

Abstract
Along with other resonance energy transfer techniques, bioluminescence resonance energy transfer (BRET) has emerged as an important method for demonstrating protein–protein interactions in cells. In the field of G protein-coupled receptors, including chemokine receptors, BRET has been widely used to investigate homo- and heterodimerization, a feature of their interactions that is emerging as integral to function and regulation. While demonstrating the existence of dimers for a given receptor proved to be fairly straightforward, quantitative comparisons of different receptors or mutants are nontrivial because of inevitable variations in the expression of receptor constructs. The uncontrollable parameters of the cellular expression machinery make amounts of transfected DNA extremely poor predictors for the expression levels of BRET donor and acceptor receptor constructs, even in relative terms. In this chapter, we show that properly accounting for receptor expression levels is critical for quantitative interpretation of BRET data. We also provide a comprehensive account of expected responses in all types of BRET experiments and propose a framework for uniform and accurate quantitative treatment of these responses. The framework allows analysis of both homodimer and heterodimer BRET data. The important caveats and obstacles for quantitative treatment are outlined, and the utility of the approach is illustrated by its application to the homodimerization of wild-type (WT) and mutant forms of the chemokine receptor CXCR4.
Keywords: G-protein coupled receptor (GPCR), chemokine receptor, CXCR4, dimerization, monomer-dimer equilibrium, bioluminescence resonance energy transfer (BRET), BRET titration, BRET saturation
1. Introduction
Although it was originally assumed and corroborated by recent in vitro studies [1, 2], that some G protein-coupled receptors (GPCRs) can function as monomers, there is now substantial evidence that many GPCRs homo- and hetero-dimerize. Further, it has been suggested that the dimer may be the minimal functional unit [3–6]. Chemokine receptors, the focus of this volume, are no exception. One of the first clues that chemokine receptors oligomerize came from the discovery of a CCR5-Δ32 mutation [7]. CCR5 is one of the two main receptors involved in HIV entry into cells during the initial infectious phase of the disease, and it was found that individuals homozygous for the mutant were resistant to infection due to retention of the mutated receptor in the endoplasmic reticulum [8]. The fact that individuals heterozygous for CCR5-Δ32 also show delayed progression was then hypothesized to be caused by oligomerization of WT CCR5 with CCR5-Δ32, resulting in abnormal trafficking of the WT receptor to the cell surface. These data led to the notion that CCR5 might function as dimer at least in some contexts, which is now well-established [8–10]. Similar phenotypic evidence for CXCR4 dimerization came from studies of the “warts, hypogammaglobulinemia, infections and myelokathexis” (WHIM) syndrome which is an immunodeficiency caused by truncation of the receptor C-terminus that results in resistance to desensitization and internalization, and thereby enhanced signaling [11, 12]. Co-expression of WT CXCR4 with WHIM CXCR4 also leads to enhanced signaling and failure of the WT receptor to internalize upon stimulation with CXCL12, and this observation has been attributed to the ability of WT CXCR4 to dimerize with the WHIM variant [13, 14].
To date, many chemokine receptors have been shown to form homo- and hetero- dimers, not only with other chemokine receptors but with GPCRs outside of the chemokine family [15, 16]. The functional consequences of these interactions have yet to be fully understood but include modulation of signaling responses such as transinhibition in ligand binding [17–20], as well changes in G protein coupling [10, 21]. Furthermore, the nature of the dimerization interfaces, the stability of the various oligomeric forms, the effects of the ligands on dimer equilibrium, conformation, and stability, and the diversity and plasticity of dimerization, are even less well understood [22–30]. For example, all five of the crystal structures of CXCR4 complexed with a small molecule antagonist or a cyclic peptide inhibitor revealed the same dimer interface involving helices V and VI [31]. Similarly the recent structure of the μ-opioid receptor bound to an irreversible morphinan antagonist revealed a dimer stabilized by a four helix bundle between helices V and VI [32], while the κ-opioid receptor bound to antagonist showed a dimer stabilized through helices I, II and VIII [33]. Nevertheless, it is not clear whether these dimer interfaces are biologically relevant interfaces or artifacts of crystallization (Figure 1), and thus biochemical approaches are needed to complement the structural studies [23, 30, 34–39]. Furthermore, higher order oligomers or array-like assemblies have been observed for some GPCRs in cryo-EM studies suggesting the existence of more than one oligomerization interface on the surface of a particular GPCR. On the other hand, studies at physiological levels of receptor expression [27, 40] only convincingly corroborate the dimer, but not the higher oligomer theory.
Figure 1.

Parallel GPCR dimer configurations observed by X-ray crystallography. The grey tubes in the middle represent a superposition of GPCR monomers from multiple X-ray structures while the peripheral blobs illustrate the orientation of their crystallographic dimer partners. The structures are viewed from the extracellular side across the membrane. In different structures, the crystallographic dimer interfaces involve helices 1 and 2, 1 and 7, 4 and 5, 5 and 6, or 3 and 4. For some receptors like CXCR4, bRho and S1PR1, more than one dimer configuration was observed. Several interface hypotheses are partially supported by biochemical studies [38, 39]. The methods described in this chapter will allow a quantitative assessment of dimer interface mutants by BRET.
In an attempt to address the relevance of the dimer observed in the crystal structures of CXCR4, we sought to introduce computationally designed mutations at strategic positions in the interfacial helices and to determine their effects on dimer stability using bioluminescence energy transfer (BRET) assay. BRET is a biophysical method that has emerged as one of the main approaches for studying protein-protein interactions in cells [3, 41–49]. This exercise revealed some limitations in the quantification of the data that is relevant to the analysis of all GPCRs, specifically when one wants to compare the relative dimerization propensity of different receptors or different mutants of the same receptor. Because BRET is a cell-based assay, a researcher has only a limited control of its input parameters (amounts of DNA for control of receptor expression), as the actual expression levels are inevitably affected by the cell machinery which in turn influences the assay readouts. Failure to take these effects into consideration may lead to misinterpretation of the assay results. Below we present the problem in more detail and propose an approach to its quantitative treatment. An analysis of both homodimer and heterodimer BRET data is presented.
2. Materials
2.1. Equipment
A 96-well plate reader or fluorimeter capable of excitation at 480 nm and detection of emission at 530 nm, or a luminometer with excitation capabilities as in 2 below. The authors use a SpectraMax M5 Molecular Devices plate reader.
A luminometer capable of detecting emission at 480 nm and 530 nm as well as unfiltered total emission. The authors use a Perkin Elmer VICTOR X Light 2030. If the luminometer is also capable of excitation 480 nm, the fluorescent plate reader or fluorimeter are not necessary; one example of a luminometer with this capability is the Perkin Elmer VICTOR X2 2030 Multilabel Plate Reader. See Note 1.
Instrumentation required for routine tissue culture research (biosafety cabinet, microscope, cell counter, centrifuge, water bath, etc).
2.2. Reagents
Appropriate growth media for the experimental cells to be used in BRET experiments.
Sterile 1 mL, 200 μL, and 20 μL micropipettes and sterile pipette tips for all three.
Any type of sterile 6-well plates commonly used for growing adherent cultured cells. (See Note 2 for discussion of alternatives).
Sterile 0.5ml–1.5ml DNA lo-bind tubes for preparation of transfection mixtures.
1.5 mL tubes for containment of cells after resuspension from the transfection plates and prior to plating in the BRET assay plate (see Note 3).
96-well BioCoat/OptiLux plates (see Note 4).
A repeating pipette capable of dispensing 10 μL volumes, with tips (see Note 5).
Transfection reagent capable of efficiently transfecting the experimental cells to be used in BRET experiments (see Note 6).
Transfection media according to the transfection protocol; for example sterile serum-free Opti-MEM media (Gibco).
Plasmid DNA encoding the proteins of interest fused to YFP and Rluc, plasmid DNA encoding the fused Rluc-YFP construct, and empty vector plasmid DNA (see Note 7).
Phosphate buffered saline (PBS): 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4, pH 7.4.
PBS with 1–5mM EDTA for detachment of adherent cells.
BRET buffer: PBS with 0.1% D-glucose.
Coelenterazine-h, 1mM in ethanol (stock concentration, working solution will be diluted just before use in the BRET experiment, see Notes 8 and 9).
2.3 Software
Data analysis software with non-linear regression capabilities and graphing functionality, e.g. ICM (MolSoft), MatLab (MathWorks), or Prism (GraphPad).
3. Methods
3.1 General description of the BRET assay for dimerization
Bioluminescence resonance energy transfer is an experimental technique for detection of spatial proximity of proteins in living cells. The proteins under question must be expressed in cells as fusion constructs with a bioluminescent donor protein and a fluorescent acceptor protein that is excited at the emission wavelength of the donor. In the process of substrate oxidation, the bioluminescent donor protein releases photons that are detected “as is” by the luminometer when the donor is in isolation. However, when appropriate acceptors are present in close proximity (50–100 Å) to the donor protein molecules, the acceptors are excited into a higher energy state and subsequently emit the photons by fluorescence at a longer wavelength. One of the widely used donor/acceptor pairs is Renilla reniformis luciferase (Rluc, max emission at 480 nm) and a genetic mutant of Aequorea victoria green fluorescent protein, yellow fluorescent protein (YFP, excitation at 480 nm, emission at 530 nm) (Figure 2). The experiment is initiated by adding a fixed amount of Rluc substrate (e.g. coelenterazine-h) to the cells co-expressing the donor/acceptor pair.
Figure 2.

The principle of the BRET experiment. (A) When the two proteins of interest do not dimerize, the luciferase emission spectrum is unchanged and the background ratio of emission intensities at 530 nm and 480 nm is observed. (B) When the proteins interact, the proximity of the Rluc and YFP molecules results in resonance energy transfer and a change in the shape of the emission spectrum with a reduction in the emission at 480 and an increase at 530nm. The resulting combined emission spectrum is characterized by a larger ratio of emission intensities at 530 nm and 480 nm, with the degree of increase being indicative of the extent of energy transfer. The degree of transfer, in turn, is affected by several parameters discussed in text.
BRET has the advantage over fluorescence resonance energy transfer (FRET) methods because it eliminates artifacts associated with photobleaching and thus allows measurements in real time over an extended period relative to FRET [50]. Because it relies upon the addition of exogenous substrate for donor emission, problems associated with sample autofluorescence as well as direct excitation of the acceptor are minimized, leading to higher signal-to-noise ratios. Cell damage from the excitation light is also minimized [51]. Unlike fluorescent techniques involving chemical labeling (e.g. [52, 53]), BRET is free of artifacts associated with labeling efficiency variations. However, BRET is prone to similar artifacts due to the presence of endogenous untagged receptor (see Section 3.4.5). Also, the presence of a bulky C-terminal fusion protein may and sometimes does affect interaction of the GPCR with downstream signaling partners and effectively alters its dynamics and membrane trafficking [54, 55] (see Note 10).
The energy transfer is quantified as the ratio of emission intensities at the acceptor emission wavelength (530 nm for YFP) and the donor emission wavelength (480 nm for Rluc). This ratio, referred to as BRET signal or BRET ratio, is but a simple numerical characteristic of the shape of the combined emission spectrum of the sample. In the absence of acceptors, this ratio has a certain background value (~0.34, Figure 2A); however the presence of acceptor molecules in energy transfer proximity of the donors changes the spectrum shape, shifting it towards longer wavelengths, thereby increasing the BRET ratio (Figure 2B). Quite importantly, the shape of the combined emission spectrum appears independent of the total emission intensity (experimentally, we observe only a small variation in the BRET ratios across a wide range of luciferase emission intensities). Consequently, the BRET signal is relatively stable over time, sometimes for as long as 1 hour, despite the fact that the absolute luminescence is subject to exponential decay as the substrate is used up.
Background BRET (BRETbkg) is the BRET ratio characterizing Rluc emission in the absence of YFP (see Note 11). Net BRET (BRETnet) is calculated as the difference between BRET and BRETbkg. The maximal achievable value of BRET, BRETmax, is defined as BRETnet in the situation when all available donor molecules are paired up with acceptor molecules. In the context of GPCR homodimerization, BRETmax depends on the donor/acceptor distance and relative orientation within the dimer. The concept of BRETmax intrinsically assumes that donors do not dimerize, which is not the case in GPCR dimerization studies; however, the relative fraction of donor-donor dimers and their contribution to the total BRET signal decreases with increasing acceptor/donor ratio. In the context of heterodimerization, apart from the standard dependencies of signal intensity on donor-acceptor distance and orientation, BRETmax is also affected by homo- vs hetero-dimerization preferences of the donors as well as their cellular distribution and availability to form dimers with acceptors (Figure 3); if donors preferentially dimerize with each other, or if they are present in some cellular compartments with no acceptors, their unchanged emission will substantially lower the overall observed BRET signal. Insufficient expression of the acceptors or a strong preference for acceptor homodimerization over heterodimerization with donors does not change the value of BRETmax in theory; however, these issues may result in the inability of the system to reach BRETmax in an experiment. In some cases, switching the donor/acceptor pair can help to clarify the true reasons behind the lowered BRETmax in heterodimerization studies.
Figure 3.
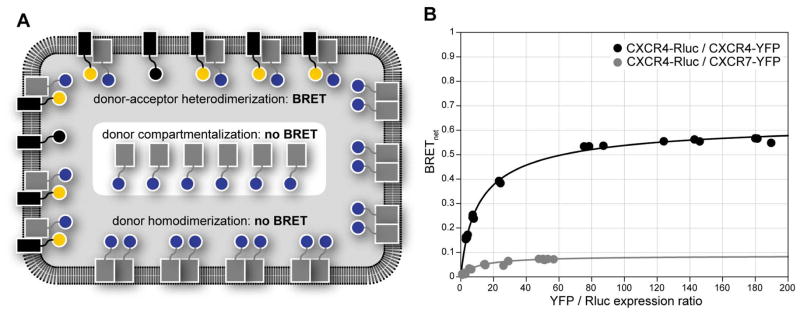
Specifics of the BRET assay for heterodimerization studies. (A) A schematic illustration of some possible reasons for lower BRETmax in a heterodimerization assay. Reasons include compartmentalization of the Rluc donor-tagged receptor, so that it is sequestered from the YFP acceptor-tagged receptor and therefore unavailable for interaction, and a preference of the donor-tagged receptor for homodimerization, which may again render the donor-tagged receptor unavailable for interaction with the acceptor-tagged receptor. While acceptor homodimerization may also have detrimental impact on the BRET signal intensity, in practice it is addressed by simply increasing the acceptor expression levels. (B) BRET titration curves for the CXCR4/CXCR7 heterodimer compared to the CXCR4 homodimer. The much smaller curve for the heterodimer likely result from one or both of the reasons mentioned above.
3.2 BRET transfection schemes and titration curves
One of the main problems with the BRET technique is that it measures spatial proximity of the molecules under study rather than their physical interaction. In experimental setups where the proteins are over-expressed in living cells, it is possible that non-zero BRETnet results from simple crowding of the proteins on the cell surface, without direct interaction between them – the so-called “bystander” BRET. Consequently, non-zero BRET in a single sample cannot be unambiguously interpreted as evidence of protein dimerization. With a goal of distinguishing the non-specific “bystander” effects from true dimerization, a rigorous BRET experiment involves a number of cell samples (7–12) transfected with varying amounts of donor- and acceptor-fused receptors followed by analysis of the resulting titration curves. Three schemes have been suggested for BRET saturation experiments [51, 56, 57] (Figure 4A):
Figure 4.
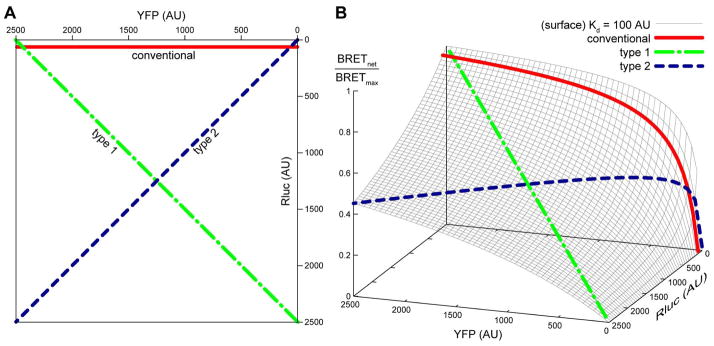
Commonly used transfection schemes in BRET titration experiments. In a conventional scheme, the Rluc expression is held constant between the samples while YFP is varied. Type 1 and type 2 experiments require maintaining either total Rluc + YFP density (type 1) or YFP/Rluc ratio (type 2) constant, while varying the other parameter (YFP/Rluc ratio for type 1 and total density for type 2). (A) A 2D representation of the assay schemes on the Rluc vs YFP plane. (B) A projection of the three BRET transfection schemes onto a 3D BRET response surface. The surface illustrates how BRET response in a system with a given dimer equilibrium dissociation constant Kd depends simultaneously on the surface densities of Rluc- and YFP-tagged receptors. Notice that a typical BRET experiment following any of the three schemes corresponds to a 2D slice of the 3D BRET response surface.
The “conventional” scheme involves sample transfection with a constant amount of donor-fused DNA and increasing amounts of acceptor-fused DNA in order to achieve variation in the acceptor/donor expression ratio. The DNA ratio is varied from 1 to as much as 250, where the higher ratio is required to fully saturate all donor molecules and obtain an accurate measurement of BRETmax. To avoid inter-sample variation due to transfection efficiency, it is important to complement the donor and acceptor DNA in all samples with empty vector for a constant total amount of DNA in each sample. Typical amounts of DNA are 0.01–0.1 μg of Rluc-tagged receptor DNA per well of a 6 well plate and up to 2.5–4 μg of YFP-tagged construct. However, as noted below, the relative expression levels of each construct must be evaluated and taken into account since expression levels are not controllable.
In the so-called “type 1” scheme, the total amount of donor + acceptor DNA is maintained constant between the samples, while the relative fraction of donors is varied. The advantage of this scheme is that, as long as the donor-tagged and acceptor-tagged receptors express equally well, any non-specific bystander effects will be equally represented in all cell samples. The decision about the optimal total DNA amount in the type 1 scheme must be made based on the signal-to-noise ratio and the goals of the experiment; in some experiments it is preferable to remain at physiological expression levels while other applications require overexpression.
In the “type 2” scheme, also sometimes described as “BRET dilution assay” [46], the acceptor/donor ratio is kept constant while varying the total amount of DNA. This allows one to study changes due to the increase in surface density. Optimal amounts of transfected DNA are calculated with a goal of sampling a wide range of expression levels: from as low as possible to get detectable expression to as high as the cells can handle without significant reduction in viability.
Additionally, a different approach to BRET titration involves competition between acceptor-tagged and untagged receptors (BRET competition experiments [43, 45, 46]), rather than saturation of donors with acceptors.
The specific BRET signal (i.e. resulting from dimer formation) is representative of two aspects of the system under study, namely: (i) the distance and relative orientation of donor and acceptor when the dimer is formed, and (ii) the fraction of donor molecules that are in dimers with acceptor molecules at the given surface densities of donors and acceptors. The experimental schemes outlined above are aimed towards controlling the latter aspect in a way that allows unambiguous analysis and interpretation of results.
3.3 A typical BRET assay
A detailed description of the BRET experimental protocol is provided elsewhere (see, for example, [47–49]). In the present protocol we outline the steps of the assay as implemented in our hands with attention given to some caveats and useful intermediate readouts. To begin the experiment, one plans a desired transfection scheme, computing the appropriate number of transfection samples, and determining positive and negative controls. Note that the protocol below is designed for measuring BRET with adherent cells that are in suspension during the assay. See Note 12 for an alternative adherent format. BRET can also be performed on permeabilized cells, cell fractions, and partially purified receptors as described in [44].
3.3.1 Day 1: seeding
Seed cells in 6-well plates (see Note 2 for discussion of alternative plates) in appropriate growth media. Perform all steps in sterile conditions in a biosafety cabinet and use aseptic techniques. The optimal seeding density must be determined for each cell type and growth conditions with the goal of obtaining 80%–95% confluency on the day of the assay.
3.3.2 Day 2: transfection
Co-transfect cells with the appropriate Rluc-tagged and YFP-tagged receptor plasmids using an optimal cationic lipid-based transfection reagent (see Notes 6, 13 and 14). Perform all steps in sterile conditions in a biosafety cabinet and use aseptic techniques.
For the conventional transfection scheme, transfect 7–12 wells as described above in Section 3.2. Additionally transfect a single well with Rluc-tagged receptor only (no YFP), using the same amount of Rluc DNA as in the assay samples for measurement of the BRETbkg. To do this, dilute the total amount of Rluc-fused receptor DNA that will be used for all of the wells into the volume of Opti-MEM (Gibco) recommended in the transfection reagent instructions. Then add the varying amounts of YFP-fused receptor DNA to each tube one by one. Finally, add whatever amount of empty vector DNA is required to each tube to obtain equal amounts of DNA for each well.
For type 1 or type 2 transfection schemes, transfect 7–12 wells as described above in Section 3.2. Additionally, include a matching well with the same amount of Rluc-tagged receptor only (no YFP) as in each Rluc/YFP sample for the determination of BRETbkg. For type 1 BRET experiments, the Rluc- and YFP-fused receptor DNAs must be added individually to all samples. For the type 2 scheme, pre-mix enough DNA for all wells to be transfected at the fixed YFP:Rluc ratio selected, then divide up the DNA into tubes in the amounts intended for transfection. Complement each tube with empty vector DNA so that the total amount of DNA in each tube is equal.
For all schemes, it is useful to include one or more wells transfected with an Rluc-YFP fusion construct (see Note 15) [49] – these wells not only serve as positive controls for BRET, but also provide means for the relative quantification of expressed Rluc-fused and YFP-fused receptors in the other samples as discussed below. 10 ng/well of the fusion DNA are typically used. However, a concentration series may also be valuable.
For all schemes, to eliminate variability in transfection efficiency between the samples, use exactly the same total amount of DNA and transfection reagent for all wells, complementing it with empty vector if necessary.
To ensure efficient co-transfection of plasmids, it is very important to combine and mix all DNA components of the transfection mixture prior to addition of the transfection reagent (see Note 13).
Once all of the DNA mixtures are adequately mixed, add transfection reagent to the tubes according to the transfection reagent instructions. After addition of the transfection reagent to the DNA, incubate at room temperature according to the reagent protocol, and then add the transfection mixture to the wells in a drop-wise manner.
Depending on the cell type and transfection conditions, the maximum protein expression may occur at any time between 12 and 72 hours post transfection; this time point should be chosen as Day 3.
3.3.3 Day 3: BRET experiment
All steps may be performed on a bench. It is advisable to keep the cells and buffers on ice and to work as fast as possible to maximize cell viability.
Rinse cells attached to the plates with PBS once. This can be done by carefully pipetting 1mL of PBS into each well of the 6-well plates, rocking the plates gently, and then aspirating the PBS.
Harvest cells in 1–2 mL of buffer and transfer to 1.5 ml tubes. Weakly adherent cells like HEK293T can be detached by gentle pipetting using BRET buffer (PBS+0.1% glucose). Other cell types require incubation with PBS+1–5 mM EDTA.
Wash twice with BRET buffer by spinning the cells down at 300 × g for 5 min, aspirating the supernatant, and resuspending in BRET buffer. Then count the cells and normalize all samples to 106 cells/ml; this number may need to be changed depending on the expression of the proteins under study (see Note 16).
Aliquot 90 ul/well of each sample in triplicate in a 96-well plate. With some luminometers, in order to avoid errors due to cross-talk between wells, it is important to leave empty spaces between wells with high expected luminescence and wells with low expected luminescence.
Measure total YFP fluorescence using excitation at 480 nm and emission at 530 nm for each well. The total fluorescence will be used to quantify protein expression.
Add 10 uL of freshly prepared 50 μM coelenterazine-h in PBS to each well of the 96-well plate containing cells, moving across the entire plate as quickly as possible, and wait for 8–10 minutes to pass the stage of rapid luminescence decrease (see Notes 8 and 9).
Using a PerkinElmer Victor X Light or comparable luminometer, measure the total luminescence with no filter selection for 0.5–1 sec/well over the entire plate and then repeat three times. The goal of the triplicate measurement is to ensure that no significant luminescence decrease occurs between the readings. If the signal is relatively stable over time, the absolute luminescence values may be considered comparable between the samples and used for quantification of Rluc-tagged construct expression levels.
Repeat the emission measurements at 480 ± 15 nm and 530 ± 15 nm 3–5 times. This filtered luminescence measurement is necessary for calculation of the BRET ratio.
3.3.4 Data analysis
Calculate the BRET ratio for each well by dividing emission at 530 nm by emission at 480 nm.
Use the BRET ratio from YFP-free wells as BRETbkg. If the experiment involves varying the amount of Rluc-tagged protein, plot BRETbkg against the total unfiltered luminescence in the YFP-free samples. If BRETbkg appears independent of the total luminescence, average the obtained BRETbkg values. If a consistent dose-dependent increase in BRETbkg is observed at low luminescence (which may happen with some luminometers), one may need to interpolate the data to obtain an accurate approximation of BRETbkg for each value of unfiltered luminescence. This issue is most relevant at low luminescence emission intensities in Type 1 and Type 2 experiments. Subtract the relevant BRETbkg from the BRET ratio in each YFP-containing well to obtain BRETnet.
-
Upon completion of the experiment, each well is characterized by the following values:
DNA transfection amounts for Rluc and YFP-tagged constructs
Total fluorescence (characterizes relative YFP-tagged construct expression)
Unfiltered luminescence measured at a time point within the slow luminescence decay stage (characterizes Rluc-tagged construct expression)
-
BRETnet
Together, these values can be manipulated to quantify surface density-dependent GPCR dimer formation. Other parameters, such as number of cells per well, luciferase substrate concentration, and time of its addition should be kept as constant as possible between the wells to eliminate unwanted variation.
3.4. Quantitative interpretation of BRET saturation assay results
In this section, we analyze the theoretical simulated shapes of titration curves in an attempt to gain a better understanding of the trends observed in each type of BRET experiment.
3.4.1 Conventional framework for the interpretation of BRET saturation data and its applicability domain
In their 2006 publication in Current Protocols in Neuroscience [49], Hamdan and co-workers suggested a simple and accurate scheme for interpretation of curves resulting from a “conventional” BRET titration experiment (Figure 5). According to this scheme, a specific interaction between the proteins labeled with donors and acceptors exhibits a saturable hyperbolic curve in coordinates of acceptor/donor ratio vs BRETnet. From such a titration curve, it is possible to derive BRETmax and the so-called BRET50, i.e. the acceptor/donor ratio at which 50% of the BRETmax is achieved. Furthermore, when comparing two titration curves, variations in the BRETmax suggest a conformational change between the binding partners while variations in BRET50 are interpreted as changes in association affinity.
Figure 5.

Interpretation of curves obtained in a “conventional” BRET titration experiment as proposed in [49]. Variations in the BRETmax suggest a conformational change between the binding partners while variations in BRET50 are interpreted as changes in association affinity. The limits of applicability of this scheme are discussed in text.
While this intuitive scheme has been instrumental for the interpretation of substantial BRET titration data, it is important to keep in mind that it is only applicable under conditions where:
all curves are constructed with a single set of transfected samples and variation is created by changing the conditions at which the measurement is taken (e.g. agonist or antagonist addition)
the samples express constant amount of donor protein and varying amounts of acceptor protein (the “conventional” scheme).
there is no competition between donors and acceptors for interaction with the same donors or the same acceptors (i.e. neither donor/donor dimerization nor acceptor-acceptor dimerization compete with donor-acceptor complex formation).
Examples of experiments in which the above conditions hold include the monitoring of agonist-induced changes in the association of a GPCR with downstream signaling molecules, such as β-arrestins or G–proteins. In these applications, it is safe to assume that total amounts, cellular distribution, and luminescence/fluorescence properties of proteins under study do not change between the titration curves, and that donor-donor or acceptor-acceptor interactions do not prevent “productive” donor-acceptor pairs from forming.
These conditions, however, do not hold for experiments involving GPCR dimerization. With GPCR dimerization, there is a considerable competition between non-productive (donor-donor or acceptor-acceptor) pairs and productive donor-acceptor pairs. Furthermore, attempts to compare the curves obtained from different transfection samples, for example between mutants and wild-type proteins [30, 43, 44], are bound to fail because of inevitable variation in receptor surface densities.
Herein we seek to develop an alternative framework that allows quantitative interpretation of BRET data while avoiding the pitfalls associated with violation of the conventional framework applicability conditions.
3.4.2 Homodimerization equilibrium equations and anatomy of BRET titration surfaces
One of the main difficulties in quantitative analysis of BRET data is its essentially three-dimensional nature in which the BRET response simultaneously and separately depends on the surface densities of Rluc- and YFP-tagged receptors in a manner that is controlled by association affinities (Figure 6A). Unlike in standard titration binding experiments, one cannot assume that one of the parameters is held constant in BRET; while every effort is made to ensure accurate and efficient transfection of the cell samples, the expressed receptor densities are ultimately determined by a variety of uncontrollable factors.
Figure 6.
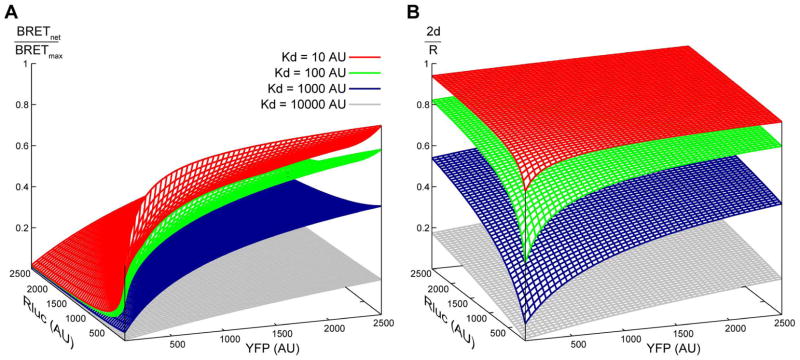
Simulated surfaces of (A) BRET response and (B) fraction of receptor found in homodimers at varying surface densities of Rluc- and YFP-tagged receptors (shown on horizontal plane axes). BRET response (A) reflects the fraction of Rluc donor-tagged receptors that are in dimers with YFP acceptor-tagged receptors and does not take into account other possible homodimers (i.e. YFP/YFP or Rluc/Rluc homodimers). In contrast, fraction of receptor in dimers (B) represents all possible dimers. Varying association kd results in changes of the surface shapes. These two types of 3D surfaces aid in understanding the pitfalls of quantitative interpretation of BRET data (see text for details).
3.4.2.1 Basic equations
In this section, theoretical BRET surfaces and curves are generated to illustrate the dependence of BRETnet readings on the input parameters of the assay. We are considering the equilibrium of five species: monomeric donor-fused and acceptor-fused receptors, and three types of dimers: donor/donor, acceptor/acceptor, and donor/acceptor. The following notation is used:
L and Y are total surface densities for Rluc-fused and YFP-fused receptors, respectively; R = L + Y is the total surface density of the receptor
l and y are surface densities of Rluc/Rluc dimers and YFP/YFP dimers; x is the density of Rluc/YFP dimers; the density of all dimers is given by d = l + y + x
L – 2l – x and Y – 2y – x are surface densities of monomeric Rluc-fused and YFP-fused receptors
kd is a two-dimensional homodimer dissociation constant measured in the same units as receptor surface densities.
We assume that C-terminal fusions do not change the dimerization propensities of the proteins under study, hence in this section, do not introduce separate dissociation constants for Rluc/Rluc, YFP/YFP, and Rluc/YFP dimers. However, because of combinatorial considerations, we need to multiply this universal kd by a factor of 2 in all equations where the dimer components are symmetric [58].
The dynamic monomer-dimer equilibrium follows the standard equilibrium equation:
| (1) |
This equation can be solved analytically to find the total dimer surface density, d, as a function of R and kd:
| (2) |
As expected, d increases with increasing R, but more importantly, the value of 2d/R representing the fraction of receptor that is in dimers also increases. 2d/R can be plotted against L and Y densities in a 3D plot as shown in Figure 6B. For transient equilibrium association, 2d/R is between 0 and 1; it increases with increasing R, and the density-dependence of this increase is controlled by the dimerization kd. For receptors that only exist in dimers, 2d/R is always equal to 1.
As for equation (1), we can write out the equilibrium equations for formation of the individual dimer types (Rluc/Rluc, YFP/YFP, and Rluc/YFP):
| (3a) |
| (3b) |
| (3c) |
By dividing (3c) by (3a) and (3b) by (3c), we obtain the following relations between monomeric and dimeric densities:
| (4) |
The two sides of equation (4) can be manipulated separately to relate the surface densities of Rluc/Rluc and YFP/YFP dimers (l and y, respectively) to that of Rluc/YFP dimers (x):
| (5) |
which, can then be plugged into the mass equation, d = l + y + x, to yield
| (6) |
Eqs. (2) and (6) link x, the density of donor-acceptor dimers, to the input parameters of the system, i.e. receptor densities L and Y and the homodimer dissociation constant kd. Because only donor-acceptor dimers manifest themselves via resonance energy transfer and a change in the emission spectrum, and because these donor-acceptor dimers co-exist in equilibrium with monomeric donor molecules and donor-donor dimers whose emission spectrum is unchanged, x is directly related to the experimental readouts of the BRET assay. The maximum achievable BRET ratio, BRETmax, occurs when the high surface density of the receptors effectively eliminates any monomeric species, and when the ratio of acceptors to donors is sufficiently high to eliminate donor-donor dimers, i.e. when d = R/2 = (L+Y)/2 and x ≈ L. At any other condition, the ratio of BRETnet/BRETmax is a reasonable approximation of x/L, i.e. of the fraction of donors that are engaged in dimers with acceptors. This relation is summarized in the simulated surface BRETnet/BRETmax dependence on L and Y densities for varying association affinities (Figure 6A).
3.4.2.2 Expected responses in different types of BRET experiments
A single BRET experiment, conventional, type 1, or type 2, essentially traces a two-dimensional slice on the three-dimensional surface of association for a particular pair of constructs in question (Figure 4B). Figure 7 provides a comprehensive account of expected responses in the three types of BRET transfection schemes in both monomer-dimer equilibrium and constitutive dimer situations. Several things should be noted:
Figure 7.

Simulated BRET responses for transient (A, C, E) and obligate (B, D, F) homodimers in a conventional (A, B), type I (C, D) and type 2 (E, F) BRET titration experiments. The 3D plots in the leftmost column illustrate BRET responses as a function of the surface density of Rluc-tagged and YFP-tagged receptors. Highlighted in thicker lines are slices taken at four Rluc densities for a conventional experiment (A, B), four total receptor densities for a type 1 experiment (C, D), and four YFP/Rluc ratios for a type 2 experiment (E, F). The second column contains projections of the same slices onto more traditional 2D coordinates. The third and fourth columns are results of translation of the corresponding BRET responses into monomer-dimer equilibrium schemes using the equations described in the text.
When a monomer-dimer equilibrium system is studied with a “conventional” experiment (Figure 7A), the shape of the curve of BRET response vs YFP/Rluc ratio depends on the density of Rluc-tagged receptors. Higher expressors manifest in faster saturating curves in these coordinates.
In contrast, if receptors only exist in dimers (Figure 7B), Rluc density has virtually no effect on the shape of BRET response vs YFP/Rluc ratio curve. This is because Rluc-tagged monomers do not exist and, consequently, do not contaminate the overall emission spectrum with unmodified luciferase emission.
When a monomer-dimer equilibrium system is studied in a “type 1” experiment (Figure 7C), the total receptor density (which is held constant between the samples) defines the shape and height of the curve of BRET response vs YFP/Rluc ratio. It is important to note that while the type 1 curves appear to plateau at higher YFP/Rluc ratios, their asymptotic value is not BRETmax; instead, it simply characterizes the monomer-dimer equilibrium at the given surface density of the receptors. The type of response shown in Figure 7C has been observed, for example, for β2 adrenergic receptor homodimers in [51] (Fig. 5 in the reference).
Studying a constitutive dimer in a type 1 experiment (Figure 7D) produces a saturation curve in the YFP/Rluc ratio vs BRET response coordinates that is independent of the total receptor density.
When a monomer-dimer equilibrium system is studied in a “type 2” experiment (Figure 7E), both the shape and the maximum value on the saturation curve in the YFP/Rluc ratio vs BRET response plane depend on the selected ratio of YFP-tagged to Rluc-tagged receptor surface densities.
With a constitutive dimer in a type 2 experiment (Figure 7F), no saturation is observed, because the fraction of donors paired with acceptors is constant and defined by the (constant) YFP/Rluc ratio in the system.
3.4.2.3 Potential BRET quantification errors
The three-dimentional interpretation of BRET response at the given surface densities of the Rluc- and YFP-tagged receptors highlights several possible reasons for errors in result quantification. For example, if for two construct pairs, the “conventional” experiment slices are taken at different donor surface densities, the resulting two-dimensional pictures may look different despite identical association affinities, or, on the contrary, look the same for pairs with different association affinities (Figure 8A, B).
Figure 8.

Potential BRET quantification errors. (A) and (B): disregarding variations in the expression levels between different constructs may lead to false conclusions about their relative homodimerization affinities. (A) Constructs 1 and 2 which have different homodimerization affinities and different overall expression levels can produce indistinguishable curves in coordinates of BRET response vs YFP/Rluc ratio. (B) On the other hand, constructs 3 and 4 with identical homodimerization affinities but different expression levels can translate into different curves on the BRET response vs YFP/Rluc ratio plane. In both (A) and (B), the two constructs produce un-matching slices of their respective (distinct) 3D BRET response surfaces; this information is lost when the responses are plotted in 2D against YFP/Rluc ratio. (C) and (D): despite the constant DNA transfection amounts, levels of Rluc-tagged receptor expression may decline (C) or increase (D) in samples with higher YFP-tagged receptor expression, as indicated by changes in the unfiltered luminescence. The plots represent data obtained in a CXCR4 homodimerization experiment (C) and in an undisclosed GPCR heterodimerization experiment (D). (E) Quantification errors due to disregarding the interdependence of expression levels of Rluc- and YFP tagged constructs. If the Rluc-tagged receptor expression changes when the YFP-tagged receptor is varied, the problem can be thought of as taking non-parallel slices from the 3D BRET response surfaces, which can lead to artifacts of interpretation.
Moreover, it often happens that the expression of the donor and acceptor proteins are interdependent. For example, despite the fact that Rluc transfection levels are kept constant between the samples in the conventional transfection scheme, in homodimerization studies one can often observe a decrease in the unfiltered luminescence of the sample with increasing YFP expression (Figure 8C). This may be due in part to the fact that the two proteins compete for the same expression and folding machinery inside the cells; loading this machinery with overwhelming amounts of YFP-tagged receptor effectively takes the resources away from the Rluc-tagged receptor. Alternatively, in conventional scheme heterodimerization experiments, one may observe the opposite effect, i.e. an increase in luminescence with increasing YFP expression (Figure 8D). A possible reason for this observation is a “chaperone” effect in which heterodimer formation makes the expression of Rluc-tagged receptor more favorable for a variety of reasons (stability, trafficking etc). Failure to relate the BRET signal to the actual expression of the proteins under study rather than to the amount of transfected DNA may lead to serious misinterpretation of the results (Figure 8E).
3.4.2.4 Interpretation of BRET data in terms of fraction receptors in dimers
With simple rearrangements, Eq. (6) provides a way to tackle these issues by converting the observable BRET signal, BRETnet/BRETmax into the standard scale of fraction of receptor found in dimers:
| (7) |
The value of 2d/R plotted against R provides a reference frame for comparing association affinities between cases not comparable in the conventional scheme, e.g. different transfection samples, including wild-type and mutant proteins. As Figure 7 illustrates, regardless of the experiment type or the expression levels, application of Eq. (7) converts the data into the coordinates of 2d/R vs R which unambiguously and reproducibly describes the dimerization equilibrium in the system (Figure 7, columns 3 and 4). According to Eq. (7), to make this conversion possible, one only needs to know the ratio of surface densities of the acceptor-fused (Y) and the total (R) receptor. Because R = L + Y, this question is equivalent to quantification of the relative densities Y and L.
3.4.2.5 A practical way for simultaneous quantification of Rluc- and YFP- tagged receptor expression
Inevitable variation in the expression between Rluc-tagged and YFP-tagged constructs makes it impossible to use the transfected DNA amounts as accurate approximations of construct expression levels. Relevant experimental readouts, i.e. sample luminescence and fluorescence, are often obtained on different instruments and are always measured in different units. However, the two quantities may be related to each other using the Rluc-YFP fusion construct and the BRET protocol readouts as follows:
Use the experimental sample fluorescence for Y (this would correspond to measurement of receptor expression in relative fluorescent units, RFU).
Determine the fluorescence-to-luminescence normalization constant c as Fluorescence (fus) / Luminescence (fus) where fus is the experimental well with the Rluc-YFP fusion construct (see Note 15). The advantage of this construct is the guaranteed 1:1 stoichiometry between Rluc and YFP. It may be useful to have several wells transfected with varying amounts of the fusion construct and build a standard curve of luminescence vs fluorescence.
Quantify the density of the donor-fused receptor as L = c × Luminescence (unfiltered) in all wells (the density L of Rluc-tagged receptors is now expressed in relative fluorescence units, similar to Y).
Quantify the total surface density as R = L + Y (also expressed in relative fluorescence units).
When applying this scheme, all densities (R, L, and Y) will be expressed in relative fluorescent units. Next, the data obtained using Eq. (7) can also be used to find kd by fitting it into Eq. (1). This kd will be expressed in the same units as R, L, and Y; in the scheme above it will be relative fluorescence units, RFU. This kd does not allow the absolute affinity quantification, but provides enough information for affinity comparison and ranking. However, if the densities are quantified by external methods in conventional units of cm−2 or mol/cm2 (e.g. [56]), the dissociation constant will be expressed in these units as well.
Note that although using unfiltered luminescence for quantifying Rluc-tagged receptor expression levels is a standard practice [13, 56], it is not always entirely accurate. In reality, when luciferase molecules are in energy transfer proximity of the acceptors, a fraction of the emitted energy is absorbed by the acceptors and then emitted at a longer wavelength, contributing to the overall unfiltered emission of the sample. However, because the YFP quantum yield is less than one (it ranges from 0.6 to 0.8 for different variants [59, 60], the resulting total unfiltered luminescence is lower than it would have been in the absence of acceptors in proximity. To the best of our knowledge, no studies have been conducted thus far to accurately quantify changes in unfiltered emission of Rluc as a result of energy transfer. One way to approach this question experimentally would be to compare the unfiltered emission of a Rluc-YFP fusion construct with that of a point mutation that renders YFP completely incapable of absorbing light in the Rluc emission spectrum.
The method of quantifying Rluc expression based on unfiltered emission relies on the assumption that the decrease in unfiltered emission due to energy transfer to the proximal YFP molecules is rather small. If the decrease is in fact significant, an additional correction factor is needed that relates the observed BRET ratio to the expected percentage decrease in unfiltered emission. Application of this factor will restore the observed unfiltered emission to its non-quenched value. The corrected emission can then be used for quantification of luciferase expression and further for BRET analysis as described above.
3.4.3 Example: monomer-dimer equilibrium of the chemokine receptor CXCR4 and its point mutants
To illustrate the utility of the framework described in Section 3.4.2, we applied it to the study of homodimerization of the chemokine receptor CXCR4 and several single-point mutants. The mutants were computationally predicted to disrupt the dimer interface observed in the five crystal structures of the receptor solved by Wu and co-workers [31]. Mutations were introduced in both YFP-tagged and Rluc-tagged receptor subcloned in pcDNA3.1 using the QuikChange site-directed mutagenesis kit (Stratagene). The constructs were transfected in HEK293T cells with TransIT LT1 (Mirus Bio) using conventional transfection protocols. Optimization of the transfection conditions led to robust and reproducible BRET signals for both WT and mutant receptors with BRETmax exceeding 0.6 for the wild type (Figure 9A). A CXCR-Rluc and GABAB-YFP receptor pair was used as a negative control and provided a linear saturation response with BRETnet never exceeding 0.025 (data not shown). The CXCR4 mutations appeared to mildly affect both BRETmax and BRET50 (according to analysis by the conventional scheme); however these changes could not be translated into association affinity differences because of (i) variations in expression and (ii) varying dependencies of luminescence (from the Rluc-tagged receptor expression signal) on YFP-tagged receptor expression between the mutant and WT constructs. By applying the equations described in Section 3.4.2, we were able to convert the BRET titration data into the common framework of dependence of fraction of receptor in dimers (2d/R) on the total receptor density (R). This exercise clearly showed a difference in homodimerization affinities between WT and selected mutants (Figure 9B). For example, about 2.5-fold higher surface density was required for the W195A mutant than for CXCR4 WT to achieve the same state on the monomer-dimer equilibrium scale. Further studies are required to elucidate whether additional mutations can be identified to further destabilize the dimer and the functional consequences of such mutations at physiological levels of receptor expression.
Figure 9.
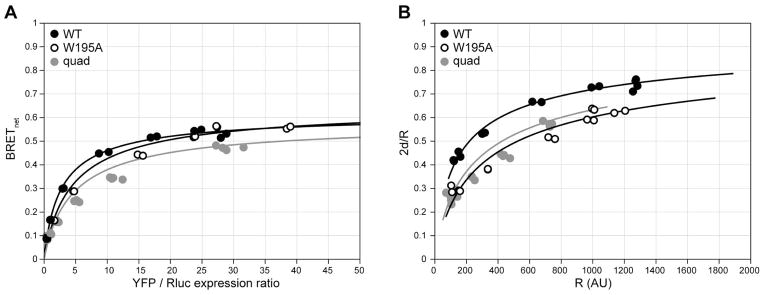
Homo-dimerization of CXCR4 and selected points mutants of the TM5-TM6 interface observed in crystallography. (A) BRETnet ratio is plotted against the YFP/Rluc expression ratio; (B) the same data converted to fraction of receptor in dimers plotted against the total surface density of the receptor. Differences between dimerization parameters have been clarified by correcting for variations in Rluc-tagged receptor expression between constructs and by converting the BRETnet/BRETmax values into 2d/R values using Eq. (7) (see text). This example illustrates the advantages of the discussed additions to the data analysis. *quad = L194A/W195A/L267A/E268A.
3.4.4 Quantifying heterodimer association affinity
When studying heterodimerization, we can no longer assume that donor-donor, acceptor-acceptor, and donor-acceptor affinities are the same. To capture this aspect of the problem, we introduce numeric coefficients, a and b, to represent fold differences between donor-acceptor dimerization affinity and the other two affinities. The equilibrium equations for formation of the individual dimer types become:
| (8a) |
| (8b) |
| (8c) |
By dividing (8c) by (8a) and (8b) by (8c), we obtain the following relations between monomeric and dimeric densities:
| (9) |
Similarly to the homodimerization case, the two sides of equation (9) can be manipulated separately to relate the surface densities of Rluc/Rluc and YFP/YFP dimers (l and y, respectively) to that of Rluc/YFP dimers (x):
| (10) |
Unfortunately, an equation analogous to Eq. (1) cannot be formulated for the heterodimerization problem because of the heterogeneity of the dimer population. Nevertheless, the experimentally derived BRETnet/BRETmax value remains a reasonable approximation of x/L. Therefore, the heterodimer association affinity can be found with the following three steps:
calculate the heterodimer surface density x from the BRET assay readouts as x = L×BRETnet/BRETmax
calculate the concurrent homodimer densities l and y using Eq. (10), and
calculate the kd from Eq. (8c).
Coefficients a and b must be optimized simultaneously with kd for the best fit with the experimental data. In the constitutive dimer system (i.e. when no monomer-dimer equilibrium is observed), the homodimer/heterodimer density ratios, l/x and y/x, are entirely dictated by the relative total surface densities of donors and acceptors. In other words, Eq. (5) holds for constitutive homodimers and constitutive heterodimers equally well.
3.4.5 Caveats and limits of the quantitative analysis
Because the BRET assay is performed on living cells, it is prone to a variety of artifacts and result variability. In this section, we outline several major limitations for quantification of BRET titration curves.
Endogenously expressed untagged receptor
If cells express the receptor of interest endogenously, the untagged receptor molecules may form dimers with Rluc-tagged or YFP-tagged receptors, thus complicating the analysis of the BRET titration data. Within the scope of BRET, it is impossible to discriminate Rluc-tagged monomers from their dimers with untagged receptor. Consequently, for quantitative BRET, it is preferable to choose a cell type with no to negligible background expression of the protein(s) under study.
Heterogeneity of protein expression
The BRET response of each individual cell is determined by receptor densities in this cell; however, the overall BRET signal is an average of signals coming from all the cells in the assay well. If the cells express varying amounts of the target proteins, the cumulative signal will represent an average of widely distributed signals from individual cells effectively lowering the signal to noise ratio. It is therefore important to choose a system (cell type and transfection method) that guarantee homogenous expression of proteins under study. To evaluate expression homogeneity, one can transfect a single sample with YFP-tagged receptor (maximal amount suggested by the transfection protocol) and test cells after 24 or 48 hours for YFP expression by flow cytometry. Untransfected cells should be used as a control. A narrow peak that is separated from the control (untransfected) peak by at least 1.5–2 logarithmic units usually corresponds to efficient homogenous transfection and expression, while multiple peaks or a wide distribution of fluorescence intensities suggest that either a transfection protocol optimization is required, or the cell type is inappropriate for quantitative BRET.
Cellular distribution of dimer partners
The inherent limitation of BRET technology is its inability to distinguish signals coming from the cell surface from those originating in the intracellular compartments. This is especially important when one attempts to quantify heterodimer association affinity. If the Rluc-tagged dimer partner is, for example, selectively retained in the endoplasmic reticulum, either inherently or as a result of the C-terminal fusion, its non-modified emission will significantly affect the overall BRETnet signal in the system, both in terms of BRETmax and the apparent association affinity. However, unequivocal discrimination of this effect from high affinity donor-donor association or from purely a consequence of heterodimer asymmetry may require at least switching the fusion partners and ultimately other experimental approaches such as fluorescence microscopy, radioligand binding, etc.
Non-dimer related BRET
The three transfection schemes described in Section 3.2 only partially address the question of specificity of the dimer interactions: they provide means to distinguishing dimer “bystander” BRET resulting from receptor over-expression and consequent crowding. However, they do not eliminate possibilities of forming higher order oligomers or receptor clustering as result of partitioning into membrane microdomains. TIRF microscopy [40], luciferase- and YFP-complementation studies [61] and other techniques must be used to ultimately resolve these uncertainties [62, 63].
Estimation of Rluc expression levels
Estimation of expression levels is important for obtaining accurate quantification of dimer densities and kd. As described in 3.4.2, while estimation of the YFP-tagged receptor expression is straightforward, determining Rluc-tagged receptor expression involves unit conversion that, in cases of high BRET, may be non-trivial. Improved methods for more direct determination of Rluc-tagged receptor expression levels are therefore desirable.
3.5 Application of the quantitative framework for analysis of dimerization mutants
Structural biology studies of GPCRs have significantly increased since 2007 with the emergence of many new structures in various activation states. Some of these such as CXCR4, the μ-opioid receptor and the κ-opioid receptor have revealed parallel dimers in their asymmetric units or in the crystallographic cells [31–33]. Characterization of the functional relevance of these dimer interfaces via site-directed mutagenesis requires methods for quantitative assessment of relative dimer stability. Our method for analytical interpretation of BRET data should facilitate such comparisons by taking into account expression levels of the BRET receptor pairs.
Acknowledgments
This work was supported by the National Institute of Health PSI:Biology grants U01 GM094612 (T.M.H and R.A) and U54 GM094618 (R.C.S) and by NIH grants R01 GM081763 (T.M.H.) and R01 GM071872 (R.A.). C.T.G and B.S. were supported by the Cellular and Molecular Pharmacology Training Grant T32 GM007752. D.H. was supported by NIH NRSA grant F32 GM083463. B.W was supported by NIH grant R01 Al100604 and the grant 11JC1414800 awarded by Science and Technology Commission of Shanghai Municipality. The authors would like to thank Jeffrey Velasquez (TSRI) for help on molecular biology, Tam Trinh and Kirk Allin (TSRI) for help on baculovirus expression, and M. Bouvier (University of Montreal) for the Rluc and YFP coding sequence containing vectors, which were used to produce all of our BRET constructs. We also thank Pascale Charest (University of Arizona) for valuable discussions regarding the BRET assays and Goran Pljevaljcic (TSRI) for constructive comments on the manuscript.
4. Notes
1. Luminometers. The choice of luminometer depends on the specifics of the assay that will be employed. The author’s laboratory utilizes a VICTOR X Light 2030 luminometer (Perkin Elmer) with a 2-channel liquid injection system for ligand addition. This instrument lacks fluorescent excitation capabilities; therefore, a separate plate reader with fluorescence reading capability is used to quantify energy acceptor expression. A liquid injection system is useful for the BRET2 assay, which recommends immediate data collection following substrate addition. The time the instrument requires to begin the assay and move from well to well may affect data collection for temporally sensitive protein-protein interactions. Lastly, temperature control options and overall instrument sensitivity should be taken into account when deciding upon a plate reader. Alternative instruments commonly used for BRET measurements include the Mithras LB 940 (Berthold Technologies) and the LUMIstar Omega (BMG Labtech) plate readers.
2. Cell culture plates. In some cases, 12- or 24-well plates may be used, rather than 6-well plates, to seed and transfect cells to be used in the BRET experiments. The plate selection should be based on the requirement of providing adequate cell numbers for the planned experiment.
3. Tubes for cell harvesting and washing. The size of the tubes used in the assay depends on the volume in which the cells are resuspended from the transfection plates, the cell numbers expected from each plate well, and the requirements of the centrifuge that is used to spin down the cells when switching buffers and normalizing cell concentrations.
4. 96-well plates for the BRET assay. The optimal plate type for BRET assays depends on the luminescence and fluorescence-detecting equipment used. For example, the authors use a fluorescent plate reader which requires clear bottom plates and a luminometer that requires non-clear bottom plates. Therefore, the authors use BioCoat/OptiLux clear bottom plates and cover the bottom with white adhesive tape between fluorescent plate reader and luminometer.
5. Dispensing coelenterazine-h into 96-well plate wells. The authors have found it useful to use a repeating pipette for this purpose, as manually pipetting the substrate into the wells one by one is too slow given the rate of coelenterazine-h oxidation by luciferase.
6. Transfection reagent suggestions. The authors have had success with TransIT LT1 (Mirus Bio) in HEK293T cells and with Lipofectamine 2000 (Invitrogen) and TransIT CHO kit (Mirus Bio) in CHO cells.
7. Working DNA concentrations. The authors use a working DNA concentration anywhere from 50 ng/μL to 500 ng/μL depending on the details of the assay. Furthermore, improved transfection efficiencies have been observed when fresh working solutions of DNA are regularly prepared from higher DNA concentration stocks. The authors have routinely used both the Marligen Biosciences PowerPrep HP Plasmid Maxiprep Kit and the Promega PureYield Plasmid Midiprep System to generate high concentration DNA stocks.
8. Long term storage of coelenterazine. Coelenterazine-h is very sensitive to decomposition. The substrate should be aliquoted into appropriate volumes to prevent repeated exposure to light and water vapor in the air. The authors have found it useful to store stock concentrations (1mM) of coelenterazine-h at −20°C, in a desiccant-filled container.
9. Recent developments in BRET technology. Improvements to the BRET assay have been made that increase signal sensitivity, reduce background signal, and extend the reliable lifetime of the measurement (see [48] and references therein). The Rluc/YFP energy donor/acceptor pair utilized in the method reported herein represents the first generation (BRET1) development of the assay. The second generation BRET (BRET2) assay utilizes a modified coelenterazine (DeepBlueC or coelenterazine-400a) substrate for Rluc which emits at ~420nm as well as a mutant GFP (GFP2 or GFP10) as the energy acceptor which emits at ~510nm. This second generation substrate/construct combination results in greater spectral separation between Rluc donor emission and YFP acceptor emission, thereby reducing background signal, but it also results in a lower overall signal and shorter time frame for reliable data collection because of substrate instability. To address the substantially weaker signal, a series of mutations have been introduced into Rluc (Rluc2 and Rluc8) that significantly increase signal intensity. For longer kinetic measurements, a chemically protected coelenterazine substrate (EnduRen) was also developed which requires the action of cellular esterases to convert the substrate before it can be oxidized. EnduRen enables measurement of the BRET ratio for up to 24 h; however, it requires the assay to be carried out at 37°C.
10. Functional effects of the donor/acceptor tags. One needs to ensure that labeling of the C-terminus of the receptor does not affect receptor function. In our experiences, addition of both small and large amino-acid motifs or proteins to the C-terminal tail can affect proper receptor trafficking and function. The inertness of the donor/acceptor labels on receptor function needs to be confirmed by ligand binding and functional assays.
11. BRETbkg. Although referred to as BRETbkg, this measurement is not actually BRET at all as it characterizes the situation with no energy transfer; it is the ratio of the luminescence emission intensities of Rluc at 530 nm and 480 nm in the absence of acceptors.
12. Suspension versus adherent format. The assay described above is intended for cells in suspension (including adherent cells in suspension). To assay cells in an adherent format, follow the same transfection protocol as described above except 24 h following transfection, lift, count and split 0.5–2×106 cells in appropriate cell growth media into a 96-well tissue-culture treated assay plate and incubate overnight. The following day, gently wash the cells without disturbing them from the bottom of the plate and replace media with 90 μL/well BRET buffer. Carry out subsequent steps as detailed in the protocol.
13. Co-transfection efficiency. At the transfection step, it is critically important that not only all cells receive the same total amount of DNA (transfection efficiency), but also that they receive the same amounts of Rluc-tagged and YFP-tagged DNA (co-transfection efficiency). Especially with liposome-forming cationic lipid reagents, one needs to make sure that all DNA components of the transfection mixture are thoroughly mixed together prior to addition of the transfection reagent: this increases the homogeneity of the liposomal particles that start forming upon reagent addition.
14. Transfection mixture preparation by serial dilutions. The authors developed a set of schemes based on serial dilutions to produce the multiple transfection mixtures for BRET experiments. They are especially useful when dealing simultaneously with a large number of samples. The schemes require that all working DNA stocks are at a concentration of 50 ng/μl. The stocks include Rluc-tagged receptor plasmid (referred to as simple Rluc in this Note), YFP-tagged receptor (a.k.a. YFP), and empty vector (authors use pcDNA3.1). The schemes are designed to deliver a total of 2.5 μg of DNA into a single well in a 6-well plate. The transfection mixtures can be prepared in 0.5 −1.5 ml DNA lo-bind tubes or in wells of a 96-well plate (see below).
Transfection scheme for a conventional BRET experiment. This scheme produces 7 samples per construct, 6 of them being for data points (in tubes 1–6) and one being a YFP-free sample for BRETbkg determination.
Aliquot 83.33 μl of YFP in tube 6, and 50 μl of pcDNA in tubes 1–3 and 7
Take 33.33 μl from tube 6, pipet it into tube 3, mix thoroughly
Take 33.33 μl from tube 3, pipet it into tube 2, mix thoroughly
Take 33.33 μl from tube 2, pipet it into tube 1, mix thoroughly
Take 22.22 μl from tube 1, pipet it into tube 4 (empty), add 27.78 μl YFP, mix thoroughly
-
Take 11.11 μl from tube 1, pipet it into tube 5 (empty), add 38.89 μl YFP, mix thoroughly
Result: 50 μl DNA at 50 ng/μl (e.g. a total of 2.5 μg DNA) in each of tubes 1–7; pure pcDNA in tube 7, pure YFP in tube 6, and decreasing YFP/pcDNA ratio in all other tubes. The obtained ratios optimally sample the range for construction of a conventional BRET response curve.
To each tube, add the desired (constant and small, no more than 5 μl) volume of Rluc, mix thoroughly. Adding 5 μl of the 50 μg/μl Rluc stock will result in a maximum YFP/Rluc ratio of 10 (in tube 7). It is usually preferable to sample higher ratios: to do so, decrease the added volume of Rluc; to reduce small volume pipetting errors dilute the 50 μg/μl working Rluc stock 10-fold or lower and pipet a proportionally larger volume.
Transfection scheme for a type 1 BRET experiment. This scheme results in 6 samples per construct (in tubes 1 through 6) and 6 samples with identical amounts of Rluc but no YFP for BRETbkg determination (tubes 1* through 6*). There will be a total of 2.5 μg receptor DNA per sample.
Aliquot 83.33 μl of Rluc in each of tubes 1 and 1*, 50 μl of YFP in tubes 2–6, and 50 μl of pcDNA in tubes 2*–6*
Take 33.33 μl from each of the tubes 1 and 1*, pipet it into tubes 2 and 2*, respectively, mix thoroughly
Take 33.33 μl from each of the tubes 2 and 2*, pipet it into tubes 3 and 3*, respectively, mix thoroughly
Take 33.33 μl from each of the tubes 3 and 3*, pipet it into tubes 4 and 4*, respectively, mix thoroughly
Take 33.33 μl from each of the tubes 4 and 4*, pipet it into tubes 5 and 5*, respectively, mix thoroughly
Take 33.33 μl from each of the tubes 5 and 5*, pipet it into tubes 6 and 6*, respectively, mix thoroughly
-
Discard 33.33 μl from tubes 6 and 6*
Result: 50 μl DNA in each of tubes 1–6 with pure Rluc in tube 1 and increasing YFP/Rluc ratios in tubes 2–6. The ratios range from 0 to almost 100 using this scheme, with more attention given to the lower part of the curve. Additionally, there will be pure Rluc in tube 1* and increasing pcDNA/Rluc ratios in tubes 2*–6* (these samples will be used for BRETbkg calculations at various luminescence intensities).
Transfection scheme for a type 2 BRET experiment. This scheme produces 6 samples per construct (in tubes 1 though 6) and 6 samples with identical amounts of Rluc but no YFP for BRETbkg determination.
Mix YFP and Rluc DNA in the desired proportion for the minimal total volume of 150 ul in a separate tube (make a little extra to account for possible pipetting errors). Repeat in another tube, using pcDNA (instead of YFP) and Rluc.
Aliquot 83.33 μl of Rluc-YFP mixture into tube 6. Aliquot 83.33 μl of Rluc-pcDNA mixture into tube 6*. Aliquot 50 μl of pcDNA in each of tubes 1–3 and 1*–3*.
Take 33.33 μl from each of tubes 6 and 6*, pipet it into tubes 3 and 3*, respectively, mix thoroughly
Take 33.33 μl from each of tubes 3 and 3*, pipet it into tubes 2 and 2*, respectively, mix thoroughly
Take 33.33 μl from each of tubes 2 and 2*, pipet it into tubes 1 and 1*, respectively, mix thoroughly
Take 22.22 μl from each of tubes 1 and 1*, pipet it into tubes 4 and 4*, respectively, add 27.78 μl Rluc-YFP mixture to tube 4 and 27.78 μl Rluc-pcDNA mixture to tube 4*, mix thoroughly
-
Take 11.11 μl from each of tubes 1 and 1*, pipet it into tubes 5 and 5*, respectively, add 38.89 μl Rluc-YFP mixture to tube 5 and 38.89 μl of Rluc-pcDNA mixture to tube 5*, mix thoroughly
Result: 50 μl DNA in each of the tubes 1–6; increasing amounts of receptor DNA from 1 to 6 with the constant YFP/Rluc ratio. Matching amounts of Rluc DNA in tubes 1* to 6* (these samples will be used for BRETbkg calculations at various luminescence intensities).
As an alternative to tubes, DNA mixtures can be prepared in a sterile untreated round- or conical-bottom 96-well plate using a 50 μl multichannel pipette. Following preparation of the DNA mixtures, one can use a multichannel pipette to add transfection media and reagents as described in the reagent protocol. In most 96-well plates, a single well can hold up to 300 μl; with the reagents that the authors use, the final volume of the transaction mixture for a single well in a 6-well plate does not exceed 250 μl, therefore, a 96-well plate is appropriate.
15. Rluc-YFP fusion construct. The Rluc-YFP fusion construct was produced in our lab by fusing the YFP coding sequence into the Rluc vector (generously provided by the Bouvier lab) downstream of the Rluc coding sequence.
16. Optimization of cell numbers. Increasing the number of cells in each well of a 96-well assay plate may improve the total luminescence/fluorescence yield from the sample, and consequently reduce signal-to-noise ratio, without affecting the BRET ratio. For example, if one or both of the proteins of interest expresses poorly, more cells may be required to provide a sufficient signal for luminometer detection. Alternatively, if the signal saturates the detector then fewer cells should be used.
17. Selection of the donor-tagged receptor in heterodimerization experiments. In all types of BRET assays, it is preferable to test a wide range of acceptor expression levels. Moreover, reliable measurement of BRETmax requires the Rluc-tagged receptor population to be saturated with YFP. A protein that expresses poorly will yield small variability in expression and result in a small YFP/Rluc expression ratio. Therefore, the lower expressing receptor should be fused to Rluc while the better expresser should be fused to YFP.
18. Temperature. Depending on the particular receptor interaction being studied, it may be advantageous to conduct the BRET assay at room temperature (as described above) or at 37°C and to pre-equilibrate the cells to that temperature prior to substrate addition. At room temperature, the fluidity of the cellular membrane, lateral receptor mobility, and the function of potentially important scaffolding proteins may be affected compared to more physiological temperatures. When measuring constitutive dimerization this may or may not affect the apparent affinity; however, when monitoring the effect of ligand on receptor dimerization the ability of receptor to diffuse within the membrane may become significant. Adjusting the assay temperature will shorten the timeframe in which reliable measurements can be made in the BRET1 and BRET2 assays due to more rapid oxidation of the coelenterazine-h substrate. On the other hand, prolonged incubation with substrate and data collection at 37°C is necessary when using the EnduRen substrate (see Note 9).
19. Effect of ligands. The effect of ligand modulation on GPCR dimerization remains unclear and thus far appears highly receptor- and/or ligand-dependent. However, the BRET assay can be a useful tool in determining whether a ligand induces a change in affinity of dimers or a change in conformation between a pre-formed dimer complex. As long as the same population of transfected cells is used, the “conventional” quantification scheme [49] can be applied to interrogate this issue because the only difference is the presence or absence of ligand; all other parameters are the same.
20. Negative controls. Another receptor that doesn’t dimerize with the receptor of interest can be useful for distinguishing between specific and bystander BRET. Many class C receptors, e.g. GABAB, form obligate homodimers and have low to no affinity for class A receptors [49, 56].
References
- 1.Whorton MR, Bokoch MP, Rasmussen SG, Huang B, Zare RN, Kobilka B, Sunahara RK. A monomeric G protein-coupled receptor isolated in a high-density lipoprotein particle efficiently activates its G protein. Proc Natl Acad Sci U S A. 2007;104:7682–7687. doi: 10.1073/pnas.0611448104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bayburt TH, Leitz AJ, Xie G, Oprian DD, Sligar SG. Transducin activation by nanoscale lipid bilayers containing one and two rhodopsins. J Biol Chem. 2007;282:14875–14881. doi: 10.1074/jbc.M701433200. [DOI] [PubMed] [Google Scholar]
- 3.Angers S, Salahpour A, Bouvier M. Dimerization: an emerging concept for G protein-coupled receptor ontogeny and function. Annu Rev Pharmacol Toxicol. 2002;42:409–435. doi: 10.1146/annurev.pharmtox.42.091701.082314. [DOI] [PubMed] [Google Scholar]
- 4.Springael JY, Urizar E, Parmentier M. Dimerization of chemokine receptors and its functional consequences. Cytokine Growth Factor Rev. 2005;16:611–623. doi: 10.1016/j.cytogfr.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 5.Terrillon S, Bouvier M. Roles of G-protein-coupled receptor dimerization. EMBO Rep. 2004;5:30–34. doi: 10.1038/sj.embor.7400052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Milligan G. G protein-coupled receptor dimerization: function and ligand pharmacology. Mol Pharmacol. 2004;66:1–7. doi: 10.1124/mol.104.000497.. [DOI] [PubMed] [Google Scholar]
- 7.Liu R, Paxton WA, Choe S, Ceradini D, Martin SR, Horuk R, MacDonald ME, Stuhlmann H, Koup RA, Landau NR. Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell. 1996;86:367–377. doi: 10.1016/s0092-8674(00)80110-5. [DOI] [PubMed] [Google Scholar]
- 8.Benkirane M, Jin DY, Chun RF, Koup RA, Jeang KT. Mechanism of transdominant inhibition of CCR5-mediated HIV-1 infection by ccr5delta32. J Biol Chem. 1997;272:30603–30606. doi: 10.1074/jbc.272.49.30603. [DOI] [PubMed] [Google Scholar]
- 9.Issafras H, Angers S, Bulenger S, Blanpain C, Parmentier M, Labbe-Jullie C, Bouvier M, Marullo S. Constitutive agonist-independent CCR5 oligomerization and antibody-mediated clustering occurring at physiological levels of receptors. J Biol Chem. 2002;277:34666–34673. doi: 10.1074/jbc.M202386200. [DOI] [PubMed] [Google Scholar]
- 10.Mellado M, Rodriguez-Frade JM, Vila-Coro AJ, Fernandez S, Martin de Ana A, Jones DR, Toran JL, Martinez AC. Chemokine receptor homo- or heterodimerization activates distinct signaling pathways. EMBO J. 2001;20:2497–2507. doi: 10.1093/emboj/20.10.2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hernandez PA, Gorlin RJ, Lukens JN, Taniuchi S, Bohinjec J, Francois F, Klotman ME, Diaz GA. Mutations in the chemokine receptor gene CXCR4 are associated with WHIM syndrome, a combined immunodeficiency disease. Nat Genet. 2003;34:70–74. doi: 10.1038/ng1149. [DOI] [PubMed] [Google Scholar]
- 12.Lagane B, Chow KY, Balabanian K, Levoye A, Harriague J, Planchenault T, Baleux F, Gunera-Saad N, Arenzana-Seisdedos F, Bachelerie F. CXCR4 dimerization and beta-arrestin-mediated signaling account for the enhanced chemotaxis to CXCL12 in WHIM syndrome. Blood. 2008;112:34–44. doi: 10.1182/blood-2007-07-102103. [DOI] [PubMed] [Google Scholar]
- 13.Percherancier Y, Berchiche YA, Slight I, Volkmer-Engert R, Tamamura H, Fujii N, Bouvier M, Heveker N. Bioluminescence resonance energy transfer reveals ligand-induced conformational changes in CXCR4 homo- and heterodimers. J Biol Chem. 2005;280:9895–9903. doi: 10.1074/jbc.M411151200. [DOI] [PubMed] [Google Scholar]
- 14.Balabanian K, Lagane B, Pablos JL, Laurent L, Planchenault T, Verola O, Lebbe C, Kerob D, Dupuy A, Hermine O, Nicolas JF, Latger-Cannard V, Bensoussan D, Bordigoni P, Baleux F, Le Deist F, Virelizier JL, Arenzana-Seisdedos F, Bachelerie F. WHIM syndromes with different genetic anomalies are accounted for by impaired CXCR4 desensitization to CXCL12. Blood. 2005;105:2449–2457. doi: 10.1182/blood-2004-06-2289. [DOI] [PubMed] [Google Scholar]
- 15.Kramp BK, Sarabi A, Koenen RR, Weber C. Heterophilic chemokine receptor interactions in chemokine signaling and biology. Exp Cell Res. 2011;317:655–663. doi: 10.1016/j.yexcr.2010.11.014. [DOI] [PubMed] [Google Scholar]
- 16.Salanga CL, O’Hayre M, Handel T. Modulation of chemokine receptor activity through dimerization and crosstalk. Cell Mol Life Sci. 2009;66:1370–1386. doi: 10.1007/s00018-008-8666-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.El-Asmar L, Springael JY, Ballet S, Andrieu EU, Vassart G, Parmentier M. Evidence for negative binding cooperativity within CCR5-CCR2b heterodimers. Mol Pharmacol. 2005;67:460–469. doi: 10.1124/mol.104.003624. [DOI] [PubMed] [Google Scholar]
- 18.Sohy D, Parmentier M, Springael JY. Allosteric transinhibition by specific antagonists in CCR2/CXCR4 heterodimers. J Biol Chem. 2007;282:30062–30069. doi: 10.1074/jbc.M705302200. [DOI] [PubMed] [Google Scholar]
- 19.Sohy D, Yano H, de Nadai P, Urizar E, Guillabert A, Javitch JA, Parmentier M, Springael JY. Hetero-oligomerization of CCR2, CCR5, and CXCR4 and the protean effects of “selective” antagonists. J Biol Chem. 2009;284:31270–31279. doi: 10.1074/jbc.M109.054809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kenakin T. Functional selectivity and biased receptor signaling. J Pharmacol Exp Ther. 2011;336:296–302. doi: 10.1124/jpet.110.173948. [DOI] [PubMed] [Google Scholar]
- 21.Contento RL, Molon B, Boularan C, Pozzan T, Manes S, Marullo S, Viola A. CXCR4-CCR5: a couple modulating T cell functions. Proc Natl Acad Sci U S A. 2008;105:10101–10106. doi: 10.1073/pnas.0804286105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bulenger S, Marullo S, Bouvier M. Emerging role of homo- and heterodimerization in G-protein-coupled receptor biosynthesis and maturation. Trends Pharmacol Sci. 2005;26:131–137. doi: 10.1016/j.tips.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 23.Hernanz-Falcon P, Rodriguez-Frade JM, Serrano A, Juan D, del Sol A, Soriano SF, Roncal F, Gomez L, Valencia A, Martinez AC, Mellado M. Identification of amino acid residues crucial for chemokine receptor dimerization. Nat Immunol. 2004;5:216–223. doi: 10.1038/ni1027. [DOI] [PubMed] [Google Scholar]
- 24.Lemay J, Marullo S, Jockers R, Alizon M, Brelot A. On the dimerization of CCR5. Nat Immunol. 2005;6:535. doi: 10.1038/ni0605-535a. author reply 535–536. [DOI] [PubMed] [Google Scholar]
- 25.Hernanz-Falcon P, Rodriguez-Frade JM, Serrano A, Martinez-A C, Mellado M. Response to “On the dimerization of CCR5”. Nat Immunol. 2005;6:535–536. [Google Scholar]
- 26.Harikumar KG, Pinon DI, Miller LJ. Transmembrane Segment IV Contributes a Functionally Important Interface for Oligomerization of the Class II G Protein-coupled Secretin Receptor. Journal of Biological Chemistry. 2007;282:30363–30372. doi: 10.1074/jbc.M702325200. [DOI] [PubMed] [Google Scholar]
- 27.Harikumar KG, Happs RM, Miller LJ. Dimerization in the absence of higher-order oligomerization of the G protein-coupled secretin receptor. Biochimica et Biophysica Acta (BBA) - Biomembranes. 2008;1778:2555–2563. doi: 10.1016/j.bbamem.2008.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gao F, Harikumar KG, Dong M, Lam PCH, Sexton PM, Christopoulos A, Bordner A, Abagyan R, Miller LJ. Functional Importance of a Structurally Distinct Homodimeric Complex of the Family B G Protein-Coupled Secretin Receptor. Molecular Pharmacology. 2009;76:264–274. doi: 10.1124/mol.109.055756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pioszak AA, Harikumar KG, Parker NR, Miller LJ, Xu HE. Dimeric Arrangement of the Parathyroid Hormone Receptor and a Structural Mechanism for Ligand-induced Dissociation. Journal of Biological Chemistry. 2010;285:12435–12444. doi: 10.1074/jbc.M109.093138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harikumar KG, Ball AM, Sexton PM, Miller LJ. Importance of lipid-exposed residues in transmembrane segment four for family B calcitonin receptor homo-dimerization. Regulatory Peptides. 2010;164:113–119. doi: 10.1016/j.regpep.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu B, Chien EY, Mol CD, Fenalti G, Liu W, Katritch V, Abagyan R, Brooun A, Wells P, Bi FC, Hamel DJ, Kuhn P, Handel TM, Cherezov V, Stevens RC. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science. 2010;330:1066–1071. doi: 10.1126/science.1194396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Manglik A, Kruse AC, Kobilka TS, Thian FS, Mathiesen JM, Sunahara RK, Pardo L, Weis WI, Kobilka BK, Granier S. Crystal structure of the micro-opioid receptor bound to a morphinan antagonist. Nature. 2012 doi: 10.1038/nature10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu H, Wacker D, Mileni M, Katritch V, Han GW, Vardy E, Liu W, Thompson AA, Huang XP, Carroll FI, Mascarella SW, Westkaemper RB, Mosier PD, Roth BL, Cherezov V, Stevens RC. Structure of the human kappa-opioid receptor in complex with JDTic. Nature. 2012 doi: 10.1038/nature10939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang J, He L, Combs CA, Roderiquez G, Norcross MA. Dimerization of CXCR4 in living malignant cells: control of cell migration by a synthetic peptide that reduces homologous CXCR4 interactions. Mol Cancer Ther. 2006;5:2474–2483. doi: 10.1158/1535-7163.MCT-05-0261. [DOI] [PubMed] [Google Scholar]
- 35.Lee B, Doranz BJ, Ratajczak MZ, Doms RW. An intricate Web: chemokine receptors, HIV-1 and hematopoiesis. Stem Cells. 1998;16:79–88. doi: 10.1002/stem.160079. [DOI] [PubMed] [Google Scholar]
- 36.de la Fuente M, Noble DN, Verma S, Nieman MT. Mapping Human Protease-activated Receptor 4 (PAR4) Homodimer Interface to Transmembrane Helix 4. Journal of Biological Chemistry. 2012;287:10414–10423. doi: 10.1074/jbc.M112.341438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gurevich VV, Gurevich EV. GPCR monomers and oligomers: it takes all kinds. Trends in Neurosciences. 2008;31:74–81. doi: 10.1016/j.tins.2007.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Khelashvili G, Dorff K, Shan J, Camacho-Artacho M, Skrabanek L, Vroling B, Bouvier M, Devi LA, George SR, Javitch JA, Lohse MJ, Milligan G, Neubig RR, Palczewski K, Parmentier M, Pin JP, Vriend G, Campagne F, Filizola M. GPCR-OKB: the G Protein Coupled Receptor Oligomer Knowledge Base. Bioinformatics. 2010;26:1804–1805. doi: 10.1093/bioinformatics/btq264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Skrabanek L, Murcia M, Bouvier M, Devi L, George SR, Lohse MJ, Milligan G, Neubig R, Palczewski K, Parmentier M, Pin JP, Vriend G, Javitch JA, Campagne F, Filizola M. Requirements and ontology for a G protein-coupled receptor oligomerization knowledge base. BMC Bioinformatics. 2007;8:177. doi: 10.1186/1471-2105-8-177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hern JA, Baig AH, Mashanov GI, Birdsall B, Corrie JET, Lazareno S, Molloy JE, Birdsall NJM. Formation and dissociation of M1 muscarinic receptor dimers seen by total internal reflection fluorescence imaging of single molecules. Proceedings of the National Academy of Sciences. 2010;107:2693–2698. doi: 10.1073/pnas.0907915107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Marullo S, Bouvier M. Resonance energy transfer approaches in molecular pharmacology and beyond. Trends Pharmacol Sci. 2007;28:362–365. doi: 10.1016/j.tips.2007.06.007. [DOI] [PubMed] [Google Scholar]
- 42.Milligan G, Bouvier M. Methods to monitor the quaternary structure of G protein-coupled receptors. FEBS J. 2005;272:2914–2925. doi: 10.1111/j.1742-4658.2005.04731.x. [DOI] [PubMed] [Google Scholar]
- 43.Ayoub MA, Pfleger KDG. Recent advances in bioluminescence resonance energy transfer technologies to study GPCR heteromerization. Current Opinion in Pharmacology. 2010;10:44–52. doi: 10.1016/j.coph.2009.09.012. [DOI] [PubMed] [Google Scholar]
- 44.Issad T, Jockers R, Ali H, Haribabu B, Walker JM. Bioluminescence Resonance Energy Transfer to Monitor Protein-Protein Interactions. In: Walker JM, editor. Transmembrane Signaling Protocols. Vol. 332. Humana Press; 2006. pp. 195–209. [DOI] [PubMed] [Google Scholar]
- 45.Achour L, Kamal M, Jockers R, Marullo S, Luttrell LM, Ferguson SSG. Using Quantitative BRET to Assess G Protein-Coupled Receptor Homo- and Heterodimerization. In: Walker JM, editor. Signal Transduction Protocols. Vol. 756. Humana Press; 2011. pp. 183–200. [DOI] [PubMed] [Google Scholar]
- 46.Kubale V, Drinovec L, Vrecl M. Quantitative Assessment of Seven Transmembrane Receptors (7TMRs) Oligomerization by Bioluminescence Resonance Energy Transfer (BRET) Technology. In: Lapota DD, editor. Bioluminescence - Recent Advances in Oceanic Measurements and Laboratory Applications. InTech; 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pfleger KDG, Eidne KA. Illuminating insights into protein-protein interactions using bioluminescence resonance energy transfer (BRET) Nat Meth. 2006;3:165–174. doi: 10.1038/nmeth841. [DOI] [PubMed] [Google Scholar]
- 48.Kocan M, Pfleger KDG, Willars GB, Challiss RAJ. Receptor Signal Transduction Protocols. Vol. 746. Humana Press; 2011. Study of GPCR–Protein Interactions by BRET; pp. 357–371. [Google Scholar]
- 49.Hamdan FF, Percherancier Y, Breton B, Bouvier M. Current Protocols in Neuroscience. John Wiley & Sons, Inc; 2006. Monitoring Protein-Protein Interactions in Living Cells by Bioluminescence Resonance Energy Transfer (BRET) [DOI] [PubMed] [Google Scholar]
- 50.Boute N, Jockers R, Issad T. The use of resonance energy transfer in high-throughput screening: BRET versus FRET. Trends in Pharmacological Sciences. 2002;23:351–354. doi: 10.1016/s0165-6147(02)02062-x. [DOI] [PubMed] [Google Scholar]
- 51.James JR, Oliveira MI, Carmo AM, Iaboni A, Davis SJ. A rigorous experimental framework for detecting protein oligomerization using bioluminescence resonance energy transfer. Nat Meth. 2006;3:1001–1006. doi: 10.1038/nmeth978. [DOI] [PubMed] [Google Scholar]
- 52.Comps-Agrar L, Maurel D, Rondard P, Pin J-P, Trinquet E, Prézeau L, Luttrell LM, Ferguson SSG. Signal Transduction Protocols. Vol. 756. Humana Press; 2011. Cell-Surface Protein–Protein Interaction Analysis with Time-Resolved FRET and Snap-Tag Technologies: Application to G Protein-Coupled Receptor Oligomerization; pp. 201–214. [DOI] [PubMed] [Google Scholar]
- 53.Albizu L, Cottet M, Kralikova M, Stoev S, Seyer R, Brabet I, Roux T, Bazin H, Bourrier E, Lamarque L, Breton C, Rives ML, Newman A, Javitch J, Trinquet E, Manning M, Pin JP, Mouillac B, Durroux T. Time-resolved FRET between GPCR ligands reveals oligomers in native tissues. Nat Chem Biol. 2010;6:587–594. doi: 10.1038/nchembio.396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ferre S, Franco R. Oligomerization of G-protein-coupled receptors: A reality. Current Opinion in Pharmacology. 2010;10:1–5. doi: 10.1016/j.coph.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McLean AJ, Milligan G. Ligand regulation of green fluorescent protein-tagged forms of the human beta(1)- and beta(2)-adrenoceptors; comparisons with the unmodified receptors. Br J Pharmacol. 2000;130:1825–1832. doi: 10.1038/sj.bjp.0703506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mercier J-Fo, Salahpour A, Angers Sp, Breit A, Bouvier M. Quantitative Assessment of beta1- and beta2-Adrenergic Receptor Homo- and Heterodimerization by Bioluminescence Resonance Energy Transfer. Journal of Biological Chemistry. 2002;277:44925–44931. doi: 10.1074/jbc.M205767200. [DOI] [PubMed] [Google Scholar]
- 57.Salahpour A, Masri B. Experimental challenge to a ‘rigorous’ BRET analysis of GPCR oligomerization. Nat Meth. 2007;4:599–600. doi: 10.1038/nmeth0807-599. [DOI] [PubMed] [Google Scholar]
- 58.Veatch W, Stryer L. The dimeric nature of the gramicidin A transmembrane channel: Conductance and fluorescence energy transfer studies of hybrid channels. Journal of Molecular Biology. 1977;113:89–102. doi: 10.1016/0022-2836(77)90042-0. [DOI] [PubMed] [Google Scholar]
- 59.Griesbeck O, Baird GS, Campbell RE, Zacharias DA, Tsien RY. Reducing the Environmental Sensitivity of Yellow Fluorescent Protein. Journal of Biological Chemistry. 2001;276:29188–29194. doi: 10.1074/jbc.M102815200. [DOI] [PubMed] [Google Scholar]
- 60.Patterson G, Day RN, Piston D. Fluorescent protein spectra. Journal of Cell Science. 2001;114:837–838. doi: 10.1242/jcs.114.5.837. [DOI] [PubMed] [Google Scholar]
- 61.Robitaille M, Héroux I, Baragli A, Hébert TE, Rich PB, Douillet C. Bioluminescence. Vol. 574. Humana Press; 2009. Novel Tools for Use in Bioluminescence Resonance Energy Transfer (BRET) Assays; pp. 215–234. [DOI] [PubMed] [Google Scholar]
- 62.Kaczor AA, Selent J. Oligomerization of G protein-coupled receptors: biochemical and biophysical methods. Curr Med Chem. 2011;18:4606–4634. doi: 10.2174/092986711797379285. [DOI] [PubMed] [Google Scholar]
- 63.Ciruela F, Vilardaga JP, Fernandez-Duenas V. Lighting up multiprotein complexes: lessons from GPCR oligomerization. Trends in Biotechnology. 2010;28:407–415. doi: 10.1016/j.tibtech.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]