

Abstract
Context
Lynch syndrome is an inherited cause of colorectal cancer caused by mutations of DNA mismatch repair genes. A number of extracolonic tumors have been associated with the disorder, including pancreatic cancer. However, the risk of pancreatic cancer in Lynch Syndrome is uncertain and not quantified.
Objective
To estimate pancreatic cancer risk in families with germline mismatch repair gene mutations.
Design, Setting, Patients
Cancer histories of probands and their relatives were evaluated in mismatch repair gene mutation carriers in the familial cancer registries of the Dana-Farber Cancer Institute and University of Michigan Comprehensive Cancer Center. Families enrolled prior to the study start date (June 2008) were eligible. Age-specific cumulative risks and hazard ratio estimates of pancreatic cancer risk were calculated and compared to the general population using modified segregation analysis, with correction for ascertainment.
Main Outcome Measures
Age-specific cumulative risks and hazard ratio estimates of pancreatic cancer risk
Results
Data on 6,342 individuals from 147 families with mismatch repair gene mutations were analyzed: 21% of families (31/147) reported at least one case of pancreatic cancer. Forty-seven pancreatic cancers were reported (21 male, 26 female) with no gender-related difference in age of diagnosis: 51.5 years v. 56.5 years for men and women respectively. The cumulative risk of pancreatic cancer in these families with gene mutations was 1.3% (95% CI: 0.31, 2.32) up to age 50 years and 3.7% (95% CI:1.45, 5.88) up to age 70 years which represents an 8.6-fold increase (95%CI:4.7, 15.7) compared to the general population.
Conclusions
Among 147 families with germline mismatch repair gene mutations, the risk of pancreatic cancer was increased compared to the U.S. population. Individuals with mismatch repair gene mutations and a family history of pancreatic cancer are appropriate to include in studies to further define the risk of pre-malignant and malignant pancreatic neoplasms and potential benefits and limitations of surveillance.
Introduction
Pancreatic cancer is the fourth leading cause of cancer deaths in the U.S.1 Though most cases are thought to be sporadic, data suggest up to 10% of ductal adenocarcinomas may be due to an inherited predisposition based on familial clustering.2,3 For most pancreatic cancer kindreds, the causative gene has not been identified. In a subset of families, pancreatic cancer may be an integral tumor in a number of familial cancer syndromes with established germline mutations. These conditions include Peutz-Jeghers Syndrome (cumulative lifetime risk of 36%),4,5 Familial Atypical Multiple Mole Melanoma Syndrome (lifetime risk = 17%),6 Hereditary Breast/Ovarian Cancer Syndrome (lifetime risks = 1.2% and 2.1%, for BRCA1 and BRCA2 carriers, respectively),7,8 Hereditary Pancreatitis (lifetime risk = 40%)9 and the newly described Familial Pancreatic Cancer due to mutations in PALB2 (risk not specified).10
Pancreatic cancer has also been observed in Lynch Syndrome, an autosomal dominant condition caused by defects in the mismatch repair (MMR) genes, MLH1, MSH2, MSH6 or PMS2. Colorectal cancer (CRC) and endometrial cancer are the most common cancers in this condition, with other specific neoplasms also occurring more frequently than in the general population. Evidence to include pancreatic cancer in the Lynch Syndrome cancer spectrum has been difficult to interpret and an elevated risk has not been convincing.11–16 Most studies examining cancer risk in Lynch Syndrome have been from families with a strong history of early-onset CRCs. This lends itself to a number of problems including the overestimation of age-specific cumulative risks of component tumors due to ascertainment bias. Additionally, many published data report on a small number of cases with incomplete testing of the full pedigree. Analyses performed exclusively on observed genotypes lack power to accurately estimate uncommon events, such as cancers with lower prevalence in a given syndrome.
The goal of our study was to quantify the risk of pancreatic cancer in families with an identified pathogenic MMR gene mutation. We have used analytic tools that correct for ascertainment and provide genotype data on subjects whose mutation status is unknown.
Methods
Selection and Description of Participants
A total of 147 families with deleterious mutations in MLH1 (GenBank: NM_000249), MSH2 (GenBank: NM_000251), and MSH6 (GenBank: NM_000179) were eligible for inclusion at the start of the study in June 2008. Families were identified from hereditary CRC registries at Dana-Farber Cancer Institute (DFCI; n=80) and University of Michigan Cancer Center (UMCC; n=67). Families presenting to our cancer genetics programs are either by self-referral or health care provider referral and are enrolled on the basis of multiple cases of CRC, CRC diagnosis at a young age, or familial association of CRC with other Lynch Syndrome-associated tumors. Patients presenting for evaluation (probands) are routinely enrolled in the registries using institutional review board–approved protocols, and personal and family cancer histories and demographic data are obtained from the proband and participating relatives. Written informed consent is provided by probands for the confirmation of cancer diagnoses and deaths by review of medical records, pathology reports, or death certificates. Clinical information is updated periodically through follow-up clinic visits or telephone encounters. For this study, we selected patients with documented deleterious MMR gene mutations who were identified prior to June 2008. Analysis of MMR germline mutations in families was performed using standard molecular techniques for full gene sequencing and conducted on either the family member with CRC (or other Lynch Syndrome-associated cancer) or an “at-risk” first- or second-degree relative. Reports of pancreatic cancer were confirmed either by pathology report or death certificate.
Mutation Analysis
Mutation Analysis Technique: DNA from white blood cells was extracted and purified from the sample of blood provided by each proband, amplified by polymerase chain reaction, and directly sequenced in forward and reverse directions. For the MLH1 gene, approximately 2300 base pairs were sequenced, comprising 19 exons and approximately 560 adjacent noncoding intronic base pairs. For the MSH2 gene, approximately 2800 base pairs were sequenced, comprising 16 exons and approximately 470 adjacent noncoding intronic base pairs. For the MSH6 gene, approximately 4080 base pairs were sequenced, comprising 10 exons and approximately 290 adjacent non-coding intronic base pairs. The non-coding intronic regions of MLH1, MSH2 and MSH6 that are analyzed by sequence analysis do not extend more than 20 base pairs proximal to the 5′ end and 10 base pairs distal to the 3′ end of each exon. The MLH1 and MSH2 genes are tested for large rearrangements that are not detected by sequence analysis. All coding exons of MLH1 and MSH2 and their respective promoters are examined for evidence of deletions and duplications by quantitative multiplexed endpoint PCR analysis.
Statistical Analysis
We used the information on diagnoses of pancreatic cancer in relatives of probands to estimate age-specific pancreatic cancer incidences in MMR mutation carriers by maximum likelihood, using a technique called modified segregation analysis. The method was implemented in MENDEL (v3.3.5).17,18 Information on genotype in relatives was included whenever available. However, mutation status was unknown for many family members (Table 1). Despite missing genotypes, these individuals do contribute important information to the analysis. The segregation analysis implemented by Mendel automatically handles missing genotype information by maximizing the marginal likelihood by summing over all possible genotype configurations in a family.19 Relatives were assumed to be followed from 20 years of age and to be censored at the age at diagnosis of pancreatic cancer, at the age of death, at the age at last follow-up, or at age 70 years, whichever occurred earlier. For individuals with missing age information the age was imputed based on relationship with proband, age of proband, deceased status at last follow-up (dead or alive). We also carried out a sensitivity analysis without imputing the age information to ensure that the age imputation did not artificially inflate estimates of penetrance and relative risk.
Table 1.
Characteristics of study population and distribution of mutations by gene
| UMCC | DFCI | Total | |
|---|---|---|---|
| Number of probands* | 67 | 80 | 147 |
| Number of FDR | 459 | 558 | 1017 |
| Total number of individuals | 2660 | 3682 | 6342 |
| Number of males | 1395 | 1900 | 3295 |
| Number of females | 1265 | 1782 | 3047 |
| (Male: Female) | 1:1 | 1:1 | 1:1 |
| Number of subjects genotyped | 200 | 232 | 432 |
| Number of mutation positive subjects | 144 | 158 | 302 |
| Mutated Gene** | |||
| MLH1 | 18 (26.9) | 37 (46.3) | 55 (37.4) |
| MSH2 | 42 (62.7) | 39 (48.8) | 81 (55.1) |
| MSH6 | 7 (10.4) | 4 (5.0) | 11 (7.5) |
UMCC=University of Michigan Cancer Center, DFCI=Dana-Farber Cancer Institute, MMR= mismatch repair; FDR=first-degree relative.
proband=index patient per family presenting for genetic evaluation
n (% of total families with MMR gene mutations)
Our study included MMR carrier families ascertained through multiple individuals with CRC. Therefore the database potentially includes a greater representation of families with multiple CRC cases and mutation-positive probands than would be identified in population-based studies. Unless appropriate statistical methods are used, this type of ascertainment (a form of selection bias) can lead to overestimation of age-specific cumulative risks of pancreatic cancer. Using a conditional likelihood allows one to correct for ascertainment bias by modeling the probability of observed disease phenotypes and genotypes of the pedigrees conditional on ascertainment. This requires a model for the ascertainment probabilities, and we present results where a family was ascertained because of phenotype and genotype status of the proband and multiple affected first-degree relatives (FDRs) with CRC. This conditioning strategy was chosen based on the typical referral pattern of families to our cancer genetics clinics, emphasizing CRC as the primary reason for the referral. We also carried out additional sensitivity analyses with different alternative ascertainment mechanisms. Results are presented in the supplementary material and provided at: http://www.sph.umich.edu/bhramar/public_html/software/supplementary.doc.
The age-specific relative risks of pancreatic cancer in carriers were obtained using a proportional hazards model. We estimated the age specific log hazard ratio (HR) parameters for two age intervals <50 and ≥50 years. In all analyses, cancer incidences in noncarriers were assumed to follow the population cohort-specific rate as obtained through the Surveillance Epidemiology and End Results (SEER) 13 database (http://seer.cancer.gov). Cumulative risk (ie. penetrance) and 95% confidence intervals (CI) were calculated from the cumulative incidence. Details of the statistical methods are provided in the supplementary materials.
Results
A total of 6,342 individuals were included in the analysis: 147 probands, 1,017 FDRs, and 5,178 other relatives from the same side of the affected family with a greater than first-degree relation (Table 1). The corresponding proportions of families carrying a mutation in one of three MMR genes were: 55/147 (37.4%) in MLH1, 81/147 (55.1%) in MSH2, and 11/147 (7.5%) in MSH6. The distribution of gene-specific mutations among both institutions is also shown in Table 1. Overall, a pathogenic gene mutation was detected in 302/6342 (8.2%) individuals and the number of subjects genotyped were similar across the two centers (individuals genotyped: 200 at UMCC v 232 at DFCI). 130 of the 432 individuals genotyped did not have an identified MMR gene mutation.
Description of pancreatic cancer cases
Forty-seven cases of pancreatic cancer were reported among 31 families. Eighteen families reported one case of pancreatic cancer, 10 families reported two cases of pancreatic cancer, and three families reported three cases of pancreatic cancer. Thirty-one pancreatic cancer cases were reported in families with an MSH2 gene mutation, 13 cases were in families with an MLH1 mutation, and 3 with an MSH6 mutation. Of the 13 families with more than one case of pancreatic cancer, 62% (n=8) had a mutation in the MSH2 gene compared to 30.8% (n=4) and 7.7% (n=1) in the MLH1 and MSH6 genes, respectively.
Forty-five percent (21/47) of pancreatic cancer cases were reported among men, who had a median age of 51.5 years at time of diagnosis (range: 19–85 years). There was no gender-related difference in age of pancreatic cancer diagnosis: 51.5 years v. 56.5 years for men and women respectively. However, 50% of men with pancreatic cancer were less than 50 years old at time of diagnosis compared to only 22% of women diagnosed at age less than 50 years (Table 2).
Table 2.
Age of diagnosis for pancreatic cancer cases stratified by gender.
| Total | Men | Women | |||||
|---|---|---|---|---|---|---|---|
| Center | # Pancreatic Cancer cases | # of cases (# cases with known ages at diagnosis) | Proportion of cancers diagnosed < age 50* (%) | Median age at diagnosis (range)* | # of cases (# cases with known ages at diagnosis) | Proportion of cancers diagnosed < age 50* (%) | Median age at diagnosis (range)* |
|
|
|||||||
| UMCC | 23 | 13 (8) | 6/8 (75.0) | 42.5 (19–75) | 10 (6) | 1/6 (16.7) | 55 (36–73) |
| DFCI | 24 | 8 (8) | 2/8 (25.0) | 65 (40–85) | 16 (12) | 3/12 (25.0) | 62 (27–79) |
| Combined | 47 | 21 (16) | 8/8 (50.0) | 51.5 (19–85) | 26 (18) | 4/18 (22.2) | 56.5 (27–79) |
UMCC=University of Michigan Cancer Center, DFCI=Dana-Farber Cancer Institute
Calculated from cases with known ages at diagnosis of pancreatic cancer
Of the 47 cases of pancreatic cancer, we were able to verify diagnoses with additional records in 3/23 from UMCC, and 7/24 from DFCI. In 20/23 of UMCC cases and 17/24 of DFCI cases, the diagnosis was based on family report alone. Pathology review was available for 2/23 UMCC cases, and 4/24 from DFCI, whereas other documentation (clinical report, death certificate) was obtained in 1/23 UMCC cases and 3 of the DFCI families. We were unable to verify the remaining pancreatic cancer cases due to patient confidentiality issues. We did not have permission to contact the next-of-kin of the reported cases of pancreatic cancer who were often biologically unrelated to the proband.
Estimates of Age-specific Cumulative Risk and Hazard Ratios
Among the 147 families with identified pathogenic MMR gene mutations, approximately 4% were diagnosed with pancreatic cancer by the age of 70 years. The increase in risk was more pronounced after age 40 years. The estimated decade-specific cumulative risks of pancreatic cancer for carriers of any of the three MMR as compared to the general population are presented in Table 3 and Figure 1.
Table 3.
Age-specific cumulative risk of pancreatic cancer*
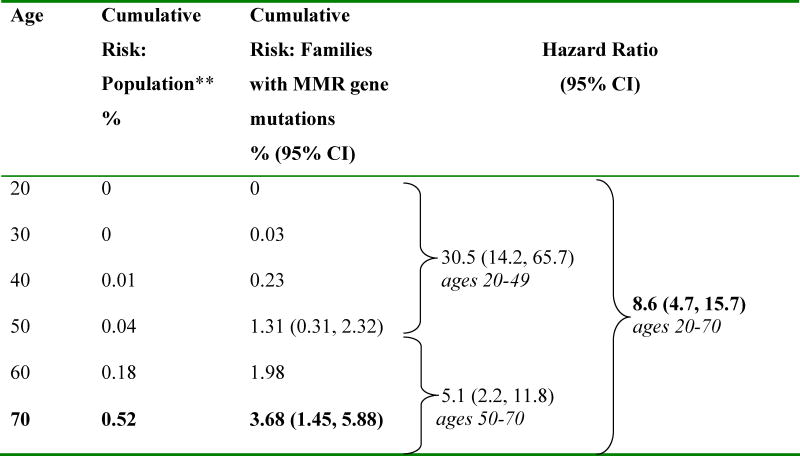
|
MMR= Mismatch repair, CI= confidence interval
Two age-specific hazard ratios in proportional hazards model (<50 years, ≥50 years), corrected for ascertainment by conditioning on genotype and phenotype of proband and phenotype of all colorectal cancer affected first-degree relatives
Surveillance Epidemiology and End Results 1992-2005 (http://seer.cancer.gov)
Figure 1. Age-specific cumulative risk of pancreatic cancer in families with pathogenic mutations in MLH1, MSH2 or MSH6 genes.
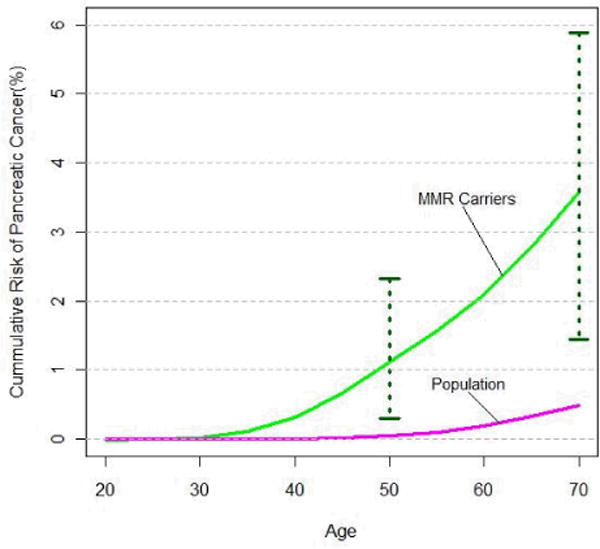
MMR Carriers=families with mismatch repair gene carriers (MLH1, MSH2 or MSH6)
The penetrance curves in Figure 1 were generated by plotting the age-specific cumulative risks of pancreatic cancer (as presented in Table 3) for a set of discrete ages from 20 to 70 years at five-year intervals and then applying a smoothing spline function. The 95% Confidence Intervals corresponding to the age-specific cumulative risk of pancreatic cancer for MMR carrier families were also plotted for ages 50 to 70 years. Population estimates of age-specific cumulative risks of pancreatic cancer are given by pancreatic cancer incidence rates reported in Surveillance Epidemiology and End Results (SEER) 13, from 1992–2005 (http://seer.cancer.gov).
There was a near 9-fold increase in risk of developing pancreatic cancer among families with pathogenic MMR gene mutations compared to the general population (HR 8.6, 95% CI: 4.7, 15.7). Table 3 also depicts the HRs for mutation carriers stratified by age (greater or less than 50 years). The estimated relative risk of pancreatic cancer was higher before age 50 years (HR 30.5 for ages 20–49 years, 95% CI: 14.2, 65.7) and then decreased with increasing age (HR 5.1 for ages 50–70 years, 95% CI: 2.2, 11.8). The absolute cumulative risk of developing pancreatic cancer in MMR gene mutation carriers at age 50 years was 1.31% (95% CI: 0.31%, 2.32%) and 3.68% (95% CI: 1.45%, 5.88%) at age 70 years. These risks are significantly higher than the population-based cumulative age-specific incidences as reported in SEER 13, which are 0.04% and 0.52% for ages 50 and 70 years respectively.
MLH1 and MSH2 mutation carriers had a similar increase in risk of developing pancreatic cancer compared to the general population. For carriers of mutations in MLH1, the overall HR was estimated at 7.5 (95% CI: 2.4, 23.0) compared to 10.9 (95% CI: 5.5, 21.9) for MSH2 carriers. Given the small number of pancreatic cancer cases among MSH6 carriers, risk estimates were not calculated for these mutation carriers. The hazard ratios were obtained from a proportional hazard model with a single log(HR) parameter across all ages. The two parameter model could not be fitted due to lack of data strength in each gene category.
Discussion
Among 147 families with germline MMR gene mutations, we found that the risk of developing pancreatic cancer was increased compared to the general population. The cumulative risk of developing pancreatic cancer was 3.68% by age 70 years, with cases among Lynch syndrome families occurring at an earlier age than sporadic cases.
The statistical methods used in this study afford many advantages.20, 21 Segregation analysis accounts for relatives who have undergone mutation analysis and those who have not. The probability of being a mutation carrier is calculated for all relatives whose mutation status is unknown and used to estimate the overall cumulative risk of cancer among family members with a germline alteration. Segregation analysis also minimizes ascertainment bias by conditioning the analysis on available phenotypic information provided for individual pedigrees. We chose a priori, to exclude from the risk estimate calculation all pancreatic cancers in probands and their FDRs with CRC. This yields a conservative estimate for pancreatic cancer risk and best corrects for how patients were ascertained at both centers. Data from the two registries provides a large sample of families who have undergone mutation analysis with identified MMR gene mutations.
Henry Lynch first reported pancreatic cancer in adenocarcinoma-prone families over 40 years ago22 and additional reports have described Lynch Syndrome families with pancreatic cancer.11–16 Pancreaticobiliary cancers are often included in the spectrum of Lynch Syndrome-associated malignancies, but data on the prevalence and risk of developing pancreatic cancer has been conflicting. A limitation of most existing data is that risk estimates were derived from families ascertained for a strong history limited to CRC.11,12,14 Additionally, studies that have not found an increased risk of pancreatic cancer were from homogeneous populations that have a preponderance of founder mutations and possibly a limited spectrum of tumors.12
However, emerging data support that pancreatic tumors are a component of Lynch Syndrome. Medullary carcinomas of the pancreas are a distinct variant of pancreatic adenocarcinoma associated with microsatellite instability (MSI), loss of protein expression of MMR genes, family history of cancer in a FDR and germline MMR gene mutation.23,24 We recently reported on a known MSH2 gene mutation carrier who developed a intraductal papillary mucinous neoplasm (IPMN) of the pancreas; the lesion showed MSI and loss of expression of MSH2 and MSH6 proteins.25 A number of small studies suggest that MSI in sporadic pancreatic cancers offers a survival benefit similar to that observed in other Lynch Syndrome tumors. To determine if long-term survival in pancreatic cancer was attributed to defective MMR, Maple et al. ascertained its prevalence in pancreatic cancer patients who survived 3 or more years after surgery.26 The data suggest that immunohistochemistry (IHC) for pancreatic cancer is both sensitive and specific for the MSI phenotype. A study of 130 families with MMR mutations reported 22 pancreatic cancers with many early-onset cases;27 however, lifetime risks of developing pancreatic cancer were not calculated.
Our study has several limitations. As in most Lynch Syndrome registries our families were ascertained through an affected proband with a classic Lynch Syndrome tumor, notably CRC. To reduce ascertainment bias, our analysis of pancreatic cancer cases specifically excluded these probands, as well as any FDRs who also had CRC. We also relied in large part on the probands’ report of pancreatic cancers in their families which may be inaccurate.28–31 The majority of pancreatic cancer patients were deceased and not available for mutation analysis. Therefore it is not possible to accurately determine those cases that may have been sporadic and whose inclusion in the analysis may have overestimated risk. Recall bias may also occur when family members without a personal history of cancer are under-reported by the proband. Although family structure was completely enumerated for a three-generation pedigree at both sites by certified genetic counselors during the retrospective review and construction of the pedigrees, it is possible the selective expansion of branches of the family with cancer might lead to this type of recall bias. In our study, this bias may be present but is likely minimal for two reasons. First, pedigree structures are routinely verified by multiple relatives who undergo genetic evaluation, increasing the chances of completely enumerating all affected and unaffected members. Second, since the majority of relatives with pancreatic cancer were identified in FDRs and second-degree relatives, the standardized construction of three-generation pedigrees is likely to reduce the magnitude of this potential recall bias. Another potential limitation reflects our choice to impute missing ages. However, this was done conservatively and is unlikely to inflate risk as shown in the supplemental sensitivity analyses. Despite these potential limitations, the elevated risk of pancreatic cancer was similar at both study sites, lending credence to the results.
Our findings have implications regarding the care of patients and families with a known MMR gene mutation. Information on cancer risk is important in planning cancer prevention and determining the efficacy of proposed prevention strategies.32–38 Several screening trials aimed at identifying early pancreatic neoplasia through radiographic and endoscopic imaging are currently underway in patients with genetic syndromes associated with high incidence of pancreatic cancer.32,33 MMR gene mutation carriers with a family history of pancreatic cancer may need to be screened in a similar manner to these individuals. Ongoing screening programs will provide information on the risks and benefits of early detection of pancreatic neoplasms, and allow further study on the spectrum of pancreatic lesions in Lynch Syndrome families.
In summary, families with an identified pathogenic MMR gene mutation have an increased lifetime risk of developing pancreatic compared to the general U.S. population. Further studies are necessary in individuals with Lynch Syndrome to further define the risk of pre-malignant and malignant pancreatic neoplasms and the potential benefits and limitations of surveillance. Pancreatic cancer is a clinically relevant component of Lynch Syndrome and quantifying this risk for gene carriers should be incorporated into clinical management.
Supplementary Material
Acknowledgments
Sources of funding and support: This work was supported in part by the National Cancer Institute (RO1 CA81488, and K24 CA 113433), the University of Michigan’s Cancer Center Support Grant (5 P30 CA46592). The funding organizations of this study had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; and preparation, review, or approval of the manuscript.
Other: We also gratefully acknowledge technical advice in the implementation of modified segregation analysis from Antonis Antoniou, PhD, Cancer Research UK Genetic Epidemiology Unit, Department of Public Health and Primary Care, University of Cambridge, United Kingdom and Mark Jenkins, PhD, Centre for Molecular, Environmental, Genetic and Analytic Epidemiology, Department of Pathology, The University of Melbourne, Australia. Drs. Antoniou and Jenkins have not been compensated for their contributions.
Footnotes
Author Contributions: Dr. Gruber had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. Drs. Kastrinos and Mukherjee contributed equally to this work. Drs. Syngal and Gruber contributed equally to this work.
(1) Study concept and design: Drs. Kastrinos, Mukherjee, Stoffel, Gruber, Syngal; Acquisition of data: Drs. Kastrinos, Mukherjee, Sparr, Bandipalliam, Stoffel, Gruber, Syngal, Victoria Raymond; Analysis and interpretation of data: Drs. Kastrinos, Mukherjee, Stoffel, Gruber, Syngal, Nabihah Tayob, Fei Wang
(2) Drafting of the manuscript: Drs. Kastrinos, Mukherjee, Sparr, Bandipalliam, Stoffel, Gruber, Syngal, Nabihah Tayob, Fei Wang, Victoria Raymond; Critical revision of the manuscript for important intellectual content: Drs. Kastrinos, Mukherjee, Gruber, Syngal
(3) Final approval for the version to be published: Drs. Kastrinos, Mukherjee, Sparr, Bandipalliam, Stoffel, Gruber, Syngal, Nabihah Tayob, Fei Wang, Victoria Raymond
Financial disclosures: Dr. Syngal served as an external advisor to Myriad Genetics Laboratories, Inc. (2007); Dr. Gruber served as an external advisor to Myriad Genetics Laboratories, Inc. (2008).
Disclosure of potential conflicts of interest: There are no conflicts of interest to report.
References
- 1.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, et al. Cancer statistics, 2008. CA Cancer J Clin. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 2.Lynch HT, Smyrk T, Kern SE, et al. Familial pancreatic cancer: a review. Semin Oncol. 1996;23:251–75. [PubMed] [Google Scholar]
- 3.Brand RE, Lynch HT. Hereditary pancreatic adenocarcinoma: a clinical perspective. Med Clin North Am. 2000;84:665–75. doi: 10.1016/s0025-7125(05)70249-2. [DOI] [PubMed] [Google Scholar]
- 4.Giardiello FM, Welsh SB, Hamilton SR, et al. Increased risk of cancer in the Peutz-Jeghers syndrome. N Engl J Med. 1987;316:1511–4. doi: 10.1056/NEJM198706113162404. [DOI] [PubMed] [Google Scholar]
- 5.Giardiello FM, Brensinger JD, Tersmette AC, et al. Very high risk of cancer in familial Peutz-Jeghers syndrome. Gastroenterology. 2000;119:1447–53. doi: 10.1053/gast.2000.20228. [DOI] [PubMed] [Google Scholar]
- 6.Vasen HFA, Gruis NA, Frants RR, van der Velden PA, Hille ETM, Bergman W. Risk of developing pancreatic cancer in families with familial atypical multiple mole melanoma associated with a specific 19 deletion of p16 (p16-Leiden) Int J Cancer. 2000;87:809–11. [PubMed] [Google Scholar]
- 7.Thompson D, Easton DF, for the Breast Cancer Linkage Consortium Cancer incidence in BRCA1 mutation carriers. J Natl Cancer Inst. 2002;94:1358–65. doi: 10.1093/jnci/94.18.1358. [DOI] [PubMed] [Google Scholar]
- 8.The Breast Cancer Linkage Consortium. Cancer risks in BRCA2 mutation carriers. J Natl Cancer Inst. 1999;91:1310–6. doi: 10.1093/jnci/91.15.1310. [DOI] [PubMed] [Google Scholar]
- 9.Lowenfels AB, Maisonneuve P, DiMagno EP, et al. Hereditary pancreatitis and the risk of pancreatic cancer. J Natl Cancer Inst. 1997;89:442–6. doi: 10.1093/jnci/89.6.442. [DOI] [PubMed] [Google Scholar]
- 10.Jones S, Hruban RH, Kamiyama M, et al. Exomic sequencing identifies PALB2 as a pancreatic cancer susceptibility gene. Science. 2009;324:217. doi: 10.1126/science.1171202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Watson P, Lynch HT. Extracolonic cancer in hereditary nonpolyposis colorectal cancer. Cancer. 1993;71:677–85. doi: 10.1002/1097-0142(19930201)71:3<677::aid-cncr2820710305>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 12.Aarnio M, Sankila R, Pukkala E, et al. Cancer risk in mutation carriers of DNA-mismatch-repair genes. Int J Cancer. 1999;81:214–8. doi: 10.1002/(sici)1097-0215(19990412)81:2<214::aid-ijc8>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 13.Vasen HFA, Stomorken A, Menko FH, et al. MSH2 mutation carriers are at higher risk of cancer than MLH1 mutation carriers: a study of hereditary nonpolyposis colorectal cancer families. J Clin Oncol. 2001;19:4074–80. doi: 10.1200/JCO.2001.19.20.4074. [DOI] [PubMed] [Google Scholar]
- 14.Goecke T, Schulmann K, Engel C, et al. Genotype-Phenotype comparison of German MLH1 and MSH2 mutation carriers clinically affected with Lynch Syndrome: a report by the German HNPCC Consortium. J Clin Oncol. 2006;24:1–8. doi: 10.1200/JCO.2005.03.7333. [DOI] [PubMed] [Google Scholar]
- 15.Aarnio M, Mecklin JP, Aaltonen LA, Nyström-Lahti M, Järvinen HJ. Life-time risk of different cancers in hereditary non-polyposis colorectal cancer (HNPCC) syndrome. Int J Cancer. 1995;64:430–3. doi: 10.1002/ijc.2910640613. [DOI] [PubMed] [Google Scholar]
- 16.Watson P, Vasen HFA, Mecklin JP, et al. The risk of extra-colonic, extra-endometrial cancer in the Lynch syndrome. Int J Cancer. 2008;123:444–9. doi: 10.1002/ijc.23508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lange K, Weeks D, Boehnke M. Programs for Pedigree Analysis: MENDEL, FISHER, and dGENE. Genet Epidemiol. 1988;5:471–2. doi: 10.1002/gepi.1370050611. [DOI] [PubMed] [Google Scholar]
- 18.Antoniou A, Pharoah PD, Narod S, et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet. 2003;72:1117–1130. doi: 10.1086/375033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Elston RC, Stewart J. A general model for the genetic analysis of pedigree data. Hum Hered. 1971;21:523–542. doi: 10.1159/000152448. [DOI] [PubMed] [Google Scholar]
- 20.Jenkins MA, Baglietto L, Dowety JG, et al. Cancer Risks for Mismatch Repair Gene Mutation Carriers: A Population-Based Early Onset Case-Family Study. Clin Gastroenterol Hepatol. 2006;4:489–508. doi: 10.1016/j.cgh.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 21.Quehenberger F, Vasen HFA, van Houwelingen HC. Risk of colorectal and endometrial cancer for carriers of mutations of the hMLH1 and hMSH2 gene: correction for ascertainment. J Med Genet. 2005;42:491–6. doi: 10.1136/jmg.2004.024299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lynch HT, Krush AJ, Larsen AL. Hereditary and multiple primary malignant neoplasms: six cancer families. Am J Med Sci. 1967;254:322–9. doi: 10.1097/00000441-196709000-00007. [DOI] [PubMed] [Google Scholar]
- 23.Banville N, Geraghty R, Fox E, et al. Medullary carcinoma of the pancreas in a man with hereditary nonpolyposis colorectal cancer due to a mutation of the MSH2 mismatch repair gene. Hum Pathol. 2006;37:1498–1502. doi: 10.1016/j.humpath.2006.06.024. [DOI] [PubMed] [Google Scholar]
- 24.Wilentz RE, Goggins M, Redston M, et al. Genetic immunohistochemical and clinical features of medullary carcinoma of the pancreas: A newly described and characterized entity. Am J Pathol. 2000;156:1641–51. doi: 10.1016/S0002-9440(10)65035-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sparr JA, Bandipalliam P, Redston MS, Syngal S. Intraductal papillary mucinous neoplasm of the pancreas with loss of mismatch repair in a patient with Lynch syndrome. Am J Surg Pathol. 2009;33:309–12. doi: 10.1097/PAS.0b013e3181882c3d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maple JT, Smyrk TC, Boardman LA, Johnson RZ, Thibodeau SN, Chari ST. Defective DNA Mismatch Repair in Long-term (>3 Years) Survivors wit Pancreatic Cancer. Pancreatology. 2005;5:220–8. doi: 10.1159/000085275. [DOI] [PubMed] [Google Scholar]
- 27.Geary J, Sasieni P, Houlston R, et al. Gene-related cancer spectrum in families with hereditary non-polyposis colorectal cancer (HNPCC) Familial Cancer. 2008;7:163–72. doi: 10.1007/s10689-007-9164-6. [DOI] [PubMed] [Google Scholar]
- 28.Murff HJ, Spigel DR, Syngal S. Does this patient have a family history of cancer? An evidence-based analysis of the accuracy of family cancer history. JAMA. 2004;292:1480–9. doi: 10.1001/jama.292.12.1480. [DOI] [PubMed] [Google Scholar]
- 29.Douglas FS, O’Dair LC, Robinson M, Evans DG, Lynch SA. The accuracy of diagnoses as reported in families with cancer: a retrospective study. J Med Genet. 1999;36:309–12. [PMC free article] [PubMed] [Google Scholar]
- 30.Love RR, Evans AM, Josten DM. The accuracy of patient reports of a family history of cancer. J Chronic Dis. 1985;38:289–93. doi: 10.1016/0021-9681(85)90074-8. [DOI] [PubMed] [Google Scholar]
- 31.Sijmons RH, Boonstra AE, Reefhuis J, et al. Accuracy of family history of cancer: clinical genetic implications. Eur J Hum Genet. 2000;8:181–6. doi: 10.1038/sj.ejhg.5200441. [DOI] [PubMed] [Google Scholar]
- 32.Brentall TA, Bronner MP, Byrd DR, Haggitt RC, Kimmey MB. Early diagnosis and treatment of pancreatic dysplasia in patients with a family history of pancreatic cancer. Ann Intern Med. 1999;131:247–55. doi: 10.7326/0003-4819-131-4-199908170-00003. [DOI] [PubMed] [Google Scholar]
- 33.Canto MI, Goggins M, Yeo CJ, et al. Screening for pancreatic neoplasia in high-risk individuals: an EUS-based approach. Clin Gastroenterol Hepatol. 2004;2:606–21. doi: 10.1016/s1542-3565(04)00244-7. [DOI] [PubMed] [Google Scholar]
- 34.Rulyak SJ, Brentall TA. Inherited pancreatic cancer: surveillance and treatment strategies for affected families. Pancreatology. 2001;1:477–85. doi: 10.1159/000055851. [DOI] [PubMed] [Google Scholar]
- 35.Canto MI, Goggins M, Hruban R, et al. Screening for early pancreatic neoplasia in high-risk individuals: A prospective controlled study. Clin Gastroenterol Hepatol. 2006;4:766–81. doi: 10.1016/j.cgh.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 36.Petersen GM, de Andrade M, Goggins M, et al. Pancreatic Cancer Gene Epidemiology Consortium. Cancer Epidemiol Biomarkers Prev. 2006;15:704–10. doi: 10.1158/1055-9965.EPI-05-0734. [DOI] [PubMed] [Google Scholar]
- 37.Hruban RH, Canto MI, Yeo CJ. Prevention of pancreatic cancer and strategies for management of familial pancreatic cancer. Dig Dis. 2001;19:76–84. doi: 10.1159/000050656. [DOI] [PubMed] [Google Scholar]
- 38.Wang W, Chen S, Brune KA, Hruban RH, Parmigiani G, Klein AP. PancPRO: Risk Assessment for Individuals With a Family History of Pancreatic Cancer. J Clin Oncol. 2007;25:1417–22. doi: 10.1200/JCO.2006.09.2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.