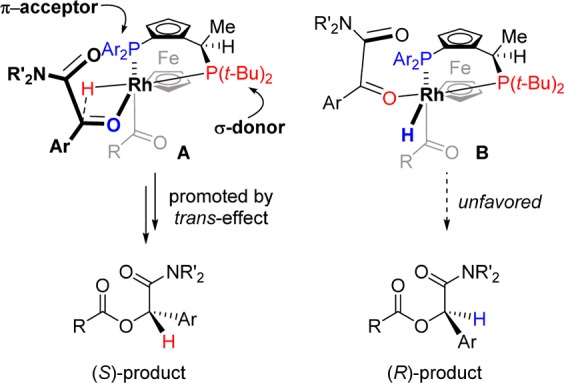
To test our hypothesis, we chose hydrocinnamaldehyde (1a) and α-ketoamide 2a as model substrates (Chart 1). A number of Rh-bis(phosphine) catalysts were examined because previous studies in our lab had demonstrated that these complexes promote intramolecular ketone hydroacylation.7,15 After studying various ligands, we found Rh(I)-Josiphos catalysts most promising. Commercially available ligand L1 provides modest yield and 13% ee. Changing to ligand L2, which contains a diphenylphosphine and a dialkylphosphine, results in a small improvement in enantioselectivity (21% ee). Josiphos L3 containing a more π-accepting difurylphosphine and a σ-donating di-tert-butylphosphine affords the desired α-acyloxyamide 3a in 46% yield and 73% ee. We reason that this C1-symmetric ligand may facilitate the product-determining hydride delivery via the trans effect; the hydride trans to the electron-rich dialkylphosphine becomes more hydridic, while the ketone trans to the π-acceptor becomes more prone to insertion (Scheme 2).16,17
Chart 1. Evaluation of Ligands for Enantioselective Ketone Hydroacylationa.
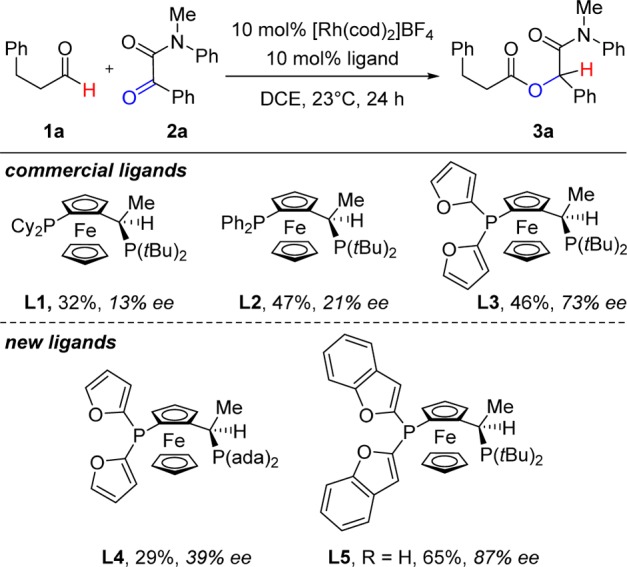
a Conditions: 1a (1.2 equiv), 2a (1 equiv), [Rh(cod)2]BF4 (0.1 equiv), ligand (0.1 equiv), DCE, rt (23 °C), 24 h.
Scheme 2. Proposed Model for Enantioinduction.
Inspired by L3, we created new Josiphos ligands to study the steric influence of the phosphine substituents. In comparison to commercial L3, ligand L4 bears bulkier adamantyl groups and shows inferior performance. In contrast, L5 with bulkier π-accepting dibenzofurylphosphines affords better results than L3 (65% yield, 87% ee). The Rh-L5 catalyst shows excellent chemoselectivity with no observable aldehyde dimerization.4,5
Scope of Intermolecular Ketone Hydroacylation
This mild Rh(I)-catalyzed method offers an attractive approach to enantioenriched esters that are structurally related to those obtained via the multicomponent Passerini reaction.19−21 A conventional route to these motifs would involve enantioselective ketone reduction followed by an additional acylation step (which generally requires stoichiometric activating agents) to obtain the α-acyloxyamides. Moreover, the asymmetric reduction of α-ketoamides has not been well-studied and remains limited in scope and/or selectivity. A method featuring Rh catalysis with bidentate amidophosphine–phosphinite ligands appears most promising and has been applied to reduce α-ketoamides with good enantioselectivity for a few aryl-substituted ketones.22 There is one example of ruthenium-catalyzed α-ketoamide hydrogenation where excellent ee was achieved following recrystallization for a bulky tert-butyl-substituted ketone.23 Biocatalytic methods have also been reported for reducing α-ketoamides, although with low efficiency.24
Thus, with Rh-L5 in hand, we studied the coupling of α-ketoamides 2 with nonchelating aliphatic aldehydes 1 (Table 1). Aryl ketones containing various substituents and substitution patterns undergo hydroacylation with hydrocinnamaldehyde (1a) (entries 1–7) and hexanal (1b) (entries 8–14) in good to high yields (53–98%) and high enantioselectivities (84–96%). Sterically demanding 2-methylphenyl (entries 2 and 9), mesityl (entries 4 and 11), and 1-naphthyl ketones (entries 5 and 12) tend to give higher conversions and selectivities even with lower (5 mol %) catalyst loadings, resulting in 73–98% yields of the α-acyloxyamides with 94–96% ee. α-Ketoamides bearing smaller aryl groups, such as 3-methylphenyl (entries 3 and 10), 4-chlorophenyl (entries 6 and 13), and 3,5-difluorophenyl groups (entries 7 and 14), require a higher catalyst loading to provide the corresponding α-acyloxyamides in 53–81% yields and 84–90% ee.
Table 1. Coupling Aldehydes with α-Ketoamidesa,b.
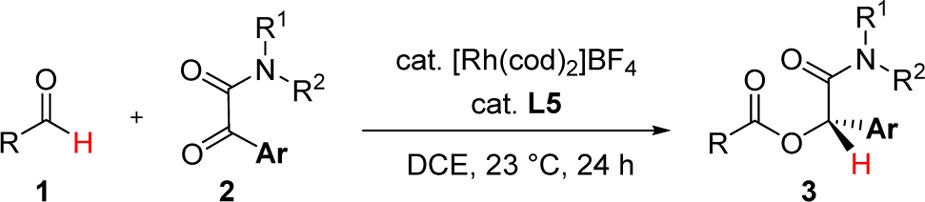
Conditions: 0.12 mmol of 1, 0.1 mmol of 2.
The catalyst was activated by hydrogenation (see Supporting Information).
Isolated yields.
Enantiomeric excess determined by chiral SFC analysis.
See ref (18).
At 60 °C.
With 1.2 mmol of 1, 1.0 mmol of 2.
At 50 °C, 4 days.
For 3 days.
L3, 7.5 h.
Sterically encumbered primary aldehydes (R = i-Bu and neopentyl, entries 15–17) are effective coupling partners, providing the desired products in high yields and ee. Using aldehyde 1d and ketone 2e, we show that the reaction can also be performed on a larger scale (1.0 mmol, 353 mg product isolated, 91% yield, 93% ee) at 5 mol % catalyst loading (entry 16). The rhodium loading can be further reduced to 3.5 mol % at elevated temperature conditions (60 °C), providing hydroacylation product 3p in 92% yield and 87% ee (entry 17). Transforming α-branched cyclohexanecarboxaldehyde (entry 18) requires elevated temperatures (50 °C) to proceed and furnishes the desired α-acyloxyamide 3q in 57% yield and 90% ee. Aryl aldehydes do not participate in hydroacylation to any appreciable extent under the current reaction conditions (<5% conversion), perhaps due to a higher barrier to C–H activation.25 Compared to the N,N′-alkylarylamide, ketones containing N,N′-diaryl- or dialkylamides are also less effective (entries 19 and 20) and provide α-acyloxyamides 3r and 3s with modest yields and enantioselectivities. A cyclic ketoamide is converted to 3-acyloxyindolinone 3t (entry 21) in 54% yield and 58% ee. The efficacy of this reaction is improved by employing L3, which provides heterocycle 3t in 88% yield and 69% ee. The amide motif impacts both reactivity and enantioselectivity, presumably due to a directing group role.
The absolute configuration of the products was assigned based on X-ray analysis of α-acyloxyamide 3c (see Supporting Information).26 The observed S-enantiomer is consistent with a proposed model that invokes the trans-effect in promoting the product-determining ketone insertion step (Scheme 2). In this model, the C1-symmetric ligand cooperatively renders the hydride more nucleophilic and the ketone more electrophilic (complex A), and thus substantially favors formation of the S-stereoisomer in the case of (SP,R)-L5. Conversely, the alternative complex B is stabilized by the trans-effect which would result in an increased barrier to migratory insertion.16,17 We propose the acyl group to be situated on the “bottom” apical position, with the less hindering N,N′-disubstituted amide carbonyl bound to the “top” apical position. This ligand arrangement minimizes unfavorable steric interactions between the acyl group and the substituents on the phosphines and is supported by DFT calculations.27
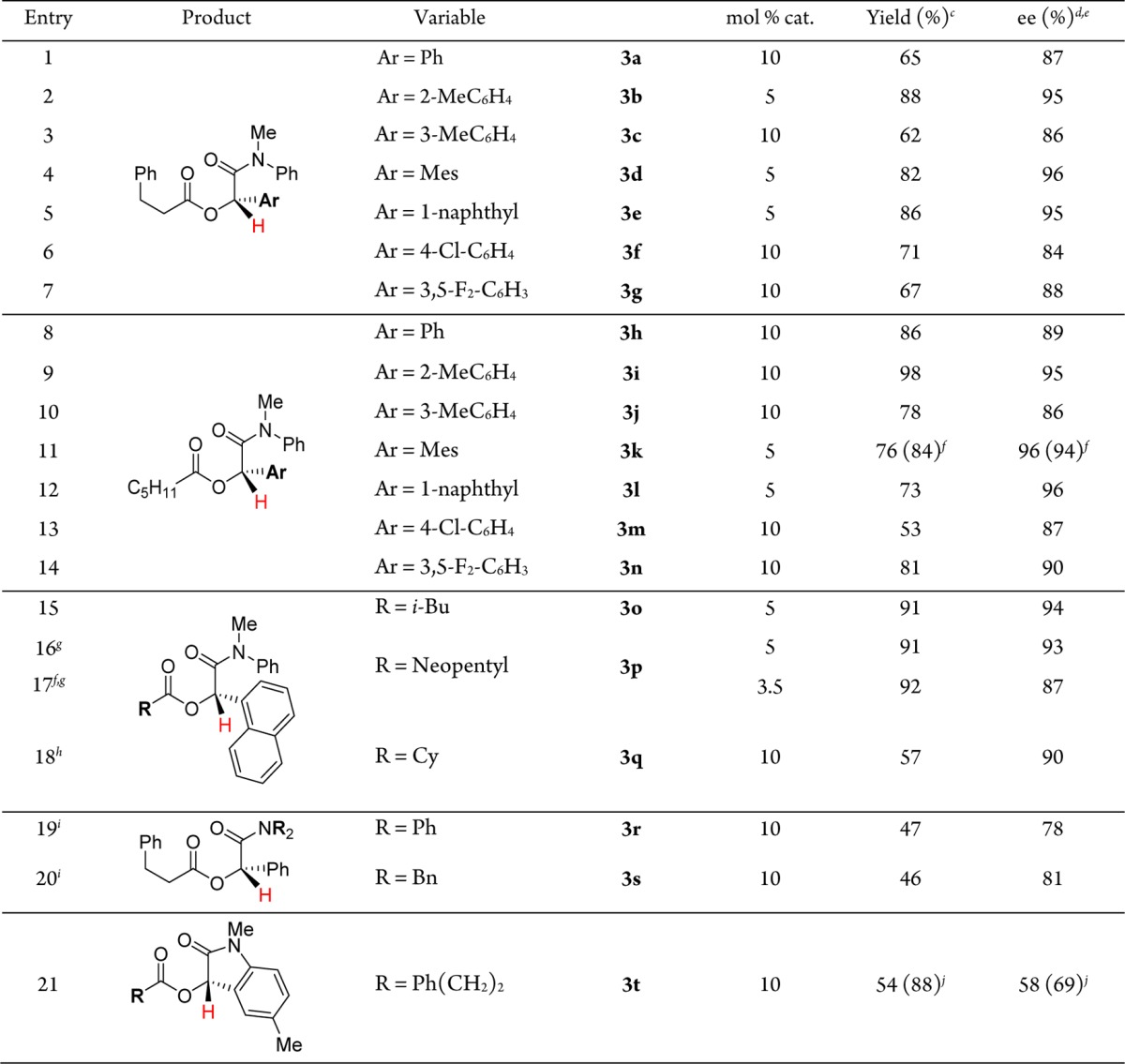
We find that morpholine amides direct hydroacylation effectively to yield cross-coupled products in good yields and enantioselectivities (Table 2). A similar trend is observed in which ketones bearing larger aryl groups perform better, both in terms of isolated yields and enantioselectivities. Phenyl ketone 4a is hydroacylated with 3,3-dimethylbutanal (1d) to furnish ester 5a in 67% yield and 80% ee, whereas larger mesityl and 1-naphthyl ketones give 5b and 5d in 94 and 75% yields, respectively (95 and 92% ee). Other aldehydes including cyclohexanecarboxaldehyde, hexanal, and hydrocinnamaldehyde are also good coupling partners. The use of the morpholine amide as a directing group provides a handle for further chemical manipulations.28
Table 2. Ketone Hydroacylation Using Morpholine Amide Directing Groupa,b.
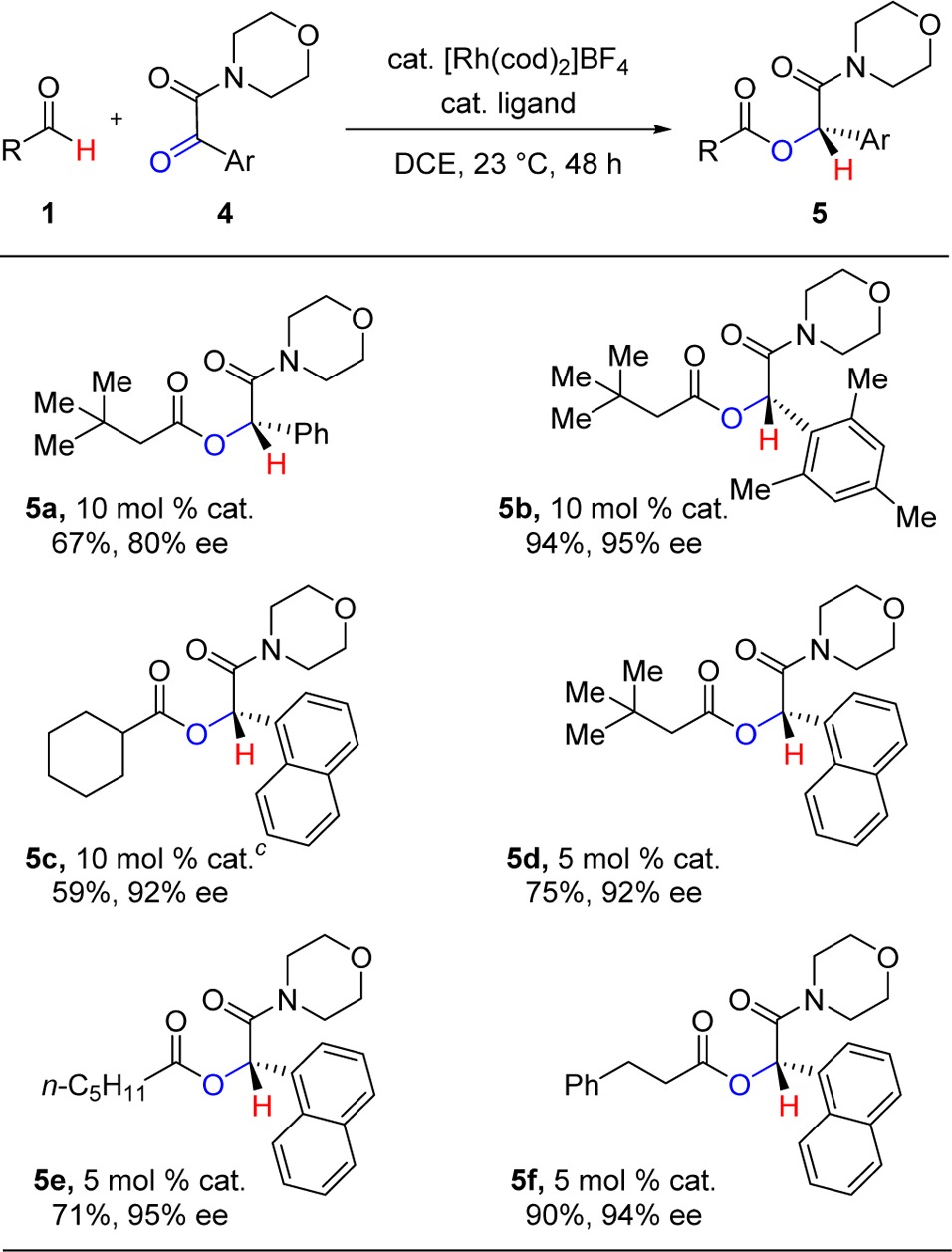
Conditions: 0.12 mmol of 1, 0.1 mmol of 2.
The catalyst was hydrogenated at rt for 45 min prior to adding substrates (see Supporting Information).
At 50 °C, 72 h.
The Rh-L5 catalyst described in this study, however, is not effective with α-keto-Weinreb amide 2k (Scheme 3). To this end, we have identified an alternative rhodium catalyst derived from methoxy-BIPHEP L6, which provides good reactivity and modest enantioselectivity for this transformation to yield ester 6 (93%, 51% ee).
Scheme 3. Ketone Hydroacylation with Weinreb Amide Directing Group.
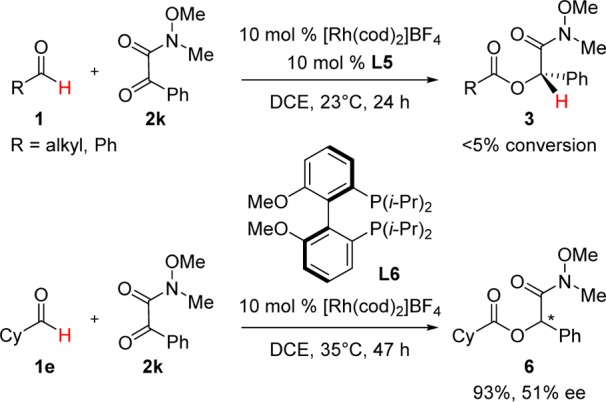
Conditions: 0.12 mmol of 1, 0.1 mmol of 2. The catalyst was hydrogenated at rt for 45 min prior to adding substrates (see Supporting Information).
Investigation of the Kinetics of Intermolecular Hydroacylation
We initiated mechanistic studies by examining the kinetics for the cross-coupling of aldehyde 1d and α-ketoamide 2e by 1H NMR. These particular substrates were chosen because their 1H NMR signals are distinct and no products of decomposition were observed over the course of reaction progress, thus simplifying data analysis. Initial rates for the hydroacylation of 2e were then measured by varying the concentrations of 1d, 2e, and rhodium catalyst. These experiments revealed a first-order dependence of the rate on both aldehyde 1d (Figure 1) and ketone 2e concentrations (Figure 2). More intriguing is the observed second-order rate dependence on catalyst concentration (Figure 3), which suggests the involvement of two rhodium species in the turnover-limiting step. A study on the correlation between the enantiomeric excess of the catalyst and that of the product yielded a small, positive nonlinear relationship (Figure 4).29 Such a phenomenon is consistent with the added degree of complexity in the reaction mechanism as indicated by the kinetic profile.
Figure 1.

Plot of initial rates (kobs) with respect to [aldehyde 1d] showing first-order dependence; [2e] = 0.17 M, [catalyst] = 0.0083 M.
Figure 2.

Plot of initial rates (kobs) with respect to [α-ketoamide 2e] showing first-order dependence; [1d] = 0.20 M, [catalyst] = 0.0083 M.
Figure 3.

Plot of initial rates (kobs) with respect to [catalyst]2 showing second-order dependence; [1d] = 0.20 M, [2e] = 0.17 M.
Figure 4.

Relationship between the enantioselectivity of the reaction and the ee of the chiral catalyst. Each data point was run in duplicate.
The rate data obtained from the kinetics experiments allowed us to calculate initial turnover frequencies. Under standard catalytic conditions using 5 mol % catalyst, 1.0 equiv of ketone, and 1.2 equiv of aldehyde at 25 °C, the initial turnover frequency is determined to be 4.8 × 10–4 s–1.
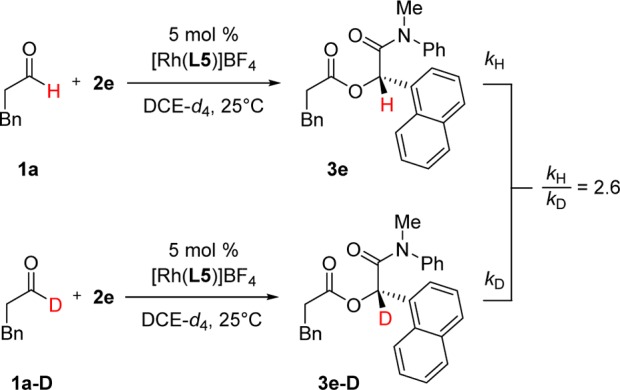
Isotope Labeling Study
To gain further insight into the mechanism, we measured the kinetic isotope effect (KIE) by running two independent, side-by-side experiments using protio-1a and deuterated 1a-D (Scheme 4).30 Aldehyde 1a-D was chosen (over 1d and others) for this study due to ease of isolation. Examination of the initial rates resulted in a KIE of 2.6 and supports C–H bond activation to be turnover-limiting. This value is larger than that previously observed for an intramolecular ketone hydroacylation where insertion was implicated as the turnover-limiting step (KIE = 1.79 ± 0.06).7b,31 The absence of a directing group on the aldehyde in the present system likely increases the barrier to oxidative addition significantly.32,33
Scheme 4. KIE Measurement from Two Parallel Reactions Using Method of Initial Rates.
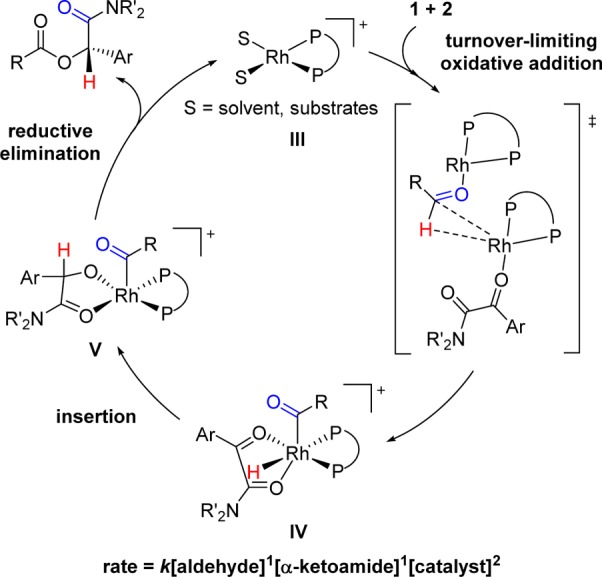
On the basis of the kinetic analysis and KIE, we propose a mechanism invoking homobimetallic activation34 of the aldehyde where one rhodium catalyst acts as a Lewis acid by coordinating the oxygen atom of the aldehyde and a second rhodium catalyst participates in the oxidative addition of the formyl C–H bond to produce acyl-Rh(III)-hydride IV (Scheme 5).35 A report of cationic rhodium complexes behaving as Lewis acid catalysts supports this proposal.36 In addition, Lewis acid additives (i.e., ZnCl2, ZnBr2, ZnI2, etc.) have been found to be beneficial in a study on intermolecular alkene hydroacylation, although their role is unclear.37 The mechanism proceeds with insertion to generate acyl-Rh(III)-alkoxide V, followed by reductive elimination to furnish the product and turnover the Rh(I) catalyst. It is possible for the reaction to occur through bimetallic intermediates following oxidative addition; however, reactive intermediates were not observed spectroscopically.38 We believe that rapid coordination and dissociation of solvent and substrate leads to broadening of the 31P NMR resonance signals and is thus consistent with [Rh(diphosphine)(solvent)2]+III being a resting state.
Scheme 5. Proposed Mechanism via Rate-Limiting Oxidative Addition Catalyzed by Two Rhodium Centers.
Conclusions
By developing a new Rh-Josiphos catalyst, we achieved the first enantioselective intermolecular ketone hydroacylation, which features the rare use of nonchelating aldehydes. Kinetic analysis reveals a second-order rate dependence on the rhodium catalyst and, together with a study on nonlinear relationship and the KIE, points to a unique mechanism involving homobimetallic oxidative addition as the turnover-limiting step. This work provides a foundation for understanding C–H activation and hydroacylation using nonactivated aldehydes which will guide future studies and applications.
Acknowledgments
We thank the National Institutes of Health (GM105938) for funding. K.G.M.K. is grateful for an NSERC CGS-D. D.N.L. is grateful for an NSF Graduate Fellowship. We thank Lauren E. Longobardi for studies using achiral catalysts, K.B. Schenthal for entry 18 (Table 1), Van M.-T. Nguyen for preparing substrate 2a, and Dr. Joseph W. Ziller for X-ray crystallographic analysis.
Supporting Information Available
Experimental procedures, X-ray crystallographic data, characterization data for new compounds, and chiral chromatographic analyses. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Trost B. M. Science 1991, 254, 1471. [DOI] [PubMed] [Google Scholar]; b Trost B. M. Angew. Chem., Int. Ed. Engl. 1995, 34, 259. [Google Scholar]
- For reviews of the Tishchenko reaction, see:; a Seki T.; Nakajo T.; Onaka M. Chem. Lett. 2006, 35, 824. [Google Scholar]; b Törmäkangas O. P.; Koskinen A. M. P. Recent Res. Dev. Org. Chem. 2001, 5, 225. [Google Scholar]; c For an industrial application of carbonyl hydroacylation (Tishchenko reaction), see: Ulrich D.; Jankowski H. Chem. Tech. 1988, 40, 393. [Google Scholar]
- For Ru catalysis, see:; a Horino H.; Ito T.; Yamamoto A. Chem. Lett. 1978, 17. [Google Scholar]; b Ozawa F.; Yamagami I.; Yamamoto A. J. Organomet. Chem. 1994, 473, 265. [Google Scholar]
- For an early example of intramolecular ketone hydroacylation catalyzed by an achiral rhodium complex, see:Bergens S. H.; Fairlie D. P.; Bosnich B. Organometallics 1990, 9, 566. [Google Scholar]
- For Rh catalysis, see:; a Slough G. A.; Ashbaugh J. R.; Zannoni L. A. Organometallics 1994, 13, 3587. [Google Scholar]; b Fuji K.; Morimoto T.; Tsutsumi K.; Kakiuchi K. Chem. Commun. 2005, 3295. [DOI] [PubMed] [Google Scholar]; c Pawley R. J.; Moxham G. L.; Dallanegra R.; Chaplin A. B.; Brayshaw S. K.; Weller A. S.; Willis M. C. Organometallics 2010, 29, 1717. [Google Scholar]
- For Ni catalysis, see:; a Ogoshi S.; Hoshimoto Y.; Ohashi M. Chem. Commun. 2010, 46, 3354. [DOI] [PubMed] [Google Scholar]; b Hoshimo Y.; Ohashi M.; Ogoshi S. J. Am. Chem. Soc. 2011, 133, 4668. [DOI] [PubMed] [Google Scholar]
- a Shen Z.; Khan H. A.; Dong V. M. J. Am. Chem. Soc. 2008, 130, 2916. [DOI] [PubMed] [Google Scholar]; b Shen Z.; Dornan P. K.; Khan H. A.; Woo T. K.; Dong V. M. J. Am. Chem. Soc. 2009, 131, 1077. [DOI] [PubMed] [Google Scholar]; c Khan H. A.; Kou K. G. M.; Dong V. M. Chem. Sci. 2011, 2, 407. [Google Scholar]
- a Sakai K.; Ide J.; Oda O.; Nakamura N. Tetrahedron Lett. 1972, 13, 1287. [Google Scholar]; b Milstein D. J. Chem. Soc., Chem. Commun. 1982, 1357. [Google Scholar]; c Milstein D. Organometallics 1982, 1, 1549. [Google Scholar]; d See ref (4).
- For examples of non-chelation-assisted C—H activation in olefin hydroacylation, see:; a Lenges C. P.; Brookhart M. J. Am. Chem. Soc. 1997, 119, 3165. [Google Scholar]; b Lenges C. P.; White P. S.; Brookhart M. J. Am. Chem. Soc. 1998, 120, 6965. [Google Scholar]; c Roy A. H.; Lenges C. P.; Brookhart M. J. Am. Chem. Soc. 2007, 129, 2082. [DOI] [PubMed] [Google Scholar]; d Tanaka K.; Shibata Y.; Suda T.; Hagiwara Y.; Hirano M. Org. Lett. 2007, 9, 1215. [DOI] [PubMed] [Google Scholar]; e Shibata Y.; Tanaka K. J. Am. Chem. Soc. 2009, 131, 12552. [DOI] [PubMed] [Google Scholar]
- For examples of non-chelation-assisted hydroacylation beyond mechanisms involving C—H activation, see:; a Leung J. C.; Krische M. J. Chem. Sci. 2012, 3, 2202. [Google Scholar]; b Hong Y.-T.; Barchuk A.; Krische M. J. Angew. Chem., Int. Ed. 2006, 45, 6885. [DOI] [PubMed] [Google Scholar]; c Shibahara F.; Bower J. F.; Krische M. J. J. Am. Chem. Soc. 2008, 130, 14120. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Omura S.; Fukuyama T.; Horiguchi J.; Murakami Y.; Ryu I. J. Am. Chem. Soc. 2008, 130, 14094. [DOI] [PubMed] [Google Scholar]; e Williams V. M.; Leung J. C.; Patman R. L.; Krische M. J. Tetrahedron 2009, 65, 5024. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Murphy S. K.; Dong V. M. J. Am. Chem. Soc. 2013, 135, 5553. [DOI] [PubMed] [Google Scholar]; g Chen Q.-A.; Kim D. K.; Dong V. M. J. Am. Chem. Soc. 2014, 136, 3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For NHC catalysis, see:; a Chan A.; Scheidt K. A. J. Am. Chem. Soc. 2006, 128, 4558. [DOI] [PubMed] [Google Scholar]; b Sreenivasulu M.; Kumar K. A.; Reddy K. S.; Kumar K. S.; Kumar P. R.; Kumar K. B.; Chandrasekhar K. B.; Pal M. Tetrahedron Lett. 2011, 52, 727. [Google Scholar]; c Du D.; Lu Y.; Jin J.; Tang W.; Lu T. Tetrahedron 2011, 67, 7557. [Google Scholar]
- Cross-Tishchenko reactions have been demonstrated with thiolate catalysis:; a Cronin L.; Manoni F.; O’Connor C. J.; Connon S. J. Angew. Chem., Int. Ed. 2010, 49, 3045. [DOI] [PubMed] [Google Scholar]; b O’Connor C. J.; Manoni F.; Curran S. P.; Connon S. J. New J. Chem. 2011, 35, 551. [Google Scholar]
- For a selenide-catalyzed cross-Tishchenko reaction, see:Curran S. P.; Connon S. J. Org. Lett. 2012, 14, 1074. [DOI] [PubMed] [Google Scholar]
- Coulter M. M.; Kou K. G. M.; Galligan B.; Dong V. M. J. Am. Chem. Soc. 2010, 132, 16330. [DOI] [PubMed] [Google Scholar]
- Phan D. H. T.; Kim B.; Dong V. M. J. Am. Chem. Soc. 2009, 131, 15608. [DOI] [PubMed] [Google Scholar]
- The trans-effect of unsymmetrical bidentate ligands in Pd-catalyzed allylic substitutions has been reported:Tu T.; Zhou Y.-G.; Hou X.-L.; Dai L.-X.; Dong X.-C.; Yu Y.-H.; Sun J. Organometallics 2003, 22, 1255. [Google Scholar]
- Hasanayn F.; Achord P.; Braunstein P.; Magnier H. J.; Krough-Jesperson K.; Goldman A. S. Organometallics 2012, 31, 4680. [Google Scholar]
- Measures were taken to ensure that the reported ee is representative of the reaction and not an artifact of self-disproportionation of enantiomers (see Supporting Information). For an example of self-disproportionation of enantiomers during achiral phase silica gel chromatography, see:Soloshonok V. A. Angew. Chem., Int. Ed. 2006, 45, 766. [DOI] [PubMed] [Google Scholar]
- a Passerini M. Gazz. Chim. Ital. 1921, 51, 126. [Google Scholar]; b Passerini M. Gazz. Chim. Ital. 1921, 51, 181. [Google Scholar]; c Passerini M.; Ragni G. Gazz. Chim. Ital. 1931, 61, 964. [Google Scholar]
- For enantioselective Passerini-type reactions, see:; a Denmark S. E.; Fan Y. J. Am. Chem. Soc. 2003, 125, 7825. [DOI] [PubMed] [Google Scholar]; b Denmark S. E.; Fan Y. J. Org. Chem. 2005, 70, 9667. [DOI] [PubMed] [Google Scholar]
- a Wang S.-X.; Wang M.-X.; Wang D.-X.; Zhu J. Angew. Chem., Int. Ed. 2008, 47, 388. [DOI] [PubMed] [Google Scholar]; b Yue T.; Wang M.-X.; Wang D.-X.; Zhu J. Angew. Chem., Int. Ed. 2008, 47, 9454. [DOI] [PubMed] [Google Scholar]; c Yue T.; Wang M.-X.; Wang D.-X.; Masson G.; Zhu J. J. Org. Chem. 2009, 74, 8396. [DOI] [PubMed] [Google Scholar]
- For Rh-catalyzed hydrogenation of α-ketoamides, see; a Carpentier J.-F.; Mortreux A. Tetrahedron: Asymmetry 1997, 8, 1083. [Google Scholar]; b Pasquier C.; Pélinski L.; Brocard J.; Mortreux A.; Agbossou-Niedercorn F. Tetrahedron Lett. 2001, 42, 2809. [Google Scholar]
- For a Ru-catalyzed reduction of α-ketoamides, see:Broger E. A.; Burkart W.; Hennig M.; Scalone M.; Schmid R. Tetrahedron: Asymmetry 1998, 9, 4043. [Google Scholar]
- For reductions of α-ketoamides via biocatalysis, see:; a Hata H.; Shimizu S.; Hattori S.; Yamada H. J. Org. Chem. 1990, 55, 4377. [Google Scholar]; b Ishihara K.; Yamamoto H.; Mitsuhashi K.; Nishikawa K.; Tsuboi S.; Tsuji H.; Nakajima N. Biosci. Biotechnol. Biochem. 2004, 68, 2306. [DOI] [PubMed] [Google Scholar]; c Ishihara K.; Nishimura M.; Nakashima K.; Machii N.; Miyake F.; Nishi M.; Yoshida M.; Masuoka N.; Nakajima N. Biochem. Insights 2010, 3, 19. [Google Scholar]; d Ishihara K.; Nagai H.; Takahashi K.; Nishiyama M.; Nakajima N. Biochem. Insights 2011, 4, 29. [Google Scholar]; e Nakayama G. R.; Schultz P. G. J. Am. Chem. Soc. 1992, 114, 780. [Google Scholar]; f Patel R. N.; Chu L.; Chidambaram R.; Zhu J.; Kant J. Tetrahedron: Asymmetry 2002, 13, 349. [Google Scholar]; g Stella S.; Chadha A. Catal. Today 2012, 198, 345. [Google Scholar]
- Wang K.; Emge T. J.; Goldman A. S.; Li C.; Nolan S. P. Organometallics 1995, 14, 4929. [Google Scholar]
- The absolute configuration of 3c was assigned by X-ray crystallography. The absolute configurations of related products are assigned by analogy. See Supporting Information for details.
- Preliminary DFT studies using N-methylisatin as a model for the α-ketoamide to simplify calculations show that having the acyl ligand on the “bottom” apical position is 5.1 kcal/mol lower in energy than having the acyl ligand on the “top” apical position (see Supporting Information).
- a Kurosu M.; Kishi Y. Tetrahedron Lett. 1998, 39, 4793. [Google Scholar]; b Badioli M.; Ballini R.; Bartolacci M.; Bosica G.; Torregiani E.; Marcantoni E. J. Org. Chem. 2002, 67, 8938. [DOI] [PubMed] [Google Scholar]; c Kochi T.; Ellman J. A. J. Am. Chem. Soc. 2004, 126, 15652. [DOI] [PubMed] [Google Scholar]
- a Kagan H. B. Adv. Synth. Catal. 2001, 343, 227. [Google Scholar]; b Girard C.; Kagan H. B. Angew. Chem., Int. Ed. 1998, 37, 2922. [DOI] [PubMed] [Google Scholar]; c Blackmond D. G. Acc. Chem. Res. 2000, 33, 402. [DOI] [PubMed] [Google Scholar]
- Simmons E. M.; Hartwig J. F. Angew. Chem., Int. Ed. 2012, 51, 3066. [DOI] [PubMed] [Google Scholar]
- Gómez-Gallego M.; Sierra M. A. Chem. Rev. 2011, 111, 4857. [DOI] [PubMed] [Google Scholar]
- Chung L. W.; Wiest O.; Wu Y. J. Org. Chem. 2008, 73, 2649. [DOI] [PubMed] [Google Scholar]
- Wang F.; Meng Q.; Li M. Mol. Simul. 2008, 34, 515. [Google Scholar]
- a Martinez L. E.; Leighton J. L.; Carsten D. H.; Jacobsen E. N. J. Am. Chem. Soc. 1995, 117, 5897. [Google Scholar]; b Hansen K. B.; Leighton J. L.; Jacobsen E. N. J. Am. Chem. Soc. 1996, 118, 10924. [Google Scholar]; c Sammis G. M.; Danjo H.; Jacobsen E. N. J. Am. Chem. Soc. 2004, 126, 9928. [DOI] [PubMed] [Google Scholar]
- For an example of rhodium behaving as a dual-role catalyst, see:Dornan P. K.; Kou K. G. M.; Houk K. N.; Dong V. M. J. Am. Chem. Soc. 2014, 136, 291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias E. L.; Brookhart M.; White P. S. Chem. Commun. 2001, 423. [DOI] [PubMed] [Google Scholar]
- Inui Y.; Tanaka M.; Imai M.; Tanaka K.; Suemune H. Chem. Pharm. Bull. 2009, 57, 1158. [DOI] [PubMed] [Google Scholar]
- Rhodium-catalyzed hydroacylation has been described as a “black box” due to difficulties in detecting reaction intermediates:; a Fairlie D. P.; Bosnich B. Organometallics 1988, 7, 946. [Google Scholar]; b See ref (4).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.