

Abstract
Spinocerebellar ataxia 7 (SCA7) is an incurable disease caused by expansion of CAG trinucleotide sequences within the Ataxin-7 gene. This elongated CAG tract results in an Ataxin-7 protein bearing an expanded polyglutamine (PolyQ) repeat. SCA7 disease is characterized by progressive neural and retinal degeneration leading to ataxia and blindness. Evidence gathered from investigating SCA7 and other PolyQ diseases strongly suggest that misregulation of gene expression contributes to neurodegeneration. In fact, Ataxin-7 is a subunit of the essential Spt-Ada-Gcn5-Acetltransferase (SAGA) chromatin modifying complex that regulates expression of a large number of genes. Here we discuss recent insights into Ataxin-7 function and, considering these findings, propose a model for how polyglutamine expansion of Ataxin-7 may affect Ataxin-7 function to alter chromatin modifications and gene expression.
Keywords: Spinocerebellar ataxia 7, SCA7, neurodegeneration, SAGA complex, chromatin, H2B ubiquitination, deubiquitinase, transcription, gene expression, acetyltransferase
Polyglutamine Expansion Diseases are Caused by Expansion of CAG/CTG Triplets
Polyglutamine-expansion diseases (or PolyQ diseases) arise from intergenerational expansion of CAG trinucleotide repeats in seemingly unrelated protein-coding genes. The expressed protein products bear expanded glutamine tracts. Nine PolyQ diseases are known, each caused by PolyQ expansion in a different protein. They include Huntington disease, spinal and bulbar muscular atrophy(SBMA)/Kennedy’s disease, Dentatorubral-pallidoluysian atrophy (DRPLA), and the spinocerebellar ataxias (SCA) 1, 2, 3, 6, 7, and 17. SCA3 is also known as Machado-Joseph disease, and intermediate expansion of Ataxin-2, the gene causing SCA2, is a predictor for amyotrophic lateral sclerosis (ALS).1
Considered a histological hallmark of this disease family, intranuclear inclusions composed of the polyglutamine-expanded protein as well as various transcriptional regulators and components of the ubiquitin proteasome were once seen as a central to disease pathology. It has since become apparent that intranuclear inclusions may not be toxic and may even play a protective role (reviewed by Michalik and Van Broeckhoven2).
Although arising from a common genetic feature, sharing some histological similarities, and occurring in an autosomal dominant manner, these diseases are not identical, suggesting that the function of the expanded protein is an important contributor to disease. In all cases progressive neural degeneration occurs, but different regions of the brain are preferentially targeted (reviewed in Taroni and DiDonato3). Unique among the PolyQ diseases, SCA7 also results in degeneration of the retina and macula, leading to blindness.
Spinocerebellar Ataxia 7 (SCA7)
Spinocerebellar ataxia 7 is caused by PolyQ expansion in the Ataxin-7 gene. The wild-type number of glutamine residues is approximately 10 and disease occurs after repeat length surpasses 34. Repeat length is highly variable and repeat lengths as high as 460 CAGs have been reported.4,5 SCA7 patients may live normally for many decades until symptoms are detected. Polyglutamine tract length is directly correlated with severity of disease, but inversely correlated with the age symptoms are first detected.6 Patients with the average repeat length of about 50 report symptoms during adolescence and lethality occurs 20–40 years later.7 Repeat lengths over 100 result in infantile SCA7 which is symptomatic in three months to three years, and results in lethality within months or years.
The classical SCA7 phenotype is often diagnosed due to declining blue-yellow color vision, which is indicative of macular degeneration.8 Continued degradation of the retina and macula eventually lead to blindness. Neurological symptoms appear later, as a result of cell death particularly in Purkinje cells, inferior olives, and cranial nerve nuclei. Lesser, but notable, degeneration occurs in retinal ganglion cells, the optic tract, and the visual cortex.8
Currently, there are no effective pharmacological treatments for SCA7. Instead, management of symptoms through use of walkers and low vision aids are recommended.
The SAGA Chromatin Modifying Complex
Ataxin-7 is a component of the highly conserved Spt-Ada-Gcn5-Acetyltransferase (SAGA) chromatin modifying complex.9 SAGA is a large multi-protein complex comprised of approximately 20 subunits (Table 1). For the purpose of this review, we will refer to the Drosophila nomenclature, unless otherwise noted. The complex possesses two enzymatic activities furnished by the acetyltransferase Gcn5 and the deubiquitinase Non-stop. The Gcn5 acetyltransferase is primarily responsible for acetylating histone H3 on lysine 9 and lysine 14. The Non-stop deubiquitinase removes ubiquitin from histone H2B and H2A10 as well as other, non-histone, substrates.11,12 SAGA is a transcriptional coactivator, playing a critical role in regulating gene expression. It is recruited to gene promoters by DNA-binding transcription factors. There it acetylates histone tails within chromatin, thereby facilitating a conformation more amenable to assembly of the transcription apparatus. As transcription begins, SAGA travels along with the moving transcription machinery and continues to acetylate histones to favor gene expression. Histone deubiquitination also occurs during this process. In yeast, deubiquitination has been shown to facilitate recruitment of the Bur1 kinase which phosphorylates RNA polymerase II (Pol II) on serine 2 of the C-terminal domain (CTD), promoting transcriptional elongation (reviewed by Koutelou, Hirsch, and Dent13).
Table 1. The SAGA chromatin modifying complex is highly conserved. Members of the SAGA complex from Human, Drosophila melanogaster, and Saccharomyces cerevisiae are listed along with their approximate molecular mass.
| SAGA chromatin modifying complex members | |||||
|---|---|---|---|---|---|
| Human | Molecular Mass (kDa) | D. melanogaster | Molecular Mass (kDa) | S. cerevisiae | Molecular Mass (kDa) |
| Gcn5/PCAF | 92.1 | Gcn5 (Pcaf) | 92.2 | Gcn5 | 51.1 |
| ADA2B | 46.2 | Ada2B (isoform B) | 62 | Ada2 | 50.5 |
| ADA3 | 47.5 | Ada3 (diskette) | 59.9 | Ada3 | 79.2 |
| SGF29 | 32.2 | Sgf29 | 37.1 | Sgf29 | 29.3 |
| SPT7L | 45.5 | Spt7 | 39.5 | Spt7 | 152.6 |
| ADA1 | 36.9 | Ada1 (Ada 1–2) | 35.4 | Ada1 | 54.4 |
| SPT3 | 34.9 | Spt3 | 43.5 | Spt3 | 38.8 |
| X | X | X | X | Spt8 | 66.1 |
| p38IP | 85.7 | Spt20 | 176.7–201 | Ada5/Spt20 | 67.7 |
| TRRAP | 421.3 | Tra1 (Nipped-A) | 436.00 | Tra1 | 433.1 |
| TAF9 | 29 | TAF9 (enhancer of yellow 1) | 29.3 | Taf9 | 17.3 |
| TAF10 | 24 | TAF10b (TBP-associated factor 10b) | 15.8 | Taf10 | 23 |
| TAF12 | 17.7 | TAF12 (TBP-associated factor 12-D) | 17.6 | Taf12 | 61 |
| X | X | WDA (will decrease acetylation) | 83.7 | X | X |
| TAF5L | 64.8 | X | X | Taf5 | 88.9 |
| SAP130 | 115.3 | X | X | X | X |
| TAF6L | 68.4 | SAF6 | 79.3 | Taf6 | 57.9 |
| USP22 | 56.4 | Nonstop | 56.4 | Ubp8 | 53.6 |
| ATXN7L3 | 38.2 | Sgf11 | 21.3 | Sgf11 | 11.2 |
| ENY2 | 11.1 | E(y)2 (enhancer of yellow 2) | 11.5 | Sus1 | 11.1 |
| ATXN7 | 98.1 | Ataxin-7 | 104.2 | Sgf73 | 72.9 |
The subunits of the SAGA complex are arranged into functional modules. The deubiquitinase module is anchored to the larger SAGA complex by Ataxin-7 (Fig. 1).14 Studies performed with yeast SAGA show that the N-terminus of Ataxin-7 protrudes into the deubiquitinase module, which is comprised of Ataxin-7, Non-stop, Eny2, and Sgf11.15,16 Crystal structures of the yeast deubiquitinase module show that the subunits are arranged so they intertwine, each touching the other three in a handshake necessary to maintain an active conformation for the deubiquitinase. Loss of any component of the yeast deubiquitinase module results in loss of activity, resulting in increased levels of H2B ubiquitination.
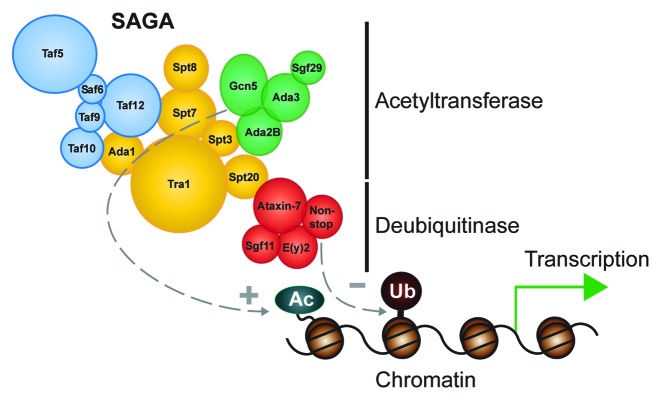
Figure 1. The SAGA complex is arranged in a modular manner, where Ataxin-7 anchors the deubiquitinase module to the larger complex.
In Drosophila, loss of Non-stop results in enhanced migration of axons from neurons in the developing eye into the optic lobe of the brain. These axons do not stop where they should—thus the gene name “Non-stop.” This defect in axonal targeting is due to the death of glial cells in Non-stop-deficient brains. These particular glial cells are critical for establishing the guiding signals retinal neurons use to arrive at their very precise final destination within the optic lobe.17 These findings indicate that regulation by the deubiquitinase is critical for many processes in the developing brain, including survival of some brain cells and neural organization. Defects in axonal targeting are also seen upon loss of other SAGA subunits, including Ada2B and Sgf11, suggesting they act as a complex to maintain neural stability.17 This role for Non-stop in regulating neural stability is conserved and, in humans, knockdown of USP22 has been shown to result in apoptosis in glioma cells.18
The Gcn5 acetyltransferase has also been implicated in regulation of neural processes. Flies lacking Gcn5 have brains approximately half the size of their wild-type counterparts (unpublished observations). In mice, loss of Gcn5 results in early embryonic lethality. However, a conditional knockout mouse model lacking Gcn5 in neural stem cells has greatly reduced brain size (microcephaly).19 Additionally, mice homozygous for an allele of Gcn5 lacking acetyltransferase activity show severe neural tube closure defects and exencephaly during embryogenesis.20
Therefore, multiple SAGA subunits have been implicated in neural regulation. Furthermore, they have been shown to affect multiple processes, suggesting that SAGA is an important player in neural development.
Ataxin-7 Function in Higher Eukaryotes
Knowledge of normal Ataxin-7 function in higher eukaryotes is limited, so models are heavily influenced by hypotheses derived from studies performed in yeast. By contrast, insight into Ataxin-7 dysfunction in PolyQ disease has mainly come from transgenic mouse, and human/mouse cell culture models in which polyglutamine-expanded Ataxin-7 is overexpressed in the presence of the wild-type alleles of Ataxin-7. In another model, human polyglutamine-expanded Ataxin-7 was expressed exogenously in Drosophila.21 In mice and in Drosophila, overexpression of PolyQ-expanded Ataxin-7 is sufficient to recapitulate major SCA7 phenotypes, including neural and retinal degeneration, ataxia, and reduced lifespan.22,23 Analysis of gene expression defects in mouse and tissue culture models, however, show a surprisingly small set of genes affected by PolyQ expansion and no “smoking gun” to suggest a critical gene causing SCA7 disease.24 A genetic modifier screen performed with the Drosophila model identified a handful of hits, mostly associated with gene expression and protein folding.21 Interestingly, genes misregulated by PolyQ expansion of Ataxin-7 do not seem to be affected by loss of Gcn5, and Gcn5 was not found to be a genetic modifier of the Ataxin-7-PolyQ phenotype in Drosophila.21,25 This lack of dependency is surprising considering that PolyQ-expanded Ataxin-7 incorporates into the SAGA complex and affects Gcn5 acetyltransferase activity.26,27 These observations suggest that more needs to be understood about how Ataxin-7 functions before we can understand how disrupting these functions might contribute to neurodegenerative disease.
Drosophila melanogaster provided the powerful genetic and biochemical tools we needed to learn more about Ataxin-7, but the gene encoding the Drosophila ortholog of Ataxin-7 was not known. To identify Drosophila Ataxin-7 we purified SAGA from Drosophila S2 cells stably expressing epitope-tagged versions of known SAGA subunits.28 Identification of the co-purifying proteins was done using MudPit shotgun proteomics. Thorough in silico analysis of the proteomics data revealed that the protein product of the uncharacterized gene CG9866 was a potential Ataxin-7 ortholog. Biochemical characterization showed that reciprocal purification through epitope-tagged CG9866 protein captured the entire SAGA complex, suggesting this was indeed a stable component of SAGA. Based on yeast results, we predicted that loss of Ataxin-7 would release the deubiquitinase module from the complex. When we used gel filtration chromatography to determine the size of SAGA in flies lacking CG9866, we found that SAGA was indeed smaller. More detailed pull-down analysis using antibodies toward endogenous Non-stop verified that the deubiquitinase was no longer stably associated with SAGA. Based upon conservation of sequence observed upon alignment of CG9866 and mammalian Ataxin-7, the stable association of CG9866 protein with the Drosophila SAGA complex, and the conservation of Ataxin-7 function in anchoring the deubiquitinase module to the larger complex, we concluded that CG9866 is the functional ortholog of Ataxin-7.
A Runaway Deubiquitinase and Neural and/or Retinal Degeneration Without PolyQ Expansion
Since loss of Ataxin-7 releases the deubiquitinase module from SAGA-mediated regulation, we were interested in understanding what activity this module might have outside of SAGA. We performed polytene chromosome squash analysis and found that Non-stop continues to bind to chromosomes in the absence of Ataxin-7. Previous analysis of the yeast deubiquitinase module suggested that the released module would be inactive without Ataxin-7, resulting in elevated levels of H2B ubiquitination. Surprisingly, when we measured bulk levels of H2B ubiquitination we found that they were reduced in mutants lacking Ataxin-7. This suggested that the deubiquitinase module might be enzymatically active in the absence of Ataxin-7. To test this hypothesis, we reassembled the deubiquitinase module from purified proteins. We found that full-length Ataxin-7 protein readily incorporated into the deubiquitinase module, and also found that the N-terminus incorporated into the deubiquitinase module as well, suggesting that the orientation of the protein was also conserved from yeast to Drosophila. When we tested the activity of the reconstituted module we found that it was indeed active without Ataxin-7.
When we examined the phenotypes resulting from of Ataxin-7 loss in flies, we were surprised to see that they recapitulated symptoms seen in mouse and Drosophila models of SCA7 in which PolyQ-expanded Ataxin-7 was exogenously expressed.21 The loss of function phenotype included neural and retinal degeneration, decreased motility, and severely decreased lifespan. To determine whether these effects were mediated by the released deubiquitinase module, we genetically reduced the copy number of Non-stop. This gene reduction resulted in partial rescue of the lethality caused by loss of Ataxin-7, suggesting that defects occurring upon loss of Ataxin-7 were partially dependent on Non-stop function.
Implications for Polyglutamine Expansion-Mediated Disruption of Ataxin-7 Function
Our results indicate that, in Drosophila, loss of Ataxin-7 causes a mutant phenotype similar to that seen upon exogenous expression of PolyQ-expanded Ataxin-7.21 Furthermore, the consequences of this loss demonstrate that the associated neural and retinal degeneration are mediated, in part, through Non-stop. We confirmed that this effect is conserved in a mammalian tissue culture model. However, the consequences of this effect and the loss-of-function phenotype in an Ataxin-7 knockout mouse model have not been described. We suggest the possibility—based on the similarity of these phenotypes—that the impact of PolyQ expansion in Ataxin-7 may partially contribute to SCA7 disease through loss of Ataxin-7 function, although this awaits further analysis. Studies in mammalian models may also be complicated by the presence of proteins that share high sequence similarity with Ataxin-728 and may have redundant functions.
Considering our observations, we hypothesize that PolyQ expansion of Ataxin-7 disrupts an important function for Ataxin-7 in regulating deubiquitination. Because Drosophila Ataxin-7 has only recently been uncovered, polyglutamine expansion of this protein has not been examined, and much is unknown. Additionally, although much has been discovered about the molecular mechanisms underlying function of the yeast SAGA deubiquitinase, less is known about deubiquitinase function in higher organisms. Furthermore, it is still unclear how misregulation of the SAGA deubiquitinase module results in neuronal phenotypes. In light of the findings described above, we propose that PolyQ expansion of Ataxin-7 might result in aberrant regulation of the SAGA deubiquitinase module, leading to neural and retinal degeneration.
PolyQ-expansion of Ataxin-7 occurs in the N-terminus of the human protein. As predicted from yeast models, this region inserts into the deubiquitinase module. Using an in vitro reconstituted deubiquitinase module, we have verified that this orientation is conserved in Drosophila.28 Therefore, we predict that PolyQ expansion of this region will affect SAGA-mediated regulation of deubiquitination. This disruption in function may occur through various mechanisms (Fig. 2). The deubiquitinase might still interact with PolyQ-expanded Ataxin-7 and be enzymatically active. However, the presence of the wild-type enzymatic activity does not mean that the module will function on chromatin if it is not brought into proximity with ubiquitinated histones. Alternatively, the deubiquitinase module may bind to the PolyQ-expanded Ataxin-7 N-terminus in a way that leaves it inactive. Lastly, it is possible that PolyQ expansion may prevent the deubiquitinase module from interacting with Ataxin-7, resulting in permanent release of the deubiquitinase module, much like the situation in which Ataxin-7 expression is lost. This last scenario would be most directly explained by the findings described above. Although we focus on chromatin, there are also non-histone substrates for the deubiquitinase module which may also be important in SCA7.

Figure 2. Model for polyglutamine-expansion-mediated disruption of SAGA function. The N-terminus of Ataxin-7 protrudes into the deubiquitinase module. It is possible that this will alter SAGA-associated deubiquitinase function.
Most likely, there are many mechanisms contributing to PolyQ expansion-mediated toxicity. For example, it has been shown that RNA toxicity contributes to neurodegeneration in Ataxin-3.29 Protein-protein interactions may also be altered. Accordingly, increased or decreased interaction between PolyQ-expanded proteins and transcription factors or transcriptional coactivators has also been demonstrated. Further investigation will allow us to determine what role a rogue deubiquitinase module might play in SCA7 disease.
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
Acknowledgments
This work was supported by funds from the Stowers Institute and the National Institutes of Health (grant GM99945-01) to S.M.A. and J.L.W.
References
- 1.Elden AC, Kim HJ, Hart MP, Chen-Plotkin AS, Johnson BS, Fang X, Armakola M, Geser F, Greene R, Lu MM, et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature. 2010;466:1069–75. doi: 10.1038/nature09320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Michalik A, Van Broeckhoven C. Pathogenesis of polyglutamine disorders: aggregation revisited. Hum Mol Genet. 2003;12:R173–86. doi: 10.1093/hmg/ddg295. [DOI] [PubMed] [Google Scholar]
- 3.Taroni F, DiDonato S. Pathways to motor incoordination: the inherited ataxias. Nat Rev Neurosci. 2004;5:641–55. doi: 10.1038/nrn1474. [DOI] [PubMed] [Google Scholar]
- 4.van de Warrenburg BP, Frenken CW, Ausems MG, Kleefstra T, Sinke RJ, Knoers NV, Kremer HP. Striking anticipation in spinocerebellar ataxia type 7: the infantile phenotype. J Neurol. 2001;248:911–4. doi: 10.1007/s004150170082. [DOI] [PubMed] [Google Scholar]
- 5.David G, Abbas N, Stevanin G, Dürr A, Yvert G, Cancel G, Weber C, Imbert G, Saudou F, Antoniou E, et al. Cloning of the SCA7 gene reveals a highly unstable CAG repeat expansion. Nat Genet. 1997;17:65–70. doi: 10.1038/ng0997-65. [DOI] [PubMed] [Google Scholar]
- 6.David G, Dürr A, Stevanin G, Cancel G, Abbas N, Benomar A, Belal S, Lebre AS, Abada-Bendib M, Grid D, et al. Molecular and clinical correlations in autosomal dominant cerebellar ataxia with progressive macular dystrophy (SCA7) Hum Mol Genet. 1998;7:165–70. doi: 10.1093/hmg/7.2.165. [DOI] [PubMed] [Google Scholar]
- 7.Michalik A, Martin JJ, Van Broeckhoven C. Spinocerebellar ataxia type 7 associated with pigmentary retinal dystrophy. Eur J Hum Genet. 2004;12:2–15. doi: 10.1038/sj.ejhg.5201108. [DOI] [PubMed] [Google Scholar]
- 8.Gouw LG, Digre KB, Harris CP, Haines JH, Ptacek LJ. Autosomal dominant cerebellar ataxia with retinal degeneration: clinical, neuropathologic, and genetic analysis of a large kindred. Neurology. 1994;44:1441–7. doi: 10.1212/WNL.44.8.1441. [DOI] [PubMed] [Google Scholar]
- 9.Weake VM, Workman JL. SAGA function in tissue-specific gene expression. Trends Cell Biol. 2012;22:177–84. doi: 10.1016/j.tcb.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang XY, Pfeiffer HK, Thorne AW, McMahon SB. USP22, an hSAGA subunit and potential cancer stem cell marker, reverses the polycomb-catalyzed ubiquitylation of histone H2A. Cell Cycle. 2008;7:1522–4. doi: 10.4161/cc.7.11.5962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Atanassov BS, Evrard YA, Multani AS, Zhang Z, Tora L, Devys D, Chang S, Dent SY. Gcn5 and SAGA regulate shelterin protein turnover and telomere maintenance. Mol Cell. 2009;35:352–64. doi: 10.1016/j.molcel.2009.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Atanassov BS, Dent SY. USP22 regulates cell proliferation by deubiquitinating the transcriptional regulator FBP1. EMBO Rep. 2011;12:924–30. doi: 10.1038/embor.2011.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koutelou E, Hirsch CL, Dent SY. Multiple faces of the SAGA complex. Curr Opin Cell Biol. 2010;22:374–82. doi: 10.1016/j.ceb.2010.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee KK, Sardiu ME, Swanson SK, Gilmore JM, Torok M, Grant PA, Florens L, Workman JL, Washburn MP. Combinatorial depletion analysis to assemble the network architecture of the SAGA and ADA chromatin remodeling complexes. Mol Syst Biol. 2011;7:503. doi: 10.1038/msb.2011.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Samara NL, Datta AB, Berndsen CE, Zhang X, Yao T, Cohen RE, Wolberger C. Structural insights into the assembly and function of the SAGA deubiquitinating module. Science. 2010;328:1025–9. doi: 10.1126/science.1190049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Köhler A, Zimmerman E, Schneider M, Hurt E, Zheng N. Structural basis for assembly and activation of the heterotetrameric SAGA histone H2B deubiquitinase module. Cell. 2010;141:606–17. doi: 10.1016/j.cell.2010.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weake VM, Lee KK, Guelman S, Lin CH, Seidel C, Abmayr SM, Workman JL. SAGA-mediated H2B deubiquitination controls the development of neuronal connectivity in the Drosophila visual system. EMBO J. 2008;27:394–405. doi: 10.1038/sj.emboj.7601966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li ZH, Yu Y, Du C, Fu H, Wang J, Tian Y. RNA interference-mediated USP22 gene silencing promotes human brain glioma apoptosis and induces cell cycle arrest. Oncol Lett. 2013;5:1290–4. doi: 10.3892/ol.2013.1188. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 19.Martínez-Cerdeño V, Lemen JM, Chan V, Wey A, Lin W, Dent SR, Knoepfler PS. N-Myc and GCN5 regulate significantly overlapping transcriptional programs in neural stem cells. PLoS One. 2012;7:e39456. doi: 10.1371/journal.pone.0039456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bu P, Evrard YA, Lozano G, Dent SY. Loss of Gcn5 acetyltransferase activity leads to neural tube closure defects and exencephaly in mouse embryos. Mol Cell Biol. 2007;27:3405–16. doi: 10.1128/MCB.00066-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Latouche M, Lasbleiz C, Martin E, Monnier V, Debeir T, Mouatt-Prigent A, Muriel MP, Morel L, Ruberg M, Brice A, et al. A conditional pan-neuronal Drosophila model of spinocerebellar ataxia 7 with a reversible adult phenotype suitable for identifying modifier genes. J Neurosci. 2007;27:2483–92. doi: 10.1523/JNEUROSCI.5453-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Duncan CE, An MC, Papanikolaou T, Rugani C, Vitelli C, Ellerby LM. Histone deacetylase-3 interacts with ataxin-7 and is altered in a spinocerebellar ataxia type 7 mouse model. Mol Neurodegener. 2013;8:42. doi: 10.1186/1750-1326-8-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Furrer SA, Waldherr SM, Mohanachandran MS, Baughn TD, Nguyen KT, Sopher BL, Damian VA, Garden GA, La Spada AR. Reduction of mutant ataxin-7 expression restores motor function and prevents cerebellar synaptic reorganization in a conditional mouse model of SCA7. Hum Mol Genet. 2013;22:890–903. doi: 10.1093/hmg/dds495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McCullough SD, Xu X, Dent SY, Bekiranov S, Roeder RG, Grant PA. Reelin is a target of polyglutamine expanded ataxin-7 in human spinocerebellar ataxia type 7 (SCA7) astrocytes. Proc Natl Acad Sci U S A. 2012;109:21319–24. doi: 10.1073/pnas.1218331110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen YC, Gatchel JR, Lewis RW, Mao CA, Grant PA, Zoghbi HY, Dent SY. Gcn5 loss-of-function accelerates cerebellar and retinal degeneration in a SCA7 mouse model. Hum Mol Genet. 2012;21:394–405. doi: 10.1093/hmg/ddr474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burke TL, Miller JL, Grant PA. Direct inhibition of Gcn5 protein catalytic activity by polyglutamine-expanded ataxin-7. J Biol Chem. 2013;288:34266–75. doi: 10.1074/jbc.M113.487538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McMahon SJ, Pray-Grant MG, Schieltz D, Yates JR, 3rd, Grant PA. Polyglutamine-expanded spinocerebellar ataxia-7 protein disrupts normal SAGA and SLIK histone acetyltransferase activity. Proc Natl Acad Sci U S A. 2005;102:8478–82. doi: 10.1073/pnas.0503493102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mohan RD, Dialynas G, Weake VM, Liu J, Martin-Brown S, Florens L, Washburn MP, Workman JL, Abmayr SM. Loss of Drosophila Ataxin-7, a SAGA subunit, reduces H2B ubiquitination and leads to neural and retinal degeneration. Genes Dev. 2014;28:259–72. doi: 10.1101/gad.225151.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li LB, Yu Z, Teng X, Bonini NM. RNA toxicity is a component of ataxin-3 degeneration in Drosophila. Nature. 2008;453:1107–11. doi: 10.1038/nature06909. [DOI] [PMC free article] [PubMed] [Google Scholar]