

Abstract
Revealing the frequency and determinants of reassortment among RNA genome segments is fundamental to understanding basic aspects of the biology and evolution of the influenza virus. To estimate the extent of genomic reassortment in influenza viruses circulating in North American swine, we performed a phylogenetic analysis of 139 whole-genome viral sequences sampled during 1998–2011 and representing seven antigenically distinct viral lineages. The highest amounts of reassortment were detected between the H3 and the internal gene segments (PB2, PB1, PA, NP, M and NS), while the lowest reassortment frequencies were observed among the H1γ, H1pdm and neuraminidase segments, particularly N1. Less reassortment was observed among specific haemagglutinin–neuraminidase combinations that were more prevalent in swine, suggesting that some genome constellations may be evolutionarily more stable.
Swine express forms of the sialic acid receptor that can be preferentially bound by both avian (N-acetylneuraminic acid-α2,3-galactose – NeuAc-α2,3Gal) and human influenza viruses (NeuAc-α2,6Gal), facilitating inter-species reassortment and the emergence of novel virus variants (Imai & Kawaoka, 2012). The recent emergence of the pandemic H1N1/09 (H1N1pdm09) influenza virus, with segments that have their ultimate origins in avian, human and multiple swine influenza viruses, exemplifies the capacity of swine to serve as a ‘mixing vessel’ (Garten et al., 2009; Smith et al., 2009). Since July 2011 novel reassortant swine-origin H3N2v influenza viruses containing M segments of H1N1pdm09 origin were identified in >100 cases of human infection in the USA, and continue to be monitored closely for pandemic potential (Centers for Disease Control and Prevention, 2012; Lindstrom et al., 2012).
Certain constellations of genome segments show evidence of conservation in swine, such as the combination of six triple reassortant internal genes (TRIG) that has remained prevalent in North American swine since 1998. The low frequency of H3N1 subtype viruses in swine similarly suggests evolutionary constraints on other genomic combinations. To assess pandemic potential, experimental studies have used reverse genetics to determine the viability of various reassortant viruses in animal models, including avian H5N1/human H3N2 (Chen et al., 2008; Li et al., 2010), avian H9N2/human H1N1pdm09 (Kimble et al., 2011) and avian H9N2/human H3N2 reassortants (Sorrell et al., 2009). To date, however, the patterns of genomic reassortment in nature have not been estimated using a phylogenetic approach for the various influenza A virus lineages that have circulated in North American swine since the late 1990s.
To determine the patterns of reassortment in North American swine, we performed an analysis of 139 whole-genome sequences collected from North American swine during 1998–2011. In particular, we compared reassortment between the neuraminidase (NA) segment, six internal gene segments and haemagglutinin (HA) segments, which are associated with seven antigenically distinct swine influenza virus lineages that have been identified previously in North American swine: namely, H1α, H1β, H1δ, H1γ, H1pdm09, H3-I and H3-IV (Vincent et al., 2008; Ducatez et al., 2011). Of the 139 genomes, 36 were collected in the USA during 2002–2010 as part of routine diagnostic investigations from samples submitted by veterinarians and were selected largely at random from the influenza virus archive at the University of Minnesota Veterinary Diagnostic Laboratory. These were sequenced as part of this study by our colleagues at the J. Craig Venter Institute using multiplex real-time PCR (Djikeng et al., 2008; Zhou et al., 2009) (Table S1, available in JGV Online). The remaining 133 genome sequences were downloaded from the Influenza Virus Resource available at NCBI’s GenBank (Bao et al., 2008), 63 of which were collected from the USA, 36 from Canada and four from Cuba. Finally, as considerably more sequence data were available for the HA and NA than for the internal gene segments, an additional analysis of the large N1 (n = 319) and N2 (n = 285) datasets was conducted (Table S2).
Sequence alignments were manually constructed for each genome segment using Se-Al (Rambaut, 2002). Phylogenetic trees were inferred separately for each segment using the Bayesian Markov chain Monte Carlo method available in the mrbayes package (version v.3.1.2; Huelsenbeck & Ronquist, 2001). This analysis incorporated a GTR model of nucleotide substitution with gamma-distributed rate heterogeneity and a proportion of invariable sites, as determined by modeltest (Posada & Crandall, 1998; parameter values available from the authors upon request). The analysis was run for 10 million generations, with two hot chains and one cold chain, until convergence was achieved, with a 25 % burn-in (for the PA segment convergence was reached after 25 million generations). For comparison, phylogenies were also inferred using the maximum-likelihood method available in paup* (Swofford, 2003), again utilizing the GTR+Γ+I model substitution model (data available upon request). Due to the deep divergence between the N1 and N2 and the H1 and H3 antigens, separate trees were inferred for these groups of sequences. Genetically distinct HA lineages were identified on the H1 (five clusters) and H3 phylogenies (two clusters) (Figs S1 and S2, respectively). The H1α, H1β, H1γ and H1pdm clusters fall within the lineage of ‘classical’ H1 influenza viruses that have circulated in North American swine since 1918. The H1δ viruses comprise two closely related lineages of human-origin seasonal H1 viruses that were both introduced into North American swine during 2002–2003 (Karasin et al., 2006; Nelson et al., 2011). The H3-I and H3-IV clusters are both in the triple reassortant lineage that was identified in North American swine in 1998 (Zhou et al., 1999). Of the 139 isolates, 14 were of the H1α lineage, 17 were H1β, 21 were H1δ, 17 were H1γ, 21 were H1pdm, 11 were H3-I and 35 were H3-IV (Table S1). In addition, three isolates exhibited mixed infections, comprising H1α/H3-IV, H1β/H3-IV and H1γ/H3-IV combinations. Although the greater genetic similarity of the H3 viruses (92–95 % sequence similarity between the H3-I and H3-IV clusters, versus 73–93 % similarity among the H1 clusters) meant that the H3-I and H3-IV clusters could have been analysed as a single H3 cluster, we conservatively considered the H3-I and H3-IV clusters separately, as combining them could have inflated the estimate of H3 reassortment. To account for potential sample bias and for uncertainties in segment assignments in three cases of mixed infection, we randomly subsampled our data and ran additional analyses (Text S1).
Reassortment events involving the HA and another genome segment were identified visually on each phylogenetic tree by colour-coding each isolate according to the HA lineage associated with that viral isolate, as identified on the H1 and H3 phylogenies: H1α, H1β, H1δ, H1γ, H1pdm, H3-I and H3-IV (for illustration, the PB2 phylogeny is presented in Fig. 1; phylogenies for the PB1, PA, NP, N1, N2, M and NS segments are presented in Figs S3–S9). To identify major reassortment events, isolates were characterized on each phylogeny by broad classifications of evolutionary origins: TRIG (avian) for PB2 and PA, TRIG (human) for PB1, TRIG (classical) for NP, M and NS; classical swine; human; pandemic; and N2-2002 and N2-1998 for NA (Table S3). This broad classification scheme is useful for identifying major reassortment events, particularly those involving segments of pandemic H1N1pdm09 origin. However, it greatly underestimates the amount of reassortment between more genetically similar viruses within these broad categories, for example the extensive intermixing of viruses of the HA types among the multiple clades positioned within the TRIG section of the PB2 phylogeny (Fig. 1). Therefore, additional reassortment events were visually identified by well-supported nodes (i.e. posterior probability ≥90 %) defining clades containing more than one HA type. Using these criteria, 22 reassortment events were identified on the PB2 phylogeny, including several cases where multiple reassortment events were defined by the same node (Fig. 1). For example, the node defining the pandemic H1N1pdm09 clade is associated with two separate reassortment events with H1β and H1δ viruses. Similarly, 22 reassortment events were identified on the PA, M and NS phylogenies; 23 on the NP tree; and 18 on the PB1 and N2 trees (Table S4, Figs S3–S9). In contrast, only six reassortment events were evident on the N1 tree (Fig. S6). The difference in the frequency of reassortment between the N1 and N2 segments cannot be explained by differences in sampling, as more N1 sequences (n = 73) were available than N2 sequences (n = 69). In fact, the addition of 246 N1 sequences, for which whole-genome sequences were not available for this study, did not result in the detection of further reassortment events on the larger N1 phylogeny (Fig. S10).
Fig. 1.

Phylogenetic relationships of the (illustrative) PB2 segment of 139 influenza viruses collected in North American swine during 1998–2011. Isolates are colour-coded and labelled by the HA lineage, as inferred from the H1 and H3 phylogenies: H1α viruses are rust, H1β are green, H1γ are blue, H1δ are turquoise, H1pdm09 are pink, H3-I are yellow and H3-IV are purple. Posterior probabilities are shown for key nodes, and nodes that define reassortment events are identified by small yellow circles (single reassortment events) and larger yellow circles (multiple reassortment events). The tree is midpoint rooted for clarity. Isolates are related to three major lineages of the PB2 segment in swine: classical swine influenza viruses, the TRIG constellation (including pandemic H1N1pdm09) and human seasonal H1 viruses.
Inferring the directionality of reassortment by the HA type is complicated when reassortment is so frequent that the HA type of internal branches is frequently ambiguous. In a few cases, the direction of reassortment is clear, such as reassortment events involving the large, well-defined clade of pandemic H1N1pdm09 viruses. But estimating the extent of reassortment of other HA types is considerably more difficult. Rather, it was feasible to estimate the extent of phylogenetic clustering of each HA type. This provided another measure of reassortment between a given segment and the HA, as phylogenies for internal genes would remain strongly clustered by the HA type in the absence of reassortment (Fig. 1). As an extreme example, the most highly clustered H1pdm segment was associated with only one reassortment event across all eight phylogenetic trees (with the NP segment, Table S4, Fig. S5). In contrast, the H3-IV segment exhibited the least evidence of phylogenetic clustering, with no monophyletic H3-IV clusters containing more than two isolates on the PB2 tree, for example Fig. 1. This clustering analysis was performed using the Bayesian Tip-Significance (BaTS) method (Parker et al., 2008), which determines whether a phenotypic trait, in this case the HA type, is more associated with the underlying phylogeny than expected by chance alone. To account for possible phylogenetic error, BaTS estimates the association between the HA type and phylogeny over a posterior distribution of trees generated by mrbayes.
To compare reassortment patterns among genomic segments, we determined the ratio of clustering by the HA type on the internal gene and the NA trees that is expected by chance alone, compared with the association that is observed in the data. These expected/observed ratios are summarized in a heat-map (Fig. 2), with the y-axis ordered by the amount of reassortment observed among the seven HA types and the x-axis ordered by the amount of reassortment between the HA and the six internal genome segments and N1 and N2. These expected/observed ratios are largely consistent with the estimates of reassortment based on well-supported phylogenetic nodes (above). Both methods identified more reassortment between the HA and the internal genome segments and relatively less between the HA and NA segments, particularly N1 (Figs 1 and 2, Table S5). The relatively high rates of reassortment observed among internal gene segments reflect how the conserved TRIG constellation has remained dominant in North American swine by continually reassorting with different HA and NA segments, displacing the internal gene segments in these other viral lineages, as has also been observed experimentally in swine (Ma et al., 2010).
Fig. 2.
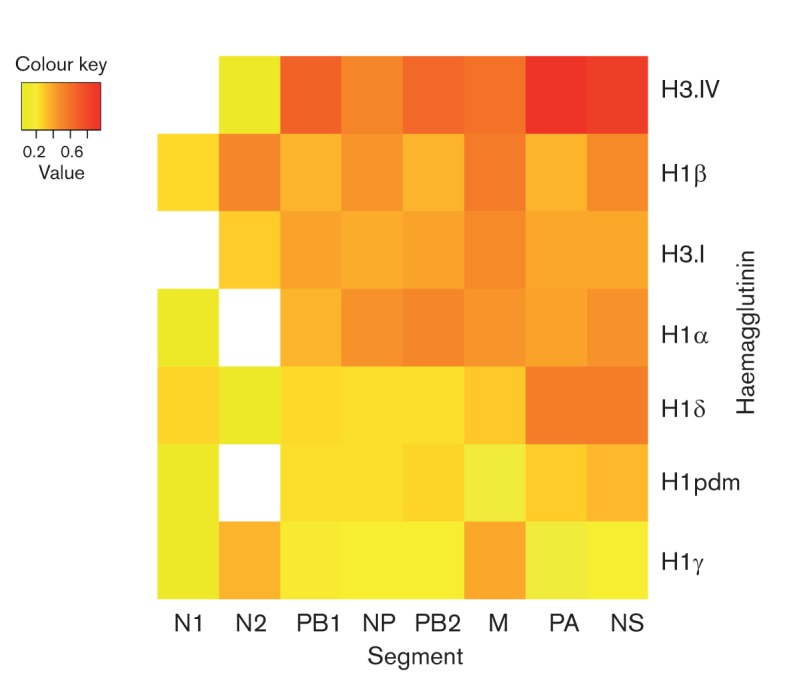
Relative frequencies of reassortment for each genome segment among seven antigenic lineages of influenza virus in North American swine, coded by colour: lower expected/observed ratios (<0.2) are yellow, middle rates (0.2–0.6) are orange and higher rates (>0.6) are red. Frequency estimates represent the ratio of association between the HA type and phylogeny as expected under the null hypothesis of a random association between the phylogeny and HA type versus the association that is observed in the data; hence, a higher number indicates higher levels of reassortment. The x-axis and y-axis are sorted in order of reassortment frequency, such that the lowest frequencies appear in the bottom left and the highest in the upper right. Blank squares indicate a viral sample number (n≤1) for which rates could not be estimated.
The BaTS analysis also provides a comparison of reassortment frequencies among the HA types. Expected/observed ratios were greatest among the H3-IV viruses, indicative of higher levels of reassortment between the H3 and internal gene segments: a ratio of 0.65 for PB2, 0.68 for PB1, 0.95 for PA, 0.52 for NP, 0.60 for M and 0.85 for NS (Table S5). In contrast, the lowest ratios were observed for the H1γ antigen, with values of 0.19, 0.20, 0.17, 0.19, 0.39 and 0.19 for the PB2, PB1, PA, NP, M and NS phylogenies, respectively. The relatively low reassortment frequency for the H1pdm segment could also reflect limited sampling during the recent emergence of these viruses in swine; indeed, reassortment is likely to increase as H1pdm09 viruses disseminate more widely in pigs and hence have more opportunities to reassort with other swine viruses.
Overall, our data indicate that the HA reassorts more frequently with internal gene segments than with NA, particularly N1. This inference is based on the relatively low numbers of reassortment events that were visually identified on the N1 tree, and the relatively high levels of phylogenetic clustering on the N1 phylogeny (Figs 2, S6 and S10, Table S5). Although only two H3N1 viruses were identified here, both isolated in 2004 (Lekcharoensuk et al., 2006), the lower reassortment frequencies of N1 and N2 are not biased by the limited sampling of some low frequency HA–NA pairings (e.g. H3N1, H1αN2 and H1pdmN2). Rather, the lowest frequencies of reassortment are observed between the most prevalent HA–NA pairings in North American swine: the H1γN1, H1pdmN1, H1δN2 and H3-IVN2 (Fig. 2, Table S5). Indeed, the HA–NA pairings associated with low sample sizes (n<50 isolates with the particular HA–NA pairing) are associated with higher frequencies of reassortment (mean expected/observed ratio = 0.33) than the more prevalent HA–NA pairs (mean expected/observed ratio = 0.08; P = 0.01, Wilcoxon non-parametric t-test) (Table S6).
Understanding how the molecular compatibility between various HA and NA segments relates to the frequency of reassortment at a population level clearly requires further data and analysis. Certain HA segments clearly have a preference for N2, particularly H3 and H1δ (~95 % of H1δ viruses were H1N2), whereas 93 % of H1γ segments were paired with N1. These observed preferences are consistent with previous studies (Lorusso et al., 2011; Nelson et al., 2012) (Tables S1 and S2). It remains unclear why the human-origin H1δ antigen might have a preference for N2 in swine when such pairings are very rare in humans. Variation in the HA–NA protein compatibility may be host-dependent (e.g. differences in receptor specificity and/or enzymic efficiency). Indeed, recent evidence that the functional balance in the HA–NA activity is higher among human H1N1pdm09 viruses than in swine influenza viruses potentially explains the greater restrictions on reassortment between the HA and NA in humans (Xu et al., 2012).
The relatively low frequencies of reassortment between HA and NA may relate to constraints on maintaining a balance between receptor binding (HA) and sialic acid cleavage (NA) activities, which are required for successful viral entry and release from host cells (Mitnaul et al., 2000). The lesser extent of N1 reassortment could reflect differences in the neuraminidase activity of the N1 protein compared with N2. HA–NA combinations that are more prevalent in North American swine (H3N2, H1γN1, H1pdmN1 and H1δN2) are characterized by lower frequencies of reassortment, which could signify a greater protein compatibility and evolutionary stability of these pairings. In contrast, less successful HA–NA combinations (H1βN2, H1δN1 and H1γN2) do not appear to transmit as efficiently in swine but are sporadically generated by reassortment, resulting in phylogenies with less clustering by the HA type.
While experimental studies have advanced our understanding of RNA packaging (Gao & Palese, 2009) and the viability of various avian–human influenza virus reassortants with pandemic potential (e.g. Kimble et al., 2011), fewer studies consider patterns of intra- and inter-subtype reassortment at the epidemiological scale. Future studies utilizing greater whole-genome influenza virus sequence data may better link the frequency of reassortment with the population dynamics of individual viral lineages and segment pairings. Such studies require systematic, population-based surveillance of influenza in swine, which will facilitate comparisons in viral dynamics across space, time and different virus lineages. Although we attempted here to account for major biases in sampling, we cannot be certain that biases in the past collection and sequencing of swine viruses have not affected our findings. Finally, it is important to determine how the reassortment patterns observed here among North American swine influenza viruses compare to frequencies of reassortment among other swine populations, particularly in regions with high genetic diversity of swine influenza such as Asia (Vijaykrishna et al., 2011), as well as with humans and avian species.
Acknowledgements
We are grateful to the pork producers and swine practitioners for participating in the USDA Swine Influenza Surveillance System through the National Animal Health Laboratory Network (NAHLN). We thank Dan Weinberger and Cécile Viboud of the Fogarty International Center for technical input. This research was conducted within the context of the Multinational Influenza Seasonal Mortality Study (MISMS), an on-going international collaborative effort to understand influenza epidemiology and evolution, led by the Fogarty International Center, NIH, with funding from the Office of Global Affairs at the Department of Health and Human Services (DHHS) (A. S., E. C. H., M. I. N. and Y. T.). This work was supported in part by funds from the National Institute of Allergy and Infectious Disease, the National Institutes of Health and the Department of Health and Human Services under contract no. HHSN272200900007C (D. E. W., R. A. H., T. B. S. and X. L.) and HHSN266200700007C (S. E. D. and M. R. G.).
Footnotes
Supplementary material is available with the online version of this paper.
References
- Bao Y., Bolotov P., Dernovoy D., Kiryutin B., Zaslavsky L., Tatusova T., Ostell J., Lipman D. (2008). The influenza virus resource at the National Center for Biotechnology Information. J Virol 82, 596–601 10.1128/JVI.02005-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention (2012). Evaluation of rapid influenza diagnostic tests for influenza A (H3N2)v virus and updated case count – United States, 2012. MMWR Morb Mortal Wkly Rep 61, 619–621 [PubMed] [Google Scholar]
- Chen L. M., Davis C. T., Zhou H., Cox N. J., Donis R. O. (2008). Genetic compatibility and virulence of reassortants derived from contemporary avian H5N1 and human H3N2 influenza A viruses. PLoS Pathog 4, e1000072 10.1371/journal.ppat.1000072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djikeng A., Halpin R., Kuzmickas R., Depasse J., Feldblyum J., Sengamalay N., Afonso C., Zhang X., Anderson N. G. & other authors (2008). Viral genome sequencing by random priming methods. BMC Genomics 9, 5 10.1186/1471-2164-9-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducatez M. F., Hause B., Stigger-Rosser E., Darnell D., Corzo C., Juleen K., Simonson R., Brockwell-Staats C., Rubrum A. & other authors (2011). Multiple reassortment between pandemic (H1N1) 2009 and endemic influenza viruses in pigs, United States. Emerg Infect Dis 17, 1624–1629 10.3201/eid1709.110338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Q., Palese P. (2009). Rewiring the RNAs of influenza virus to prevent reassortment. Proc Natl Acad Sci U S A 106, 15891–15896 10.1073/pnas.0908897106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garten R. J., Davis C. T., Russell C. A., Shu B., Lindstrom S., Balish A., Sessions W. M., Xu X., Skepner E. & other authors (2009). Antigenic and genetic characteristics of swine-origin 2009 A(H1N1) influenza viruses circulating in humans. Science 325, 197–201 10.1126/science.1176225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huelsenbeck J. P., Ronquist F. (2001). mrbayes: Bayesian inference of phylogenetic trees. Bioinformatics 17, 754–755 10.1093/bioinformatics/17.8.754 [DOI] [PubMed] [Google Scholar]
- Imai M., Kawaoka Y. (2012). The role of receptor binding specificity in interspecies transmission of influenza viruses. Curr Opin Virol 2, 160–167 10.1016/j.coviro.2012.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karasin A. I., Carman S., Olsen C. W. (2006). Identification of human H1N2 and human-swine reassortant H1N2 and H1N1 influenza A viruses among pigs in Ontario, Canada (2003 to 2005). J Clin Microbiol 44, 1123–1126 10.1128/JCM.44.3.1123-1126.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimble J. B., Sorrell E., Shao H., Martin P. L., Perez D. R. (2011). Compatibility of H9N2 avian influenza surface genes and 2009 pandemic H1N1 internal genes for transmission in the ferret model. Proc Natl Acad Sci U S A 108, 12084–12088 10.1073/pnas.1108058108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lekcharoensuk P., Lager K. M., Vemulapalli R., Woodruff M., Vincent A. L., Richt J. A. (2006). Novel swine influenza virus subtype H3N1, United States. Emerg Infect Dis 12, 787–794 10.3201/eid1205.051060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C., Hatta M., Nidom C. A., Muramoto Y., Watanabe S., Neumann G., Kawaoka Y. (2010). Reassortment between avian H5N1 and human H3N2 influenza viruses creates hybrid viruses with substantial virulence. Proc Natl Acad Sci U S A 107, 4687–4692 10.1073/pnas.0912807107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindstrom S., Garten R., Balish A., Shu B., Emery S., Berman L., Barnes N., Sleeman K., Gubareva L. & other authors (2012). Human infections with novel reassortant influenza A(H3N2)v viruses, United States, 2011. Emerg Infect Dis 18, 834–837 10.3201/eid1805.111922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorusso A., Vincent A. L., Harland M. L., Alt D., Bayles D. O., Swenson S. L., Gramer M. R., Russell C. A., Smith D. J. & other authors (2011). Genetic and antigenic characterization of H1 influenza viruses from United States swine from 2008. J Gen Virol 92, 919–930 10.1099/vir.0.027557-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma W., Lager K. M., Lekcharoensuk P., Ulery E. S., Janke B. H., Solórzano A., Webby R. J., García-Sastre A., Richt J. A. (2010). Viral reassortment and transmission after co-infection of pigs with classical H1N1 and triple-reassortant H3N2 swine influenza viruses. J Gen Virol 91, 2314–2321 10.1099/vir.0.021402-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitnaul L. J., Matrosovich M. N., Castrucci M. R., Tuzikov A. B., Bovin N. V., Kobasa D., Kawaoka Y. (2000). Balanced hemagglutinin and neuraminidase activities are critical for efficient replication of influenza A virus. J Virol 74, 6015–6020 10.1128/JVI.74.13.6015-6020.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson M. I., Lemey P., Tan Y., Vincent A., Lam T. T., Detmer S., Viboud C., Suchard M. A., Rambaut A. & other authors (2011). Spatial dynamics of human-origin H1 influenza A virus in North American swine. PLoS Pathog 7, e1002077 10.1371/journal.ppat.1002077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson M. I., Vincent A. L., Kitikoon P., Holmes E. C., Gramer M. R. (2012). Evolution of novel reassortant A/H3N2 influenza viruses in North American swine and humans, 2009–2011. J Virol 86, 8872–8878 10.1128/JVI.00259-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker J., Rambaut A., Pybus O. G. (2008). Correlating viral phenotypes with phylogeny: accounting for phylogenetic uncertainty. Infect Genet Evol 8, 239–246 10.1016/j.meegid.2007.08.001 [DOI] [PubMed] [Google Scholar]
- Posada D., Crandall K. A. (1998). modeltest: testing the model of DNA substitution. Bioinformatics 14, 817–818 10.1093/bioinformatics/14.9.817 [DOI] [PubMed] [Google Scholar]
- Rambaut, A. (2002). Sequence alignment editor, version 2.0. Available: http://tree.bio.ed.ac.uk/software/seal/ Accessed 11 December 2011.
- Smith G. J., Vijaykrishna D., Bahl J., Lycett S. J., Worobey M., Pybus O. G., Ma S. K., Cheung C. L., Raghwani J. & other authors (2009). Origins and evolutionary genomics of the 2009 swine-origin H1N1 influenza A epidemic. Nature 459, 1122–1125 10.1038/nature08182 [DOI] [PubMed] [Google Scholar]
- Sorrell E. M., Wan H., Araya Y., Song H., Perez D. R. (2009). Minimal molecular constraints for respiratory droplet transmission of an avian-human H9N2 influenza A virus. Proc Natl Acad Sci U S A 106, 7565–7570 10.1073/pnas.0900877106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swofford D. L. (2003). paup*: Phylogenetic analysis using parsimony (and other methods), version 4. Sunderland, MA: Sinauer Associates [Google Scholar]
- Vijaykrishna D., Smith G. J., Pybus O. G., Zhu H., Bhatt S., Poon L. L., Riley S., Bahl J., Ma S. K. & other authors (2011). Long-term evolution and transmission dynamics of swine influenza A virus. Nature 473, 519–522 10.1038/nature10004 [DOI] [PubMed] [Google Scholar]
- Vincent A. L., Ma W., Lager K. M., Janke B. H., Richt J. A. (2008). Swine influenza viruses a North American perspective. Adv Virus Res 72, 127–154 10.1016/S0065-3527(08)00403-X [DOI] [PubMed] [Google Scholar]
- Xu R., Zhu X., McBride R., Nycholat C. M., Yu W., Paulson J. C., Wilson I. A. (2012). Functional balance of the hemagglutinin and neuraminidase activities accompanies the emergence of the 2009 H1N1 influenza pandemic. J Virol 86, 9221–9232 10.1128/JVI.00697-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou N. N., Senne D. A., Landgraf J. S., Swenson S. L., Erickson G., Rossow K., Liu L., Yoon K., Krauss S., Webster R. G. (1999). Genetic reassortment of avian, swine, and human influenza A viruses in American pigs. J Virol 73, 8851–8856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B., Donnelly M. E., Scholes D. T., St George K., Hatta M., Kawaoka Y., Wentworth D. E. (2009). Single-reaction genomic amplification accelerates sequencing and vaccine production for classical and swine origin human influenza A viruses. J Virol 83, 10309–10313 10.1128/JVI.01109-09 [DOI] [PMC free article] [PubMed] [Google Scholar]