

Abstract
Neutrophilic dermatoses are a spectrum of autoinflammatory skin disorders that are characterized by extensive infiltration of neutrophils into the epidermis and dermis. The underlining biological pathways that are responsible for this heterogeneous group of cutaneous diseases have remained elusive. However, recent work from our laboratory and other groups has shown that missense mutations in Ptpn6, which encodes for the non-receptor protein tyrosine phosphatase Src homology region 2 (SH2) domain-containing phosphatase-1 (SHP-1), results in a skin disease with many of the major histopathological and clinical features that encompass neutrophilic dermatoses in humans. In particular, we found that loss-of-function mutation in Ptpn6 results in unremitting footpad swelling, suppurative inflammation, and neutrophilia. Dysregulated wound healing responses were discovered to contribute to chronic inflammatory skin disease in SHP-1 defective mice and genetic abrogation of interleukin-1 receptor (IL-1R) protected mice from cutaneous inflammation, suggesting that IL-1-mediated events potentiate disease. Surprisingly, inflammasome activation and IL-1β-mediated events were dispensable for Ptpn6spin-mediated footpad disease. Instead, RIP1-mediated regulation of IL-1α was identified to be the major driver of inflammation and tissue damage.
Keywords: neutrophilic dermatosis, SHP-1, Ptpn6, inflammasome, NOD-like receptor, caspase-1, interleukin, RIP1, RIP3, inflammation
Introduction
Neutrophilic dermatoses are a heterogeneous group of autoinflammatory skin disorders that include Sweet syndrome, pyoderma gangrenosum, and subcorneal pustular dermatosis.1-3 Neutrophilia and cutaneous inflammatory lesions that are dominated by neutrophils are defining clinical features of all neutrophilic dermatoses. Afflicted individuals develop erythematous skin lesions (papules, nodules, and plaques) that are both painful and unsightly. Current strategies to treat inflammatory flare-ups include the prescription of strong immunosuppressive regimens such as systemic administration of corticosteroids. Although such therapies have proven efficacious in the control of immediate inflammation, numerous drawbacks exist. These include the inability to prevent future disease and increased susceptibility to infection that results from global immunosuppression. Complete characterization of neutrophilic dermatosis etiology has been hampered by the lack of an experimental model system to elucidate critical biological players. However, our recent findings and work from other groups demonstrates that missense mutations in the gene that encodes SHP-1 (Ptpn6) promotes the development of an inflammatory skin disease in mice that closely resembles neutrophilic dermatosis in humans.4,5
SHP-1 is a protein tyrosine phosphatase that has been described to function as a negative regulator of signal transduction in a variety of cell types.6-10 Alterations in SHP-1 activity have been linked to multiple human diseases including leukemia, psoriatic arthritis and multiple sclerosis.11-15 In addition, pivotal roles for SHP-1 in the regulation of autoinflammatory disease came from the discovery that loss-of-function mutations in Ptpn6 provoke severe granulocytic skin lesions and pneumonitis in mice.16-19 Persistent cutaneous inflammation in SHP-1 deficient mice results in a motheaten phenotype that is characterized by hair loss and runted appearance. Elucidation of the overarching biological role of SHP-1 in autoinflammatory disease pathogenesis has been hindered by the fact that SHP-1 null mice are immunodeficient at birth and succumb to a devastating pneumonitis and glomerulonephritis by 2–9 wk of age.
Recently, a new strain of SHP-1 mutant mice that carry an Y208N amino acid substitution in the C-terminal SH2 domain of SHP-1 (referred to as Ptpn6spin mice) was generated.20 Mice homozygous for this mutant allele develop a less aggressive disease phenotype, which is attributed to 70–80% reductions in SHP-1 phosphatase activity. Importantly, Ptpn6spin mutation provokes the development of a spontaneous chronic inflammatory disorder at 8–16 wk of age that is characterized by persistent footpad swelling and suppurative inflammation. Characterization of disease pathogenesis revealed that Ptpn6spin mice exhibit many of the prominent clinical and histopathological hallmarks that define neutrophilic dermatoses.4 These include neutrophilia, the formation of intraepidermal pustules and cutaneous tissue damage that is associated with marked infiltration of neutrophils. Interestingly, splice variants and coding mutations in Ptpn6 are closely associated with multiple neutrophilic dermatoses including Sweet syndrome and pyoderma gangrenosum in humans.21 This association between mutations in Ptpn6 and clinic cases of neutrophilic dermatoses highlights the value of Ptpn6 mutant mice in the study of physiological autoinflammatory skin disorders. Indeed, utilization of SHP-1 deficient mice has significantly aided in the elucidation of the cellular and molecular players that contribute to tissue damage and inflammation in neutrophilic dermatosis and other autoinflammatory skin disorders.5,22
IL-1 in Neutrophilic Dermatosis
The IL-1 family cytokines IL-1α and IL-1β have emerged as principal mediators involved in both the initiation and perpetuation of multiple inflammatory diseases.23,24 Both IL-1α and IL-1β are potent immunological mediators that trigger proinflammatory signaIing through the engagement of the IL-1 receptor (IL-1R). IL-1β is generated in an inactive pro-form that requires cleavage to elicit its biological activity and secretion. The most well characterized mechanism for IL-1β processing is via activated caspase-1 in the inflammasome complex; although additional inflammasome-independent cleavage pathways have recently been proposed.23 Inflammasomes are multi-protein complexes that consist of a sensor molecule such as a NOD-like receptor (NLR), the adaptor protein ASC, and caspase-1. The recognition of pathogen- or danger-associated molecular patterns (PAMPs and DAMPs, respectively) by NLRs promotes the recruitment of ASC and caspase-1 into the inflammasome complex, which is required to correctly orient caspase-1 for auto-cleavage and activation. Importantly, recent studies have identified pivotal roles for inflammasome-derived IL-1β in a variety of infectious, metabolic, autoimmune, and inflammatory diseases.25,26
In comparison to IL-1β, our understanding of the roles and regulation of IL-1α in disease is severely limited. IL-1α exists in three biological active forms (precursor, propiece, and mature form), however their discrete contributions to inflammation and the molecular pathways that promote their production remain to be formally elucidated. Aberrant forms of cell death, such as necrosis, that result from cellular stress, trauma, and hypoxia are believed to provoke the passive release of IL-1α. Furthermore, IL-1α has been implicated in wound healing responses and sterile inflammation.27,28
Previous work demonstrated that IL-1R is required for the development of persistent cutaneous inflammation in Ptpn6spin mutant mice,20 which suggests that dysregulated IL-1 signaling directly contributes to disease pathogenesis. In our studies, we set out to characterize the upstream biological events and players that are responsible for neutrophilic dermatosis-related disease in Ptpn6spin mice. We initially hypothesized that unchecked regulation of NLRP3/inflammasome-induced IL-1β production was responsible for autoinflammatory skin disease. This was based on emerging data suggesting pivotal roles for inflammasome activation in multiple IL-1-mediated inflammatory disorders. Surprisingly, genetic deletion of critical inflammasome components (NLRP3 and caspase-1) did not provide any protection against Ptpn6spin-associated footpad inflammation. In addition, targeted ablation of IL-1β also did not rescue disease in this model. Rather, IL-1α was discovered to be the major driver of autoinflammation and skin damage in Ptpn6spin mutant mice.4 Genetic abrogation of IL-1α in Ptpn6spin mice was found to prevent disease progression by limiting the numbers of circulating neutrophils and dampening downstream proinflammatory cytokine production (Fig. 1). These findings help to delineate unique roles for IL-1α in autoinflammatory disease that are distinct from inflammasomes and IL-1β.
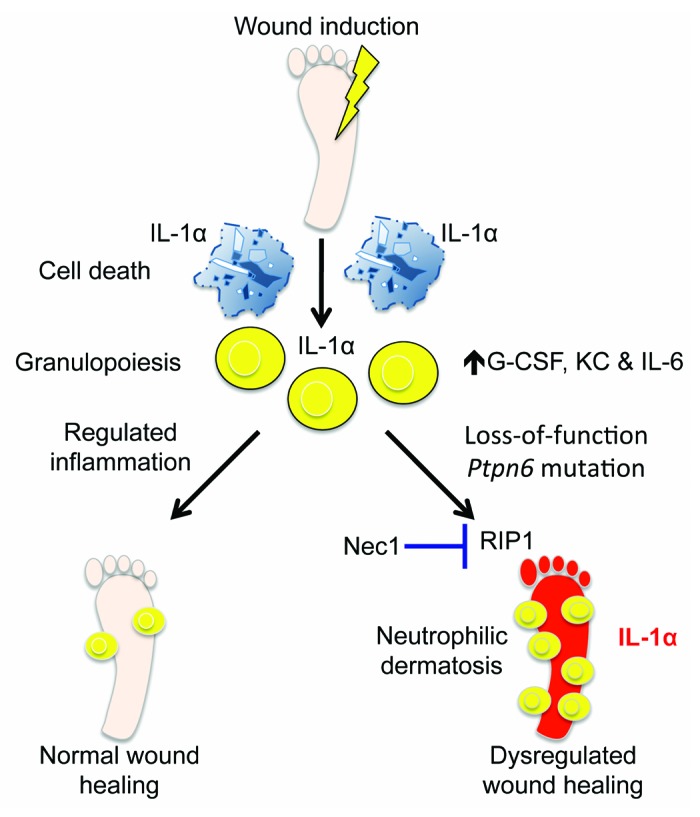
Figure 1. Dysregulated IL-1α provokes neutrophilic dermatosis. Tissue that is abruptly damaged following trauma, hypoxia, or extreme cellular stress promotes the generation of proinflammatory cytokines (G-CSF, KC, and IL-6) that are associated with granulopoiesis and the mobilization of neutrophils. Neutrophils are recruited to the site of tissue damage where they help to contain the insult and engulf damaged tissue. Regulated mobilization of immune cells and controlled inflammation are required to initiate proper wound healing responses and tissue regeneration. However, unchecked wound-associated immune responses can adversely cause autoinflammation and deleterious tissue destruction. Loss-of-function mutations in the gene that encodes for the tyrosine phosphatase SHP-1 (Ptpn6) results in aberrant wound healing responses and the development of a chronic autoinflammatory skin disorder that is characterized by suppurative cutaneous lesions, severe edema, and marked infiltration of neutrophils into the skin. IL-1α instigates the inflammatory cascade that is responsible for the development of neutrophilic dermatosis in Ptpn6 mutant mice. Pharmacological inhibition of RIP1 with necrostatin-1 (Nec1) treatment ameliorates altered IL-1α production and provides protection against excessive immune-mediated wound healing responses in Ptpn6 mutant mice.
IL-1α is an alarmin molecule that orchestrates beneficial wound healing responses following injury, hypoxia, and other inducers of tissue necrosis. However, unchecked IL-1α release can provoke inflammation and tissue destruction, and thus dysregulated IL-1α secretion is believed to play deleterious roles in multiple sterile inflammatory disorders including stroke and atherosclerosis.29-31 To elucidate whether aberrant wound healing responses were responsible for driving neutrophil-associated inflammatory skin disease in Ptpn6spin mutant mice we induced microabrasion injuries on the plantar surfaces of wild-type and asymptomatic Ptpn6spin mice. Loss-of-function mutation in Ptpn6 caused exacerbated proinflammatory cytokine production following cutaneous tissue damage. Moreover, Ptpn6spin mutant mice were unable to resolve the inflammation that was brought upon by microabrasion-induced trauma and as a result developed an aggravated state of footpad inflammation that was characterized by severe pustular dermatosis, and edema. Interestingly, genetic abrogation of IL-1α protected against excessive proinflammatory cytokine production and conferred normal wound healing responses in Ptpn6spin mice. Collectively, these results identify SHP-1 and IL-1α as central regulators of wound-induced inflammation. Furthermore, they suggest that dysregulated wound healing responses may be involved in the pathogenesis of neutrophilic dermatoses.
To identify whether defective SHP-1 signaling in hematopoietic cells or radioresistant skin cells is required to promote persistent autoinflammatory skin disease in Ptpn6spin mice, reciprocal bone marrow chimera studies were conducted. Confining Ptpn6spin mutation only to radioresistant cells by transplanting wild-type bone marrow cells into irradiated Ptpn6spin mice provided complete protection from skin disease. In contrast, loss-of-function mutation in Ptpn6 in the bone marrow compartment alone was sufficient to induce chronic autoinflammatory skin disease. This suggests that impaired SHP-1 function specifically in bone marrow-derived immune cells drives this IL-1α-mediated cutaneous disorder.
RIP1 Controls IL-1α Driven Autoinflammatory Skin Disease
The molecular pathways that contribute to IL-1α secretion remain poorly defined. The prevailing paradigm is that IL-1α is passively released following cellular catastrophe and necrosis.23,28,32 Until recently, it was thought that necrosis was an unprogrammed form of cell death that ensues following devastating and rapid cellular demise. However in the last few years, significant progress has been made in the identification of signaling molecules that coordinate necrosis. For instance, RIP3 downstream of RIP1 activation has emerged as a critical mediator of one form of necrosis referred to as necroptosis.33-35 In these studies, it was shown that the RIP1 kinase inhibitor necrostatin-1 (Nec1) could block RIP3-driven necroptosis. To elucidate whether RIP3-induced necroptosis contributes to IL-1α release and Ptpn6spin-mediated autoinflammatory skin disease, wild-type, and Ptpn6spin mutant mice were treated with Nec1 and then subjected to microabrasion trauma on their footpads. Inhibition of RIP1 with Nec1 protected against excessive proinflammatory cytokine production in Ptpn6spin mutant mice. Genetic deletion of RIP1 in fetal liver chimera mice also rescued Ptpn6spin-induced autoinflammatory skin disease, confirming a critical downstream role for RIP1 in disease progression. Surprisingly, targeted deletion of RIP3 did not provide protection, and Ptpn6spinxRip3−/− mice developed severe neutrophilic dermatosis, suggesting that RIP1 controls autoinflammatory skin disease independently of its role in RIP3-driven necroptosis. Rather, we found that RIP1-mediated regulation of inflammatory NFκB signaling contributes to inflammatory syndrome in Ptpn6spin mutant mice.
Future Perspectives and Conclusions
Although our recent findings and work by other groups have firmly established that dysregulated immune responses are responsible for driving neutrophilic dermatoses, numerous important questions still remain. Identification of the pathogenic role of unchecked IL-1α-mediated signaling in cutaneous autoinflammation has provided critical insight into the etiology of neutrophilic dermatoses. IL-1α exists in 3 biological active forms (precursor, propiece, and mature form), however which species trigger inflammatory skin disease still remains to be formally elucidated. Furthermore, whether IL-1α is released passively as a result of cellular catastrophe or requires specific cleavage by a protease to potentiate cutaneous pathology is still unresolved. Our understanding of IL-1α biology in comparison to IL-1β is severely limited, and we have only recently begun to dissect the discrete roles of IL-1α in disease pathogenesis. Thus, additional investigation is greatly needed to further advance this field. Unique IL-1α-dependent functions have recently been described in multiple disease models including atherosclerosis,31 DNA damage-induced senescence and tumorigenesis,36 Legionella pneumophila infection37, and inflammation associated with necrotic cell death.27 However the discrete roles of IL-1α and IL-1β in the etiology of most IL-1R-mediated diseases still remain to be defined.
Blockade of IL-1 signaling with the IL-1R antagonist Anakinra has proven effective in the treatment of various human disorders including cryopyrin-associated periodic syndromes (CAPS) and type 2 diabetes.38,39 However, such treatment strategies have significant downsides, including high costs and serious side effects that are associated with aggressive immunosuppression. Improved therapeutics that only target the specific pathogenic IL-1 molecules, while sparing other potentially beneficial IL-1-mediated functions (e.g., protective anti-pathogen and -tumor responses), are needed. Our work highlighting a critical role for RIP1-dependent IL-1α secretion in autoinflammatory skin disease suggests that therapeutic neutralization of IL-1α may prove beneficial for treating autoinflammatory diseases, while not interfering with IL-1β-mediated responses. In addition, we provide in vivo evidence that the RIP1 kinase inhibitor necrostatin-1 (Nec1) can be utilized to limit IL-1α-mediated inflammation and ameliorate exacerbated wound healing responses. It is possible that Nec1 can also be exploited to treat additional IL-1α-driven disorders, thus in the future it will be important to formally test this attractive treatment avenue.
Currently, the roles of RIP1 in inflammatory disease progression also remain poorly defined. The perinatal lethality that is associated with RIP1 deficiency in mice has severely hampered our ability to fully uncover the physiological functions of RIP1 in the regulation of inflammation and immune responses. In vitro studies, however, have positioned RIP1 as a master regulator of various cellular events that have been associated with inflammatory and autoimmune disease. For instance, RIP1 has been described to centrally function in the control of NFκB inflammatory signaling, apoptosis, and RIP3-associated necroptosis.40-43 Thus, it is conceivable that RIP1 is an important in vivo regulator of autoinflammation. Our discovery that RIP1 is essential for the development of persistent inflammatory skin disease in Ptpn6spin mutant mice provides direct evidence that RIP1 can contribute to inflammatory disease pathogenesis. Interestingly, we found that RIP1 controls inflammation through the regulation of NFκB activation and not through its role in RIP3-mediated necroptosis. Future studies that investigate the ability of RIP1 to influence immune responses and inflammation in other disease models are needed to illuminate the physiological functions of RIP1. Utilization of RIP1 deficient fetal liver chimeras and conditional RIP1 deletion mice will facilitate such studies and aid in the characterization of the SHP-1/RIP1/IL-1α inflammatory axis.
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
Acknowledgments
We apologize to authors whose work could not be referenced in this review due to space limitations. This work was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases, part of the National Institutes of Health, under Award Number AR056296 (T.-D.K); the National Cancer Institute, part of the National Institutes of Health, under Award Number CA163507 (T.-D.K); the National Institute of Allergy and Infectious Diseases, part of the National Institutes of Health, under Award Number AI101935 (T.-D.K); and ALSAC.
Glossary
Abbreviations:
- IL
interleukin
- Nec-1
necrostatin-1
- NLR
NOD-like receptor
- RIP
Receptor-Interacting Protein
- SH2
Src Homology 2
- SHP-1
SH2 domain-containing phosphatase-1
- CAPS
cryopyrin-associated periodic syndromes
- PAMPs
pathogen-associated molecular patterns
- and DAMPs
danger-associated molecular patterns
References
- 1.Berk DR, Bayliss SJ. Neutrophilic dermatoses in children. Pediatr Dermatol. 2008;25:509–19. doi: 10.1111/j.1525-1470.2008.00765.x. [DOI] [PubMed] [Google Scholar]
- 2.Anzalone CL, Cohen PR. Acute febrile neutrophilic dermatosis (Sweet’s syndrome) Curr Opin Hematol. 2013;20:26–35. doi: 10.1097/MOH.0b013e32835ad132. [DOI] [PubMed] [Google Scholar]
- 3.Cohen PR. Neutrophilic dermatoses: a review of current treatment options. Am J Clin Dermatol. 2009;10:301–12. doi: 10.2165/11310730-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 4.Lukens JR, Vogel P, Johnson GR, Kelliher MA, Iwakura Y, Lamkanfi M, Kanneganti TD. RIP1-driven autoinflammation targets IL-1α independently of inflammasomes and RIP3. Nature. 2013;498:224–7. doi: 10.1038/nature12174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nesterovitch AB, Szanto S, Gonda A, Bardos T, Kis-Toth K, Adarichev VA, Olasz K, Ghassemi-Najad S, Hoffman MD, Tharp MD, et al. Spontaneous insertion of a b2 element in the ptpn6 gene drives a systemic autoinflammatory disease in mice resembling neutrophilic dermatosis in humans. Am J Pathol. 2011;178:1701–14. doi: 10.1016/j.ajpath.2010.12.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang J, Somani AK, Siminovitch KA. Roles of the SHP-1 tyrosine phosphatase in the negative regulation of cell signalling. Semin Immunol. 2000;12:361–78. doi: 10.1006/smim.2000.0223. [DOI] [PubMed] [Google Scholar]
- 7.Lorenz U. SHP-1 and SHP-2 in T cells: two phosphatases functioning at many levels. Immunol Rev. 2009;228:342–59. doi: 10.1111/j.1600-065X.2008.00760.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johnson DJ, Pao LI, Dhanji S, Murakami K, Ohashi PS, Neel BG. Shp1 regulates T cell homeostasis by limiting IL-4 signals. J Exp Med. 2013;210:1419–31. doi: 10.1084/jem.20122239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pao LI, Badour K, Siminovitch KA, Neel BG. Nonreceptor protein-tyrosine phosphatases in immune cell signaling. Annu Rev Immunol. 2007;25:473–523. doi: 10.1146/annurev.immunol.23.021704.115647. [DOI] [PubMed] [Google Scholar]
- 10.Mauldin IS, Tung KS, Lorenz UM. The tyrosine phosphatase SHP-1 dampens murine Th17 development. Blood. 2012;119:4419–29. doi: 10.1182/blood-2011-09-377069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cao H, Hegele RA. Identification of polymorphisms in the human SHP1 gene. J Hum Genet. 2002;47:445–7. doi: 10.1007/s100380200062. [DOI] [PubMed] [Google Scholar]
- 12.Christophi GP, Hudson CA, Gruber RC, Christophi CP, Mihai C, Mejico LJ, Jubelt B, Massa PT. SHP-1 deficiency and increased inflammatory gene expression in PBMCs of multiple sclerosis patients. Lab Invest. 2008;88:243–55. doi: 10.1038/labinvest.3700720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tibaldi E, Brunati AM, Zonta F, Frezzato F, Gattazzo C, Zambello R, Gringeri E, Semenzato G, Pagano MA, Trentin L. Lyn-mediated SHP-1 recruitment to CD5 contributes to resistance to apoptosis of B-cell chronic lymphocytic leukemia cells. Leukemia. 2011;25:1768–81. doi: 10.1038/leu.2011.152. [DOI] [PubMed] [Google Scholar]
- 14.Wu C, Sun M, Liu L, Zhou GW. The function of the protein tyrosine phosphatase SHP-1 in cancer. Gene. 2003;306:1–12. doi: 10.1016/S0378-1119(03)00400-1. [DOI] [PubMed] [Google Scholar]
- 15.Eriksen KW, Woetmann A, Skov L, Krejsgaard T, Bovin LF, Hansen ML, Grønbaek K, Billestrup N, Nissen MH, Geisler C, et al. Deficient SOCS3 and SHP-1 expression in psoriatic T cells. J Invest Dermatol. 2010;130:1590–7. doi: 10.1038/jid.2010.6. [DOI] [PubMed] [Google Scholar]
- 16.Green MC, Shultz LD. Motheaten, an immunodeficient mutant of the mouse. I. Genetics and pathology. J Hered. 1975;66:250–8. doi: 10.1093/oxfordjournals.jhered.a108625. [DOI] [PubMed] [Google Scholar]
- 17.Shultz LD, Coman DR, Bailey CL, Beamer WG, Sidman CL. “Viable motheaten,” a new allele at the motheaten locus. I. Pathology. Am J Pathol. 1984;116:179–92. [PMC free article] [PubMed] [Google Scholar]
- 18.Shultz LD, Schweitzer PA, Rajan TV, Yi T, Ihle JN, Matthews RJ, Thomas ML, Beier DR. Mutations at the murine motheaten locus are within the hematopoietic cell protein-tyrosine phosphatase (Hcph) gene. Cell. 1993;73:1445–54. doi: 10.1016/0092-8674(93)90369-2. [DOI] [PubMed] [Google Scholar]
- 19.Tsui HW, Siminovitch KA, de Souza L, Tsui FW. Motheaten and viable motheaten mice have mutations in the haematopoietic cell phosphatase gene. Nat Genet. 1993;4:124–9. doi: 10.1038/ng0693-124. [DOI] [PubMed] [Google Scholar]
- 20.Croker BA, Lawson BR, Rutschmann S, Berger M, Eidenschenk C, Blasius AL, Moresco EM, Sovath S, Cengia L, Shultz LD, et al. Inflammation and autoimmunity caused by a SHP1 mutation depend on IL-1, MyD88, and a microbial trigger. Proc Natl Acad Sci U S A. 2008;105:15028–33. doi: 10.1073/pnas.0806619105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nesterovitch AB, Gyorfy Z, Hoffman MD, Moore EC, Elbuluk N, Tryniszewska B, Rauch TA, Simon M, Kang S, Fisher GJ, et al. Alteration in the gene encoding protein tyrosine phosphatase nonreceptor type 6 (PTPN6/SHP1) may contribute to neutrophilic dermatoses. Am J Pathol. 2011;178:1434–41. doi: 10.1016/j.ajpath.2010.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abram CL, Roberge GL, Pao LI, Neel BG, Lowell CA. Distinct roles for neutrophils and dendritic cells in inflammation and autoimmunity in motheaten mice. Immunity. 2013;38:489–501. doi: 10.1016/j.immuni.2013.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lukens JR, Gross JM, Kanneganti TD. IL-1 family cytokines trigger sterile inflammatory disease. Front Immunol. 2012;3:315. doi: 10.3389/fimmu.2012.00315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lukens JR, Dixit VD, Kanneganti TD. Inflammasome activation in obesity-related inflammatory diseases and autoimmunity. Discov Med. 2011;12:65–74. [PMC free article] [PubMed] [Google Scholar]
- 25.Kanneganti TD. Central roles of NLRs and inflammasomes in viral infection. Nat Rev Immunol. 2010;10:688–98. doi: 10.1038/nri2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kanneganti TD, Dixit VD. Immunological complications of obesity. Nat Immunol. 2012;13:707–12. doi: 10.1038/ni.2343. [DOI] [PubMed] [Google Scholar]
- 27.Chen CJ, Kono H, Golenbock D, Reed G, Akira S, Rock KL. Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat Med. 2007;13:851–6. doi: 10.1038/nm1603. [DOI] [PubMed] [Google Scholar]
- 28.Cohen I, Rider P, Carmi Y, Braiman A, Dotan S, White MR, Voronov E, Martin MU, Dinarello CA, Apte RN. Differential release of chromatin-bound IL-1alpha discriminates between necrotic and apoptotic cell death by the ability to induce sterile inflammation. Proc Natl Acad Sci U S A. 2010;107:2574–9. doi: 10.1073/pnas.0915018107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Allan SM, Tyrrell PJ, Rothwell NJ. Interleukin-1 and neuronal injury. Nat Rev Immunol. 2005;5:629–40. doi: 10.1038/nri1664. [DOI] [PubMed] [Google Scholar]
- 30.Boutin H, LeFeuvre RA, Horai R, Asano M, Iwakura Y, Rothwell NJ. Role of IL-1alpha and IL-1beta in ischemic brain damage. J Neurosci. 2001;21:5528–34. doi: 10.1523/JNEUROSCI.21-15-05528.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Freigang S, Ampenberger F, Weiss A, Kanneganti TD, Iwakura Y, Hersberger M, Kopf M. Fatty acid-induced mitochondrial uncoupling elicits inflammasome-independent IL-1α and sterile vascular inflammation in atherosclerosis. Nat Immunol. 2013;14:1045–53. doi: 10.1038/ni.2704. [DOI] [PubMed] [Google Scholar]
- 32.Dinarello CA. Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood. 2011;117:3720–32. doi: 10.1182/blood-2010-07-273417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C, Hakem R, Salvesen GS, Green DR. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature. 2011;471:363–7. doi: 10.1038/nature09852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kaiser WJ, Upton JW, Long AB, Livingston-Rosanoff D, Daley-Bauer LP, Hakem R, Caspary T, Mocarski ES. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature. 2011;471:368–72. doi: 10.1038/nature09857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dillon CP, Oberst A, Weinlich R, Janke LJ, Kang TB, Ben-Moshe T, Mak TW, Wallach D, Green DR. Survival function of the FADD-CASPASE-8-cFLIP(L) complex. Cell Rep. 2012;1:401–7. doi: 10.1016/j.celrep.2012.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Orjalo AV, Bhaumik D, Gengler BK, Scott GK, Campisi J. Cell surface-bound IL-1alpha is an upstream regulator of the senescence-associated IL-6/IL-8 cytokine network. Proc Natl Acad Sci U S A. 2009;106:17031–6. doi: 10.1073/pnas.0905299106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barry KC, Fontana MF, Portman JL, Dugan AS, Vance RE. IL-1α signaling initiates the inflammatory response to virulent Legionella pneumophila in vivo. J Immunol. 2013;190:6329–39. doi: 10.4049/jimmunol.1300100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hoffman HM, Rosengren S, Boyle DL, Cho JY, Nayar J, Mueller JL, Anderson JP, Wanderer AA, Firestein GS. Prevention of cold-associated acute inflammation in familial cold autoinflammatory syndrome by interleukin-1 receptor antagonist. Lancet. 2004;364:1779–85. doi: 10.1016/S0140-6736(04)17401-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Larsen CM, Faulenbach M, Vaag A, Vølund A, Ehses JA, Seifert B, Mandrup-Poulsen T, Donath MY. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N Engl J Med. 2007;356:1517–26. doi: 10.1056/NEJMoa065213. [DOI] [PubMed] [Google Scholar]
- 40.Thapa RJ, Nogusa S, Chen P, Maki JL, Lerro A, Andrake M, Rall GF, Degterev A, Balachandran S. Interferon-induced RIP1/RIP3-mediated necrosis requires PKR and is licensed by FADD and caspases. Proc Natl Acad Sci U S A. 2013;110:E3109–18. doi: 10.1073/pnas.1301218110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vandenabeele P, Declercq W, Van Herreweghe F, Vanden Berghe T. The role of the kinases RIP1 and RIP3 in TNF-induced necrosis. Sci Signal. 2010;3:re4. doi: 10.1126/scisignal.3115re4. [DOI] [PubMed] [Google Scholar]
- 42.Zhang H, Zhou X, McQuade T, Li J, Chan FK, Zhang J. Functional complementation between FADD and RIP1 in embryos and lymphocytes. Nature. 2011;471:373–6. doi: 10.1038/nature09878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kelliher MA, Grimm S, Ishida Y, Kuo F, Stanger BZ, Leder P. The death domain kinase RIP mediates the TNF-induced NF-kappaB signal. Immunity. 1998;8:297–303. doi: 10.1016/S1074-7613(00)80535-X. [DOI] [PubMed] [Google Scholar]