

Abstract
CHARGE syndrome is a rare, autosomal dominant condition caused by mutations in the CHD7 gene. Although central nervous system defects have been reported, the detailed description and analysis of these anomalies in CHARGE syndrome patients lag far behind the description of other, more easily observed defects. We recently described cerebellar abnormalities in CHARGE syndrome patients and used mouse models to identify the underlying causes. Our studies identified altered expression of the homeobox genes Otx2 and Gbx2 in the developing neural tube of Chd7−/− embryos. Furthermore, we showed that the expression of Fgf8 is sensitive to Chd7 gene dosage and demonstrated an epistatic relationship between these genes during cerebellar vermis development. These findings provided, for the first time, an example of cerebellar vermis hypoplasia in a human syndrome that can be linked to deregulated FGF signaling. I discuss some of these observations and their implications for CHARGE syndrome.
Keywords: CHARGE syndrome, Chd7, Fgf8, cerebellum, embryo, mid-hindbrain, isthmus organizer, brain, development, mouse
Epistasis in Human Disease
In the context of disease-susceptibility alleles, interactions between more than one risk allele that alters the incidence of disease to a significantly greater extent than expected from the simple addition of the individual allelic effects can be defined as epistatic interactions.1 It is becoming increasingly clear that epistatic effects are of fundamental importance to understand the genetic basis of complex diseases that may be caused by the combination of many genetic risk factors, each with a relatively small contribution to disease susceptibility. In addition, conditions with relatively “simple” genetics but pleiotropic phenotypic effects, e.g., syndromes caused by autosomal dominant gene effects, might also be subject to strong epistatic effects. For example, 22q11del (DiGeorge) syndrome exhibits large clinical variability.2 The existence of so-called “modifier” genes, i.e., protective or susceptibility alleles that alter the effect of the dominant disease-associated gene on disease incidence, has been postulated for many years. Studies in model organisms, typically making use of loss-of-function alleles, have been useful in identifying candidate genetic modifiers of disease.2 However, it has proven rather difficult to identify modifier genes in the human population.
CHARGE Syndrome
CHARGE (coloboma of the eye, heart defects, atresia of the nasal choanae, retardation of growth and/or development, genital and/or urinary abnormalities, and ear abnormalities and deafness) syndrome is an autosomal dominant disorder with an estimated prevalence of 1 per 10 000.3-5 Most patients (60–70%) have mutations in the CHD7 (Chromodomain helicase DNA-binding protein 7) gene.6-8 A CHARGE syndrome clinical diagnosis is normally made when three to four major abnormalities (coloboma, choanal atraesia, ear defects, and cranial nerve dysfunction) are seen or two to three major and two to three minor criteria (e.g., genital hypoplasia, retarded growth, cardiovascular defects, orofacial cleft, or tracheo-esophageal fistula) are met.9-11 However, CHARGE syndrome is characterized by high phenotypic variability, making diagnosis and the definition of key clinical characteristics difficult. Interestingly, CHD7 mutations have also been reported in other conditions, most notably Kallmann syndrome and idiopathic hypogonadotropic hypogonadism with hearing loss12,13 and velocardiofacial and/or DiGeorge syndrome.14,15 Loss-of-function mutations in Chd7 and Tbx1 (a DiGeorge syndrome gene) interact during aortic arch development in mouse embryos, suggesting that the function of these genes intersect at some point.15
A key signaling pathway linked to Tbx1 function is the FGF signaling pathway. Tbx1 is required for normal Fgf8 expression in the endoderm, and Tbx1 and Fgf8 loss-of-function alleles are in epistasis during pharyngeal development.16 We therefore postulated that Chd7 might also function upstream of the FGF signaling pathway. However, we did not detect a statistically significant interaction between Chd7 and Fgf8 loss-of-function alleles during aortic arch development.15 There are several possible explanations for this result, including: (1) Our hypothesis is incorrect and Chd7 haplo-insufficiency does not affect Fgf8 gene expression or signaling and there is no epistatic relationship between these genes; (2) CHD7 can function upstream of FGF signaling, but these effects are highly context-dependent and not relevant during aortic arch development; or (3) Chd7 and Fgf8 interactions are modified, i.e., in epistasis with additional genetic factors and the genetic background used in this study masked any interactions. We turned to central nervous system (CNS) development in an attempt to resolve these questions.
CNS Defects are Prevalent in CHARGE Syndrome
CNS defects like arhinencephaly, hypoplasia of the cerebellum, and brainstem and cerebellar heterotopia are detected in 70–80% of CHARGE syndrome cases.17-20 This incidence is in the same range as other more regularly reported clinical features of CHARGE syndrome: coloboma (82%), atraesia of the choanae (57%), ear abnormalities and deafness (95%), cranial nerve dysfunction (61%), heart defects (80%), retardation of growth/development (90%), and genito-urinary anomalies (60%). Thus, CNS defects are likely to represent a significant component of the clinical spectrum that typifies CHARGE syndrome. However, the exact scope and penetrance of CNS defects associated with CHARGE syndrome and CHD7 deficiency remain to be fully defined.
Cerebellar Defects in CHARGE Syndrome
Until recently, reports on cerebellar anomalies in CHARGE syndrome were rare, with the most convincing evidence being described in pre-natal fetuses.19,21,22 This has led to the erroneous assumption by some that cerebellar defects may not be a significant feature characteristic of CHARGE syndrome. However, there are two alternative explanations: (1) MRI examinations capable of detecting cerebellar defects in CHARGE syndrome are not routinely performed and (2) these defects are associated with the most severe cases of CHARGE syndrome that tend to be incompatible with life. Indeed, one study reports a correlation between patient survival and Atrioventricular septal defects (AVSD) with cerebellar and brainstem defects.20
We recently reported the first systematic analysis of cerebellar structure in MRI scans from a cohort of patients with CHD7 mutations and diagnosed with CHARGE syndrome.23 Approximately 50% of patients showed some cerebellar abnormality. These included cerebellar vermis hypoplasia (35%) and foliation defects (25%). The former defect was of particular interest, as our previous studies in mouse models have shown that inappropriate levels of FGF signaling in the embryonic isthmus organizer (IsO, a key signaling center located at the embryonic mid-hindbrain boundary) dramatically affected the size of the cerebellar vermis, with little effect on the size of the cerebellar hemispheres.24 Thus, it appears that expansion of the vermis progenitor zone is critically dependent on high levels of FGF signals from the adjacent IsO. The identification of cerebellar vermis hypoplasia in CHARGE syndrome suggested a potential link between CHD7 and FGF signaling.
The examination of mouse models uncovered compelling evidence for epistatic interactions between Chd7 and Fgf8. Whereas Chd7+/− and Fgf8+/− mouse embryos exhibited no discernable cerebellar defects, Chd7+/−;Fgf8+/− embryos presented with cerebellar vermis aplasia. This epistatic interaction between Chd7 and Fgf8 loss-of-function alleles identified a functionally important link between CHD7 and FGF signaling.
Having uncovered strong genetic evidence in support of a link between Chd7 and Fgf8, we searched for a mechanistic explanation for these genetic observations. The most obvious prediction was that CHD7 was required for the normal expression of Fgf8, or other FGF signaling components. Indeed, we could show that Fgf8 expression levels at the IsO directly correlated with Chd7 genotype.23 These findings placed CHD7 upstream of Fgf8.
These findings predict that hypomorphic mutations in the FGF signaling pathway will strongly enhance cerebellar vermis defects in CHARGE syndrome patients. It is of considerable interest to note that mutations in multiple genes in the FGF pathway have been reported in patients with Kallmann's syndrome, consistent with a model whereby these gene mutations can interact epistatically to determine the incidence and perhaps severity of disease.25 This raises the possibility that mutations or polymorphisms that affect the function and/or expression of FGF8 and other FGF signaling components might affect the penetrance and expressivity of cerebellar defects in CHARGE syndrome. Sequencing of the FGF8 gene in a cohort of CHARGE syndrome patients with CHD7 mutations has not identified any obviously damaging mutations in patients with or without cerbellar vermis defects yet (personal communication, Conny van Ravenswaaij-Arts). Additional targeted or exome sequencing studies in CHARGE syndrome patients, with careful analyses of correlations between genetic features and particular phenotypes are likely to yield substantial insights into the etiology and genetic complexities of CHARGE syndrome.
Our identification of a link between CHD7 and FGF signaling at the IsO provides an explanation for the cerebellar vermis hypoplasia present in some CHARGE syndrome patients. Several patients had additional cerebellar defects, including abnormal foliation patterns.23 Previous studies have also reported cellular heterotopia, the mis-localization of neurons in the cerebellum.19,22 These observations imply additional roles for CHD7 in controlling postnatal cerebellar development, when granule cell precursor (GCp) proliferation in the external granule cell layer and subsequent migration of GCps into the cerebellar cortex drive cerebellar foliation.
Identification of CHD7 as a Critical Regulator of Homeobox Gene Expression
CHD7 shares significant homology in amino acid sequence (44%) and domain organization with the Drosophila chromodomain factor kismet. The kismet gene was originally identified as a member of the Trithorax group of regulators and kismet mutant flies shows evidence of homeotic transformations and corresponding alterations in homeobox gene expression.26 Given these similarities it is tempting to speculate that some of the developmental anomalies typical of Chd7 deficiency might be caused by deregulated homeobox gene expression. Certainly, some evidence in support of this notion has been reported. For example, the expression of Krox20, a homeobox gene expressed in two specific segments of the embryonic hindbrain, rhombomere 3 (r3) and r5, is reduced in Chd7-deficient embryos.23,27 Defects in r3 and r5 identity might be responsible for some of the cranial nerve paralysis observed in a high proportion of CHARGE syndrome patients, with the VIIth and VIIIth cranial nerves most commonly affected.18
The homeobox genes Otx2 and Gbx2 impart positional identity in the early anterior neural tube. The IsO forms at the sharp boundary between the anterior Otx2 and posterior Gbx2 expression domains that arises as a result of cross-repressive interactions between these transcription factors. Studies in various model organisms have accumulated a large body of evidence indicating that OTX2 functions as a potent repressor of Fgf8 expression at the IsO.28-30 Studies in the mouse have shown that transgenic Otx2 misexpression could re-position the Fgf8-expressing IsO to more posterior positions in the embryo.30 The intriguing links between CHD7/kismet and homeobox gene regulation led us to examine whether CHD7 depletion might affect Fgf8 expression through changes in Otx2 and Gbx2 gene expression. We found that Otx2 was de-repressed and Gbx2 lost in the region normally fated to become r1. Intriguingly, we found that Fgf8 expression was initialised at the correct position in Chd7−/− embryos and was therefore located in an Otx2-expressing region. Thus, rather than shifting the Fgf8 expression domain as in transgenic Otx2 misexpression experiments,30 Fgf8 expression was reduced, consistent with the role of OTX2 as a repressor of Fgf8. This finding has several important implications for understanding the potential genetic causes of cerebellar vermis hypoplasia in humans. Mutations that affect the activity of other endogenous repressors of OTX2 and mutations in critical OTX2 regulatory elements might be responsible for some cases of cerebellar vermis hypoplasia. This observation might also help explain the fact that mutations in the FGF8 coding sequence have not as yet been described in patients with cerebellar vermis hypoplasia. Given the many critical developmental roles performed by FGF8 in development, mutations with potent enough effects on FGF8 function are likely to be incompatible with life. However, one might predict that mutations in non-coding regions that specifically affect FGF8 expression in the IsO might predispose an individual to cerebellar vermis hypoplasia (Fig. 1).
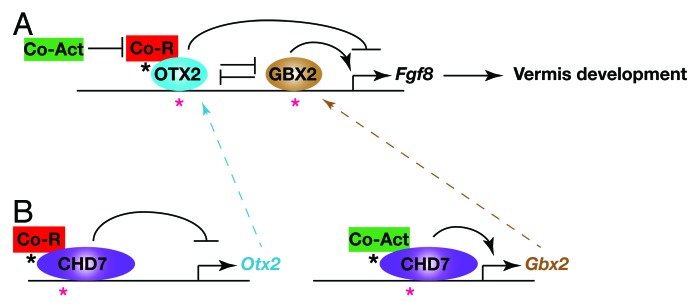
Figure 1. Gene regulatory interactions implicated in cerebellar vermis hypoplasia in CHARGE syndrome. The diagram depicts a model of the homeobox gene-regulatory network that impacts on Fgf8 expression at the IsO. Possible regulatory mutations that might result in de-regulated Fgf8 expression and cerebellar vermis hypoplasia are indicated by asterisks. (A) OTX2 represses Fgf8 expression by interacting with transcriptional co-repressors, such as Groucho (Co-R, Tle4/Grg4), whereas GBX2 acts as a positive regulator of Fgf8 expression, presumably through interacting with transcriptional co-activators (Co-Act). OTX2 and GBX2 cross-repress each other’s expression. Mutations in OTX2 or GBX2 regulatory regions (red asterisks) or mutations that affect the ability of these factors to recruit or activate co-activators or repressors (black asterisk) might affect Fgf8 expression levels and result in cerebellar vermis hypoplasia. (B) CHD7 functions as an Otx2 repressor and GBX2 activator in the mid-hindbrain region, possibly also through interacting with additional chromatin remodelling and transcriptional regulators that contribute to repressive or activator activities. Mutations in Otx2 or Gbx2 regulatory elements to which CHD7 are recruited (red asterisks) or mutations affecting the activity or recruitment of additional co-activating or repressing factors (black asterisks) are predicted to affect Otx2 or Gbx2 expression, which in turn will affect Fgf8 expression and cause cerebellar vermis abnormalities.
Implications for Other CHARGE Syndrome Phenotypes
An obvious question that arises from our work on the cerebellum is whether the regulation of homeobox gene expression and FGF signaling by CHD7 is unique to this part of the embryo, or whether it might also explain other phenotypes associated with CHD7 deficiency. Fgf8 and Otx2 are involved in the development of several other organs and structures affected in CHARGE syndrome, including the eye, ear, olfactory placode and the forebrain. Intriguingly, Otx2 expression is reduced in the otic vesicle and olfactory placode of Chd7−/− embryos, suggesting that CHD7 can regulate Otx2 expression in other embryonic regions, but that the effect on Otx2 expression is context-dependent.31,32
Concluding Remarks
Elucidating the developmental basis of CNS defects caused by CHD7 deficiency is important for understanding the etiology of cognitive impairments associated with CHARGE syndrome. In addition to its developmental roles, CHD7 functions as a key regulator of neural progenitor cell differentiation in the adult hippocampus (Feng et al.33). To what extent CHD7 deficiency in the hippocampus affects cognitive ability of adults with CHD7 mutations remains to be determined.
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
Acknowledgments
I acknowledge funding from the Wellcome Trust (091475) and the Medical Research Council (MR/K022377/1) who supported this work and thank Kieran Jones for comments on the manuscript.
Glossary
Abbreviations:
- AVSD
Atrioventricular septal defects
- FGF
Fibroblast Growth Factor
- CHD7
Chromodomain Helicase DNA binding factor 7
- Otx2
Orthodenticle homeobox 2
- Gbx2
Gastrulation brain homeobox 2
- Tbx1
T box transcription factor 1
- Krox20
Egr2, early growth response gene 2
- CNS
Central Nervous System
- MRI
Magnetic Resonance Imaging
- ChIP-Seq
Chromatin Immunoprecipitation-sequencing
- GCp
Granule cell precursor
- r1
rhomobomere 1
- r3
rhombomere 3
- r5
rhombomere 5
- IsO
Isthmus Organizer
References
- 1.Cordell HJ. Epistasis: what it means, what it doesn’t mean, and statistical methods to detect it in humans. Hum Mol Genet. 2002;11:2463–8. doi: 10.1093/hmg/11.20.2463. [DOI] [PubMed] [Google Scholar]
- 2.Aggarwal VS, Morrow BE. Genetic modifiers of the physical malformations in velo-cardio-facial syndrome/DiGeorge syndrome. Dev Disabil Res Rev. 2008;14:19–25. doi: 10.1002/ddrr.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hall BD. Choanal atresia and associated multiple anomalies. J Pediatr. 1979;95:395–8. doi: 10.1016/S0022-3476(79)80513-2. [DOI] [PubMed] [Google Scholar]
- 4.Hittner HM, Hirsch NJ, Kreh GM, Rudolph AJ. Colobomatous microphthalmia, heart disease, hearing loss, and mental retardation--a syndrome. J Pediatr Ophthalmol Strabismus. 1979;16:122–8. doi: 10.3928/0191-3913-19790301-10. [DOI] [PubMed] [Google Scholar]
- 5.Pagon RA, Graham JM, Jr., Zonana J, Yong SL. Coloboma, congenital heart disease, and choanal atresia with multiple anomalies: CHARGE association. J Pediatr. 1981;99:223–7. doi: 10.1016/S0022-3476(81)80454-4. [DOI] [PubMed] [Google Scholar]
- 6.Janssen N, Bergman JE, Swertz MA, Tranebjaerg L, Lodahl M, Schoots J, Hofstra RM, van Ravenswaaij-Arts CM, Hoefsloot LH. Mutation update on the CHD7 gene involved in CHARGE syndrome. Hum Mutat. 2012;33:1149–60. doi: 10.1002/humu.22086. [DOI] [PubMed] [Google Scholar]
- 7.Vissers LE, van Ravenswaaij CM, Admiraal R, Hurst JA, de Vries BB, Janssen IM, van der Vliet WA, Huys EH, de Jong PJ, Hamel BC, et al. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat Genet. 2004;36:955–7. doi: 10.1038/ng1407. [DOI] [PubMed] [Google Scholar]
- 8.Bilan F, Legendre M, Charraud V, Manière B, Couet D, Gilbert-Dussardier B, Kitzis A. Complete screening of 50 patients with CHARGE syndrome for anomalies in the CHD7 gene using a denaturing high-performance liquid chromatography-based protocol: new guidelines and a proposal for routine diagnosis. J Mol Diagn. 2012;14:46–55. doi: 10.1016/j.jmoldx.2011.08.003. [DOI] [PubMed] [Google Scholar]
- 9.Lalani SR, Safiullah AM, Fernbach SD, Harutyunyan KG, Thaller C, Peterson LE, McPherson JD, Gibbs RA, White LD, Hefner M, et al. Spectrum of CHD7 mutations in 110 individuals with CHARGE syndrome and genotype-phenotype correlation. Am J Hum Genet. 2006;78:303–14. doi: 10.1086/500273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blake KD, Davenport SL, Hall BD, Hefner MA, Pagon RA, Williams MS, Lin AE, Graham JM., Jr. CHARGE association: an update and review for the primary pediatrician. Clin Pediatr (Phila) 1998;37:159–73. doi: 10.1177/000992289803700302. [DOI] [PubMed] [Google Scholar]
- 11.Verloes A. Updated diagnostic criteria for CHARGE syndrome: a proposal. Am J Med Genet A. 2005;133A:306–8. doi: 10.1002/ajmg.a.30559. [DOI] [PubMed] [Google Scholar]
- 12.Jongmans MC, van Ravenswaaij-Arts CM, Pitteloud N, Ogata T, Sato N, Claahsen-van der Grinten HL, van der Donk K, Seminara S, Bergman JE, Brunner HG, et al. CHD7 mutations in patients initially diagnosed with Kallmann syndrome--the clinical overlap with CHARGE syndrome. Clin Genet. 2009;75:65–71. doi: 10.1111/j.1399-0004.2008.01107.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim HG, Kurth I, Lan F, Meliciani I, Wenzel W, Eom SH, Kang GB, Rosenberger G, Tekin M, Ozata M, et al. Mutations in CHD7, encoding a chromatin-remodeling protein, cause idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. Am J Hum Genet. 2008;83:511–9. doi: 10.1016/j.ajhg.2008.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Corsten-Janssen N, Saitta SC, Hoefsloot LH, McDonald-McGinn DM, Driscoll DA, Derks R, Dickinson KA, Kerstjens-Frederikse WS, Emanuel BS, Zackai EH, et al. More clinical overlap between 22q11.2 deletion syndrome and CHARGE syndrome than often anticipated. Mol Syndromol. 2013;4:235–45. doi: 10.1159/000351127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Randall V, McCue K, Roberts C, Kyriakopoulou V, Beddow S, Barrett AN, Vitelli F, Prescott K, Shaw-Smith C, Devriendt K, et al. Great vessel development requires biallelic expression of Chd7 and Tbx1 in pharyngeal ectoderm in mice. J Clin Invest. 2009;119:3301–10. doi: 10.1172/JCI37561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vitelli F, Taddei I, Morishima M, Meyers EN, Lindsay EA, Baldini A. A genetic link between Tbx1 and fibroblast growth factor signaling. Development. 2002;129:4605–11. doi: 10.1242/dev.129.19.4605. [DOI] [PubMed] [Google Scholar]
- 17.Lin AE, Siebert JR, Graham JM., Jr. Central nervous system malformations in the CHARGE association. Am J Med Genet. 1990;37:304–10. doi: 10.1002/ajmg.1320370303. [DOI] [PubMed] [Google Scholar]
- 18.Tellier AL, Cormier-Daire V, Abadie V, Amiel J, Sigaudy S, Bonnet D, de Lonlay-Debeney P, Morrisseau-Durand MP, Hubert P, Michel JL, et al. CHARGE syndrome: report of 47 cases and review. Am J Med Genet. 1998;76:402–9. doi: 10.1002/(SICI)1096-8628(19980413)76:5<402::AID-AJMG7>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 19.Sanlaville D, Etchevers HC, Gonzales M, Martinovic J, Clément-Ziza M, Delezoide AL, Aubry MC, Pelet A, Chemouny S, Cruaud C, et al. Phenotypic spectrum of CHARGE syndrome in fetuses with CHD7 truncating mutations correlates with expression during human development. J Med Genet. 2006;43:211–7. doi: 10.1136/jmg.2005.036160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Issekutz KA, Graham JM, Jr., Prasad C, Smith IM, Blake KD. An epidemiological analysis of CHARGE syndrome: preliminary results from a Canadian study. Am J Med Genet A. 2005;133A:309–17. doi: 10.1002/ajmg.a.30560. [DOI] [PubMed] [Google Scholar]
- 21.Becker R, Stiemer B, Neumann L, Entezami M. Mild ventriculomegaly, mild cerebellar hypoplasia and dysplastic choroid plexus as early prenatal signs of CHARGE association. Fetal Diagn Ther. 2001;16:280–3. doi: 10.1159/000053928. [DOI] [PubMed] [Google Scholar]
- 22.Legendre M, Gonzales M, Goudefroye G, Bilan F, Parisot P, Perez MJ, Bonnière M, Bessières B, Martinovic J, Delezoide AL, et al. Antenatal spectrum of CHARGE syndrome in 40 fetuses with CHD7 mutations. J Med Genet. 2012;49:698–707. doi: 10.1136/jmedgenet-2012-100926. [DOI] [PubMed] [Google Scholar]
- 23.Yu T, Meiners LC, Danielsen K, Wong MT, Bowler T, Reinberg D, Scambler PJ, van Ravenswaaij-Arts CM, Basson MA. Deregulated FGF and homeotic gene expression underlies cerebellar vermis hypoplasia in CHARGE syndrome. Elife. 2013;2:e01305. doi: 10.7554/eLife.01305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Basson MA, Echevarria D, Ahn CP, Sudarov A, Joyner AL, Mason IJ, Martinez S, Martin GR. Specific regions within the embryonic midbrain and cerebellum require different levels of FGF signaling during development. Development. 2008;135:889–98. doi: 10.1242/dev.011569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miraoui H, Dwyer AA, Sykiotis GP, Plummer L, Chung W, Feng B, Beenken A, Clarke J, Pers TH, Dworzynski P, et al. Mutations in FGF17, IL17RD, DUSP6, SPRY4, and FLRT3 are identified in individuals with congenital hypogonadotropic hypogonadism. Am J Hum Genet. 2013;92:725–43. doi: 10.1016/j.ajhg.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Daubresse G, Deuring R, Moore L, Papoulas O, Zakrajsek I, Waldrip WR, Scott MP, Kennison JA, Tamkun JW. The Drosophila kismet gene is related to chromatin-remodeling factors and is required for both segmentation and segment identity. Development. 1999;126:1175–87. doi: 10.1242/dev.126.6.1175. [DOI] [PubMed] [Google Scholar]
- 27.Alavizadeh A, Kiernan AE, Nolan P, Lo C, Steel KP, Bucan M. The Wheels mutation in the mouse causes vascular, hindbrain, and inner ear defects. Dev Biol. 2001;234:244–60. doi: 10.1006/dbio.2001.0241. [DOI] [PubMed] [Google Scholar]
- 28.Acampora D, Gulisano M, Broccoli V, Simeone A. Otx genes in brain morphogenesis. Prog Neurobiol. 2001;64:69–95. doi: 10.1016/S0301-0082(00)00042-3. [DOI] [PubMed] [Google Scholar]
- 29.Heimbucher T, Murko C, Bajoghli B, Aghaallaei N, Huber A, Stebegg R, Eberhard D, Fink M, Simeone A, Czerny T. Gbx2 and Otx2 interact with the WD40 domain of Groucho/Tle corepressors. Mol Cell Biol. 2007;27:340–51. doi: 10.1128/MCB.00811-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Broccoli V, Boncinelli E, Wurst W. The caudal limit of Otx2 expression positions the isthmic organizer. Nature. 1999;401:164–8. doi: 10.1038/43670. [DOI] [PubMed] [Google Scholar]
- 31.Layman WS, Hurd EA, Martin DM. Reproductive dysfunction and decreased GnRH neurogenesis in a mouse model of CHARGE syndrome. Hum Mol Genet. 2011;20:3138–50. doi: 10.1093/hmg/ddr216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hurd EA, Poucher HK, Cheng K, Raphael Y, Martin DM. The ATP-dependent chromatin remodeling enzyme CHD7 regulates pro-neural gene expression and neurogenesis in the inner ear. Development. 2010;137:3139–50. doi: 10.1242/dev.047894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Feng W, Khan MA, Bellvis P, Zhu Z, Bernhardt O, Herold-Mende C, Liu HK. The chromatin remodeler CHD7 regulates adult neurogenesis via activation of SoxC transcription factors. Cell Stem Cell. 2013;13:62–72. doi: 10.1016/j.stem.2013.05.002. [DOI] [PubMed] [Google Scholar]