

Abstract
This study compared the complete genome sequences of 16 NL63 strain human coronaviruses (hCoVs) from respiratory specimens of paediatric patients with respiratory disease in Colorado, USA, and characterized the epidemiology and clinical characteristics associated with circulating NL63 viruses over a 3-year period. From 1 January 2009 to 31 December 2011, 92 of 9380 respiratory specimens were found to be positive for NL63 RNA by PCR, an overall prevalence of 1 %. NL63 viruses were circulating during all 3 years, but there was considerable yearly variation in prevalence and the month of peak incidence. Phylogenetic analysis comparing the genome sequences of the 16 Colorado NL63 viruses with those of the prototypical hCoV-NL63 and three other NL63 viruses from the Netherlands demonstrated that there were three genotypes (A, B and C) circulating in Colorado from 2005 to 2010, and evidence of recombination between virus strains was found. Genotypes B and C co-circulated in Colorado in 2005, 2009 and 2010, but genotype A circulated only in 2005 when it was the predominant NL63 strain. Genotype C represents a new lineage that has not been described previously. The greatest variability in the NL63 virus genomes was found in the N-terminal domain (NTD) of the spike gene (nt 1–600, aa 1–200). Ten different amino acid sequences were found in the NTD of the spike protein among these NL63 strains and the 75 partial published sequences of NTDs from strains found at different times throughout the world.
Introduction
Respiratory tract infections cause the greatest burden of disease worldwide, surpassing that of human immunodeficiency virus infection, malaria, cancer and heart disease. The World Health Organization estimates that respiratory tract infections are the leading cause of death in low-income countries (WHO, 2003, 2008). Even in the USA, respiratory tract infections cause more disease and death than any other type of infection, and over the past 50 years there has been little decrease in mortality associated with lung infections (Mizgerd, 2006, 2008). Over the past decade, eight novel respiratory viral pathogens have been discovered (Allander et al., 2005, 2007; Dominguez et al., 2008; Gaynor et al., 2007; Lamson et al., 2006; Lau et al., 2007; McErlean et al., 2007; van den Hoogen et al., 2001), including three new human coronaviruses (hCoVs) (Drosten et al., 2003; van der Hoek et al., 2004; Woo et al., 2005).
There are now five known hCoV species, exemplified by the prototypical strains hCoV-229E, hCoV-OC43, hCoV-HKU1, severe acute respiratory syndrome (SARS)-CoV and hCoV-NL63. Strains within each of these hCoV species, except for SARS-CoV, cause worldwide seasonal epidemics of respiratory disease every year. With the possible exception of hCoV-OC43, humans are the only known host for these CoVs. The antigenicity, clinical presentations and disease severity of CoVs of many mammalian and avian species can be affected by mutations that cause amino acid substitutions, deletions or insertions, and by recombination between viral RNA genomes during co-infections. The prototype hCoV-NL63 strain (NL63/AMS/2004/1) was identified in 2004, and NL63 viruses have subsequently been shown to have a worldwide distribution (Fouchier et al., 2004; van der Hoek et al., 2004). Retrospective epidemiological studies by our laboratory and others suggest that the prevalence of NL63 viruses in children with respiratory illness ranges from 2 to 9 %, and that these NL63 viruses are associated with asymptomatic infections and mild upper respiratory tract infections, as well as with pneumonia, bronchiolitis and croup (Arden et al., 2005; Bastien et al., 2005a, b; Chiu et al., 2005; Dominguez et al., 2009; Ebihara et al., 2005; Esper et al., 2005; Kaiser et al., 2005; Kuypers et al., 2007; Prill et al., 2012; Suzuki et al., 2005; Vabret et al., 2005; van der Hoek et al., 2005). Until recently, it has been difficult or impossible to isolate NL63 viruses from clinical specimens in continuous cell lines, although the prototype NL63 strain has been propagated on LLC-MK2 cells (van der Hoek et al., 2004) and in primary, differentiated human bronchial–tracheal respiratory epithelial cells cultured at the air–liquid interface (Banach et al., 2009).
To date, only four full-genome sequences of NL63 viruses have been deposited in GenBank, and all of these sequences are from clinical specimens collected in the Netherlands (Fouchier et al., 2004; Pyrc et al., 2006; van der Hoek et al., 2004). Analysis of these genome sequences identified two distinct genotypes, A and B, of NL63 viruses with an overall genome identity of 99 %, and one recombinant between these strains (Pyrc et al., 2006). Here, we report the epidemiology and disease association of NL63 viruses over a 3-year period (2009–2011) at the Children’s Hospital Colorado (CO, USA). Sequencing the NL63 genomes from 23 of our clinical specimens yielded 16 full-length genomes (>99 % coverage) and identified genomic signatures of at least three distinct circulating genotypes of NL63 and one recombinant. These genomes are the first NL63 genomes from the western hemisphere and broaden our understanding of the complexity of NL63 hCoVs circulating globally.
Results
Clinical epidemiology
In order to determine the yearly and seasonal variations in the prevalence of NL63 virus infection in our paediatric patient population, specimens were analysed over a 3-year period. From 1 January 2009 to 31 December 2011, 92 of 9380 respiratory specimens were found to be positive for NL63 viral RNA by multiplex PCR (see Methods), an overall prevalence of 1 %. NL63 viruses were circulating in the paediatric population during all three of the study years (Fig. 1), primarily during the winter months from December to March. However, there was considerable yearly variation in prevalence and the month of peak incidence. During December 2010 and January 2011, there was a much higher incidence of NL63, with 29 and 21 positive specimens, accounting for 10 and 6 % of all specimens tested, respectively. Of the 92 respiratory specimens positive for NL63 viral RNA during this study period, 33 % were also positive for an additional virus. The most common co-infections were human rhinoviruses (13 %), respiratory syncytial virus (8 %), adenoviruses (5 %) and hCoV-229E (5 %).
Fig. 1.
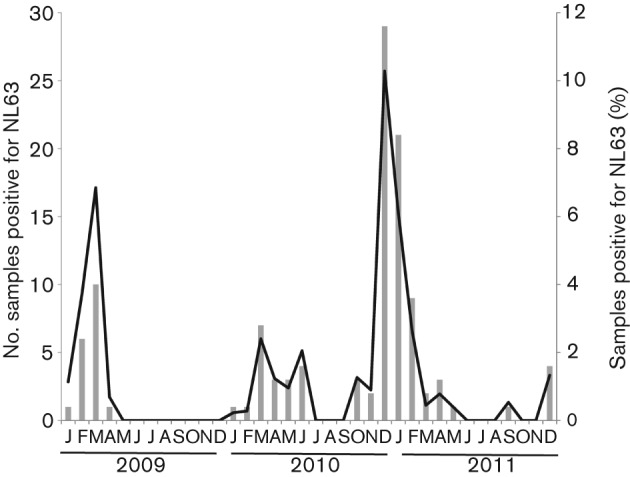
Number and percentage of NL63-positive respiratory specimens detected each month during the 3-year study period.
Of the children positive for NL63 viruses, 42 % were under 12 months of age and 56 % were under 2 years of age. Seventy-one per cent of the NL63-positive patients were admitted to the hospital and 51 % had an underlying medical condition. To characterize the clinical syndromes associated with NL63 viruses, we analysed the primary discharge diagnoses given by the care providers to the patients with NL63-positive respiratory specimens. The most common diagnosis were viral syndrome or upper respiratory tract infection (n = 27), fever and neutropenia (n = 9), croup (n = 9), bronchiolitis (n = 8), rule out sepsis/fever in the neonate (n = 6) and pneumonia (n = 5). The most common symptoms in the NL63-positive patients were fever (73 %), cough (41 %) and hypoxia (34 %). The clinical characteristics of the patients associated with the NL63 genomes that were fully sequenced are presented in Table 1.
Table 1. Clinical characteristics of sequenced Denver NL63 viruses.
The genotypes of the 2010 strains and NL63/DEN/2005/291 are based on the S sequence only. ALTE, Acute life-threatening event; CLD, chronic lung disease; CF, cystic fibrosis; F&N, fever and neutropenia; KD, Kawasaki disease; PCKD, polycystic kidney disease; Unk, unknown; VSD, ventricular septal defect.
| NL63 isolate | NL63genotype | Sampledate | Sex | Admitted | Symptom(s) | Underlyingmedicalcondition(s) | Dischargediagnosis | Age(months) |
| NL63/DEN/2005/193 | A | 1/11/2005 | M | Y | Apnoea andhypoxia | None | Rule out sepsis | 1 |
| NL63/DEN/2005/271 | A | 1/23/2005 | F | N | Fever, rash | None | Viral syndrome | 24 |
| NL63/DEN/2005/347 | A | 2/1/2005 | M | N | Fever | None | Viral syndrome | 53 |
| NL63/DEN/2005/449 | A | 2/9/2005 | F | N | Fever, sore throat | PCKD | Viral syndrome | 90 |
| NL63/DEN/2005/1062 | A | 4/12/2005 | M | Y | Fever, apnoea,sore throat | None | ALTE | 7 |
| NL63/DEN/2005/1120 | A | 4/25/2005 | M | Y | Apnoea,hypoxia,seizures | None | ALTE | 2 |
| NL63/DEN/2005/1876 | A | 11/21/2005 | F | Y | Apnoea,congestion | None | ALTE | 5 |
| NL63/DEN/2005/232 | B | 1/18/2005 | M | N | Fever, seizures,cough, congestion | None | Seizures and viral syndrome | 221 |
| NL63/DEN/2005/235 | B | 1/19/2005 | M | Y | Fever, cough | Hepatoblastoma | F&N | 5 |
| NL63/DEN/2009/9 | B | 3/16/2009 | M | Y | Cough, emesis | VSD | Viral syndrome | 12 |
| NL63/DEN/2009/14 | B | 3/1/2009 | M | Y | Fever, cough,congestion | None | KD | 36 |
| NL63/DEN/2009/15 | B | 2/13/2009 | M | Y | Fever, emesis,diarrhoea | Immunodeficiency | Viral syndrome | 22 |
| NL63/DEN/2009/22 | B | 3/3/2009 | M | N | Unk | Unk | Unk | Unk |
| NL63/DEN/2010/25 | B | 12/23/2010 | M | Y | Hypoxia,tachypnea | CF | CF exacerbation | 7 |
| NL63/DEN/2010/36 | B | 12/30/2010 | M | N | Fever, cough,congestion | None | Bronchiolitis | 3 |
| NL63/DEN/2005/291 | B | 1/26/2005 | M | Y | Fever, cough,hypoxia | Genetic syndrome | Pneumonia | 54 |
| NL63/DEN/2005/1862 | C | 11/1/2005 | M | N | Unk | Unk | Unk | Unk |
| NL63/DEN/2008/16 | C | 1/8/2008 | F | N | Unk | Unk | Unk | Unk |
| NL63/DEN/2009/6 | C | 2/25/2009 | M | Y | Difficulty breathing | Laryngomalacia | Mucous plugging | 97 |
| NL63/DEN/2009/20 | C | 3/12/2009 | F | Y | Hypoxia,tachypnea | None | Viral syndrome | 8 |
| NL63/DEN/2010/20 | C | 12/15/2010 | M | Y | Fever, cough,congestion | None | Rule out sepsis | 1 |
| NL63/DEN/2010/28 | C | 12/3/2010 | F | Y | Fever, hypoxia | Prematurity, CLD | Viral syndrome | 10 |
| NL63/DEN/2010/31 | C | 12/11/2010 | M | Y | Fever, cough,stridor | None | Croup | 32 |
| NL63/DEN/2010/35 | C | 12/14/2010 | F | Y | Fever, cough,anorexia | Hypotonia | Viral syndrome | 7 |
| NL63/DEN/2009/31 | R | 2/21/2009 | M | Y | Respiratorydistress | Tonsillectomy | Viral syndrome | 33 |
Two patients had multiple specimens positive for NL63 viral RNA over an extended period of time. Both of these patients were immunocompromised with underlying oncological diagnoses. The first patient had six NL63-positive respiratory specimens over a 5-month period and the second had nine NL63-positive specimens over a 10-month period.
Genome sequences, phylogenetic analysis and genotypes
Complete or nearly complete genome sequences were obtained for 19 of the 23 samples submitted for sequencing. All samples submitted for sequencing were positive only for NL63 and no other viruses were detected in these samples. The sequences ranged from 27 456 to 27 495 nt, lacking only short sequences at the 3′ and 5′ termini (the original NL63 isolate was 27 553 nt) and with some having one or more small gaps within the final pseudomolecule. Poor template quality precluded further sequence analysis of four samples. The nearly complete genome sequences consisted of 2–18 contigs after two rounds of directed closure, and these were listed as ‘draft’. However, only those in two or fewer contigs (n = 16) were included in the whole-genome analysis. All full-length genomes assembled into single contigs, except for NL63/DEN/2009/6 and NL63/DEN/2009/31, which remained in two contigs with small intervening gaps. The absence of ambiguous bases in these contigs strongly suggested (but does not prove) that each of the samples sequenced contained one predominant NL63 genome and thus that the two contigs arose from different parts of the same genome. The G+C content of the 16 complete genomes was 34.5 mol%, considerably lower than that of other alpha-CoVs, which range from 39 to 42 mol% G+C (Woo et al., 2010).
The overall genome organization was identical for all 16 NL63 virus specimens and identical to the four known NL63 sequences from the Netherlands, and was similar to that of other alpha-CoVs. The putative core sequence (CS) of the transcription regulatory sequence (TRS) motif 5′-CUAAAC-3′ (Sola et al., 2005; Woo et al., 2009, 2010; Zuñiga et al., 2004) was found at the 3′ end of the leader sequence and preceded each of the translated ORFs. The CS upstream of the spike (S) and envelope (E) coding regions, however, was slightly modified to 5′-CUAAUC-3′ and 5′-CUAUAC-3′, respectively. The CSs of the membrane (M) and nucleocapsid (N) genes were found in duplicate, with the terminal C of the first sequence serving as the starting C of the second sequence (5′-CUAAACUAAAC-3′). The CS and TRS sequences in all of the genes were conserved across all the full-length NL63 genomes.
A phylogenetic tree comparing the genome sequences of the 16 Colorado NL63 strains combined with the four full-length NL63 genomes found in the Netherlands is shown in Fig. 2. These 20 NL63 strains fell into three genotypes, designated genotypes A (seven strains), B (six strains) and C (four strains). Genotypes A and B were identified previously in European clinical specimens (Fouchier et al., 2004; Pyrc et al., 2006; van der Hoek et al., 2004). Two of the strains previously sequenced from the Netherlands and one of the Colorado strains formed a fourth group, which probably represents recombinants of the other genotypes (Fig. 2). vista analysis using the prototypical NL63 isolate from 2004 as the reference showed that the greatest areas of dissimilarity between the genotypes were from 1.0 to 3.5 kb [1a and non-structural protein 3 (nsp3) genes] and from 21.5 to 23.5 kb (S gene) (Fig. 3 and Figs S1 and S2, available in JGV Online). Fig. 3 clearly illustrates regions of the NL63 genome that had large areas of divergent sequences, shows how these contribute to the genotype clusters (A, B and C) of the NL63 full-genome sequences and illustrates how these markers can be useful for identifying potential recombinants between these genotypes. (The single thin peak near the 5′ end of the NL63/DEN/2009/31 genome and the peak near the beginning of the S gene of NL63/DEN/2009/6 represent areas of poor sequence coverage due to the presence of two contigs for these sequences.) The degree of phylogenetic differences between each ORF of the 20 NL63 genomes is pictured schematically in a heat map (Fig. 4). This provides another way of visualizing recombination along the length of the genomes. It shows a pairwise comparison of the shapes of the phylogenetic trees constructed separately for each coding sequence. Where the pairwise comparisons were highly similar (blue), these genes had the same phylogenetic relationship, and where different (yellow), the corresponding regions of the genomes had different phylogenetic relationships. The data suggested that, during the evolution of NL63 viruses, recombination events have contributed to the diversity of circulating virus strains.
Fig. 2.
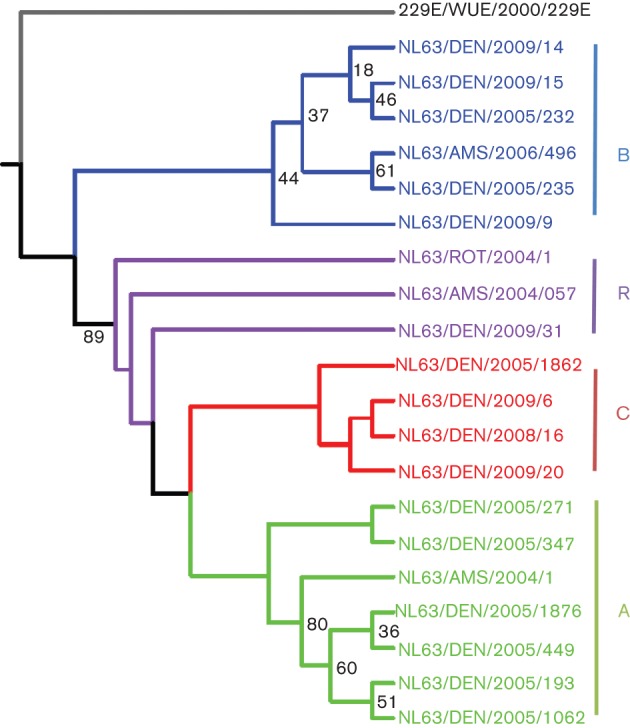
Phylogenetic analysis of all known full genomes of NL63 viruses isolated in different years from Denver (DEN), Amsterdam (AMS) and Rotterdam (ROT). The viral sequences fell into three clusters (genotypes A, B and C). The viruses shown in purple (R) probably represent recombinants. Bootstrap percentage values are shown on some of the nodes. The 229E reference strain (grey) from Wuerzberg (WUE) was used as an outlier (GenBank accession no. NC_002645).
Fig. 3.
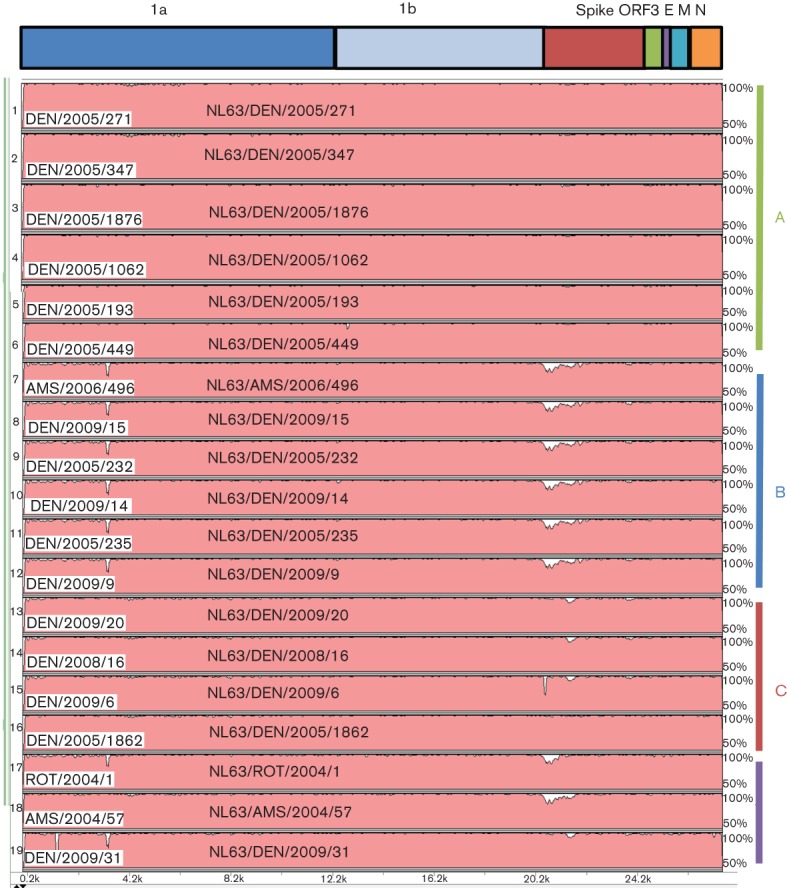
vista plot of NL63-related viral genomes using the original Netherland isolate (NL63/AMS/2004/1) as the comparison strain. Genomes numbers 1–6 correspond to genotype A (green in Fig. 2), numbers 7–12 correspond to genotype B (blue in Fig. 2), numbers 13–16 correspond to genotype C (red in Fig. 2) and numbers 17–19 correspond to the purple group in Fig. 2, most likely representing recombinants.
Fig. 4.
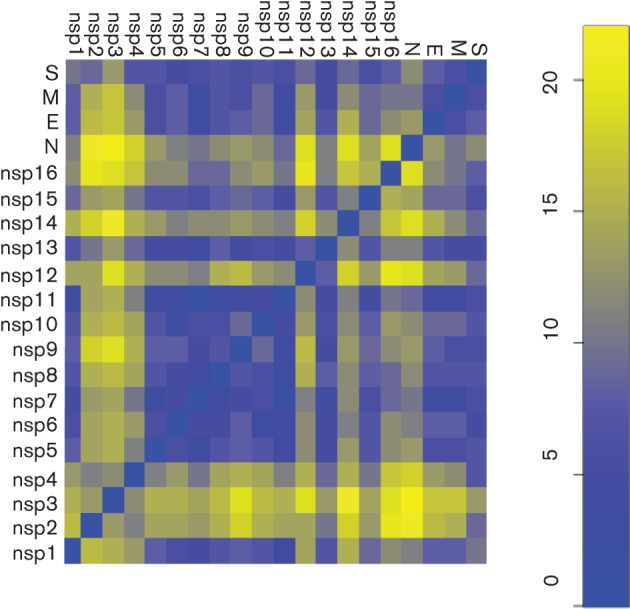
Comparison of phylogenetic tree topologies of genes of the 20 NL63 viruses. Gene tree topologies were compared in a pairwise fashion using the Robinson–Foulds topological distance DRF(a,b) calculation. The distance matrix DRF was visualized and converted into a colour representation using the r statistical package. Dark blue indicates identical phylogenetic trees. The scale on the right indicates the relative degree of phylogenetic dissimilarity, increasing from blue to yellow.
As reported previously for the Netherlands NL63 sequences (Pyrc et al., 2006), we found two signature deletions in our multiple sequence alignments, one in-frame deletion of 15 nt located between nt 3310 and 3350 in gene 1a (nsp3) and a 3 nt deletion located between nt 20 798 and 28 000 within the S gene (S1 subunit). These signature deletions segregated with the different clades. In Fig. 2, the strains coloured in purple differed from this overall pattern (some had both the 1a and S gene deletions, whilst others had neither), indicating that these markers may have swapped between genomes after strain divergence.
Based on the vista analysis, we examined the nucleotide sequences of the nsp3 and S genes for potential recombination sites. Looking at representative sequences from each of the genotypes, genotype A (NL63/DEN/2005/1876) and genotype C (NL63/DEN/2009/20) were similar in sequence, with genotype B (NL63/DEN/2009/14) exhibiting a slightly divergent sequence until around nt 1030 of nsp3 (nt 4014 in the whole genome of reference strain NL63/AMS/2004/1). After nt 1030, genotypes B and C had similar sequences, with genotype A having a more divergent sequence. Similarly, in the S gene, genotypes A and C had similar sequences until around nt 920 (nt 21 392 in the whole genome of reference strain NL63/AMS/2004/1) where their sequences diverged. From nt 920 to 1710, the three genotypes had relatively divergent sequences. Following nt 1710, genotypes B and C had relatively similar sequences. This segregation of nucleotide sequences was maintained at the amino acid level in nsp3 but only in the first part of the S gene.
Associations between the date of detection, age, sex, type and severity of clinical disease, and the presence of underlying medical conditions among the NL63 genotypes were analysed (Table 1). Included in this analysis were six additional respiratory specimens from the 2010–2011 season for which the entire S gene was sequenced. Genotypes B and C co-circulated in Colorado in 2005, 2009 and 2010, but genotype A was detected only in 2005 when it was the predominant NL63 strain. Unexpectedly, all eight of the patients with genotype B were male. Whether this represents a true association or a statistical anomaly will require sequencing of additional NL63 specimens. Genotype A was more commonly associated with a diagnosis of apnoea or acute life-threatening events than the other NL63 genotypes.
Bootscan analysis
From the clustering identified in Fig. 2, we created four groups, labelled A–C and R. Fig. 5 shows the bootscans for these four groups using the unique Denver genotype C group as reference (actually called query by the Simplot program). Deviations from the mean (spikes up or down) showed regions of the genome where the query genomes were more or less similar to the reference genome.
Fig. 5.

Bootscan analysis of the three NL63 viral genotypes and possible recombinants. Bootstrapped phylogenetic trees were produced for overlapping window segments of 200 bp. The mean bootstrap value across all members of the query group segment versus the remaining three groups was plotted against window position. Comparisons are shown by colour, as represented in Fig. 2 (genotype A, green; genotype B, blue; recombinants, purple) compared with the unique Denver genotype C as the query group.
Using all of the available NL63 genome sequences for analysis, the ratios of synonymous (Ka) to non-synonymous (Ks) amino acids for the various coding regions were calculated (Table 2). The highest Ka : Ks ratio in the NL63 genomes was observed in the S gene (0.272), followed by the nsp3 (0.250) and nsp2 (0.194) genes, strongly indicating that these gene products are under the strongest selection pressure.
Table 2. Estimation of amino acid substitution rates (Ka and Ks) in known full-length genomes of NL63 viruses.
| ORF | No. sequences | Ka | Ks | Ka : Ks |
| nsp1 | 24 | 0.005 | 0.041 | 0.122 |
| nsp2 | 21 | 0.006 | 0.031 | 0.194 |
| nsp3 | 22 | 0.005 | 0.020 | 0.250 |
| nsp4 | 23 | 0.002 | 0.020 | 0.100 |
| nsp5 | 23 | 0.001 | 0.009 | 0.111 |
| nsp6 | 24 | 0.001 | 0.012 | 0.083 |
| nsp7 | 24 | 0.001 | 0.010 | 0.100 |
| nsp8 | 24 | 0.001 | 0.015 | 0.067 |
| nsp9 | 24 | 0.000 | 0.018 | 0.000 |
| nsp10 | 24 | 0.000 | 0.040 | 0.000 |
| nsp11 | 24 | 0.000 | 0.016 | 0.000 |
| nsp12 | 21 | 0.000 | 0.011 | 0.000 |
| nsp13 | 23 | 0.000 | 0.008 | 0.000 |
| nsp14 | 22 | 0.001 | 0.016 | 0.063 |
| nsp15 | 22 | 0.001 | 0.013 | 0.077 |
| nsp16 | 24 | 0.001 | 0.028 | 0.036 |
| S | 19 | 0.041 | 0.151 | 0.272 |
| ORF3 | 22 | 0.001 | 0.021 | 0.048 |
| E | 24 | 0.000 | 0.019 | 0.000 |
| M | 23 | 0.001 | 0.012 | 0.083 |
| N | 24 | 0.002 | 0.016 | 0.125 |
Comparative sequence analysis of the S gene among NL63 viruses
Because the S gene was identified as the most variable region of the genome, we analysed more closely the S gene from our NL63-positive samples and sequenced this gene from six additional Colorado clinical specimens for further comparison. As reported previously, the greatest variability in the S gene was found in the N-terminal domain (NTD) of the S gene (nt 1–600, aa 1–200). We found ten different amino acid sequences in the NTD of our NL63 sequences and the 75 NL63 NTD S sequences in GenBank (Fig. S1). Although sequences outside the S gene were not available for the majority of these specimens, five of the ten different NTD sequences were most similar to genotype A. Among these ten different NTD sequences, there were 29 amino acid differences (15 %) (Fig. 6). Within the NTD, there were eight potential N-linked glycosylation sites (NXS or NXT), five of which were conserved in all NL63 strains. NL63 viruses containing each of the ten NTD sequence variants have been detected at different times throughout the world in the Netherlands, Belgium, Sweden, Hong Kong and the USA (Denver) (Chiu et al., 2005; Fouchier et al., 2004; Koetz et al., 2006; Leung et al., 2009; Moës et al., 2005; Pyrc et al., 2006; van der Hoek et al., 2004).
Fig. 6.

Amino acid sequence alignment of the NTDs of the S proteins (aa 10–177 shown) of representatives of all published NL63 S genes. In addition to the NTDs from the full genomes shown in Figs 2 and 3, NTDs from partial NL63 sequences from GenBank are shown (black). Potential N-glycosylation sites are underlined.
Discussion
This is the first study to describe 16 complete NL63 genome sequences from clinical respiratory specimens in North America. We found that there have been at least three distinct genotypes of NL63 viruses (A, B and C) circulating in Colorado, USA. A few ‘unusual strains’ (NL63/DEN/2009/31, NL63/ROT/2004/1 and NL63/AMS/2004/057) displayed incongruent phylogenetic positions, with some regions more like genotype B and others more like genotype C. These strains probably represent recombinants between genotypes.
In agreement with other studies (Arden et al., 2005; Bastien et al., 2005a, b; Chiu et al., 2005; Dominguez et al., 2009; Ebihara et al., 2005; Esper et al., 2005; Kaiser et al., 2005; Kuypers et al., 2007; Prill et al., 2012; Suzuki et al., 2005; Vabret et al., 2005; van der Hoek et al., 2005), only a small percentage (1 %) of all of the samples submitted for respiratory testing to the clinical virology laboratory during our 3-year study period were positive for NL63 RNA. The prevalence of NL63 infection, however, varied markedly from year to year, with a peak/mini-outbreak during the winter of 2010–2011 when 10 % of respiratory specimens submitted during December and January were positive for NL63 RNA. Our previous study also detected a high prevalence of NL63 viruses circulating during the 2004–2005 respiratory season, with 4 % of all samples positive for NL63 viruses during the months of January–March (Dominguez et al., 2009). These data are in agreement with other multi-year studies and support the notion that the prevalence of NL63, like other human CoVs, varies markedly from year to year (Dare et al., 2007; Gaunt et al., 2010; Gerna et al., 2007; Lau et al., 2006; Talbot et al., 2009; Vabret et al., 2008). Similarly, serological studies have shown yearly variations in the prevalence of hCoV-299E and hCoV-OC43 infection (Monto & Lim, 1974). Interestingly, it was shown recently that infection with one hCoV may elicit cross-protective immunity from subsequent infection with a different CoV in the same antigenic group (Dijkman et al., 2012).
We examined the epidemiology and clinical presentations associated with the different genotypes of NL63 viruses in Colorado. Genotype A was identified only in clinical samples from 2005 and was the predominant NL63 strain circulating in Colorado in that year. Although detected in all of the years studied, genotypes B and C were the predominant NL63 strains circulating in 2009 and 2010. These data suggest that there is annual variation in the prevalence of NL63 and suggest antigenic variation similar to that seen for other respiratory viruses, most notably influenza. We did not find a clear association of clinical presentation with particular genotypes of NL63, although genotype A was more commonly detected in patients with apnoea or acute life-threatening events.
It is unclear how long CoV shedding can persist in the human respiratory tract following natural infection. We previously detected HCoV-229E RNA in respiratory specimens from an immunocompromised child for 11 weeks (Dominguez et al., 2007). Here, we have reported prolonged shedding of NL63 for at least 5 and 10 months in two immunocompromised patients, suggesting that persistent infection of immunocompromised patients may provide a reservoir for adaptation and/or further spread of NL63.
CoV transcription proceeds via a discontinuous RNA synthesis step guided by the TRS. The TRS contains a CS within a hairpin structure essential for efficient production of subgenomic mRNAs (Dufour et al., 2011; Sola et al., 2005; Zuñiga et al., 2004). The CSs are conserved among all unique sequences of NL63 genomes and within all subgenomic RNAs, except for the E and S genes. The CSs of the E and S genes may alter the RNA secondary structures, affecting the production of subgenomic mRNAs. This might explain why the levels of expression of subgenomic mRNAs encoding S and E are lower than expression of ORF3 (Pyrc et al., 2004). In contrast, the M and N genes are preceded by tandem duplicates of CSs, which could affect the production of subgenomic mRNAs. As the M protein is essential for virus budding and the N protein is necessary for packaging viral RNA, this most likely represents a mechanism for increased production.
Due to their discontinuous RNA replication and their very long (~30 kb) RNA genomes, in vivo recombination occurs in many CoVs and can lead to the generation of new recombinant strains or genotypes, and may alter their virulence, immunogenicity and viral host range. For example, feline coronavirus (FCoV) type II apparently originated from recombination between FCoV type I strains and canine CoVs (Herrewegh et al., 1998). Similarly, recombination between genomes of different bat CoVs has resulted in the emergence of novel SARS-related bat and civet CoVs (Lau et al., 2010). Through whole-genome sequencing of 22 hCoV-HKU1 specimens, researchers in Hong Kong detected a third, novel genotype that had probably arisen from recombination of the two other known strains (Woo et al., 2006). Analysis of hCoV-OC43 genomes has suggested that a novel genotype of hCoV-OC43 has arisen through natural recombination and has become the dominant circulating strain (Lau et al., 2011). A study of NL63 genomes in the Netherlands provided the first evidence of recombination for NL63 (Pyrc et al., 2006). The present study provides an additional example of natural recombination between NL63 genomes and illustrates that recombination utilized by CoVs occurs in humans and could result in the emergence of novel strainsand/or rapid dissemination of antigenic variants that escape immune recognition or have other selective advantages.
The most variable region in the NL63 genome is the S gene, particularly the NTD (nt 1–600). Comparison of the nucleotide and amino acid sequences of the NL63 S NTDs in our study with the NL63 sequences in GenBank confirmed the extensive amino acid sequence variability in NL63 strains circulating throughout the world and at different times. The functions of the NTD and the reasons for its variability are not yet known. One possibility is that the NTDs may exhibit antigenic variation, providing a mechanism to evade neutralization by host immune responses whilst maintaining the more highly conserved, receptor-binding C-domain of the S protein. This mechanism is utilized by other respiratory viruses including respiratory syncytial virus and influenza virus (Plotkin et al., 2002; Smith et al., 2004; Sullender, 2000). The presence of multiple potential glycosylation sites in the NTD, with the deletion of up to three of them in certain NL63 samples, supports this possibility. The NL63 NTD may also play a role in virus infection in several ways: it may aid in virus attachment by recognizing cell-surface sugars, it may facilitate binding of the C-domain to the protein receptor human angiotensin-converting enzyme 2 (hACE2) or it may play a critical role in entry into cells by influencing a conformation change required for fusion of the viral envelope with the cell membrane. These possibilities are supported by studies showing that, although the NTD was not essential for binding to hACE2 or for hACE2-dependent membrane fusion of lentiviruses pseudotyped with NL63 S protein, deletion of the NTD markedly reduced both binding and membrane fusion (Hofmann et al., 2006). The core of the NTD of the CoV mouse hepatitis virus S protein has the same β-sandwich fold as human galectins (S-lectins), which are sugar-binding proteins that modulate immune responses. Most alpha-, beta- and gamma-CoV S glycoproteins contain NTDs, and their biological significance is an important topic for future investigation.
The results presented here highlight the utility of genome sequencing of viruses in clinical specimens from different parts of the world. We found that the same NL63 genotypes (A and B) circulate in different parts of the world. We also detected a new NL63 genotype (C) and have provided further evidence for circulation of naturally occurring NL63 recombinants, which has important evolutionary implications. In our paediatric population, the predominant genotype of NL63 varied from year to year, suggesting yearly antigenic variation. Sequence analysis demonstrated high sequence diversity in the NTD of the S gene. Future structural and functional studies are needed to elucidate the role this domain may play in infection, immunity, virus entry and pathogenesis of NL63 viruses.
Methods
Clinical specimens.
Beginning in January 2009, the Clinical Virology Laboratory of the Children’s Hospital Colorado began using a multiplex PCR [xTag Respiratory Virus Panel (RVP); Luminex Molecular Diagnostics] in combination with direct fluorescent antibody (DFA) assays to identify pathogens in respiratory specimens (nasopharyngeal washes, tracheal aspirates and bronchoalveolar lavages) from children with respiratory symptoms. Clinicians had the option of testing specimens by DFA or RVP only, DFA followed by RVP if the DFA testing was negative, or DFA and RVP concurrently. Nucleic acids were extracted from specimens submitted for RVP testing using Virus Minikits version 2.0 on BioRobot EZ1 extractors (Qiagen), following the manufacturer’s instructions, and tested by RVP. This assay detects 12 types of respiratory viruses (including influenza A and B viruses, parainfluenza viruses 1–4, adenoviruses, respiratory syncytial virus A and B, human metapneumovirus, rhinoviruses/enteroviruses and hCoV-229E, hCoV-OC43, hCoV-HKU1 and hCoV-NL63). RVP is not cleared by the US Food and Drug Administration for routine reporting of hCoV results, but the ability of this test to identify hCoVs has been reported (Mahony et al., 2007; Wong et al., 2008) and was confirmed by our clinical laboratory prior to this study. As part of an ongoing investigation of the aetiology of paediatric viral respiratory infections, all clinical respiratory specimens positive for any hCoV were archived at −70 °C for further analysis. Medical chart review of NL63-positive specimens was performed using a standardized form. Use of the banked specimens and clinical data for this study was approved by the Colorado Multiple Institutional Review Board.
Subsets of NL63-positive clinical specimens collected from 2009 to 2011, as well as samples collected in 2005 from our previously reported study (Dominguez et al., 2009), were selected for genome sequencing as follows. First, specimens identified initially as positive for NL63 RNA by RVP were confirmed using a modified quantitative real-time RT-PCR (qRT-PCR) assay that detected RNAs from all four non-SARS hCoVs (Dominguez et al., 2007; Kuypers et al., 2007). Specimens positive in the consensus CoV qRT-PCR assay were then analysed using NL63-specific conventional RT-PCR assays using SuperScript III reverse transcriptase (Invitrogen) with random primers, followed by PCR. Primer sets were used that bound regions unique to the NL63 CoV S and N genes (Bastien et al., 2005b; Moës et al., 2005).
RT-PCR and full-genome sequencing.
A 96-well plate of degenerate NL63-specific primers was designed from a consensus sequence of the four NL63 reference genomes from GenBank (accession nos DQ445912, DQ445911, NC_005831 and AY567487) using a PCR primer design pipeline developed at the J. Craig Venter Institute (JCVI) (Li et al., 2008). This produced tiled amplicons with an optimal length of 550 bp, with a 100 bp overlap and at least twofold amplicon coverage at every base. An M13 sequence tag added to the 5′ end of each primer was used for sequencing.
RNA extracted from the clinical samples was subjected to RT-PCR using a Qiagen One-step kit to produce cDNA amplicons from each of the primer pairs in the 96-well plate. For each clinical specimen, two independent sets of reactions were performed to produce sequence coverage consistent with JCVI standards for finished Sanger sequences. PCR products were treated with a mixture of exonuclease and shrimp alkaline phosphatase to remove primers and dNTPs, and then subjected to Sanger sequencing using the common M13 primers at the 5′ end of each primer.
Sequencing reads were trimmed to remove amplicon primer-linker sequences as well as low-quality sequences, and assembled using minimus, part of the open-source AMOS project (http://sourceforge.net/apps/mediawiki/amos/index.php?title=AMOS). Initial assemblies were inspected using an in-house application, Cloe (Closure Editor, http://cloe.sourceforge.net), and directed PCR-based sequencing reactions were conducted to improve the sequence. Assembled sequences for each viral sample were run through the vigor annotation pipeline. vigor (Viral Genome ORF Reader, http://www.jcvi.org/vigor; Wang et al., 2010) was developed at JCVI to decode sequences from many classes of virus by taking into account virus-specific features such as alternative splicing, internal ORFs and ribosomal slippage, and was used to validate the newly assembled NL63 genomes during the finishing process. After sequences were annotated and validated by vigor, the gene predictions were subjected to manual inspection and quality control before loading into the JCVI annotation database and submission to GenBank. To predict mature peptides, annotated polyproteins and ORFs were searched by blast against a reference set of known CoV mature peptides. The edges of each blast hit were scored by proximity to other blast hits and the quality of the hits. The hits with the highest scoring edges were used to cover the polyprotein. A final step resolved gaps and overlaps by using known cleavage motifs in the polyprotein whilst maximizing the homology of each predicted mature peptide to the reference mature peptide (Djikeng et al., 2008; Li et al., 2008; Wang et al., 2010).
Sequencing methods for the 2010 amplicons.
For the samples collected in 2010, only the region spanning the S gene was sequenced. Extracted RNA was reverse transcribed in an oligo(dT)-primed reaction using SuperScript III (Invitrogen). An amplicon of ~5.6 kb covering the S region was generated by PCR using Accuprime (Invitrogen) and the following primer pair: forward, 5′-TTGCAGTCTGCTGAATGGAAGTGTGG-3′, and reverse, 5′-ACCAACAACAGGCTCTTCGGCAAA-3′. Each amplicon was gel purified and then simultaneously amplified and bar-coded in two separate reactions using a modified sequence-independent single-primer amplification (SISPA) approach (Djikeng et al., 2008). A pool of these and other viral samples were used to construct an Illumina library and sequenced on a HiSeq platform. After sequencing, reads from each sample were deconvoluted by bar code and trimmed for quality and to remove the SISPA hexamer primer and barcode sequences. The Illumina reads were then assembled de novo using the clc_novo_assemble program (CLC Bio). The resulting de novo contigs were then used to pick a best reference, which was then used for a mapping assembly using the clc_ref_assemble_long program (CLC Bio). The assembled amplicons were annotated using vigor as described above.
Sequence analysis.
The viral genome sequences were multiply aligned with muscle version 3.7 (Edgar, 2004), using the sum-of-pairs profile score (VTML240) option over the course of five iterations. From this alignment, a distance-based neighbour-joining phylogenetic tree was constructed using the Kimura two-parameter distance (Kimura, 1980), with gamma-distributed rates across sites (α = 0.04) with the phylip package version 3.69 (Felsenstein, 1989). Bootstrap analysis of the genome alignments was performed using seqboot and consense with 1000 pseudo-replicates. To analyse possible recombination events, bootscan analyses were undertaken on the alignment using Simplot version 3.5.1 (Lole et al., 1999). Bootstrapped phylogenetic trees were built for overlapping window segments of 200 bp. The mean bootstrap value across all members of the query group segment versus the remaining three groups was plotted versus window position. For comparison of ORFs across the genomes, a heat map was constructed. Coding sequences of each viral gene were multiply aligned with muscle using a maximum of 30 iterations and the diagonal expansion option. Maximum-parsimony trees for each coding sequence were constructed with the phylip utility dnapars. The collection of most parsimonious trees for a given coding sequence was then consolidated into a single consensus tree using phylip consense. The resulting gene tree topologies were compared in a pairwise fashion using the Robinson–Foulds topological distance, DRF(a,b), with phylip dnadist. During calculation of DRF(a,b), in cases where one or more samples were missing a coding sequence in topology a, the missing genes were temporarily pruned from the tree topology b to achieve the same coding sequence in both topologies (using retree). The distance matrix DRF was visualized as a heat map using the r statistical package.
Nucleotide sequence accession numbers.
The clinical specimens positive for NL63 viruses were named according to the following nomenclature: virus/location/year of collection/specimen number (e.g. NL63/DEN/2009/6).
Acknowledgements
This project was funded in part with Federal funds from the National Institute of Allergy and Infectious Disease, the National Institutes of Health, and the Department of Health and Human Services under contract number HHSN272200900007C and by an NIH grant to S. R. D (1-K08AI073525). We thank Jonathan Motley, Christina Osborne, Andrew Berglund, the staff of the Children’s Hospital Colorado Clinical Virology Laboratory and members of the JCVI viral sequencing and informatics groups for their assistance with these studies, and Zhaohui Qian for critical review of this manuscript.
Footnotes
Three supplementary figures are available with the online version of this paper.
References
- Allander T., Tammi M. T., Eriksson M., Bjerkner A., Tiveljung-Lindell A., Andersson B. (2005). Cloning of a human parvovirus by molecular screening of respiratory tract samples. Proc Natl Acad Sci U S A 102, 12891–12896 10.1073/pnas.0504666102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allander T., Andreasson K., Gupta S., Bjerkner A., Bogdanovic G., Persson M. A., Dalianis T., Ramqvist T., Andersson B. (2007). Identification of a third human polyomavirus. J Virol 81, 4130–4136 10.1128/JVI.00028-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arden K. E., Nissen M. D., Sloots T. P., Mackay I. M. (2005). New human coronavirus, HCoV-NL63, associated with severe lower respiratory tract disease in Australia. J Med Virol 75, 455–462 10.1002/jmv.20288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banach B. S., Orenstein J. M., Fox L. M., Randell S. H., Rowley A. H., Baker S. C. (2009). Human airway epithelial cell culture to identify new respiratory viruses: coronavirus NL63 as a model. J Virol Methods 156, 19–26 10.1016/j.jviromet.2008.10.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastien N., Anderson K., Hart L., Van Caeseele P., Brandt K., Milley D., Hatchette T., Weiss E. C., Li Y. (2005a). Human coronavirus NL63 infection in Canada. J Infect Dis 191, 503–506 10.1086/426869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastien N., Robinson J. L., Tse A., Lee B. E., Hart L., Li Y. (2005b). Human coronavirus NL-63 infections in children: a 1-year study. J Clin Microbiol 43, 4567–4573 10.1128/JCM.43.9.4567-4573.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu S. S., Chan K. H., Chu K. W., Kwan S. W., Guan Y., Poon L. L., Peiris J. S. (2005). Human coronavirus NL63 infection and other coronavirus infections in children hospitalized with acute respiratory disease in Hong Kong, China. Clin Infect Dis 40, 1721–1729 10.1086/430301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dare R. K., Fry A. M., Chittaganpitch M., Sawanpanyalert P., Olsen S. J., Erdman D. D. (2007). Human coronavirus infections in rural Thailand: a comprehensive study using real-time reverse-transcription polymerase chain reaction assays. J Infect Dis 196, 1321–1328 10.1086/521308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dijkman R., Jebbink M. F., Gaunt E., Rossen J. W., Templeton K. E., Kuijpers T. W., van der Hoek L. (2012). The dominance of human coronavirus OC43 and NL63 infections in infants. J Clin Virol 53, 135–139 10.1016/j.jcv.2011.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djikeng A., Halpin R., Kuzmickas R., Depasse J., Feldblyum J., Sengamalay N., Afonso C., Zhang X., Anderson N. G. & other authors (2008). Viral genome sequencing by random priming methods. BMC Genomics 9, 5 10.1186/1471-2164-9-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez S. R., O’Shea T. J., Oko L. M., Holmes K. V. (2007). Detection of group 1 coronaviruses in bats in North America. Emerg Infect Dis 13, 1295–1300 10.3201/eid1309.070491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez S. R., Briese T., Palacios G., Hui J., Villari J., Kapoor V., Tokarz R., Glodé M. P., Anderson M. S. & other authors (2008). Multiplex MassTag-PCR for respiratory pathogens in pediatric nasopharyngeal washes negative by conventional diagnostic testing shows a high prevalence of viruses belonging to a newly recognized rhinovirus clade. J Clin Virol 43, 219–222 10.1016/j.jcv.2008.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez S. R., Robinson C. C., Holmes K. V. (2009). Detection of four human coronaviruses in respiratory infections in children: a one-year study in Colorado. J Med Virol 81, 1597–1604 10.1002/jmv.21541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drosten C., Günther S., Preiser W., van der Werf S., Brodt H. R., Becker S., Rabenau H., Panning M., Kolesnikova L. & other authors (2003). Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med 348, 1967–1976 10.1056/NEJMoa030747 [DOI] [PubMed] [Google Scholar]
- Dufour D., Mateos-Gomez P. A., Enjuanes L., Gallego J., Sola I. (2011). Structure and functional relevance of a transcription-regulating sequence involved in coronavirus discontinuous RNA synthesis. J Virol 85, 4963–4973 10.1128/JVI.02317-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebihara T., Endo R., Ma X., Ishiguro N., Kikuta H. (2005). Detection of human coronavirus NL63 in young children with bronchiolitis. J Med Virol 75, 463–465 10.1002/jmv.20289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R. C. (2004). muscle: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32, 1792–1797 10.1093/nar/gkh340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esper F., Weibel C., Ferguson D., Landry M. L., Kahn J. S. (2005). Evidence of a novel human coronavirus that is associated with respiratory tract disease in infants and young children. J Infect Dis 191, 492–498 10.1086/428138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J. (1989). phylip – Phylogeny Inference Package (version 3.2). Cladistics 5, 164–166 [Google Scholar]
- Fouchier R. A., Hartwig N. G., Bestebroer T. M., Niemeyer B., de Jong J. C., Simon J. H., Osterhaus A. D. (2004). A previously undescribed coronavirus associated with respiratory disease in humans. Proc Natl Acad Sci U S A 101, 6212–6216 10.1073/pnas.0400762101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaunt E. R., Hardie A., Claas E. C., Simmonds P., Templeton K. E. (2010). Epidemiology and clinical presentations of the four human coronaviruses 229E, HKU1, NL63, and OC43 detected over 3 years using a novel multiplex real-time PCR method. J Clin Microbiol 48, 2940–2947 10.1128/JCM.00636-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaynor A. M., Nissen M. D., Whiley D. M., Mackay I. M., Lambert S. B., Wu G., Brennan D. C., Storch G. A., Sloots T. P., Wang D. (2007). Identification of a novel polyomavirus from patients with acute respiratory tract infections. PLoS Pathog 3, e64 10.1371/journal.ppat.0030064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerna G., Percivalle E., Sarasini A., Campanini G., Piralla A., Rovida F., Genini E., Marchi A., Baldanti F. (2007). Human respiratory coronavirus HKU1 versus other coronavirus infections in Italian hospitalised patients. J Clin Virol 38, 244–250 10.1016/j.jcv.2006.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrewegh A. A., Smeenk I., Horzinek M. C., Rottier P. J., de Groot R. J. (1998). Feline coronavirus type II strains 79-1683 and 79-1146 originate from a double recombination between feline coronavirus type I and canine coronavirus. J Virol 72, 4508–4514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann H., Simmons G., Rennekamp A. J., Chaipan C., Gramberg T., Heck E., Geier M., Wegele A., Marzi A. & other authors (2006). Highly conserved regions within the spike proteins of human coronaviruses 229E and NL63 determine recognition of their respective cellular receptors. J Virol 80, 8639–8652 10.1128/JVI.00560-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser L., Regamey N., Roiha H., Deffernez C., Frey U. (2005). Human coronavirus NL63 associated with lower respiratory tract symptoms in early life. Pediatr Infect Dis J 24, 1015–1017 10.1097/01.inf.0000183773.80217.12 [DOI] [PubMed] [Google Scholar]
- Kimura M. (1980). A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16, 111–120 10.1007/BF01731581 [DOI] [PubMed] [Google Scholar]
- Koetz A., Nilsson P., Lindén M., van der Hoek L., Ripa T. (2006). Detection of human coronavirus NL63, human metapneumovirus and respiratory syncytial virus in children with respiratory tract infections in south-west Sweden. Clin Microbiol Infect 12, 1089–1096 10.1111/j.1469-0691.2006.01506.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuypers J., Martin E. T., Heugel J., Wright N., Morrow R., Englund J. A. (2007). Clinical disease in children associated with newly described coronavirus subtypes. Pediatrics 119, e70–e76 10.1542/peds.2006-1406 [DOI] [PubMed] [Google Scholar]
- Lamson D., Renwick N., Kapoor V., Liu Z., Palacios G., Ju J., Dean A., St George K., Briese T., Lipkin W. I. (2006). MassTag polymerase-chain-reaction detection of respiratory pathogens, including a new rhinovirus genotype, that caused influenza-like illness in New York State during 2004–2005. J Infect Dis 194, 1398–1402 10.1086/508551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau S. K., Woo P. C., Yip C. C., Tse H., Tsoi H.-W., Cheng V. C., Lee P., Tang B. S., Cheung C. H. & other authors (2006). Coronavirus HKU1 and other coronavirus infections in Hong Kong. J Clin Microbiol 44, 2063–2071 10.1128/JCM.02614-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau S. K., Yip C. C., Tsoi H.-W., Lee R. A., So L.-Y., Lau Y.-L., Chan K.-H., Woo P. C., Yuen K.-Y. (2007). Clinical features and complete genome characterization of a distinct human rhinovirus (HRV) genetic cluster, probably representing a previously undetected HRV species, HRV-C, associated with acute respiratory illness in children. J Clin Microbiol 45, 3655–3664 10.1128/JCM.01254-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau S. K., Li K. S., Huang Y., Shek C.-T., Tse H., Wang M., Choi G. K., Xu H., Lam C. S. & other authors (2010). Ecoepidemiology and complete genome comparison of different strains of severe acute respiratory syndrome-related Rhinolophus bat coronavirus in China reveal bats as a reservoir for acute, self-limiting infection that allows recombination events. J Virol 84, 2808–2819 10.1128/JVI.02219-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau S. K., Lee P., Tsang A. K., Yip C. C., Tse H., Lee R. A., So L.-Y., Lau Y.-L., Chan K.-H. & other authors (2011). Molecular epidemiology of human coronavirus OC43 reveals evolution of different genotypes over time and recent emergence of a novel genotype due to natural recombination. J Virol 85, 11325–11337 10.1128/JVI.05512-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung T. F., Li C. Y., Lam W. Y., Wong G. W., Cheuk E., Ip M., Ng P. C., Chan P. K. (2009). Epidemiology and clinical presentations of human coronavirus NL63 infections in Hong Kong children. J Clin Microbiol 47, 3486–3492 10.1128/JCM.00832-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li K., Brownley A., Stockwell T. B., Beeson K., McIntosh T. C., Busam D., Ferriera S., Murphy S., Levy S. (2008). Novel computational methods for increasing PCR primer design effectiveness in directed sequencing. BMC Bioinformatics 9, 191 10.1186/1471-2105-9-191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lole K. S., Bollinger R. C., Paranjape R. S., Gadkari D., Kulkarni S. S., Novak N. G., Ingersoll R., Sheppard H. W., Ray S. C. (1999). Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J Virol 73, 152–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahony J., Chong S., Merante F., Yaghoubian S., Sinha T., Lisle C., Janeczko R. (2007). Development of a respiratory virus panel test for detection of twenty human respiratory viruses by use of multiplex PCR and a fluid microbead-based assay. J Clin Microbiol 45, 2965–2970 10.1128/JCM.02436-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McErlean P., Shackelton L. A., Lambert S. B., Nissen M. D., Sloots T. P., Mackay I. M. (2007). Characterisation of a newly identified human rhinovirus, HRV-QPM, discovered in infants with bronchiolitis. J Clin Virol 39, 67–75 10.1016/j.jcv.2007.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizgerd J. P. (2006). Lung infection – a public health priority. PLoS Med 3, e76 10.1371/journal.pmed.0030076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizgerd J. P. (2008). Acute lower respiratory tract infection. N Engl J Med 358, 716–727 10.1056/NEJMra074111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moës E., Vijgen L., Keyaerts E., Zlateva K., Li S., Maes P., Pyrc K., Berkhout B., van der Hoek L., Van Ranst M. (2005). A novel pancoronavirus RT-PCR assay: frequent detection of human coronavirus NL63 in children hospitalized with respiratory tract infections in Belgium. BMC Infect Dis 5, 6 10.1186/1471-2334-5-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monto A. S., Lim S. K. (1974). The Tecumseh study of respiratory illness. VI. Frequency of and relationship between outbreaks of coronavirus infection. J Infect Dis 129, 271–276 10.1093/infdis/129.3.271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotkin J. B., Dushoff J., Levin S. A. (2002). Hemagglutinin sequence clusters and the antigenic evolution of influenza A virus. Proc Natl Acad Sci U S A 99, 6263–6268 10.1073/pnas.082110799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prill M. M., Iwane M. K., Edwards K. M., Williams J. V., Weinberg G. A., Staat M. A., Willby M. J., Talbot H. K., Hall C. B. & other authors (2012). Human coronavirus in young children hospitalized for acute respiratory illness and asymptomatic controls. Pediatr Infect Dis J 31, 235–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyrc K., Jebbink M. F., Berkhout B., van der Hoek L. (2004). Genome structure and transcriptional regulation of human coronavirus NL63. Virol J 1, 7 10.1186/1743-422X-1-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyrc K., Dijkman R., Deng L., Jebbink M. F., Ross H. A., Berkhout B., van der Hoek L. (2006). Mosaic structure of human coronavirus NL63, one thousand years of evolution. J Mol Biol 364, 964–973 10.1016/j.jmb.2006.09.074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith D. J., Lapedes A. S., de Jong J. C., Bestebroer T. M., Rimmelzwaan G. F., Osterhaus A. D., Fouchier R. A. (2004). Mapping the antigenic and genetic evolution of influenza virus. Science 305, 371–376 10.1126/science.1097211 [DOI] [PubMed] [Google Scholar]
- Sola I., Moreno J. L., Zúñiga S., Alonso S., Enjuanes L. (2005). Role of nucleotides immediately flanking the transcription-regulating sequence core in coronavirus subgenomic mRNA synthesis. J Virol 79, 2506–2516 10.1128/JVI.79.4.2506-2516.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullender W. M. (2000). Respiratory syncytial virus genetic and antigenic diversity. Clin Microbiol Rev 13, 1–15 . 10.1128/CMR.13.1.1-15.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A., Okamoto M., Ohmi A., Watanabe O., Miyabayashi S., Nishimura H. (2005). Detection of human coronavirus-NL63 in children in Japan. Pediatr Infect Dis J 24, 645–646 10.1097/01.inf.0000168846.71517.ee [DOI] [PubMed] [Google Scholar]
- Talbot H. K., Crowe J. E., Jr, Edwards K. M., Griffin M. R., Zhu Y., Weinberg G. A., Szilagyi P. G., Hall C. B., Podsiad A. B. & other authors (2009). Coronavirus infection and hospitalizations for acute respiratory illness in young children. J Med Virol 81, 853–856 10.1002/jmv.21443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vabret A., Mourez T., Dina J., van der Hoek L., Gouarin S., Petitjean J., Brouard J., Freymuth F. (2005). Human coronavirus NL63, France. Emerg Infect Dis 11, 1225–1229 10.3201/eid1108.050110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vabret A., Dina J., Gouarin S., Petitjean J., Tripey V., Brouard J., Freymuth F. (2008). Human (non-severe acute respiratory syndrome) coronavirus infections in hospitalised children in France. J Paediatr Child Health 44, 176–181 10.1111/j.1440-1754.2007.01246.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Hoogen B. G., de Jong J. C., Groen J., Kuiken T., de Groot R., Fouchier R. A., Osterhaus A. D. (2001). A newly discovered human pneumovirus isolated from young children with respiratory tract disease. Nat Med 7, 719–724 10.1038/89098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Hoek L., Pyrc K., Jebbink M. F., Vermeulen-Oost W., Berkhout R. J., Wolthers K. C., Wertheim-van Dillen P. M., Kaandorp J., Spaargaren J., Berkhout B. (2004). Identification of a new human coronavirus. Nat Med 10, 368–373 10.1038/nm1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Hoek L., Sure K., Ihorst G., Stang A., Pyrc K., Jebbink M. F., Petersen G., Forster J., Berkhout B., Uberla K. (2005). Croup is associated with the novel coronavirus NL63. PLoS Med 2, e240 10.1371/journal.pmed.0020240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S., Sundaram J. P., Spiro D. (2010). vigor, An annotation program for small viral genomes. BMC Bioinformatics 11, 451 10.1186/1471-2105-11-451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO (2003). The World Health Report 2003: Shaping the Future. Geneva, Switzerland: World Health Organization [Google Scholar]
- WHO (2008). The Global Burden of Disease: 2004 Update. Geneva, Switzerland: World Health Organization [Google Scholar]
- Wong S., Pabbaraju K., Pang X. L., Lee B. E., Fox J. D. (2008). Detection of a broad range of human adenoviruses in respiratory tract samples using a sensitive multiplex real-time PCR assay. J Med Virol 80, 856–865 10.1002/jmv.21136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo P. C., Lau S. K., Chu C.-M., Chan K.-H., Tsoi H.-W., Huang Y., Wong B. H., Poon R. W., Cai J. J. & other authors (2005). Characterization and complete genome sequence of a novel coronavirus, coronavirus HKU1, from patients with pneumonia. J Virol 79, 884–895 10.1128/JVI.79.2.884-895.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo P. C., Lau S. K., Yip C. C., Huang Y., Tsoi H.-W., Chan K.-H., Yuen K.-Y. (2006). Comparative analysis of 22 coronavirus HKU1 genomes reveals a novel genotype and evidence of natural recombination in coronavirus HKU1. J Virol 80, 7136–7145 10.1128/JVI.00509-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo P. C., Lau S. K., Huang Y., Yuen K.-Y. (2009). Coronavirus diversity, phylogeny and interspecies jumping. Exp Biol Med (Maywood) 234, 1117–1127 10.3181/0903-MR-94 [DOI] [PubMed] [Google Scholar]
- Woo P. C., Huang Y., Lau S. K., Yuen K.-Y. (2010). Coronavirus genomics and bioinformatics analysis. Viruses 2, 1804–1820 10.3390/v2081803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zúñiga S., Sola I., Alonso S., Enjuanes L. (2004). Sequence motifs involved in the regulation of discontinuous coronavirus subgenomic RNA synthesis. J Virol 78, 980–994 10.1128/JVI.78.2.980-994.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]