

Abstract
Background
Multiple endocrine neoplasia type 1 (MEN1) is a rare inherited disorder characterized by the simultaneous occurrence of endocrine tumors in target tissues (mainly the pituitary, endocrine pancreas, and parathyroid glands). MEN1 is caused by mutations in the MEN1 gene, which functions as a tumor suppressor and consists of one untranslated exon and nine exons encoding the menin protein. This condition is usually suspected when we encounter patients diagnosed with tumors in multiple endocrine organs, as mentioned above.
Methods
A 65-year-old woman who underwent surgery for a pancreatic tumor (serous cystadenoma) 5 years previously was referred to our hospital due to neurologic symptoms of diplopia and left ptosis. Brain magnetic resonance imaging revealed a 3.4-cm lesion originating from the cavernous sinus wall and extending into the sellar region. It was thought to be a nonfunctioning tumor from the results of the combined pituitary function test. Incidentally, we found that she also had a pancreatic tumor, indicating the necessity of genetic analysis for MEN1.
Results
Genomic analysis using peripheral leukocytes revealed a heterozygous c.1621G>A mutation in the MEN1 gene that was previously reported to be either a pathogenic mutation or a simple polymorphism. We pursued a stereotactic approach to the pituitary lesion, and microscopic findings of the tumor revealed it to be an intrasellar cavernous hemangioma, a rare finding in the sellar region and even rarer in relation to oculomotor palsy. The patient recovered well from surgery, but refused further evaluation for the pancreatic lesion.
Conclusion
There is great emphasis placed on genetic testing in the diagnosis of MEN1, but herein we report a case where it did not assist in diagnosis, hence, further discussion on the role of genetic testing in this disease is needed. Also, in cases of pituitary tumor with cranial nerve palsy, despite its low prevalence, intrasellar cavernous hemangioma could be suspected.
Keywords: Multiple endocrine neoplasia type 1, MEN1 gene, Intrasellar cavernous hemangioma
INTRODUCTION
Multiple endocrine neoplasia type 1 (MEN1) is an autosomal dominant genetic disease involving more than two tumors occurring simultaneously among the pituitary, islet cell, and parathyroid gland. Adrenocortical tumors, lipoma, goiter, carcinoidosis, and pheochromocytoma may also be present [1], and constitutive disease may occur that requires long-term follow-up, since it is a rare disease with complicated manifestations resulting from multiple endocrine tumors.
The MEN1 gene was first discovered by Chandrasekharappa et al. [2] in 1997, and, since then, greater attention has been paid to tests for and diagnosis involving molecular genetics. MEN1 occurs due to a germline mutation of the MEN1 gene located at 11q13, and the MEN1 gene encodes the menin protein.
The authors identified pituitary and pancreatic tumors in a patient who was admitted complaining of diplopia and exhibiting left ptosis. Molecular genetic testing was performed on the MEN1 gene, which indicated a mutation and a possible diagnosis of MEN1. A genetic mutation was found at exon number 10, but, in previous studies, this mutation was reported either a missense mutation [3] or a single nucleotide polymorphism [4], hence a confirmed diagnosis was deferred. To obtain an accurate diagnosis, a surgical approach and biopsy were conducted. The resulting diagnosis was intrasellar cavernous hemangioma.
METHODS
Case study
A 65-year-old female patient presented complaining of diplopia and left ptosis. She had a Whipple procedure performed in another hospital after incidental discovery of a pancreatic tumor in 2008, was diagnosed with serous cystadenoma and was under follow-up care. She also complained of intermittent headaches for 14 days prior to hospital admission.
Physical examination revealed blood pressure 120/70 mm Hg, pulse 78 beats per minute, respiratory rate 18/minute, body temperature 36.6℃, and there was no significant change in blood pressure or pulse rate over the course of hospitalization. During cervical inspection and palpation, the thyroid was not enlarged and no mass was felt. Abdominal inspection revealed signs of surgery in the lower part of the upper abdomen, but no tenderness was found. Auscultation of the lungs and heart were normal. The diplopia worsened on left gaze and a visual field defect was noted. Axillar and pubic hairs were normal and there was no sign of lactation.
Laboratory test results of peripheral blood were: white blood cell 8,310/mm3, hemoglobin 10.5 g/dL, platelets 527,000/mm3, and urinalysis was normal. All results from serum chemistry examination were within normal limits: blood urea nitrogen 15.5 mg/dL, creatinine 0.6 mg/dL, Na 139 mEq/L, K 5.2 mEq/L, Cl 105 mEq/L, total protein 6.9 g/dL, albumin 3.9 g/dL, phosphorous 4.4 mg/dL, uric acid 3.6 mg/dL, and calcium 8.4 mg/dL. Serum parathyroid hormone was 60 pg/mL (range, 10 to 65) and, according to thyroid fuction test, thyroid stimulating hormone was 2.67 µIU/mL (range, 0.55 to 4.78), T3 was 83 ng/dL (range, 60 to 180), and free T4 was 0.9 ng/dL (range, 0.89 to 1.78). Baseline pituitary hormone tests were all within normal limits: prolactin 42.34 ng/mL, luteinizing hormone 2.6 mIU/mL, follicle stimulating hormone 17.5 mIU/mL, growth hormone 1.31 ng/mL, adrenocortical hormone 42.9 pg/mL, and cortisol 8.13 µg/dL. Fasting plasma insulin was 2.96 µU/mL and fasting blood glucose was 83 mg/dL. Serum glucagon and gastrin were 45 pg/mL (range, 59 to 177) and 85.7 pg/mL (<110), respectively. Twenty-four-hour urinalysis results were all within normal limits: vanillylmandelic acid 0.6 mg/day (range, 0 to 8), epinephrine 3.4 µg/day (range, 0 to 20), norepinephrine 7.7 µg/day (range, 15 to 80), and metanephrine 16 mg/day (range, 52 to 341).
On magnetic resonance imaging (MRI), a 3.4-cm contrast-enhanced mass was observed from the cavernous sinus wall and extending into the sellar region, which indicated a pituitary tumor (Fig. 1A, B). No significant findings were noted on chest computed tomography (CT) apart from a small ganglion on the left mediastinum; on abdominal CT, a 2.6-cm contrast-enhanced mass was noted on the body of the pancreas (Fig. 2). On cervical ultrasound, multiple cysts and calcifications were noted on both thyroid lobes, but no tumors were found on the parathyroid. No abnormal findings were found on PET scan.
Fig. 1.

(A, B) Preoperative magnetic resonance imaging. A 3.4-cm enhanced mass (arrows) with lobulated contour at the cavernous sinus extending into the sellar region (A, axial view; B, coronal view). (C, D) Three months after radiosurgery, the enhanced mass exhibits a decrease in the extent of the lobulated contour at the cavernous sinus extending into the sellar region (C, axial view; D, coronal view).
Fig. 2.

A 2.5-cm arterial wall enhancing mass with distal p-duct dilatation in the pancreas body (arrows).
After hospitalization, the patient was treated conservatively and a pre-surgical examination was conducted for biopsy of the pituitary tumor. The combined pituitary stimulation test presented a normal pituitary function (Table 1). Microneedle aspiration was performed under endoscopic ultrasound on the tumorous body of the pancreas found on abdominal CT. The cytology specimen exhibited benign epithelial cells. The patient underwent a craniotomy and tumor biopsy and was diagnosed with cavernous hemangioma (Fig. 3). It was determined to be a benign brain tumor, which was located on difficult site to operate, and was treated with stereotactic radiation therapy. The size and range of the tumor were reduced, as determined from a brain MRI conducted 3 months later, and diplopia and left ptosis were improved (Fig. 1C, D). We considered surgical treatment for the pancreatic tumor found during abdominal CT, but the patient refused further treatment.
Table 1.
Combined Pituitary Stimulation Test (Cocktail Test)
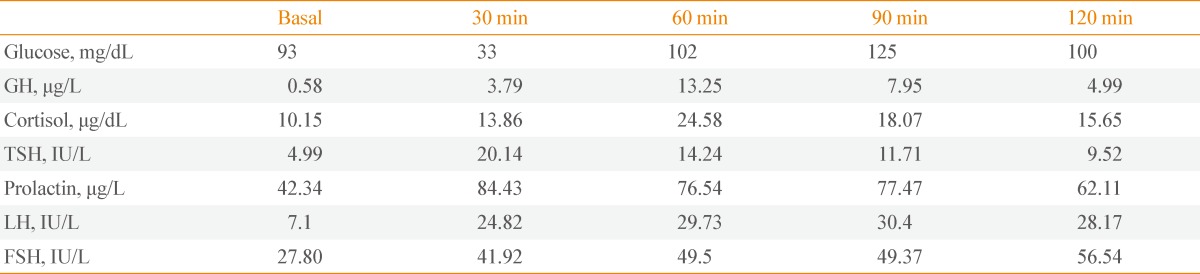
GH, growth hormone; TSH, thyroid stimulating hormone; LH, luteinizing hormone; FSH, follicle stimulating hormone.
Fig. 3.

(A, B) On histologic examination, the mass was composed of dilated vessels occasionally containing thrombi (A, H&E stain, ×100; B, H&E stain, ×400). (C) Immunohistochemically, these endothelial cells were fully reactive in CD31 (CD31, ×400). These findings were indicative of cavernous hemangioma.
Methods
Genetic testing was conducted after receiving patient consent to investigate the possibility of MEN1 with involvement of pituitary and pancreatic tumors.
Specimen preparation
Genomic DNA was extracted with a QiaAmp DNA blood mini kit (Qiagene, Valencia, CA, USA) from leukocytes of the patient's peripheral blood.
Polymerase chain reaction
Polymerase chain reaction (PCR) was conducted on nine exons, transcript exon 2 to exon 10 of 10 exons, and the primers used are listed in Table 2. The PCR reaction solution consisted of 12.5 µL GC I buffer, 4 µL dNTP mixture solution, 0.2 µL LA-Taq DNA polymerase, and 2 µL of each primer, and was combined with 6.3 µL extracted DNA for a total mixed solution of 25 µL. Denaturation was conducted for 1 minute at 95℃, for 30 seconds at 96℃ and 60℃, for 40 seconds at 72℃ 35 times, and then finally for 5 minutes at 68℃. The reacted product underwent electrophoresis on 1.5% agarose gel and the identified DNA bands were then purified with an AccuPrep PCR Purification Kit (Bioneer, Daejeon, Korea).
Table 2.
Polymerase Chain Reaction Primers Used for MEN1 Somatic Mutation Analysis

Direct DNA sequencing
For direct DNA sequencing, an automated sequencing machine, the Genetic Analyzer 3100 (Applied Biosystems, Foster City, CA, USA), was used. In direct sequencing, forward and reverse sequencing analysis were conducted for each primer to reduce analysis errors, and mutation and polymorphism were identified by comparing the DNA sequencing results to data on dbSNP (http://www.ncbi.nlm.nih.gov/projects/SNP/).
Prediction of the effects of amino acid change
A polymorphism phenotyping program, PolyPhen [5], was used to predict the effects of amino acid change on protein function and to hypothesize the clinical significance, which was analyzed in silico. This program supplied three categories according to changes in proteins: probable damage>possible damage>benign.
RESULTS
Pathological diagnosis of tumors
The mass contained dilated blood vessels and a vessel with thrombus. Endothelial cells indicated a strong positivity to CD 31, which corresponds to hemangioma (Fig. 3).
Somatic mutation of the MEN1 gene
After testing nine exon parts with PCR and direct DNA sequencing, we found a heterozygous c.1621G>A (Ala541Thr) mutation at exon 10 of the MEN1 gene (Fig. 4). This mutation was found to match rs2959656 after searching the dbSNP database, and a probable benign result was reported after in silico analysis of the effects of amino acid change (Fig. 5).
Fig. 4.

Polymerase chain reaction-direct sequencing demonstrates the tumor's heterogeneous pattern and variant allele, suggesting that a change occurred to the nucleotide sequence from the GCA to ACA (alanine to threonine).
Fig. 5.

PolyPhen-2 is a tool for prediction of the possible impact of amino acid substitutions on the structure and function of human proteins. The PolyPhen-2 report indicates that the heterogeneous c.1621G>A (Ala541Thr) mutation is predicted to be benign.
DISCUSSION
MEN1 is a rare disorder characterized by the simultaneous occurrence of endocrine tumors in target tissues (pituitary, endocrine pancreas, and parathyroid glands and islet cells), and the prevalence is approximately 1 in 30,000 to 50,000 people [6]. MEN1 is reported very rarely in Korea.
The clinical findings of MEN1 are very similar to those of primary tumors that affect the same tissues, but it does have some distinctive characteristics [7]. First, as MEN1 simultaneously occurs in various endocrine tissues, the recurrence rate is high after treatment. Some MEN1tumors occur at an early age and tend to be malignant when compared to primary tumors of the same tissues, and are the major cause of mortality in MEN1 patients [8]. Therefore, distinguishing combinations of tumors in different tissues related to other diseases from MEN1 is very important for prognosis and appropriate treatment. It is recommended that specific gene mutations be identified through genetic testing.
The causative gene for MEN1 is the MEN1 gene, a tumor suppressor gene that codes for the menin protein. The MEN1 gene is comprised of one untranslated exon and nine exons [9]. The menin protein controls cell growth and the cell cycle, so a mutation in this gene generally induces a production of immature proteins [10,11]. More than 500 germline and somatic cell mutations were reported in MEN1, and these mutations occur simultaneously across the coding area. No correlation between the MEN1 genotype and phenotype has yet been clearly identified [9]. In this patient, a heterozygous c.1621G>A (Ala541Thr) mutation was found on exon 10, and two conflicting opinions exist for this mutation. In 1998, Shan et al. [3] investigated menin protein mutations in relation to the MEN1 gene, and classified this same mutation in a parathyroid tumor patient as a missense mutation. In 2008, however, Bazzi et al. [4] discovered the same mutation in patients with pancreatic tumors and determined the mutation to be an SNP. Based on the conflicting opinions on these mutations, there are limitations to diagnosis of MEN1 solely based on gene mutation results that affected our patient in particular.
In this patient, there was no sign of parathyroid tumor, hyperproliferation, or increase in serum calcium. Microneedle aspiration was performed under endoscopic ultrasound on the pancreatic tumor found by abdominal CT. Epithelial cells appeared benign and the hormone test was normal; thus, MEN1 was ruled out. For pituitary tumors that appear as a contrast-enhanced mass on brain MRI, the possibility of meningioma or hemangioma should be kept in mind and an octerotide scan should be considered in the differential diagnosis of nonfunctioning pituitary tumors.
In this patient, pituitary cavernous hemangioma presented in a pathologically similar manner to meningioma, in terms of presentation of contrast-enhanced imaging with low or identical signals on the T1-focused image and presentation of consistent contrast-enhancement after the administration of gadolinium. However, cavernous hemangioma can be distinguished by contrast-enhancement of a T2-focused image as presenting with clearer high-strength contrast-enhancement than a meningioma. Also, a pituitary tumor can be distinguished by its clear boundaries on high-strength consistent contrast-enhancement on T2-focused images. The frequency of cavernous hemangioma is low, but due to the possibility of hemorrhage or neurological abnormalities, the risk cannot be underestimated. Intrasellar cavernous hemangioma is a particularly rare disease; there were only seven previous cases reported overseas and only one prior case reported in Korea; each of these cases is presented in Table 3 [12,13,14,15,16,17,18,19]. Intrasellar or cranial cavernous hemangioma was introduced by Lombardi et al. [12] in 1994, and was classified into three types: hemangioma of the cavernous venous sinus growing lateral or medial to the lumen of the venous sinus, or growing outside of the cavernous venous sinus. In the case of intrasellar cavernous hemangioma, there is a minority opinion that a cavernous hemangioma grows on primarily the intrasellar region toward the cavernous venous sinus [20], but this has not been firmly established. Also, the optic nerve palsy that was reported in this intrasellar cavernous hemangioma case has rarely been described outside of a report by Chuang et al. [20] in 2006, and there have been no cases in Korea. However, the authors were able to identify a correlation between optic nerve palsy and the position of the brain tumor in the present case.
Table 3.
Summary of the Clinical Profile of Previously Reported Intrasellar Cavernous Hemangiomas
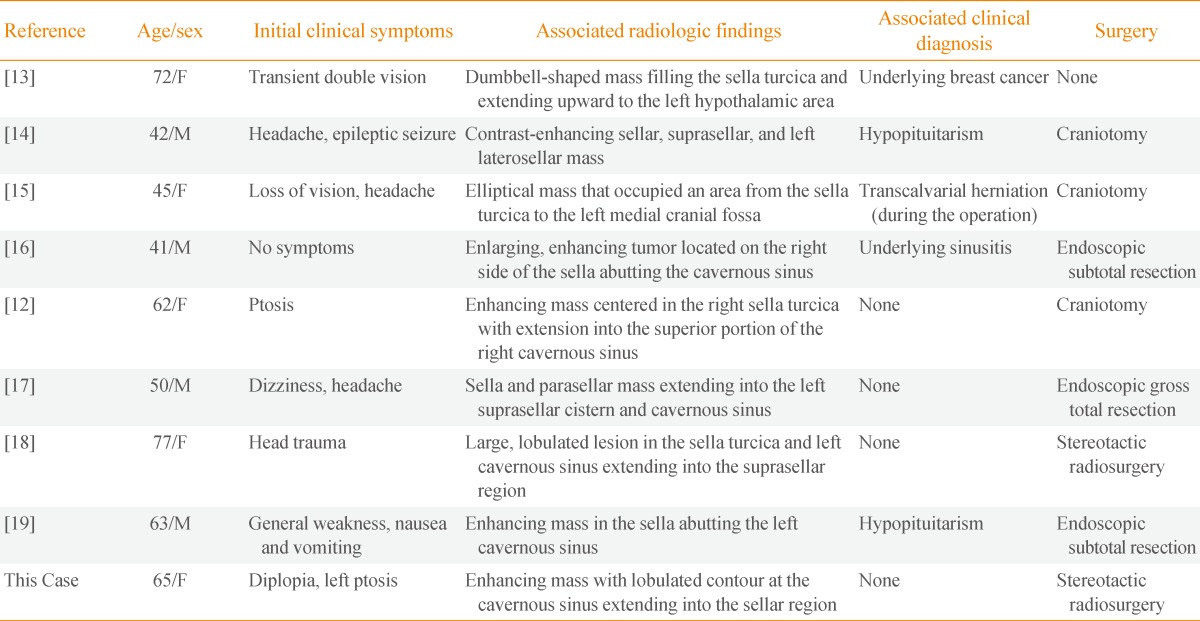
F, female; M, male.
Cavernous hemangioma can be fully cured by total resection, but studies are still being conducted on the surgical results for highly angiogenic tumors in the intrasellar region, due to the risk of cerebral hemorrhage. If cavernous hemangioma is suspected, obtaining a lyophilized specimen is important for diagnosis and treatment, as is control of bleeding during resection of the tumor [12]. There are a number of conflicting opinions on the efficacy of radiosurgery for the treatment of cavernous hemangioma; according to a recent large-scale study, the risk and prevalence of postsurgical hemorrhage was clearly reduced after radiosurgery [21,22]. Considering this recent study and the risk of hemorrhage during surgery, stereotactic radiation therapy was performed in this patient and the clinical response was observed through brain MRI. The patient's symptoms were improved during follow-up examination 3 months after surgery.
In summary, through radiological, chemical, and hormonal examinations, genetic tests and biopsy, the authors were able to diagnose a case of intrasellar cavernous hemangioma in a patient admitted to the hospital complaining of diplopia and left ptosis. We investigated the possibility of MEN1 due to the pituitary and pancreatic cell tumors identified from the patient's medical history and radiological findings, and identified a genetic mutation of the MEN1 gene, but a firm diagnosis was difficult, as there were conflicting opinions on this particular mutation. Biopsy of the pituitary tumor led to a diagnosis of cavernous hemangioma expanding to within the intrasellar region due to medial growth from the wall of the cavernous venous sinus. Stereotactic radiation therapy was conducted. Somatic mutation tests on the genes found in hereditary tumors may influence the prognosis and help to determine treatment option for a sporadic tumor. There is great emphasis placed on genetic testing in the diagnosis of MEN1, but herein we report a case where it did not assist in diagnosis, hence, further discussion on the role of genetic testing in this disease is needed. Also, in cases of pituitary tumor with cranial nerve palsy, despite its low prevalence, intrasellar cavernous hemangioma could be suspected, and stereotactic radiation therapy considered as the treatment of choice.
ACKNOWLEDGMENTS
The authors thank Professors Seon-Yong Jeong and Hyun-Seok Jin for their genetic analysis. This work was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT and Future Planning, Korea (NRF-2013R1A1A1A05005629 to S.L).
Footnotes
No potential conflict of interest relevant to this article was reported.
References
- 1.Trump D, Farren B, Wooding C, Pang JT, Besser GM, Buchanan KD, Edwards CR, Heath DA, Jackson CE, Jansen S, Lips K, Monson JP, O'Halloran D, Sampson J, Shalet SM, Wheeler MH, Zink A, Thakker RV. Clinical studies of multiple endocrine neoplasia type 1 (MEN1) QJM. 1996;89:653–669. doi: 10.1093/qjmed/89.9.653. [DOI] [PubMed] [Google Scholar]
- 2.Chandrasekharappa SC, Guru SC, Manickam P, Olufemi SE, Collins FS, Emmert-Buck MR, Debelenko LV, Zhuang Z, Lubensky IA, Liotta LA, Crabtree JS, Wang Y, Roe BA, Weisemann J, Boguski MS, Agarwal SK, Kester MB, Kim YS, Heppner C, Dong Q, Spiegel AM, Burns AL, Marx SJ. Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science. 1997;276:404–407. doi: 10.1126/science.276.5311.404. [DOI] [PubMed] [Google Scholar]
- 3.Shan L, Nakamura Y, Nakamura M, Yokoi T, Tsujimoto M, Arima R, Kameya T, Kakudo K. Somatic mutations of multiple endocrine neoplasia type 1 gene in the sporadic endocrine tumors. Lab Invest. 1998;78:471–475. [PubMed] [Google Scholar]
- 4.Bazzi W, Renon M, Vercherat C, Hamze Z, Lacheretz-Bernigaud A, Wang H, Blanc M, Roche C, Calender A, Chayvialle JA, Scoazec JY, Cordier-Bussat M. MEN1 missense mutations impair sensitization to apoptosis induced by wild-type menin in endocrine pancreatic tumor cells. Gastroenterology. 2008;135:1698–1709. doi: 10.1053/j.gastro.2008.07.031. [DOI] [PubMed] [Google Scholar]
- 5.Ramensky V, Bork P, Sunyaev S. Human non-synonymous SNPs: server and survey. Nucleic Acids Res. 2002;30:3894–3900. doi: 10.1093/nar/gkf493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sakurai A, Shirahama S, Fujimori M, Katai M, Itakura Y, Kobayashi S, Amano J, Fukushima Y, Hashizume K. Novel MEN1 gene mutations in familial multiple endocrine neoplasia type 1. J Hum Genet. 1998;43:199–201. doi: 10.1007/s100380050070. [DOI] [PubMed] [Google Scholar]
- 7.Schussheim DH, Skarulis MC, Agarwal SK, Simonds WF, Burns AL, Spiegel AM, Marx SJ. Multiple endocrine neoplasia type 1: new clinical and basic findings. Trends Endocrinol Metab. 2001;12:173–178. doi: 10.1016/s1043-2760(00)00372-6. [DOI] [PubMed] [Google Scholar]
- 8.Wilkinson S, Teh BT, Davey KR, McArdle JP, Young M, Shepherd JJ. Cause of death in multiple endocrine neoplasia type 1. Arch Surg. 1993;128:683–690. doi: 10.1001/archsurg.1993.01420180085016. [DOI] [PubMed] [Google Scholar]
- 9.Lemos MC, Thakker RV. Multiple endocrine neoplasia type 1 (MEN1): analysis of 1336 mutations reported in the first decade following identification of the gene. Hum Mutat. 2008;29:22–32. doi: 10.1002/humu.20605. [DOI] [PubMed] [Google Scholar]
- 10.Chandrasekharappa SC, Teh BT. Functional studies of the MEN1 gene. J Intern Med. 2003;253:606–615. doi: 10.1046/j.1365-2796.2003.01165.x. [DOI] [PubMed] [Google Scholar]
- 11.Kim H, Lee JE, Cho EJ, Liu JO, Youn HD. Menin, a tumor suppressor, represses JunD-mediated transcriptional activity by association with an mSin3A-histone deacetylase complex. Cancer Res. 2003;63:6135–6139. [PubMed] [Google Scholar]
- 12.Lombardi D, Giovanelli M, de Tribolet N. Sellar and parasellar extra-axial cavernous hemangiomas. Acta Neurochir (Wien) 1994;130:47–54. doi: 10.1007/BF01405502. [DOI] [PubMed] [Google Scholar]
- 13.Sansone ME, Liwnicz BH, Mandybur TI. Giant pituitary cavernous hemangioma: case report. J Neurosurg. 1980;53:124–126. doi: 10.3171/jns.1980.53.1.0124. [DOI] [PubMed] [Google Scholar]
- 14.Buonaguidi R, Canapicci R, Mimassi N, Ferdeghini M. Intrasellar cavernous hemangioma. Neurosurgery. 1984;14:732–734. doi: 10.1227/00006123-198406000-00014. [DOI] [PubMed] [Google Scholar]
- 15.Mitsuhashi T, Hashimoto R, Nagahama S, Nagata Y. Intrasellar cavernous angioma in neurofibromatosis. Hum Pathol. 1991;22:623–624. doi: 10.1016/0046-8177(91)90244-j. [DOI] [PubMed] [Google Scholar]
- 16.Cobbs CS, Wilson CB. Intrasellar cavernous hemangioma. Case report. J Neurosurg. 2001;94:520–522. doi: 10.3171/jns.2001.94.3.0520. [DOI] [PubMed] [Google Scholar]
- 17.Fraser JF, Mass AY, Brown S, Anand VK, Schwartz TH. Transnasal endoscopic resection of a cavernous sinus hemangioma: technical note and review of the literature. Skull Base. 2008;18:309–315. doi: 10.1055/s-0028-1086059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hori S, Hayashi N, Nomoto K, Sato H, Hayashi T, Nagai S, Nishikata M, Endo S. Cavernous sinus cavernous hemangioma largely extending into the sella turcica and mimicking pituitary adenoma: case report. Neurol Med Chir (Tokyo) 2010;50:330–332. doi: 10.2176/nmc.50.330. [DOI] [PubMed] [Google Scholar]
- 19.Jeon SC, Yi JS, Yang JH, Lee IW. Intrasellar cavernous hemangioma. J Korean Neurosurg Soc. 2004;36:163–165. [Google Scholar]
- 20.Chuang CC, Jung SM, Yang JT, Chang CN, Pai PC. Intrasellar cavernous hemangioma. J Clin Neurosci. 2006;13:672–675. doi: 10.1016/j.jocn.2005.08.017. [DOI] [PubMed] [Google Scholar]
- 21.Hasegawa T, McInerney J, Kondziolka D, Lee JY, Flickinger JC, Lunsford LD. Long-term results after stereotactic radiosurgery for patients with cavernous malformations. Neurosurgery. 2002;50:1190–1197. doi: 10.1097/00006123-200206000-00003. [DOI] [PubMed] [Google Scholar]
- 22.Liu KD, Chung WY, Wu HM, Shiau CY, Wang LW, Guo WY, Pan DH. Gamma knife surgery for cavernous hemangiomas: an analysis of 125 patients. J Neurosurg. 2005;102(Suppl):81–86. doi: 10.3171/jns.2005.102.s_supplement.0081. [DOI] [PubMed] [Google Scholar]