

Abstract
Huntington disease is a rare neurodegenerative disease resulting from insertion and/or expansion of a polyglutamine repeats close to the N-terminal of the huntingtin protein. Although unequivocal genetic tests have been available for about 20 years, current pharmacological treatments do not prevent or slow down disease progression. Recent basic research identified potential novel drug targets for the treatment of Huntington disease. However, there are clear challenges in translating these discoveries into treatment strategies for these patients. The following is a brief discussion of these challenges using our recent experience as an example.
Keywords: Huntington disease, neurodegeneration, mitochondria, polyglutamine, animal model, protein-protein interactions, P110 peptide inhibitor, Drp1
Introduction
Huntington disease (HD), a dominantly inherited fatal neurodegenerative disorder, is caused by expansion of polyglutamine (polyQ, CAG) repeat within the first exon of the protein huntingtin (Htt).2 HD prevalence is 5–10 per 100 000 individuals in Western Europe and North America, and the age of disease onset is inversely proportional to the length of the CAG repeat; affected individuals usually live for 15–20 years after the initial onset of symptoms.3 Clinical characteristics of HD are devastating and include progressive cognitive impairment, abnormal movements, psychiatric disturbances, dementia, and death.2 Even though the genetic mutation of HD has been identified 20 years ago, a cure or even disease-modifying treatments remain elusive.
At least three factors contribute to the challenges in identifying drug targets and developing new treatments for HD.
The Critical Cellular and Molecular Pathological Pathways that Should Be Targeted For HD Therapy Development are Still Unclear
Many cellular and molecular pathological pathways have been suggested to contribute to HD. These include a role of polyQ aggregates and misfolded mutant huntingtin (mHtt) in neurotoxicity4; a role of a variety of mHtt fragments in the pathology3; and a role of mHtt in mitochondrial dysfunctions, including imbalanced fusion and/or fission (also called mitochondrial dynamics), increased mitochondrial immobility in the neurons, inhibition of mitochondrial biogenesis, and dysregulated mitochondrial bioenergetics.5 Genomic analysis also demonstrates the association between HD and changes in the expression of many genes,6 suggesting that mHtt may regulate transcription. Which of these pathways and molecular events have a pivotal role in the pathology? How many of these processes should be blocked to prevent HD pathology? What molecular tools can be used to test these possibilities? Current studies in many laboratories examine these issues, but a consensus has not yet been formed; even the role of wild type huntingtin protein is far from being fully understood.
Wild type Htt is a very large multi-domain protein of 3144 amino acids.2 Recent proteomic studies demonstrate that wild type Htt interacts directly or indirectly with hundreds of proteins.7,8 Which of the interacting protein(s) is dysregulated in HD is not known. Further, it may be a challenge to affect the dysregulated interaction(s) without disturbing other roles of Htt. It is also unclear whether HD pathology represents a gain of function or a loss of function of the Htt protein. Specifically, insertion of a long polyQ repeat in Htt may result in a gain of function due to, e.g., polyQ-dependent release of huntingtin-interacting protein (HIP) from Htt and its activation of apoptosis.9 Expansion of a polyQ repeat may also cause a loss of function, e.g., due to mislocalization of Htt and the Htt-binding proteins, increased proteolysis of Htt and/or of Htt-binding proteins, etc.10 PolyQ repeat-containing proteins, like mHtt, form polymers, fibrils, and large inclusion bodies, which are thought to contribute to or trigger neurotoxicity.4 Lack of information on which of the above molecular events is required and sufficient to induce the HD-associated pathology makes it difficult to use a rational search for strategies to prevent or inhibit the progression of the disease.
Lack of Optimal Animal Models to Test Therapeutic Approaches for HD
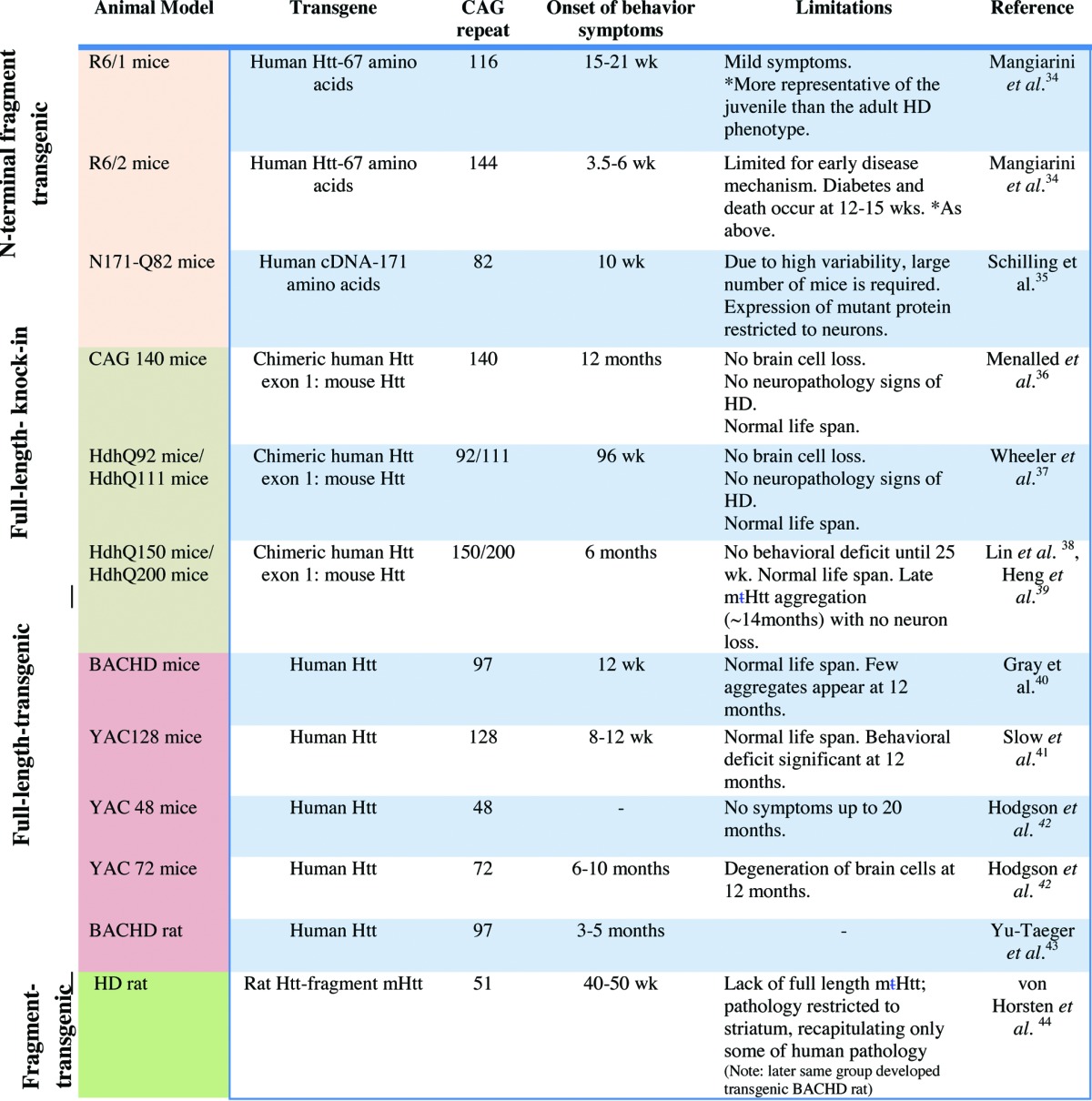
It is difficult to create a proper transgenic model for HD because of the size of the mutated gene, the difference in the number of polyQ repeats between patients and the finding that mHtt creates pathology in only selected areas in the brain.11 Further, the slow progression, age-dependency, and the cognitive pathologies of the disease are difficult to recapitulate and assess in animal models.11 Multiple models of HD have been created with fragments and full-length mHtt.12,13 These models vary in the type of pathologies that they mimic and the rate of disease progression (e.g., see Table 1 and references within). Expression of full-length and N-terminal fragments (which contain the polyQ repeat) of mHtt with or without untranslated upstream regions of the gene induces only some of the HD pathologies as determined in a variety of animal models (11). Some of the animal models exhibit cognitive and behavioral abnormalities, sleep pattern abnormalities, and a shorter lifespan.12,13 The extent of regional or general neurodegeneration varies between models. In discussing which of the models is most suitable to study HD, it is apparent that each of the models has its limitations. Thus, in agreement with others,12-14 utilization of multiple HD models to test preclinical drug candidates is likely required to compensate for the limitations of each of these models.
Table 1. List of commonly used rodent models in Huntington disease.
The Challenge in Developing Clinical Trials in a Rare Chronic Progressive Neurodegenerative Disease
Some of the challenges in developing clinical trials for HD are typical for any clinical trials in patients with a rare disease: limited number of patients, geographical challenge for patients to access trial sites, the number of competing clinical trials for the same patient populations, etc.15 Other challenges relate to the nature of HD. HD progresses over a long period (years2). There are differences in the severity and in the rate of disease progression.16 The clinical assessment for HD is not sufficiently quantitative, objective and robust and there are no surrogate markers to assess disease progression and therapeutic benefit over a short trial time.15,16
Basic research in academia thrives to help address this unmet clinical need. In the following, we reflect on our recent experience in addressing the above challenges.
What Target to Be Chosen for HD Drug Development?
In academia, the choice of testing the validity of a particular drug target often relates to current basic research and strength of a particular laboratory rather than a result of a systematic assessment of all potential drug targets. Our laboratory was intrigued by the findings that mHtt interacts with the mitochondrial fission machinery and specifically with dynamin-related protein 1 (Drp1;17,18). Further evidence that mitochondrial fission and fusion are dysregulated10,17,18 peaked our interest. Because we developed expertise and tools to examine the role of abnormal mitochondrial dynamics and excessive mitochondrial fission in neurodegeneration,19,20 we opted to focus on determining whether inhibition of excessive fission, by inhibiting excessive Drp1 activation, would reduce mHtt-induced pathology.
When Identifying a Possible Therapeutic Strategy, What Models of HD Should Be Used?
We recently identified an inhibitor of excessive Drp1 function, which inhibited pathology and neuronal cell death in models of another neurodegenerative disease, Parkinson disease.19,20 Using six models of HD,1 we then tested whether mitochondrial excessive fission occurs and the effect of treatment with our Drp1 inhibitor. Each of the models that we used and its limitations is listed below.
Mouse striatal cells that express a full-length human mHtt containing 111 CAG repeats (HdhQ111), which were compared with mouse striatal cells carrying 7 CAG repeats (HdhQ7; wild-type). This model21 measures direct neurotoxicity of mHtt. However, mHtt is overexpressed and its expression is not regulated as it would be in patients. Further, the cultured striatal-like cells lack the influence of other brain cells on the pathology, and the extent of protein aggregation in this model is limited.
We also used HEK293T cells expressing a fragment of the human Htt gene containing 73 polyglutamine repeats (73Q). If some of the toxicity of mHtt relates to domains other than the one encoded by the first exon, these will not be represented in this cell culture model. Further, since HEK293T are not neuronal cells, in addition to the limitation of the use of culture models mentioned above, the cytotoxicity measured in these cells may not mimic neurotoxicity.
We next used dermal fibroblasts obtained from HD patients and compared their mitochondrial abnormalities and response to treatment with the Drp1 peptide inhibitor, P110, with that of dermal fibroblasts from control (healthy) subjects. This model has the same limitations as the HEK293 cells have in cell culture studies of non-neuronal cells. However, since the dermal cells are derived from human patients, the regulation of mHtt gene expression, and thus potential contribution of changes in expression of mHtt and other genes, were undisturbed.
We also used neurons derived from control and HD patient-iPS cells, which conserve the same number of CAG repeats in patients and all the other benefits of model 3, above,1 but better represent the organ where HD pathology is noted.
Further, we selected HD patient-derived GABAergic neurons and medium spiny neurons (MSNs) that are most affected in HD patients and are largely degenerated in the late stage of the disease. These patient-derived neuronal cells represent a unique human disease-specific cellular system to elucidate disease mechanisms underlying HD; they were also suggested to be a useful platform of patient cells for screening therapeutic candidates.22 However, this cultured model exhibits only some of the HD neuropathology, and cannot replicate the overall pathology as seen in intact brains of HD patients.
Finally, we used the R6/2 mice, which express exon 1 of the human mHtt gene carrying more than 120 CAG repeats.23 This mouse model allowed us to test the importance of the pathway for HD pathology in vivo. However, since only a part of the mHtt protein is expressed, it has many limitations, including the presence of wild-type Htt, the lack of regulated expression, and the use of human mHtt that might not interact optimally with mouse Htt-interacting partners. (Table 1 describes other transgenic rodent models for HD and lists some of their advantages and limitations).
Is Inhibition of Excessive Drp1 Activation a New HD Drug Target?
Using the six models listed above, we found mHtt-induced Drp1 activation (as measured by its association with the mitochondria) relative to cells that do not express mHtt.10,17,18 Importantly, we showed that inhibition of Drp1 hyper-activation by P110 rescued mHtt-induced mitochondrial fragmentation, corrected defects in mitochondrial function, and reduced cell death in multiple HD cell culture models derived from either mice or patients, including patient GABAergic striatal neurons differentiated from patient-induced pluripotent stem cells (HD-iPS cells).1 Moreover, in HD transgenic R6/2 mice, sustained treatment with the Drp1 peptide inhibitor corrected mitochondrial fragmentation, cristae disruption and respiratory inactivity. These, in turn, were associated with reduced HD-related motor deficits, striatal neuronal loss, and mHtt aggregate accumulation, and increased animal survival.1 An important feature of the Drp1 peptide inhibitor is that its effect is specific for activated Drp1 during excessive fission, as shown in R6/2 mice; the inhibitor has no effect on basal level Drp1 activity and non-pathological mitochondrial fission as demonstrated in cultured cell studies and in vivo, using wild type mice. 1 Together, our data provide a proof-of-concept for the use of the Drp1 inhibitor, such as P110, to inhibit or slow down HD progression.
Why Would Inhibition of One Aspect of HD Pathology (Mitochondrial Excessive Fragmentation) Be Effective?
It is surprising that inhibition of mHtt-induced Drp1-mediated excessive mitochondrial fragmentation appears to be sufficient to suppress HD-related cell death in culture and to prevent selective neuronal loss in the striatum in vivo. We suggest that cell-type-specific mitochondrial characteristics might help explain the efficacy of the above treatment. The striatum is particularly sensitive to defects in mitochondrial oxidative phosphorylation (OXPHOS) due to an increased reliance on OXPHOS function in this area.24 Thus, one can imagine that striatal neurons can be preferentially affected by functional decline in mitochondrial activity relative to that in neurons residing in other brain regions. The loss of PGC-1alpha, a master regulator of mitochondrial biogenesis, in medium spiny neurons and not in other neurons (e.g., interneurons)25 may also explain the causal role of mitochondrial damage in selective loss of striatal neurons. Further, mitochondrial Ca2+ buffering affects striatal medium spiny neurons more severely than that in other areas, such as the cortex.26 Finally, medium spiny neurons have long projections, which might render them more vulnerable to trafficking and mitochondrial fission and fusion defects.27
It is also possible that inhibition of mitochondrial fission impairment can re-balance the fusion and fission cycle and improve the overall quality of mitochondria, ensuring proper mitochondrial transportation to nerve terminals for optimal energy supply. Thus, selective protection of mitochondria might preferentially protect striatal neurons from various toxicities directly or indirectly induced by mHtt. Indeed, we found that preventing mHtt-induced excessive mitochondrial fission by treatment with the Drp1 peptide inhibitor reduced the formation of dysfunctional mitochondria (as evidenced by reduced mitochondrial depolarization and mitochondrial reactive oxygen species) and improved overall mitochondrial functions (as measured by for example ATP content).1 Healthier mitochondria can provide more ATP to the neurons, and fewer dysfunctional mitochondria can reduce the burden of oxidative stress within the cells. Supporting this possibility is our finding that treatment with the Drp1 peptide inhibitor improved neuronal morphology as evidenced by increased neurite outgrowth and inhibited cell death rate in patient-derived neurons. It is likely that increasing the cellular energy reserves may enable the neurons to withstand other injurious effects of mHtt. These in turn can translate to inhibition of mHtt-induced neurological symptoms and lethality, in vivo, that we observed.1
Whether a Drp1 inhibitor or inhibitors of other molecular events associated with HD pathology are sufficient to alleviate the burden of HD disease in patients will need to be tested in clinical trials. This aspect of drug development is often driven and funded by industry. However, academic research can help identify surrogate biomarkers for HD.
Lack of Biomarkers for HD to Assist in Patient Assessments during Clinical Trials Hinders Drug Development for the Disease
Identification of biomarkers for diagnosis prior to clinical symptoms and for use during the development of therapeutics should be considered as a high priority for the success of translational research in this field. (Biomarkers are cellular, biochemical, or molecular indicators that can be used to evaluate pharmacological responses to a therapeutic treatment.28) So far only clinical examinations, which are not sensitive enough to detect small changes over short period of times, are used to assess disease progression.28,29 Sensitive surrogate HD biomarkers will allow following the beneficial effect of a tested drug prior to the manifestation of clinical symptoms.
Some biomarkers have been suggested. These include plasma creatine kinase (CK-BB isozyme), whose levels are reduced in R6/2 mice and in humans with HD.30 Other biomarkers include 8-hydroxy-2-deoxyguanosine (8-OH-2-dG), an indicator of oxidative injury to DNA, which was found to be highly elevated in HD patients; this marker can be detected in the urine and blood and brain tissue of patients.31 Measurements of oxidized molecules in blood and urine, levels of endogenous anti-oxidants, and branch chain amino acids as products of metabolism have also been suggested as potential biomarkers.32 Another potential biomarker to monitor HD is the overexpression of H2AFY, a member of the H2A histone family.33 An unbiased gene expression profiling identified it to be elevated in the blood of HD patients. The authors suggest H2AFY as a potential biomarker associated with disease activity and pharmacodynamic response. Whether any of the above biomarkers is indeed predictive of disease progression and is a useful surrogate for treatment efficacy remains to be determined in future studies. Further unbiased studies as well as rational studies, using patient samples and animal models are underway; these are likely to shorten the length (and cost) of clinical trials for HD patients and will contribute an important component to ensuring the success of developing effective HD treatments.
Perspectives
HD and other aging-related neurodegenerative diseases, such as Parkinson disease and Alzheimer disease, share the major features of late-onset and selective neuronal loss, as well as accumulation of protein aggregates.5 Because of its well-characterized mutation in a single gene, the Htt gene, HD was expected to be an “easier” disease among neurodegenerative diseases, to elucidate the pathology and to identify the pathway that can be modulated for treatment. Yet the research in the past 20 years suggests that the task is far from being completed. Several recent preclinical studies, including our own, suggest that pharmacological agents that protect mitochondrial functions may provide effective means to slow down or inhibit the progression of HD and possibly other neurological diseases. We described some of the challenges in developing new therapeutics for HD and believe that academic research can greatly assist in addressing these challenges.
Disclosure of Potential Conflicts of Interest
A patent on the design and application of mitochondrial fission peptide inhibitor has been filed. The authors declare no conflicts of interest.
References
- 1.Guo X, Disatnik MH, Monbureau M, Shamloo M, Mochly-Rosen D, Qi X. Inhibition of mitochondrial fragmentation diminishes Huntington’s disease-associated neurodegeneration. J Clin Invest. 2013;123:5371–88. doi: 10.1172/JCI70911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.The Huntington’s Disease Collaborative Research Group A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 1993;72:971–83. doi: 10.1016/0092-8674(93)90585-E. [DOI] [PubMed] [Google Scholar]
- 3.Imarisio S, Carmichael J, Korolchuk V, Chen CW, Saiki S, Rose C, Krishna G, Davies JE, Ttofi E, Underwood BR, et al. Huntington’s disease: from pathology and genetics to potential therapies. Biochem J. 2008;412:191–209. doi: 10.1042/BJ20071619. [DOI] [PubMed] [Google Scholar]
- 4.Poirier MA, Jiang H, Ross CA. A structure-based analysis of huntingtin mutant polyglutamine aggregation and toxicity: evidence for a compact beta-sheet structure. Hum Mol Genet. 2005;14:765–74. doi: 10.1093/hmg/ddi071. [DOI] [PubMed] [Google Scholar]
- 5.Knott AB, Perkins G, Schwarzenbacher R, Bossy-Wetzel E. Mitochondrial fragmentation in neurodegeneration. Nat Rev Neurosci. 2008;9:505–18. doi: 10.1038/nrn2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sugars KL, Rubinsztein DC. Transcriptional abnormalities in Huntington disease. Trends Genet. 2003;19:233–8. doi: 10.1016/S0168-9525(03)00074-X. [DOI] [PubMed] [Google Scholar]
- 7.Li SH, Li XJ. Huntingtin-protein interactions and the pathogenesis of Huntington’s disease. Trends Genet. 2004;20:146–54. doi: 10.1016/j.tig.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 8.Shirasaki DI, Greiner ER, Al-Ramahi I, Gray M, Boontheung P, Geschwind DH, Botas J, Coppola G, Horvath S, Loo JA, et al. Network organization of the huntingtin proteomic interactome in mammalian brain. Neuron. 2012;75:41–57. doi: 10.1016/j.neuron.2012.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bhattacharyya NP, Banerjee M, Majumder P. Huntington’s disease: roles of huntingtin-interacting protein 1 (HIP-1) and its molecular partner HIPPI in the regulation of apoptosis and transcription. FEBS J. 2008;275:4271–9. doi: 10.1111/j.1742-4658.2008.06563.x. [DOI] [PubMed] [Google Scholar]
- 10.Costa V, Giacomello M, Hudec R, Lopreiato R, Ermak G, Lim D, Malorni W, Davies KJ, Carafoli E, Scorrano L. Mitochondrial fission and cristae disruption increase the response of cell models of Huntington’s disease to apoptotic stimuli. EMBO Mol Med. 2010;2:490–503. doi: 10.1002/emmm.201000102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bibb JA, Yan Z, Svenningsson P, Snyder GL, Pieribone VA, Horiuchi A, Nairn AC, Messer A, Greengard P. Severe deficiencies in dopamine signaling in presymptomatic Huntington’s disease mice. Proc Natl Acad Sci U S A. 2000;97:6809–14. doi: 10.1073/pnas.120166397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Trancikova A, Ramonet D, Moore DJ. Genetic mouse models of neurodegenerative diseases. Prog Mol Biol Transl Sci. 2011;100:419–82. doi: 10.1016/B978-0-12-384878-9.00012-1. [DOI] [PubMed] [Google Scholar]
- 13.William Yang X, Gray M. Mouse Models for Validating Preclinical Candidates for Huntington's Disease. 2011. [PubMed]
- 14.Switonski PM, Szlachcic WJ, Gabka A, Krzyzosiak WJ, Figiel M. Mouse models of polyglutamine diseases in therapeutic approaches: review and data table. Part II. Mol Neurobiol. 2012;46:430–66. doi: 10.1007/s12035-012-8316-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Munoz-Sanjuan I, Bates GP. The importance of integrating basic and clinical research toward the development of new therapies for Huntington disease. J Clin Invest. 2011;121:476–83. doi: 10.1172/JCI45364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ross CA, Tabrizi SJ. Huntington’s disease: from molecular pathogenesis to clinical treatment. Lancet Neurol. 2011;10:83–98. doi: 10.1016/S1474-4422(10)70245-3. [DOI] [PubMed] [Google Scholar]
- 17.Shirendeb UP, Calkins MJ, Manczak M, Anekonda V, Dufour B, McBride JL, Mao P, Reddy PH. Mutant huntingtin’s interaction with mitochondrial protein Drp1 impairs mitochondrial biogenesis and causes defective axonal transport and synaptic degeneration in Huntington’s disease. Hum Mol Genet. 2012;21:406–20. doi: 10.1093/hmg/ddr475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Song W, Chen J, Petrilli A, Liot G, Klinglmayr E, Zhou Y, Poquiz P, Tjong J, Pouladi MA, Hayden MR, et al. Mutant huntingtin binds the mitochondrial fission GTPase dynamin-related protein-1 and increases its enzymatic activity. Nat Med. 2011;17:377–82. doi: 10.1038/nm.2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Qi X, Qvit N, Su YC, Mochly-Rosen D. A novel Drp1 inhibitor diminishes aberrant mitochondrial fission and neurotoxicity. J Cell Sci. 2013;126:789–802. doi: 10.1242/jcs.114439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Su YC, Qi X. Inhibition of excessive mitochondrial fission reduced aberrant autophagy and neuronal damage caused by LRRK2 G2019S mutation. Hum Mol Genet. 2013;22:4545–61. doi: 10.1093/hmg/ddt301. [DOI] [PubMed] [Google Scholar]
- 21.Trettel F, Rigamonti D, Hilditch-Maguire P, Wheeler VC, Sharp AH, Persichetti F, Cattaneo E, MacDonald ME. Dominant phenotypes produced by the HD mutation in STHdh(Q111) striatal cells. Hum Mol Genet. 2000;9:2799–809. doi: 10.1093/hmg/9.19.2799. [DOI] [PubMed] [Google Scholar]
- 22.Robinton DA, Daley GQ. The promise of induced pluripotent stem cells in research and therapy. Nature. 2012;481:295–305. doi: 10.1038/nature10761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cha JH, Kosinski CM, Kerner JA, Alsdorf SA, Mangiarini L, Davies SW, Penney JB, Bates GP, Young AB. Altered brain neurotransmitter receptors in transgenic mice expressing a portion of an abnormal human huntington disease gene. Proc Natl Acad Sci U S A. 1998;95:6480–5. doi: 10.1073/pnas.95.11.6480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pickrell AM, Pinto M, Hida A, Moraes CT. Striatal dysfunctions associated with mitochondrial DNA damage in dopaminergic neurons in a mouse model of Parkinson’s disease. J Neurosci. 2011;31:17649–58. doi: 10.1523/JNEUROSCI.4871-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cui L, Jeong H, Borovecki F, Parkhurst CN, Tanese N, Krainc D. Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell. 2006;127:59–69. doi: 10.1016/j.cell.2006.09.015. [DOI] [PubMed] [Google Scholar]
- 26.Brustovetsky N, Brustovetsky T, Purl KJ, Capano M, Crompton M, Dubinsky JM. Increased susceptibility of striatal mitochondria to calcium-induced permeability transition. J Neurosci. 2003;23:4858–67. doi: 10.1523/JNEUROSCI.23-12-04858.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bossy-Wetzel E, Petrilli A, Knott AB. Mutant huntingtin and mitochondrial dysfunction. Trends Neurosci. 2008;31:609–16. doi: 10.1016/j.tins.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hersch SM, Rosas HD. Biomarkers to Enable the Development of Neuroprotective Therapies for Huntington's Disease. 2011. [PubMed]
- 29.Weir DW, Sturrock A, Leavitt BR. Development of biomarkers for Huntington’s disease. Lancet Neurol. 2011;10:573–90. doi: 10.1016/S1474-4422(11)70070-9. [DOI] [PubMed] [Google Scholar]
- 30.Kim J, Amante DJ, Moody JP, Edgerly CK, Bordiuk OL, Smith K, et al. Reduced creatine kinase as a central and peripheral biomarker in Huntington's disease. Biochimica et biophysica acta 2010; 1802:673-81. [DOI] [PMC free article] [PubMed]
- 31.Hersch SM, Gevorkian S, Marder K, Moskowitz C, Feigin A, Cox M, Como P, Zimmerman C, Lin M, Zhang L, et al. Creatine in Huntington disease is safe, tolerable, bioavailable in brain and reduces serum 8OH2’dG. Neurology. 2006;66:250–2. doi: 10.1212/01.wnl.0000194318.74946.b6. [DOI] [PubMed] [Google Scholar]
- 32.Fonteh AN, Harrington RJ, Huhmer AF, Biringer RG, Riggins JN, Harrington MG. Identification of disease markers in human cerebrospinal fluid using lipidomic and proteomic methods. Dis Markers. 2006;22:39–64. doi: 10.1155/2006/202938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hu Y, Chopra V, Chopra R, Locascio JJ, Liao Z, Ding H, Zheng B, Matson WR, Ferrante RJ, Rosas HD, et al. Transcriptional modulator H2A histone family, member Y (H2AFY) marks Huntington disease activity in man and mouse. Proc Natl Acad Sci U S A. 2011;108:17141–6. doi: 10.1073/pnas.1104409108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, et al. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. doi: 10.1016/S0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- 35.Schilling G, Becher MW, Sharp AH, Jinnah HA, Duan K, Kotzuk JA, Slunt HH, Ratovitski T, Cooper JK, Jenkins NA, et al. Intranuclear inclusions and neuritic aggregates in transgenic mice expressing a mutant N-terminal fragment of huntingtin. Hum Mol Genet. 1999;8:397–407. doi: 10.1093/hmg/8.3.397. [DOI] [PubMed] [Google Scholar]
- 36.Menalled LB, Sison JD, Dragatsis I, Zeitlin S, Chesselet MF. Time course of early motor and neuropathological anomalies in a knock-in mouse model of Huntington’s disease with 140 CAG repeats. J Comp Neurol. 2003;465:11–26. doi: 10.1002/cne.10776. [DOI] [PubMed] [Google Scholar]
- 37.Wheeler VC, White JK, Gutekunst CA, Vrbanac V, Weaver M, Li XJ, Li SH, Yi H, Vonsattel JP, Gusella JF, et al. Long glutamine tracts cause nuclear localization of a novel form of huntingtin in medium spiny striatal neurons in HdhQ92 and HdhQ111 knock-in mice. Hum Mol Genet. 2000;9:503–13. doi: 10.1093/hmg/9.4.503. [DOI] [PubMed] [Google Scholar]
- 38.Lin CH, Tallaksen-Greene S, Chien WM, Cearley JA, Jackson WS, Crouse AB, Ren S, Li XJ, Albin RL, Detloff PJ. Neurological abnormalities in a knock-in mouse model of Huntington’s disease. Hum Mol Genet. 2001;10:137–44. doi: 10.1093/hmg/10.2.137. [DOI] [PubMed] [Google Scholar]
- 39.Heng MY, Tallaksen-Greene SJ, Detloff PJ, Albin RL. Longitudinal evaluation of the Hdh(CAG)150 knock-in murine model of Huntington’s disease. J Neurosci. 2007;27:8989–98. doi: 10.1523/JNEUROSCI.1830-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gray M, Shirasaki DI, Cepeda C, André VM, Wilburn B, Lu XH, Tao J, Yamazaki I, Li SH, Sun YE, et al. Full-length human mutant huntingtin with a stable polyglutamine repeat can elicit progressive and selective neuropathogenesis in BACHD mice. J Neurosci. 2008;28:6182–95. doi: 10.1523/JNEUROSCI.0857-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Slow EJ, van Raamsdonk J, Rogers D, Coleman SH, Graham RK, Deng Y, Oh R, Bissada N, Hossain SM, Yang YZ, et al. Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Hum Mol Genet. 2003;12:1555–67. doi: 10.1093/hmg/ddg169. [DOI] [PubMed] [Google Scholar]
- 42.Hodgson JG, Agopyan N, Gutekunst CA, Leavitt BR, LePiane F, Singaraja R, Smith DJ, Bissada N, McCutcheon K, Nasir J, et al. A YAC mouse model for Huntington’s disease with full-length mutant huntingtin, cytoplasmic toxicity, and selective striatal neurodegeneration. Neuron. 1999;23:181–92. doi: 10.1016/S0896-6273(00)80764-3. [DOI] [PubMed] [Google Scholar]
- 43.Yu-Taeger L, Petrasch-Parwez E, Osmand AP, Redensek A, Metzger S, Clemens LE, Park L, Howland D, Calaminus C, Gu X, et al. A novel BACHD transgenic rat exhibits characteristic neuropathological features of Huntington disease. J Neurosci. 2012;32:15426–38. doi: 10.1523/JNEUROSCI.1148-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.von Hörsten S, Schmitt I, Nguyen HP, Holzmann C, Schmidt T, Walther T, Bader M, Pabst R, Kobbe P, Krotova J, et al. Transgenic rat model of Huntington’s disease. Hum Mol Genet. 2003;12:617–24. doi: 10.1093/hmg/ddg075. [DOI] [PubMed] [Google Scholar]