

Abstract
Accumulating evidence suggests that activated pancreatic stellate cells (PSC) play an important role in chronic pancreatitis (CP), and inhibition of the activated PSC is considered as a potential strategy for the treatment and prevention of CP. Herein, we disclose our findings that apigenin and its novel analogues suppress the proliferation and induce apoptosis in PSC which reduce the PSC-mediated fibrosis in CP. Chemical modifications of apigenin have been directed to build a focused library of O-alkylamino-tethered apigenin derivatives at 4′-O position of the ring C with the attempt to enhance the potency and drug-like properties including aqueous solubility. A number of compounds such as 14, 16, and 24 exhibited potent antiproliferative effects as well as improved aqueous solubility. Intriguingly, apigenin, new analogues 23 and 24 displayed significant efficacy to reduce pancreatic fibrosis even at a low dose of 0.5 mg/kg in our proof-of-concept study using a preclinical in vivo mouse model of CP.
Keywords: Chronic pancreatitis, Pancreatic stellate cells, Fibrosis, Apigenin, Apigenin analogues, Therapy
1. Introduction
Chronic pancreatitis (CP) is a progressive, non-curable disorder of the pancreas.1 Pathologically, both the endocrine and exocrine pancreas undergo progressive and often irreversible morphological changes, including glandular fibrosis.2–5 In the United States, disorders of the exocrine pancreas affect over 1 million patients and result in a cost over $3.7 billion annually.6,7 Current treatment options for CP are limited to supportive and palliative care; patients with advanced disease can be managed with endoscopic and/or surgical pancreatic decompression, denervation, resection, bypass or transplantation.8,9 Overall, patients have a poor quality of life, and are burdened by chronic abdominal pain, increased hospitalizations, impaired digestion, diarrhea, weight loss, diabetes, complications like pseudocysts and an increased risk of pancreatic cancer.9 Therefore, the development of effective, safe and affordable therapeutic agents remains a critical need.
Within the last two decades, it has been well-established that pancreatic stellate cells (PSC) are responsible for the fibrotic component of CP, and suppressing PSC is therefore a potential therapeutic target for the disease.10–12 In the normal pancreas, PSC are inactive/quiescent, whereas during tissue injury, PSC display an activated, myofibroblastic phenotype, with increased proliferation, motility, and secretion of extracellular matrix proteins.10–12 CP favors the perpetual activation of PSC.13 Consequently, a promising strategy for the prevention and treatment of CP involves limiting the proliferation and inducing apoptosis of activated PSC.13–16
The sentinel acute pancreatitis event (SAPE) hypothesis provides a unified model for the pathogenesis of CP.17 After studying cases of hereditary pancreatitis, Whitcomb et al. found that 50% of patients with gain-of-function trypsinogen mutations experienced repeated episodes of acute pancreatitis (AP) that later developed into CP.1,17 Regardless of the inciting etiology(s) of the sentinel event of AP, recurrent episodes of AP cause CP. AP is initiated with acinar cell injury, characterized by premature acinar zymogen activation, recruitment of inflammatory cells, auto-digestion and necrosis of acinar and ductal cells, subsequent anti-inflammatory response, and PSC-dependent scarring.1,5,17–19 Recurrent pancreatic injury overwhelms normal repair mechanisms, favoring progressive irreversible fibrosis.1,5,17 We have directed our efforts to develop novel compounds that would limit repeated pancreatic injury, and thus minimize the progression of CP.
Natural products, especially common dietaries consumed on a daily basis, continue to serve as a valuable source in developing drug-like candidates for chemoprevention and chemotherapy.20–22 Flavonoids, which are ubiquitously distributed in many dietary plant materials, have received a great deal of attention because some of them have been shown to exert various beneficial effects on human health.23 Apigenin (Figure 1) is abundantly present in common fruits and vegetables, and has gained particular interest in recent years as a beneficial and health-promoting agent because of its low intrinsic toxicity.24 Apigenin has been demonstrated to possess various clinically relevant properties such as anti-inflammatory, anti-oxidant, antiproliferative and pro-apoptotic activities likely through multiple mechanisms.25 Our research efforts on this natural product with low intrinsic toxicity have led to the discovery that apigenin can ameliorate the stromal fibrosis characteristic of CP. Despite its promising anti-fibrotic property, apigenin suffers from poor aqueous solubility and low metabolic stability like most flavonoids, limiting its clinical potential. While many research groups focused on developing novel compounds based on the apigenin structure as anticancer agents, most of these apigenin derivatives suffered from unfavorable physicochemical properties, including limited aqueous solubility, and none of them has been approved for clinical investigation.26,27 Herein, we report our design and synthesis of novel apigenin analogues that suppress the proliferation and promote apoptosis in activated PSC with improved potency and favorable physicochemical properties. More importantly, we also disclose that at a low dose, new analogues 23 and 24 are as efficacious as apigenin in reducing pancreatic fibrosis in a preclinical animal model, providing the proof-of-concept as potential therapeutics for CP.
Figure 1. Chemical structure of apigenin.
2. Results and discussion
2.1. Design
Considerable efforts on the modifications of apigenin as the lead compound in anticancer drug design have been made. Reported structural-activity relationship (SAR) studies indicate that the A ring of apigenin as well as its C ring are suitable for diverse modifications.28–31 In addition, increasing studies have demonstrated that the polymethoxylated flavones or flavone analogues with nonpolar and hydrophobic substituents on A ring generally exhibit more potent antiproliferative activities against various human cancer cell lines.28,32 Moreover, methylated flavones have dramatically higher intestinal permeability and higher metabolic stability.33–35 Combined with the structural features of the SAR trends, we directed our initial optimization effort to discover novel apigenin derivatives by introducing aqueous solubility-enhancing moieties at 4′-O position of apigenin with 5,7-dimethoxy groups on the A ring.
2.2. Chemistry
The synthesis of new apigenin derivatives with chemical optimizations on 4′-hydroxyl group is outlined in Scheme 1. The key intermediate 5 was prepared in a three-step synthesis starting with 1-(2-hydroxy-4,6-dimethoxyphenyl)ethanone (2) and 4-allyloxybenzaldehyde (3) according to a literature procedure.36 As shown in Scheme 1, base-catalyzed aldol condensation of 2 with 3 afforded the chalcone 4 in a yield of 76% with a simple purification. The chalcone was cyclized in the presence of catalytic iodine in dimethyl sulfoxide at 140 °C to provide the flavone 5 in high yield. The allyl protecting group of flavones 5 was cleaved with a catalytic amount of Pd(PPh3)4 in the presence of K2CO3 in MeOH at reflux for 4 h to obtain the key intermediate 4′-hydroxyflavone 6 for direct use without further purification.
Scheme 1a.

aReagents and conditions: (a) 4-allyloxybenzaldehyde (3), 50% NaOH/H2O, EtOH, rt, 16 h, 76%; (b) cat. I2, DMSO, 140 °C, 4 h, 91%; (c) cat. Pd(PPh3)4, K2CO3, MeOH, 90 °C, 4 h, 95%; (d) R1OH, Ph3P, DIAD, THF, rt, 16 h, 81–94%; (e) R2H, KI, K2CO3, acetone, reflux, 18 h, 66–99%; (f) Boc-R3OH, Ph3P, DIAD, THF, rt, 2 h; (g) TFA, CH2Cl2, 0 °C to rt, 3 h, 59–90% (two steps).
New analogues 7–13 were conveniently synthesized by Mitsunobu reaction of the key intermediate 6 with appropriate substituted alcohols in high yields (81–94%). Alkylation of the bromide intermediate 7 with the corresponding amine in the presence of K2CO3 and KI in acetone introduced basic functionalities providing final compounds 14–18 in 66–99% yields. Mitsunobu coupling of 6 with N-Boc-protected amino alcohols followed by the Boc-deprotection with the treatment of TFA in CH2Cl2 afforded analogues 19–21 with diversified O-alkylamino side chains in 59–90% yields (two steps).
The synthetic route to 3-amino-2-hydroxypropoxy-flavonesis outlined in Scheme 2. The reaction of the key intermediate 6 with excessive epichlorohydrin (22) and successive treatment of the intermediate epoxide (23) with appropriate amines under reflux afforded the desired derivatives 24–26 in yields of 71–94%. The Mitsunobu coupling of 6 with (2,2-dimethyl-1,3-dioxolan-4-yl)methanol (solketal, 27) generated an intermediate which was subjected to cleavage conditions using 0.5% HCl (aq.) in EtOH to produce the 1,2-diol derivative 28. For the synthesis of diversified apigenin analogues on side chain of the ring C at para-position as depicted in Scheme 3, the starting material 2 was condensed with 4-bromobenzaldehyde (29) to give chalcone 30, which was then cyclized to give the key intermediate 31. Compound 33 was obtained by Suzuki coupling reaction of 31 with 2-fluoropyridine-5-boronic acid (32) in the presence of Pd(dppf)Cl2 catalyst in a yield of 85%. Palladium-catalyzed Buchwald-Hartwig amination reaction of 31 with N,N-dimethylethylenediamine (34) or 2-(pyrrolidin-1-yl)ethanamine (35) afforded the targeted compounds 36 and 37 in yield of 64% and 57%, respectively.
Scheme 2a.

aReagents and conditions: (a) epichlorohydrin (22), K2CO3, acetone, reflux, 24 h, 67%; (b) R4H, K2CO3, EtOH, reflux, 3 h, 71–94%; (c) Ph3P, DIAD, THF, (2,2-dimethyl-1,3-dioxolan-4-yl)methanol (27), rt, 4 h; (d) 0.5 N HCl (aq.), EtOH, reflux, 2 h, 71% (two steps).
Scheme 3a.

aReagents and conditions: (a) 4-bromobenzaldehyde (29), 50% NaOH/H2O, EtOH, rt, 16 h, 65%; (b) cat. I2, DMSO, 140 °C, 4 h, 92%; (c) 2-fluoropyridine-5-boronic acid (32), Pd(dppf)Cl2, KOAc, THF/EtOH/H2O, 80 °C, 18 h, 85%; (d) NH2R5 [N,N-dimethylethylenediamine (34) for 36 or 2-(pyrrolidin-1-yl)ethanamine (35) for 37], Pd2(dba)3, NaOtBu, (±)-BINAP, toluene, 80 °C, 48 h, 57–64%.
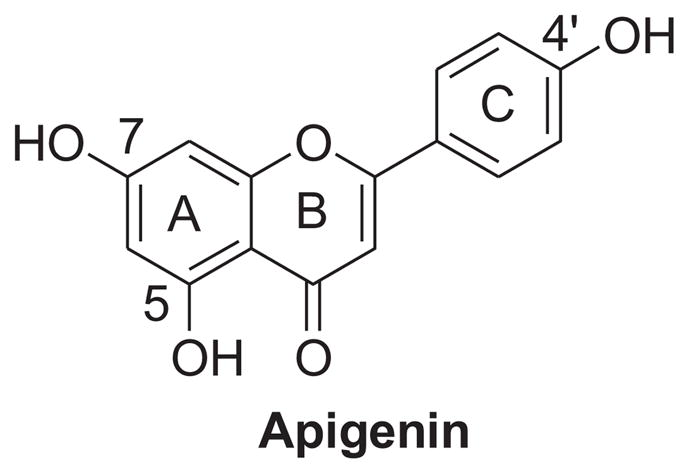
Scheme 4 outlines the synthesis of demethylated derivatives 38 and 39. Generation of 38 was achieved in 79% yield by a mono-demethylation of 16 using 2 equiv of boron tribromide at room temperate for 2 h. Both methyl groups on A-ring of 16 were successfully removed by treatment with 3 equiv of boron tribromide for 24 h, leading to the demethylated analogue 39 in a yield of 73%.
Scheme 4a.
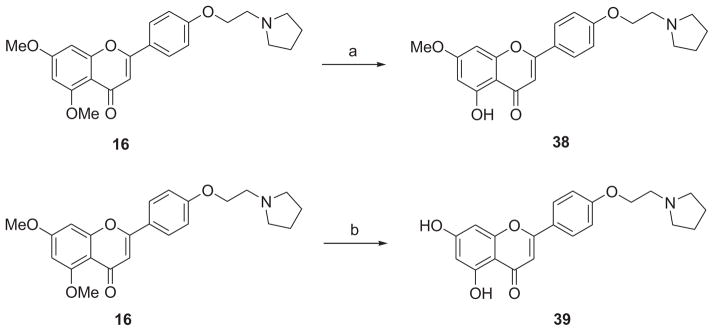
aReagents and conditions: (a) 1 N BBr3 (in CH2Cl2), CH2Cl2, rt, 2h, 79%; (b) 1 N BBr3 (in CH2Cl2), CH2Cl2, rt, 24 h, 73%.
2.3. Biology
The calculated lipophilicity (cLogP) and topological polar surface area (tPSA) values of all newly synthesized analogues are listed in Table 1. The results indicate that all these new compounds meet the criteria of Lipinski’s “Rule of Five” and may have favorable physicochemical properties. To explore a meaningful SAR and examine how the substitutions on the key moieties affect biological activities of new apigenin derivatives, we first evaluated the in vitro antiproliferative effects of these analogues on transformed PSC using AlamarBlue Cell Viability Assay (Life Technologies) as described in the Experimental Section. AlamarBlue is a non-toxic reagent that is converted to a highly fluorescent end product by viable cells. The capabilities of these new analogues to inhibit the proliferation of transformed PSC are summarized in Table 1.
Table 1.
Effects of apigenin and newly synthesized apigenin analogues on PSC proliferation.
| Entry | cLogPa | tPSAb | Inhibitory Effect (%)c | ||
|---|---|---|---|---|---|
| 5 μM | 10 μM | 20 μM | |||
| Apigenin (1) | 2.33 | 90.9 | 11% | 34% | 64% |
| 6 | 2.73 | 68.9 | NEd | NE | NE |
| 7 | 3.86 | 57.9 | NE | NE | 20% |
| 8 | 3.35 | 57.9 | NE | NE | 21% |
| 9 | 3.29 | 61.2 | 28% | 49% | NDe |
| 10 | 2.73 | 61.2 | 19% | 51% | 65% |
| 11 | 2.45 | 70.4 | NE | NE | NE |
| 12 | 3.06 | 61.2 | 36% | 59% | 86% |
| 13 | 2.69 | 78.2 | 7% | 14% | 33% |
| 14 | 3.84 | 61.2 | 45% | 64% | 97% |
| 15 | 2.66 | 64.4 | 22% | 42% | 67% |
| 16 | 3.48 | 61.2 | 48% | 66% | 98% |
| 17 | 3.54 | 61.2 | 33% | 60% | 72% |
| 18 | 3.22 | 87.5 | 22% | 37% | 67% |
| 19 | 3.08 | 69.9 | NE | 21% | ND |
| 20 | 1.93 | 83.9 | 6% | 35% | 70% |
| 21 | 2.43 | 83.9 | 22% | 39% | ND |
| 23 | 2.50 | 70.4 | 69% | 89% | 93% |
| 24 | 3.03 | 81.4 | 34% | 65% | 95% |
| 25 | 2.66 | 81.4 | 21% | 37% | 57% |
| 26 | 2.13 | 81.4 | 42% | 60% | 71% |
| 28 | 1.77 | 98.4 | NE | 11% | 16% |
| 31 | 3.80 | 48.7 | 12% | 10% | 22% |
| 33 | 4.05 | 61.6 | NE | 6% | 19% |
| 36 | 2.72 | 63.9 | 30% | 60% | 84% |
| 37 | 3.13 | 63.9 | 21% | 51% | 80% |
| 38 | 3.28 | 72.1 | NE | 39% | 99% |
| 39 | 2.98 | 83.1 | 2% | 71% | 90% |
Values are mean of at least three independent experiments.
NE: No effect.
ND: Not determined.
The key intermediate 6 was found to display no significant antiproliferative effect even at 20 μM. After introduction of an O-bromoalkyl moiety or an O-fluoroalkyl moiety into the derivative 6 at 4′-O position, compounds 7 and 8 showed a slightly increased antiproliferative effects in comparison with 6, indicating that appropriate modifications on 4′-O position may regain the antiproliferative activity. O-Alkylamino-tethered derivatives 9, 10 and 12 exhibited a moderate antiproliferative activity at 10 μM with inhibitory effects of 49%, 51% and 59%, respectively. This finding suggests that optimizations with a nitrogen-containing hydrophilic moiety at 4′-O position appears to be a viable strategy to yield more potent compounds with a better aqueous solubility. To this end, compounds 14 with a piperidinyl moiety and 16 with a pyrrolidinyl group displayed potent antiproliferative activity at 10 μM with inhibitory effects of 64% and 66%, respectively. Meanwhile, we found that the tertiary amines with alkylated amino groups appeared to be more favorable than secondary or primary amines with free amino groups at the terminal of the side chains. For instance, in comparison with compounds 14 and 16, analogues 19–21 only exhibited a moderate to low inhibitory effects. The similar trend of SAR was also observed for derivatives 24–26 and 28. Compound 28 with a terminal OH group at the tail resulted in a dramatic loss of activities compared with its according analogues 24–26 with a terminal amino moiety. Interestingly, compound 23 with an epoxide was identified as a highly potent inhibitor suppressing PSC proliferation.
Structural modifications of para-position on C-ring with diversified substitutions were also investigated. Compound 33 with a pyridinyl group on the para-position resulted in a substantial loss of the activity. In contrast, introduction of 2-dimethylamino-ethylamino moiety or 2-pyrrolidin-1-yl-ethylamino group displayed significantly better antiproliferative activities at 10 μM with inhibitory effects of 60% and 51%, respectively. Demethylation of compound 16 resulted in generation of two O-demethylated compounds 38 and 39. We found that neither mono-demethylation nor full demethylation of the methoxy groups on the A-ring is favorable for the enhancement of activity, and thus more extensive SAR study on the A-ring was not pursued.
Since one goal of our drug discovery effort was to identify new apigenin derivatives with improved water-solubility and oral bioavailability, aqueous solubility of several selected analogues with enhanced antiproliferative effects was determined by an HPLC analysis method.37 As depicted in Figure 2, compounds 14, 16 and 24 (in the form of HCl salt) have demonstrated to possess more favorable aqueous solubility, with a saturated concentration of 17.4, 13.7 and 84.1 mg/mL, respectively, while that of apigenin is only 2.16 μg/mL.26
Figure 2. Aqueous solubility of apigenin and selected apigenin derivatives.
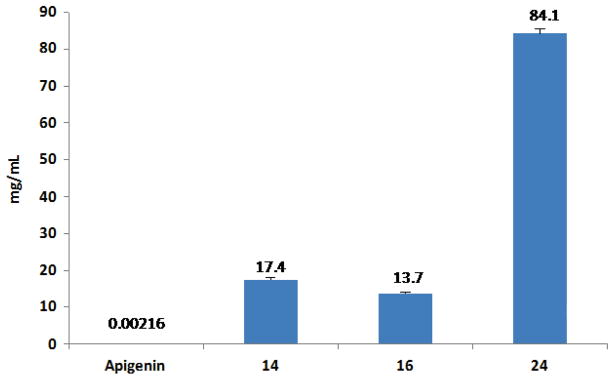
Compounds 14, 16 and 24 (in the form of HCl salt) showed significantly improved solubility as compared with apigenin.
To investigate more detailed information about the antiproliferative effects of this series of apigenin derivatives against human PSC, compounds 23 (HJC0561) and 24 (HJC05100) were selected based on their potency or aqueous solubility for further evaluations using cell proliferation and cell death assays, as well as an in vivo model of CP. As shown in Figure 3A, both analogues 23 and 24 inhibited PSC proliferation at lower doses than apigenin, indicating their enhanced potency in vitro. With logarithmic transformation and nonlinear regression, a best-fit curve was generated, allowing determination of their IC50, representing the concentration at which the compound causes 50% inhibition of PSC proliferation (Figure 3B). Compound 23 was identified as a highly potent analogue with an IC50 value of 2.5 ± 0.6 μM. Compound 24 was slightly less potent with an IC50 value of 8.0 ± 1.8 μM, and that of apigenin was 18.6 ± 1.6 μM. The irreversible fibrosis that defines CP is mediated by activated PSC, which produce and remodel the extracellular matrix (ECM).10 Therefore, the potent antiproliferative effect of these new analogues on PSC supports their further development as anti-fibrotic agents for CP.
Figure 3. Effect of apigenin and its analogues on PSC proliferation.
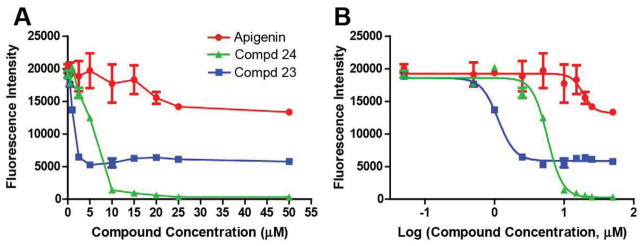
Human PSC were treated with apigenin or analogues at various doses for 24 h. (A) Representative data from one experiment: cell viability was measured using the AlamarBlue colorimetric assay. (B) The IC50 values of apigenin, 23, and 24 are 18.6 ± 1.6 μM, 2.5 ± 0.6 μM, and 8.0 ± 1.8 μM, respectively. The calculated IC50 values are derived from the mean ± SEM of at least three independent experiments.
These two representative analogues were also investigated to determine whether the growth inhibition induced by compounds 23 and 24 in PSC was attributed to apoptosis. Apoptosis was determined using the Cell Death Detection ELISAPLUS assay. This sandwich-enzyme-immunoassay involved antibodies binding to cytoplasmic nucleosomes, which were specific to the process of apoptosis rather than necrosis. Analogue 23 was found to be quite potent, inducing greater PSC cell death at lower concentrations than apigenin (Figure 4A). At low concentrations, compound 24 failed to induce apoptosis; however, between the concentrations of 25–35 μM, it produced a steep dose-response curve, and concentrations beyond 35 μM induced significant cell death. Next, the dose-response curve was generated (Figure 4B). The EC50 represented the concentration at which the compound yielded half of the maximal amount of cell death. The most potent analogue 23 exhibited the lowest EC50 value of 9.6 ± 1.8 μM, while the EC50 value of apigenin was 24.5 ± 2.5 μM, and that of 24 was 35.2 ± 5.5 μM. The capacity of the compounds to induce cell death in activated PSC would most likely translate into reduced fibrosis in CP. Analogue 23 more potently induced PSC apoptosis than apigenin, while 24 appeared to act through a different cellular mechanism, inducing stellate cell toxicity at concentrations greater than 25 μM.
Figure 4. Effect of apigenin and its analogues on PSC apoptosis.
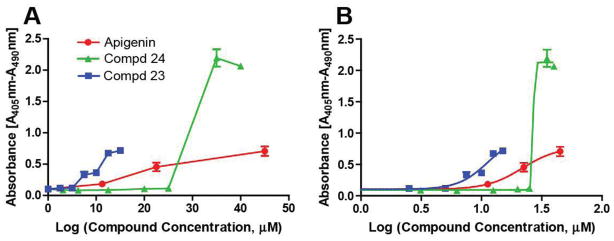
Human PSC were treated with apigenin or analogues at various doses for 14 h. (A) Representative data from one experiment: Apoptosis was measured using the Cell Death Detection ELISAPLUS assay. (B) The EC50 values of apigenin, 23, and 24 are 24.5 ± 2.5 μM, 9.6 ± 1.8 μM, and 35.2 ± 5.5 μM, respectively. The calculated EC50 values are derived from the mean ± SEM of at least three independent experiments.
These encouraging results prompted us to further test 23 and 24 as lead compounds for the development of a new series of anti-fibrotic agents which may be useful for treating CP.38 In order to evaluate the anti-fibrotic effect of these new compounds as a proof-of-concept, we tested the compounds in a preclinical animal model of CP. CP was induced in mice using the well-established model of repeated cerulean (CR) injections.39,40 Treatment with vehicle, apigenin or analogues (0.5 mg/kg daily gavage) was started the second week of the experiment and continued with CP induction. After four weeks, the pancreata were processed and stained for fibronectin, which is a major component of the ECM. The 400 × microscope image in Figure 5A showed a large amount of periacinar and perilobular fibrosis (brown color), edema (the space between lobules), and atrophic, irregularly shaped acini.41 Treatment with apigenin and analogues 23 and 24 significantly decreased the stromal fibrosis of CP, reduced tissue edema, and limited acinar cell damage (Figures 5B–D). ImageJ analysis of the slides quantified the significant decrease in fibrosis (p < 0.001) (Figure 5E). Despite no statistical difference between apigenin and compounds 23 or 24 at the low dose of 0.5 mg/kg, these data demonstrated that apigenin and new analogues significantly reduced fibrosis in the pre-clinical animal model of CP. The present investigation has provided a proof-of-concept study that apigenin and analogues have the potential to function as anti-fibrotic agents in CP. The relevant mechanistic studies and further dose-response investigation of in vivo efficacy are currently ongoing.
Figure 5. Effect of apigenin and its analogues in preclinical animal model of CP.

CP was induced using repeated cerulean (CR) injections as outlined in the methods. Control mice received PBS (n = 5), and mice injected with CR developed CP (n = 10–11). Treatment with vehicle (0.5% methylcellulose + 0.025% Tween 20 in ddH2O), apigenin or analogues (0.5 mg/kg, daily gavage) started the second week and was continued 3 additional weeks while continuing CP induction. Pancreata were stained for fibronectin by IHC, and counter-stained with hematoxylin. Representative 400 × images of each group are shown: (A) CR + Vehicle; (B) CR + Apigenin; (C) CR + 24; and (D) CR + 23. ImageJ analysis of the slides quantified the percent area of brown fibronectin stain in (E). *** represents p < 0.001, comparing to the effect from apigenin at the same dosage.
3. Conclusion
Accumulating evidence suggests that activated PSC play an important role in CP. We have proposed a potential strategy for the prevention and treatment of CP by decreasing the proliferation and inducing apoptosis of PSC. While a few flavone-based compounds have previously been reported to possess antiproliferative effects for cancer, we are the first to show that apigenin and its novel analogues suppress the proliferation of PSC and reduce the associated fibrosis. Chemical modifications of apigenin have been directed to build a focused library of O-alkylamino-tethered apigenin derivatives at 4′-O position of the ring C with the attempt to enhance the potency and drug-like properties including aqueous solubility. A series of novel apigenin analogues have been synthesized from the key intermediate 6, and a number of compounds such as 14, 16, and 24 have been identified to exhibit potent antiproliferative effects as well as improved aqueous solubility. Even at a low dose of 0.5 mg/kg, apigenin and new analogues 23 and 24 significantly reduced the fibrotic response in a preclinical animal model of CP, providing a proof-of-concept study that supports their development as promising therapeutics for CP.
4. Experimental section
4.1. Chemistry
4.1.1. General
All commercially available starting materials and solvents were reagent grade, and used without further purification. Reactions were performed under a nitrogen atmosphere in dry glassware with magnetic stirring. Preparative column chromatography was performed using silica gel 60, particle size 0.063–0.200 mm (70–230 mesh, flash). Analytical TLC was carried out employing silica gel 60 F254 plates (Merck, Darmstadt). Visualization of the developed chromatograms was performed with detection by UV (254 nm). NMR spectra were recorded on a Bruker-600 (1H, 600 MHz; 13C, 150 MHz) spectrometer. 1H and 13C NMR spectra were recorded with TMS as an internal reference. Chemical shifts were expressed in ppm, and J values were given in Hz. High-resolution mass spectra (HRMS) were obtained from Thermo Fisher LTQ Orbitrap Elite mass spectrometer. Parameters include the following: Nano ESI spray voltage was 1.8 kV; Capillary temperature was 275 °C and the resolution was 60,000; Ionization was achieved by positive mode. Melting points were measured on a Thermo Scientific Electrothermal Digital Melting Point Apparatus and uncorrected. Purity of final compounds was determined by analytical HPLC, which was carried out on a Shimadzu HPLC system (model: CBM-20A LC-20AD SPD-20A UV/VIS). HPLC analysis conditions: Waters μBondapak C18 (300 × 3.9 mm); flow rate 0.5 mL/min; UV detection at 270 and 254 nm; linear gradient from 30% acetonitrile in water [0.1%, trifluoroacetic acid (TFA)] to 100% acetonitrile (0.1% TFA) in 20 min followed by 30 min of the last-named solvent. All biologically evaluated compounds are > 95% pure.
4.1.2. 2-(4-Hydroxy-phenyl)-5,7-dimethoxy-chromen-4-one (6)
Starting from the commercially available 4-allyloxybenzaldehyde (3) and 1-(2-hydroxy-4,6-dimethoxy-phenyl)-ethanone (2), the key intermediate 6 was prepared in three steps according to literature procedures.36 HPLC purity 97.0% (tR = 16.94 min). 1H NMR (600 MHz, DMSO-d6) δ 7.88 (d, 2H, J = 9.0 Hz), 6.90 (d, 2H, J = 9.0 Hz), 6.83 (d, 1H, J = 2.4 Hz), 6.58 (s, 1H), 6.49 (d, 1H, J = 2.4 Hz), 3.89 (s, 3H), 3.82 (s, 3H). HRMS (ESI) calcd for C17H15O5 299.0914 (M + H)+, found 299.0917.
4.1.3. 2-(4-(2-Bromoethoxy)phenyl)-5,7-dimethoxy-chromen-4-one (7)
To a solution of 6 (200 mg, 0.67 mmol) and Ph3P (351 mg, 1.34 mmol) in THF (10 mL) was added 2-bromoethanol (168 mg, 1.34 mmol) and diisopropylazodicarboxylate (DIAD, 271 mg, 1.34 mmol). The mixture was stirred at r.t. for 16 h. The reaction mixture was diluted with ethyl acetate (EtOAc, 200 mL) and extracted with H2O (40 mL). The organic layer was washed with brine (10 mL), dried over anhydrous Na2SO4, and concentrated to give the crude product. This residue was purified with silica gel column (EtOAc) to provide 7 (240 mg, 88%) as a white solid (mp 202–203 °C). HPLC purity 99.6% (tR = 21.17 min). 1H NMR (600 MHz, DMSO-d6) δ 8.00 (d, 2H, J = 8.4 Hz), 7.12 (d, 2H, J = 9.0 Hz), 6.86 (d, 1H, J = 2.4 Hz), 6.68 (s, 1H), 6.50 (d, 1H, J = 1.8 Hz), 4.43 (t, 2H, J = 5.4 Hz), 3.90 (s, 3H), 3.85(t, 2H, J = 5.4 Hz), 3.83 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 175.6, 163.6, 160.4, 160.2, 159.5, 159.1, 127.8, 123.5, 115.1, 108.3, 106.9, 96.2, 93.4, 68.0, 56.1, 56.0, 31.2. HRMS (ESI) calcd for C19H18BrO5 405.0332 (M + H)+, found 405.0334.
4.1.4. 2-(4-(2-Fluoroethoxy)phenyl)-5,7-dimethoxy-chromen-4-one (8)
8 was prepared in 90% yield by a procedure similar to that used to prepare 7. The title compound was obtained as a white solid (mp 187–188 °C). HPLC purity 97.3% (tR = 19.56 min). 1H NMR (600 MHz, CDCl3) δ 7.82 (d, 2H, J = 10.2 Hz), 7.03 (d, 2H, J = 10.2 Hz), 6.59 (s, 1H), 6.55 (d, 1H, J = 2.4 Hz), 6.37 (d, 1H, J = 1.8 Hz), 4.82–4.84 (m, 1H), 4.74–4.76 (m, 1H), 4.30–4.32 (m, 1H), 4.26–4.27 (m, 1H), 3.95 (s, 3H), 3.91 (s, 3H). 13C NMR (150 MHz, CDCl3) δ 177.8, 164.1, 161.0, 160.9, 160.6, 160.0, 127.8, 124.6, 115.1, 109.4, 108.0, 96.2, 93.0, 81.9 (d, J = 171.5 Hz), 67.4 (d, J = 20.3 Hz), 56.6, 55.9. HRMS (ESI) calcd for C19H18FO5 345.1133 (M + H)+, found 345.1135.
4.1.5. 5,7-Dimethoxy-2-(4-(1-methylpiperidin-4-yloxy)phenyl)-chromen-4-one (9)
9 was prepared in 94% yield by a procedure similar to that used to prepare 7. The title compound was obtained as a white solid (mp 143–144 °C). HPLC purity 99.3% (tR = 15.86 min). 1H NMR (600 MHz, CDCl3) δ 7.80 (d, 2H, J = 9.0 Hz), 6.99 (d, 2H, J = 9.0 Hz), 6.58 (s, 1H), 6.55 (d, 1H, J = 2.4 Hz), 6.37 (d, 1H, J = 3.0 Hz), 4.44 (s, 1H), 3.95 (s, 3H), 3.91 (s, 3H), 2.72 (s, 2H), 2.38 (s, 2H), 2.34 (s, 3H), 2.04–2.08 (m, 2H), 1.88–1.92 (m, 2H). 13C NMR (150 MHz, CDCl3) δ 177.8, 164.0, 161.0, 160.8, 160.1, 160.0, 127.8, 123.9, 116.2, 109.4, 107.8, 96.2, 93.0, 56.6, 55.9, 52.4, 46.1, 30.6. HRMS (ESI) calcd for C23H26NO5 396.1806 (M + H)+, found 396.1808.
4.1.6. 2-(4-(2-Dimethylaminoethoxy)phenyl)-5,7-dimethoxy-chromen-4-one (10)
10 was prepared in 81% yield by a procedure similar to that used to prepare 7. The title compound was obtained as a pale yellow solid (mp 153–154 °C). HPLC purity 98.8% (tR = 15.25 min). 1H NMR (600 MHz, CDCl3) δ 7.81 (d, 2H, J = 9.0 Hz), 7.02 (d, 2H, J = 9.0 Hz), 6.59 (s, 1H), 6.55 (d, 1H, J = 1.8 Hz), 6.37 (d, 1H, J = 1.8 Hz), 4.14 (t, 2H, J = 6.0 Hz), 3.95 (s, 3H), 3.91 (s, 3H), 2.78 (t, 2H, J = 6.0 Hz), 2.36 (s, 6H). 13C NMR (150 MHz, CDCl3) δ 177.8, 164.0, 161.4, 161.0, 160.8, 160.0, 127.7, 124.1, 115.1, 109.4, 107.9, 96.2, 92.9, 66.3, 58.2, 56.6, 55.9, 46.0. HRMS (ESI) calcd for C21H24NO5 370.1649 (M + H)+, found 370.1652.
4.1.7. 5,7-Dimethoxy-2-(4-(2-morpholin-4-yl-ethoxy)phenyl)-chromen-4-one (11)
11 was prepared in 82% yield by a procedure similar to that used to prepare 7. The title compound was obtained as a white solid (mp 149–150 °C). HPLC purity 99.8% (tR = 15.26 min). 1H NMR (600 MHz, CDCl3) δ 7.80 (d, 2H, J = 9.0 Hz), 6.99 (d, 2H, J = 9.0 Hz), 6.58 (s, 1H), 6.55 (s, 1H), 6.37 (s, 1H), 4.18 (t, 2H, J = 6.0 Hz), 3.95 (s, 3H), 3.90 (s, 3H), 3.74 (t, 4H, J = 3.0 Hz), 2.83 (t, 2H, J = 6.0 Hz), 2.59 (s, 4H). 13C NMR (150 MHz, CDCl3) δ 177.8, 164.1, 161.3, 161.0, 160.7, 160.0, 127.7, 124.2, 115.1, 109.4, 107.9, 96.2, 93.0, 67.0, 66.2, 57.6, 56.6, 55.9, 54.3. HRMS (ESI) calcd for C23H26NO6 412.1755 (M + H)+, found 412.1757.
4.1.8. 2-(4-(3-Dimethylaminopropoxy)phenyl)-5,7-dimethoxy-chromen-4-one (12)
12 was prepared in 91% yield by a procedure similar to that used to prepare 7. The title compound was obtained as a white solid (mp 105–106 °C). HPLC purity 98.2% (tR = 15.76 min). 1H NMR (600 MHz, CDCl3) δ 7.81 (d, 2H, J = 9.0 Hz), 6.99 (d, 2H, J = 9.0 Hz), 6.59 (s, 1H), 6.56 (d, 1H, J = 2.4 Hz), 6.37 (d, 1H, J = 2.4 Hz), 4.09 (t, 2H, J = 9.0 Hz), 3.95 (s, 3H), 3.91 (s, 3H), 2.48 (t, 2H, J = 7.2 Hz), 2.27 (s, 6H), 1.98–2.00 (m, 2H). 13C NMR (150 MHz, CDCl3) δ 177.8, 164.0, 161.7, 161.1, 160.9, 160.0, 127.7, 123.9, 115.0, 109.4, 108.0, 96.2, 93.0, 66.6, 56.6, 56.4, 55.9, 45.6, 27.6. HRMS (ESI) calcd for C22H26NO5 384.1806 (M + H)+, found 384.1808.
4.1.9. 1-(2-(4-(5,7-Dimethoxy-4-oxo-4H-chromen-2-yl)phenoxy)ethyl)-pyrrolidin-2-one (13)
13 was prepared in 85% yield by a procedure similar to that used to prepare 7. The title compound was obtained as a white solid (mp 115–116 °C). HPLC purity 99.3% (tR = 18.44 min). 1H NMR (600 MHz, CDCl3) δ 7.81 (d, 2H, J = 8.4 Hz), 6.97 (d, 2H, J = 8.4 Hz), 6.58 (s, 1H), 6.55 (d, 1H, J = 2.4 Hz), 6.37 (d, 1H, J = 1.8 Hz), 4.18 (t, 2H, J = 5.4 Hz), 3.95 (s, 3H), 3.91 (s, 3H), 3.71 (t, 2H, J = 5.4 Hz), 3.58 (t, 2H, J = 7.2 Hz), 2.39 (t, 2H, J = 8.4 Hz), 2.01–2.08 (m, 2H). 13C NMR (150 MHz, CDCl3) δ 177.8, 175.6, 164.1, 161.0, 160.9, 160.7, 160.0, 127.8, 124.4, 114.9, 109.3, 107.9, 96.2, 92.9, 66.8, 56.6, 55.9, 49.1, 42.4, 30.9, 18.3. HRMS (ESI) calcd for C23H24NO6 410.1598 (M + H)+, found 410.1601.
4.1.10. 5,7-Dimethoxy-2-(4-(2-piperidin-1-yl-ethoxy)phenyl)-chromen-4-one (14)
To a solution of 7 (30 mg, 0.074 mmol), KI (25 mg, 0.15 mmol) and K2CO3 (102 mg, 0.74 mmol) in acetone (5 mL) was added piperidine (31 mg, 0.37 mmol) at 0 °C. The mixture was stirred at 75 °C for 18 h. The solution was diluted with EtOAc (100 mL), washed with 0.1 N HCl (aq.) (10 mL) and brine (10 mL). The organic layer was dried over anhydrous Na2SO4, and then concentrated under reduced pressure. The residue was purified by silica gel column chromatography (EtOAc) to give the desired product 14 (30 mg, 99%) as a white solid (mp 79–80 °C). HPLC purity 95.1% (tR = 16.24 min). 1H NMR (600 MHz, CDCl3) δ 7.83 (d, 2H, J = 6.6 Hz), 7.02 (d, 2H, J = 6.6 Hz), 6.62 (s, 1H), 6.58 (d, 1H, J = 2.4 Hz), 6.39 (d, 1H, J = 2.4 Hz), 4.22 (t, 2H, J = 6.0 Hz), 3.98 (s, 3H), 3.94 (s, 3H), 2.85 (t, 2H, J = 6.0 Hz), 2.57–2.59 (m, 4H), 1.65–1.67 (m, 4H), 1.50–1.52 (m, 2H). 13C NMR (150 MHz, CDCl3) δ 177.8, 164.1, 161.4, 161.0, 160.8, 160.0, 127.7, 124.0, 115.1, 109.4, 107.8, 96.2, 93.0, 66.3, 57.8, 56.6, 55.9, 55.2, 25.9, 24.2. HRMS (ESI) calcd for C24H28NO5 410.1962 (M + H)+, found 410.1964.
4.1.11. 5,7-Dimethoxy-2-(4-(2-(4-methylpiperazin-1-yl)ethoxy)phenyl)-chromen-4-one(15)
15 was prepared in 80% yield by a procedure similar to that used to prepare 14. The title compound was obtained as a white solid (mp 144–145 °C). HPLC purity 98.7% (tR = 16.43 min). 1H NMR (600 MHz, CDCl3) δ 7.79–7.81 (m, 2H), 6.98–7.00 (m, 2H), 6.58 (s, 1H), 6.55 (d, 1H, J = 2.4 Hz), 6.36 (d, 1H, J = 2.4 Hz), 4.17 (t, 2H, J = 6.0 Hz), 3.95 (s, 3H), 3.90 (s, 3H), 2.85 (t, 2H, J = 6.0 Hz), 2.50–2.65 (m, 8H), 2.30 (s, 3H). 13C NMR (150 MHz, CDCl3) δ 177.8, 164.0, 161.4, 161.0, 160.8, 160.0, 127.7, 124.1, 115.1, 109.4, 107.8, 96.2, 92.9, 66.3, 57.1, 56.6, 55.9, 55.1, 53.7, 46.1. HRMS (ESI) calcd for C24H29N2O5 425.2071 (M + H)+, found 425.2070.
4.1.12. 5,7-Dimethoxy-2-(4-(2-pyrrolidin-1-yl-ethoxy)phenyl)-chromen-4-one (16)
16 was prepared in 85% yield by a procedure similar to that used to prepare 14. The title compound was obtained as a pale brown solid (mp 114–115 °C). HPLC purity 99.2% (tR = 15.91 min). 1H NMR (600 MHz, CDCl3) δ 7.78(d, 2H, J = 6.6 Hz), 6.99 (d, 2H, J = 6.6 Hz), 6.55 (d, 1H, J = 3.6 Hz), 6.52 (d, 1H, J = 2.4 Hz), 6.34 (d, 1H, J = 2.4 Hz), 4.17 (t, 2H, J = 9.0 Hz), 3.92 (s, 3H), 3.88 (s, 3H), 2.93 (t, 2H, J = 9.0 Hz), 2.65–2.66 (m, 4H), 1.81–1.82 (m, 4H). 13C NMR (150 MHz, CDCl3) δ 177.8, 164.0, 161.4, 161.0, 160.8, 160.0, 127.7, 123.9,115.0, 109.3, 107.7, 96.2, 93.0, 67.3, 56.5, 55.8, 54.9, 54.8, 23.6. HRMS (ESI) calcd for C23H26NO5 396.1806 (M + H)+, found 396.1809.
4.1.13. 2-(4-(2-Diethylaminoethoxy)-phenyl)-5,7-dimethoxy-chromen-4-one (17)
17 was prepared in 85% yield by a procedure similar to that used to prepare 14. The title compound was obtained as a pale yellow solid (mp 116–117 °C). HPLC purity 99.5% (tR = 16.09 min). 1H NMR (600 MHz, CDCl3) δ 7.80 (d, 2H, J = 9.0 Hz), 7.00 (d, 2H, J = 9.0 Hz), 6.59 (d, 1H, J = 2.4 Hz), 6.56 (d, 1H, J = 1.8 Hz), 6.37 (d, 1H, J = 1.8 Hz), 4.12 (t, 2H, J = 6.0 Hz), 3.95 (s, 3H), 3.91 (s, 3H), 2.91 (t, 2H, J = 6.0 Hz), 2.65–2.68 (m, 4H), 1.09 (t, 6H, J = 7.2 Hz). 13C NMR (150 MHz, CDCl3) δ 177.8, 164.0, 161.5, 161.0, 160.8, 160.0, 127.7, 124.0, 115.1, 109.4, 107.8, 96.2, 93.0, 67.0, 56.6, 55.9, 51.8, 48.0, 12.0. HRMS (ESI) calcd for C23H28NO5 398.1962 (M + H)+, found 398.1964.
4.1.14. 1-(2-(4-(5,7-Dimethoxy-4-oxo-4H-chromen-2-yl)phenoxy)ethyl)-pyrrolidine-2-carboxylic acid methyl ester (18)
18 was prepared in 66% yield by a procedure similar to that used to prepare 14. The title compound was obtained as a white solid (mp 121–122 °C). HPLC purity 98.4% (tR = 16.15 min). 1H NMR (600 MHz, CDCl3) δ 7.83 (d, 2H, J = 9.0 Hz), 7.00 (d, 2H, J = 9.0 Hz), 6.61 (s, 1H), 6.58 (d, 1H, J = 2.4 Hz), 6.40 (d, 1H, J = 1.8 Hz), 4.18–4.24 (m, 2H), 3.98 (s, 3H), 3.94 (s, 3H), 3.66 (s, 3H), 3.39–3.41 (m, 1H), 3.32–3.35 (m, 1H), 3.11–3.14 (m, 1H), 3.04–3.07 (m, 1H), 2.59–2.62 (m, 1H), 2.20–2.22 (m, 1H), 1.96–2.00 (m, 2H), 1.86–1.88 (m, 1H). 13C NMR (150 MHz, CDCl3) δ 177.8, 174.9, 164.0, 161.2, 161.0, 160.8, 160.0, 127.7, 124.1, 115.0, 109.4, 107.9, 96.2, 93.0, 67.5, 66.2, 56.6, 55.9, 55.1, 53.6, 52.0, 29.9, 23.5. HRMS (ESI) calcd for C25H28NO7 454.1860 (M + H)+, found 454.1865.
4.1.15. 5,7-Dimethoxy-2-(4-(piperidin-4-yloxy)-phenyl)-chromen-4-one (19)
To a solution of 6 (60 mg, 0.2 mmol) and Ph3P (210 mg, 0.8 mmol) in THF (5 mL) was added 4-hydroxy-piperidine-1-carboxylic acid tert-butyl ester (121 mg, 0.6 mmol) in THF (5 mL) and DIAD (121 mg, 0.6 mmol). The mixture was stirred at r.t. for 2 h, and was then concentrated to give the crude product. This residue was purified with silica gel column (EtOAc) to afford 80 mg of the intermediate as a white solid. To the solution of the intermediate (80 mg) in CH2Cl2 (3 mL) was added TFA (1 mL) at 0 °C. The mixture was stirred at r.t. for 3 h, and was then concentrated. The residue was partitioned between EtOAc (250 mL) and 1 N NaHCO3 (aq., 10 mL). The organic layer was washed with H2O (10 mL) and dried over Na2SO4. The organic layer was concentrated. The residue was purified with silica gel column (EtOAc) to afford 19 (60 mg, 78%, two steps) as a pale yellow solid (mp 205–206 °C). HPLC purity 98.6% (tR = 15.43 min). 1H NMR (600 MHz, DMSO-d6) δ 7.98–8.00 (m, 2H), 7.15–7.17 (m, 2H), 6.85 (d, 1H, J = 1.8 Hz), 6.68 (s, 1H), 6.51 (d, 1H, J = 2.4 Hz), 4.77 (t, 1H, J = 4.2 Hz), 3.90 (s, 3H), 3.83 (s, 3H), 3.21–3.25 (m, 4H), 3.03–3.07 (m, 2H), 2.09–2.12 (m, 2H), 1.78–1.81 (m, 2H). 13C NMR (150 MHz, DMSO-d6) δ 178.6, 164.5, 161.3, 160.9, 160.0, 159.4, 128.1, 124.4, 116.1, 108.9, 107.4, 96.4, 93.0, 69.2, 56.3, 55.9, 40.6, 28.0. HRMS (ESI) calcd for C22H24NO5 382.1649 (M + H)+, found 382.1651.
4.1.16. 2-(4-(2-Aminoethoxy)-phenyl)-5,7-dimethoxy-chromen-4-one (20)
20 was prepared in 59% yield (two steps) by a procedure similar to that used to prepare 19. The title compound was obtained as a white solid (mp 165–166 °C). HPLC purity 98.4% (tR = 14.50 min). 1H NMR (600 MHz, CD3OD) δ 7.87 (d, 2H, J = 9.0 Hz), 7.05 (d, 2H, J = 8.4 Hz), 6.61 (d, 2H, J = 9.0 Hz), 6.42 (s, 1H), 4.12 (t, 2H, J = 4.8 Hz), 3.94 (d, 6H, J = 2.4 Hz), 3.14 (s, 2H). 13C NMR (150 MHz, CD3OD) δ 178.9, 164.7, 161.7, 161.6, 161.0, 160.1, 128.1, 124.0, 115.1, 108.8, 107.2, 96.5, 93.1, 69.1, 56.2, 56.0, 40.7. HRMS (ESI) calcd for C19H20NO5 342.1336 (M + H)+, found 342.1337.
4.1.17. 2-(4-(3-Aminopropoxy)-phenyl)-5,7-dimethoxy-chromen-4-one (21)
21 was prepared in 90% yield (two steps) by a procedure similar to that used to prepare 19. The title compound was obtained as a white solid (mp 170–171 °C). HPLC purity 98.4% (tR = 15.07 min). 1H NMR (600 MHz, CD3OD) δ 7.88 (d, 2H, J = 9.0 Hz), 7.05 (d, 2H, J = 9.0 Hz), 6.66 (d, 1H, J = 1.8 Hz), 6.60 (s, 1H), 6.45 (d, 1H, J = 1.8 Hz), 4.18 (t, 2H, J = 6.0 Hz), 3.95 (d, 6H, J = 3.0 Hz), 3.35 (s, 2H), 3.09 (t, 2H, J = 7.2 Hz), 2.14 (t, 2H, J = 6.0 Hz). 13C NMR (150 MHz, CD3OD) δ 179.3, 165.2, 162.2, 161.9, 161.1, 160.4, 128.3, 124.1, 115.3, 108.9, 107.1, 96.8, 93.4, 65.9, 56.3, 56.2, 38.2, 29.4. HRMS (ESI) calcd for C20H22NO5 356.1493 (M + H)+, found 356.1496.
4.1.18. 5,7-Dimethoxy-2-(4-oxiranylmethoxyphenyl)-chromen-4-one (23)
To a solution of 6 (200 mg, 0.67 mmol) and epichlorohydrin (22) (617 mg, 6.7 mmol) in acetone (10 mL) was added K2CO3 (462 mg, 3.35 mmol). The mixture was stirred at 80 °C for 24 h, and was then concentrated to give the crude product. The residue was diluted with EtOAc (100 mL), and washed with 0.1 N HCl (aq.) (10 mL) followed by brine (10 mL). The organic layer was dried over anhydrous Na2SO4, and then concentrated under reduced pressure. The residue was purified by silica gel column chromatography (CH2Cl2/MeOH = 20/1) to give the desired product 23 (160 mg, 67%) as a pale yellow solid (mp 191–192 °C). HPLC purity 98.9% (tR = 18.99 min). 1H NMR (600 MHz, CDCl3) δ 7.80–7.82 (m, 2H), 7.00–7.03 (m, 2H), 6.58 (s, 1H), 6.55 (d, 1H, J = 2.4 Hz), 6.36 (d, 1H, J = 2.4 Hz), 4.31–4.33 (m, 1H), 3.99–4.02 (m, 1H), 3.95 (s, 3H), 3.90 (s, 3H), 3.37–3.39 (m, 1H), 2.93–2.94 (m, 1H), 2.78–2.79 (m, 1H). 13C NMR (150 MHz, CDCl3) δ 177.7, 164.1, 161.0, 161.0, 160.6, 160.0, 127.8, 124.6, 115.1, 109.4, 108.0, 96.2, 93.0, 69.1, 56.6, 55.9, 50.1, 44.7. HRMS (ESI) calcd for C20H19O6 355.1176 (M + H)+, found 355.1180.
4.1.19. 2-(4-(2-Hydroxy-3-piperidin-1-yl-propoxy)phenyl)-5,7-dimethoxy-chromen-4-one (24)
To a solution of 23 (30 mg, 0.085 mmol) and piperidine (72 mg, 0.85 mmol) in EtOH (5 mL) was added K2CO3 (117 mg, 0.85 mmol). The mixture was stirred at 100 °C for 3 h, and was then concentrated to give the crude product. The residue was diluted with EtOAc (100 mL), and washed with 0.1 N HCl (aq.) (10 mL) followed by brine (10 mL). The organic layer was dried over anhydrous Na2SO4, and then concentrated under reduced pressure. The residue was purified by silica gel column chromatography (CH2Cl2/MeOH = 10/1) to give the desired product 24 (35 mg, 94%) as a white solid (mp 109–110 °C). HPLC purity 98.5% (tR = 15.78 min). 1H NMR (600 MHz, CDCl3) δ 7.79–7.81 (m, 2H), 7.00–7.03 (m, 2H), 6.58 (s, 1H), 6.54 (d, 1H, J = 3.0 Hz), 6.36 (d, 1H, J = 1.8 Hz), 4.11–4.15 (m, 1H), 4.02–4.06 (m, 2H), 3.95 (s, 3H), 3.90 (s, 3H), 2.66 (bs, 1H), 2.54–2.56 (m, 2H), 2.50–2.55 (m, 2H), 2.41–2.43 (m, 2H), 1.58–1.65 (m, 4H), 1.46–1.48 (m, 2H). 13C NMR (150 MHz, CDCl3) δ 177.8, 164.0, 161.4, 161.0, 160.8, 160.0, 127.7, 124.2, 115.1, 109.3, 107.8, 96.2, 92.9, 70.7, 65.3, 61.1, 56.6, 55.9, 54.9, 26.0, 24.2. HRMS (ESI) calcd for C25H30NO6 440.2068 (M + H)+, found 440.2071.
4.1.20. 2-(4-(2-Hydroxy-3-pyrrolidin-1-yl-propoxy)phenyl)-5,7-dimethoxy-chromen-4-one (25)
25 was prepared in 94% yield by a procedure similar to that used to prepare 24. The title compound was obtained as a pale yellow solid (mp 137–138 °C). HPLC purity 99.9% (tR = 15.42 min). 1H NMR (600 MHz, CDCl3) δ 7.82 (d, 2H, J = 9.0 Hz), 7.03 (d, 2H, J = 9.0 Hz), 6.59 (s, 1H), 6.55 (d, 1H, J = 2.4 Hz), 6.36 (d, 1H, J = 2.4 Hz), 4.12–4.15 (m, 1H), 4.06 (d, 2H, J = 5.4 Hz), 3.95 (s, 3H), 3.91 (s, 3H), 3.10–3.30 (bs, 1H), 2.86–2.89 (m, 1H), 2.76–2.79 (m, 2H), 2.58–2.62 (m, 2H), 1.82–1.85 (m, 4H). 13C NMR (150 MHz, CDCl3) δ 177.8, 164.1, 161.3, 161.0, 160.7, 160.0, 127.7, 124.3, 115.1, 109.4, 107.9, 96.2, 93.0, 70.7, 67.2, 58.6, 56.6, 55.9, 54.4. HRMS (ESI) calcd for C24H28NO6 426.1911 (M + H)+, found 426.1915.
4.1.21. 2-(4-(3-Dimethylamino-2-hydroxypropoxy)-phenyl)-5,7-dimethoxy-chromen-4-one (26)
26 was prepared in 71% yield by a procedure similar to that used to prepare 24. The title compound was obtained as a white solid (mp 92–93 °C). HPLC purity 99.6% (tR = 10.65 min). 1H NMR (600 MHz, CDCl3) δ 7.83 (d, 2H, J = 9.0 Hz), 7.04 (d, 2H, J = 9.0 Hz), 6.61 (s, 1H), 6.58 (d, 1H, J = 2.4 Hz), 6.39 (d, 1H, J = 2.4 Hz), 4.12–4.15 (m, 1H), 4.11–4.12 (m, 2H), 3.98 (s, 3H), 3.93 (s, 3H), 2.61–2.65 (m, 1H), 2.42–2.48 (m, 2H), 2.39 (s, 6H). 13C NMR (150 MHz, CDCl3) δ 177.8, 164.1, 161.3, 161.0, 160.8, 160.0, 127.7, 124.2, 115.1, 109.3, 107.9, 96.2, 93.0, 70.7, 66.1, 61.8, 56.6, 55.9, 45.6. HRMS (ESI) calcd for C22H26NO6 400.1755 (M + H)+, found 400.1756.
4.1.22. 2-(4-(2,3-Dihydroxypropoxy)phenyl)-5,7-dimethoxy-chromen-4-one (28)
To a solution of 6 (50 mg, 0.17 mmol) and Ph3P (88 mg, 0.34 mmol) in THF (5 mL) was added (2,2-dimethyl-1,3-dioxolan-4-yl)methanol (27) (44 mg, 0.34 mmol) in THF (5 mL) and DIAD (68 mg, 0.34 mmol). The mixture was stirred at r.t. for 4 h, and was then concentrated to give the crude product. This residue was purified with silica gel column (CH2Cl2/MeOH = 20/1 to 10/1) to afford 70 mg of the intermediate as a white solid. To the solution of the intermediate (70 mg) in EtOH (5 mL) was added 0.5 N HCl (aq., 0.5 mL) at 0 °C. The mixture was stirred at 100 °C for 2 h, and was then concentrated. The residue was partitioned between EtOAc (50 mL) and 1 N NaHCO3 (aq., 10 mL). The organic layer was washed with H2O (10 mL) and dried over Na2SO4. The organic layer was concentrated. The residue was purified with silica gel column (CH2Cl2/MeOH = 10/1) to afford 28 (45 mg, 71%, two steps) as a white solid (mp 203–204 °C). HPLC purity 97.6% (tR = 15.51 min). 1H NMR (600 MHz, DMSO-d6) δ 7.97–8.00 (m, 2H), 7.08–7.11 (m, 2H), 6.86 (d, 1H, J = 2.4 Hz), 6.66 (s, 1H), 6.50 (d, 1H, J = 2.4 Hz), 5.00 (d, 1H, J = 5.4 Hz), 4.70 (t, 1H, J = 6.0 Hz), 4.09–4.11 (m, 1H), 3.95–3.98 (m, 1H), 3.90 (s, 3H), 3.83 (s, 3H), 3.80–3.83 (m, 1H), 3.46 (t, 2H, J = 6.0 Hz). 13C NMR (150 MHz, DMSO-d6) δ 175.6, 163.6, 161.4, 160.2, 159.7, 159.1, 127.7, 122.9, 115.0, 108.3, 106.7, 96.2, 93.4, 69.9, 69.8, 62.6, 56.1, 56.0. HRMS (ESI) calcd for C20H21O7 373.1282 (M + H)+, found 373.1284.
4.1.23. 2-(4-Bromophenyl)-5,7-dimethoxy-chromen-4-one (31)
Starting from the commercially available 4-bromobenzaldehyde (29) and 1-(2-hydroxy-4,6-dimethoxy-phenyl)ethanone (2), 31 was prepared in two steps according to literature procedures.36 HPLC purity 96.5% (tR = 21.53 min). 1H NMR (600 MHz, DMSO-d6) δ7.99 (d, 2H, J = 8.4 Hz), 7.75 (d, 2H, J = 9.0 Hz), 6.87 (d, 1H, J = 2.4 Hz), 6.82 (s, 1H), 6.51 (d, 1H, J = 1.8 Hz), 3.90 (s, 3H), 3.83 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 175.6, 163.9, 160.3, 159.1, 158.5, 132.0, 130.2, 127.9, 125.1, 108.6, 108.4, 96.4, 93.4, 56.1, 56.0. HRMS (ESI) calcd for C17H14BrO4 361.0070 (M + H)+, found 361.0070.
4.1.24. 2-(4-(6-Fluoropyridin-3-yl)phenyl)-5,7-dimethoxy-chromen-4-one (33)
To a solution of 31 (90 mg, 0.25 mmol) and 2-fluoropyridine-5-boronic acid (32) (42 mg, 0.3 mmol) in THF/EtOH/H2O (2 mL/2 mL/2 mL) was added KOAc (94 mg, 0.75 mmol) and then Pd(dppf)Cl2 (20 mg, 0.025 mmol). The resulting mixture was deoxygenated via five vacuum/N2-refill cycles. The mixture was stirred at 80 °C for 18 h, and was then concentrated under vacuum. The residue was partitioned between EtOAc (100 mL) and H2O (20 mL). The organic layer was separated and washed with brine (10 mL), dried over anhydrous Na2SO4, filtrated and concentrated to give an oily residue. This residue was purified with silica gel column (CH2Cl2/MeOH = 10/1) to obtain 33 (80 mg, 85%) as a pale red solid (mp 209–210 °C). HPLC purity 98.4% (tR = 20.91 min). 1H NMR (600 MHz, CDCl3) δ 8.48 (d, 1H, J = 1.8 Hz), 8.01–8.04 (m, 1H), 7.98 (s, 1H), 7.97 (s, 1H), 7.04–7.06 (m, 1H), 6.72 (s, 1H), 6.59 (d, 1H, J = 2.4 Hz), 6.40 (d, 1H, J = 2.4 Hz), 3.97 (s, 3H), 3.93 (s, 3H). 13C NMR (150 MHz, CDCl3) δ 177.5, 164.5, 164.4, 162.9, 161.2, 160.1, 160.0, 146.2, 146.1, 139.8, 139.5, 133.8, 131.6, 127.6, 126.9, 110.0, 109.8, 109.6, 96.5, 93.1, 56.6, 55.9. HRMS (ESI) calcd for C22H17FNO4 378.1136 (M + H)+, found 378.1138.
4.1.25. 2-(4-(2-Dimethylaminoethylamino)phenyl)-5,7-dimethoxy-chromen-4-one (36)
NaOtBu (43 mg, 0.45 mmol), Pd2(dba)3 (28 mg, 0.03 mmol) and (±)-BINAP (19 mg, 0.03 mmol) were placed into a flask and dissolved into distilled toluene (10 mL). To this solution was added 31 (108 mg, 0.3 mmol) and N,N-dimethylethylenediamine (34) (40 mg, 0.45 mmol) dropwise with stirring at room temperature and the mixture was refluxed at 80 °C for 48 h. After the mixture was cooled, 20 mL of H2O was added and extracted with EtOAc (100 mL). The organic layer was dried over anhydrous Na2SO4, filtered and concentrated in vacuo. The residue was purified by silica gel column chromatography (CH2Cl2/MeOH = 10/1) to give the desired product (65 mg, 64%) as a pale yellow solid (mp 159–160 °C). HPLC purity 98.8% (tR = 15.10 min). 1H NMR (600 MHz, CDCl3) δ 7.70 (d, 2H, J = 9.0 Hz), 6.66 (d, 2H, J = 9.0 Hz), 6.54 (d, 1H, J = 1.8 Hz), 6.53 (s, 1H), 6.35 (d, 1H, J = 1.8 Hz), 4.84 (s, 1H), 3.95 (s, 3H), 3.90 (s, 3H), 3.22 (s, 2H), 2.60 (t, 1H, J = 6.0 Hz), 2.28 (s, 6H). 13C NMR (150 MHz, CDCl3) δ 177.9, 163.8, 161.7, 161.0, 160.0, 151.1, 127.7, 119.4, 112.5, 109.4, 106.2, 96.0, 93.0, 57.7, 56.6, 55.8, 45.2, 40.6, 31.1. HRMS (ESI) calcd for C21H25N2O4 369.1809 (M + H)+, found 369.1811.
4.1.26. 5,7-Dimethoxy-2-(4-(2-pyrrolidin-1-yl-ethylamino)phenyl)-chromen-4-one (37)
37 was prepared in 57% yield by a procedure similar to that used to prepare 36. The title compound was obtained as a pale yellow solid (mp 148–149 °C). HPLC purity 98.2% (tR = 15.73 min). 1H NMR (600 MHz, CDCl3) δ 7.69 (d, 2H, J = 9.0 Hz), 6.66 (d, 2H, J = 8.4 Hz), 6.53 (d, 1H, J = 2.4 Hz), 6.52 (s, 1H), 6.34 (d, 1H, J = 2.4 Hz), 4.91 (s, 1H), 3.94 (s, 3H), 3.89 (s, 3H), 3.27 (d, 2H, J = 2.4 Hz), 2.78 (t, 1H, J = 6.0 Hz), 2.59 (s, 4H), 1.81 (s, 4H). 13C NMR (150 MHz, CDCl3) δ 177.9, 163.8, 161.7, 161.0, 160.0, 151.1, 127.6, 119.3, 112.5, 109.4, 106.1, 96.0, 93.0, 56.5, 55.8, 54.6, 54.0, 41.8, 23.6. HRMS (ESI) calcd for C23H27N2O4 395.1965 (M + H)+, found 395.1968.
4.1.27. 5-Hydroxy-7-methoxy-2-(4-(2-pyrrolidin-1-yl-ethoxyphenyl)-chromen-4-one (38)
To a solution of 16 (30 mg, 0.076 mmol) in 5 mL of CH2Cl2 was added 1 N BBr3 in CH2Cl2 (0.15 mL, 0.15 mmol) at 0 °C. The resulting mixture was stirred at r.t. for 2 h. The solution was diluted with CH2Cl2/MeOH (10/1, 50 mL), washed with H2O (15 mL) and brine (10 mL). The organic layer was dried over anhydrous Na2SO4 and then concentrated under reduced pressure. The residue was purified by silica gel column chromatography (CH2Cl2/MeOH = 10/1) to give the desired product (23 mg, 79%) as a pale yellow solid (mp 127–129 °C). HPLC purity 99.7% (tR = 18.02 min). 1H NMR (600 MHz, CDCl3) δ 12.81 (s, 1H), 7.83 (d, 2H, J = 9.0 Hz), 7.03 (d, 2H, J = 9.0 Hz), 6.60 (s, 1H), 6.48 (d, 1H, J = 2.4 Hz), 6.36 (d, 1H, J = 2.4 Hz), 4.21 (t, 2H, J = 9.0 Hz), 3.88 (s, 3H), 2.97 (t, 2H, J = 9.0 Hz), 2.67–2.69 (m, 4H), 1.83–1.85 (m, 4H). 13C NMR (150 MHz, CDCl3) δ 182.6, 165.6, 164.2, 162.3, 162.0, 157.9, 128.2, 123.8, 115.2, 105.7, 104.5, 98.2, 92.8, 67.4, 55.9, 54.9, 54.8, 23.6. HRMS (ESI) calcd for C22H24NO5 382.1649 (M + H)+, found 386.1652.
4.1.28. 5,7-Dihydroxy-2-(4-(2-pyrrolidin-1-yl-ethoxy)phenyl)-chromen-4-one (39)
To a solution of 16 (22 mg, 0.056 mmol) in 5 mL of CH2Cl2 was added 1 N BBr3 in CH2Cl2 (0.17 mL, 0.17 mmol) at 0 °C. The resulting mixture was stirred at r.t. for 24 h. The solution was diluted with CH2Cl2/MeOH (10/1, 50 mL), washed with H2O (5 mL) and brine (5 mL). The organic layer was dried over anhydrous Na2SO4 and then concentrated under reduced pressure. The residue was purified by silica gel column chromatography (CH2Cl2/MeOH = 10/1) to give the desired product (15 mg, 73%) as a pale yellow solid (mp 228–229 °C). HPLC purity 97.5% (tR = 16.04 min). 1H NMR (600 MHz, CDCl3/CD3OD 2:1) δ 7.80 (d, 2H, J = 9.0 Hz), 6.98 (d, 2H, J = 9.0 Hz), 6.50 (s, 1H), 6.39 (d, 1H, J = 2.4 Hz), 6.22 (d, 1H, J = 2.4 Hz), 4.16 (t, 2H, J = 6.0 Hz), 2.94 (t, 2H, J = 6.0 Hz), 2.65–2.67 (m, 4H), 1.80–1.83 (m, 4H). 13C NMR (150 MHz, CDCl3/CD3OD 2:1) δ 182.6, 164.4, 164.3, 161.8, 161.7, 158.1, 128.2, 123.8, 115.1, 104.7, 103.9, 99.4, 94.5, 66.8, 54.8, 54.7, 23.4. HRMS (ESI) calcd for C21H22NO5 368.1493 (M + H)+, found 368.1495.
4.2. Biology
4.2.1. Human pancreatic stellate cell culture and reagents
Under an IRB-approved tissue protocol, discarded human resected pancreatic tissue (500 mm3) was attained fresh from the operating room at the University of Texas Medical Branch. The tissue was minced and plated on a collagen-coated flask (15 μg/mL, Invitrogen) with DMEM (VWR; Radnor, PA) supplemented with penicillin 200 U/mL, streptomycin 200 μg/mL, amphotericin B 0.25 μg/mL and gentimicin 50 μg/mL (Invitrogen, Carlsbad CA), 10% fetal bovine serum (Lonza, Walkersville, MD), 1% insulin-transferrin, selenium-ethanolamine (Gibco, Grand Island, NY), and 1% non-essential amino acids (Sigma-Aldrich, St. Louis, MO). The human PSC were isolated by the outgrowth method.41–43 The purity of PSC culture was confirmed by immunohistochemical staining for vimentin, α-smooth muscle actin, glial fibrillar acidic protein, or Oil red O staining. Primary PSC were transformed and immortalized for use in experiments using lentiviral vectors containing SV40 Large T antigen and human telomerase (plasmid # 12245 and 12246; Addgene, Cambridge, MA). Cultures were maintained in DMEM with 10% FBS, at 37 °C in a humidified 95% O2/5% CO2 atmosphere.
4.2.2. Cell proliferation assay
Transformed PSC (3 × 103) were plated in 96-well plates, in sextuplicate. The next day, the media was changed to 1% FBS with apigenin or analogues for 24–48h. AlamarBlue reagent (10% sample volume, DAL1025) was added to each well per Invitrogen’s protocol. Fluorescence was recorded using excitation/emission wavelengths of 544/590 nm and the SpectraMax M2 Microplate Reader (Molecular Devices, Sunnyvale CA).
4.2.3. Cell death assay
Transformed PSC (8 × 103) were plated in 96-well plates in triplicate. The next day, the media was changed to 1% FBS with apigenin or analogues for a 14 h incubation. The Cell Death Detection ELISAPLUS assay (Roche Applied Science, Indianapolis, IN) was used and protocol followed (Version 11.0). Absorbance was measured at 405 nm, using the ELx800 Automated Microplate Reader (Bio-TEK Instruments, Inc., Winooski, VT).
4.2.4. Chronic pancreatitis (CP) animal model
Animal experiments were conducted under an Institutional Animal Care and Use Committee-approved protocol. CP was induced in C57BL/6 mice using supraoptimal pancreatic stimulation with cerulein (CR), a cholecystokinin analogue (Bachem, Torrance, CA). CP (50 μg CR/kg mouse weight) was administered via intraperitoneal route hourly for 5 h for 3 d/week for a total of 4 weeks. Control mice received phosphate buffered saline (PBS) injections following the same schedule. Starting Week 2 of the experiment, apigenin, analogue, or vehicle (0.5% methylcellulose + 0.025% Tween 20 in ddH20) were administered (0.5 mg/kg/d, oral gavage, once daily, 6 d/week, for 3 week) while continuing CR injections. At the end of Week 4, the mice were anesthestized with isoflurane and sacrificed per protocol. The pancreata were quickly harvested and processed.
4.2.5. Immunohistochemistry (IHC) and image analysis
Pancreata were formalin-fixed and paraffin-embedded. Prior to staining, sections (5 μm) were deparaffinized with xylene, dehydrated with ethanol, and subjected to heat-mediated antigen retrieval (DAKO, Carpinteria, CA) to optimize antigen immunoreactivity. Fibronectin antibody (1:600; Santa Cruz Biotechnology, Dallas, TX), and biotinylated anti-goat IgG (1:400; Vector Laboratories Inc., Burlingame, CA) were used. IHC staining was completed with the VECTASTAIN Elite ABC kit (Vector Lab), color development with DAB (DAKO), and counterstaining with hematoxylin 7211 (Thermo Scientific, Kalamazoo, MI). Five non-overlapping images of each pancreas (400 ×) were taken using an Olympus BX51 microscope connected to a DP71 Olympus digital camera. The percent area of brown fibronectin staining was quantified using the Image Processing and Analysis in Java (ImageJ) 1.46r software (NIH, Bethesda, MD) and a color deconvolution plug-in.44,45
4.2.6. Statistical analysis
Dose-response curves were generated by plotting fluorescence or absorbance versus log (compound concentration). A best-fit curve was created using nonlinear regression, and the IC50 or EC50 determined from the graph (GraphPad Prism 5; GraphPad Software Inc., La Jolla, CA). To analyze continous variables, unpaired student t-tests and one-way ANOVA with Tukey-Kramer multiple comparisons post-test (GraphPad Prism 5). Significance was set at p < 0.05.
Acknowledgments
This work was supported by grants P50 CA097007, P30 DA028821, R21 MH093844 (JZ), T32 DK007639-21 (MRH) and K08 CA125209 (CC) from the National Institutes of Health, R. A. Welch Foundation Chemistry and Biology Collaborative Grant from the Gulf Coast Consortia (GCC), John Sealy Memorial Endowment Fund, Institute for Translational Sciences (ITS), and the Center for Addiction Research (CAR) at UTMB. We thank Drs. Lawrence C. Sowers, Jacob A. Theruvathu, and Tianzhi Wang for the NMR spectroscopy assistance, and Dr. Carol Nilsson for mass spectrometry assistance.
Footnotes
The authors declare no competing financial interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Schneider A, Whitcomb DC. Best Pract Res Clin Gastroenterol. 2002;16:347. doi: 10.1053/bega.2002.0311. [DOI] [PubMed] [Google Scholar]
- 2.Bordalo O, Goncalves D, Noronha M, Cristina ML, Salgadinho A, Dreiling DA. Am J Gastroenterol. 1977;68:278. [PubMed] [Google Scholar]
- 3.Shimizu K. J Gastroenterol. 2008;43:823. doi: 10.1007/s00535-008-2249-7. [DOI] [PubMed] [Google Scholar]
- 4.Sahel J, Sarles H. Dig Dis Sci. 1979;24:897. doi: 10.1007/BF01311942. [DOI] [PubMed] [Google Scholar]
- 5.Witt H, Apte MV, Keim V, Wilson JS. Gastroenterology. 2007;132:1557. doi: 10.1053/j.gastro.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 6.Yang AL, Vadhavkar S, Singh G, Omary MB. Arch Intern Med. 2008;168:649. doi: 10.1001/archinte.168.6.649. [DOI] [PubMed] [Google Scholar]
- 7.Everhart JE, Ruhl CE. Gastroenterology. 2009;136:376. doi: 10.1053/j.gastro.2008.12.015. [DOI] [PubMed] [Google Scholar]
- 8.Trikudanathan G, Navaneethan U, Vege SS. Gastroenterol Clin North Am. 2012;41:63. doi: 10.1016/j.gtc.2011.12.009. [DOI] [PubMed] [Google Scholar]
- 9.Forsmark CE. Gastroenterology. 2013;144:1282. doi: 10.1053/j.gastro.2013.02.008. [DOI] [PubMed] [Google Scholar]
- 10.Erkan M, Adler G, Apte MV, Bachem MG, Buchholz M, Detlefsen S, Esposito I, Friess H, Gress TM, Habisch HJ, Hwang RF, Jaster R, Kleeff J, Kloppel G, Kordes C, Logsdon CD, Masamune A, Michalski CW, Oh J, Phillips PA, Pinzani M, Reiser-Erkan C, Tsukamoto H, Wilson J. Gut. 2012;61:172. doi: 10.1136/gutjnl-2011-301220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jaster R, Emmrich J. Best Pract Res Clin Gastroenterol. 2008;22:17. doi: 10.1016/j.bpg.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 12.Masamune A, Watanabe T, Kikuta K, Shimosegawa T. Clin Gastroenterol Hepatol. 2009;7:S48. doi: 10.1016/j.cgh.2009.07.038. [DOI] [PubMed] [Google Scholar]
- 13.Madro A, Slomka M, Celinski K. Adv Med Sci. 2011;56:132. doi: 10.2478/v10039-011-0023-1. [DOI] [PubMed] [Google Scholar]
- 14.Gao X, Cao Y, Yang W, Duan C, Aronson JF, Rastellini C, Chao C, Hellmich MR, Ko TC. Am J Physiol Gastrointest Liver Physiol. 2013;304:G804. doi: 10.1152/ajpgi.00306.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Spector I, Honig H, Kawada N, Nagler A, Genin O, Pines M. Pancreas. 2010;39:1008. doi: 10.1097/MPA.0b013e3181da8aa3. [DOI] [PubMed] [Google Scholar]
- 16.He J, Sun X, Qian KQ, Liu X, Wang Z, Chen Y. Biochim Biophys Acta. 2009;1792:56. doi: 10.1016/j.bbadis.2008.10.010. [DOI] [PubMed] [Google Scholar]
- 17.Whitcomb DC. Gut. 1999;45:317. doi: 10.1136/gut.45.3.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kloppel G, Maillet B. Pancreas. 1993;8:659. doi: 10.1097/00006676-199311000-00001. [DOI] [PubMed] [Google Scholar]
- 19.Comfort MW, Gambill EE, Baggenstoss AH. Gastroenterology. 1946;6:376. [PubMed] [Google Scholar]
- 20.Koehn FE, Carter GT. Nat Rev Drug Discov. 2005;4:206. doi: 10.1038/nrd1657. [DOI] [PubMed] [Google Scholar]
- 21.Harvey AL. Drug Discov Today. 2008;13:894. doi: 10.1016/j.drudis.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 22.Li JW, Vederas JC. Science. 2009;325:161. doi: 10.1126/science.1168243. [DOI] [PubMed] [Google Scholar]
- 23.Srinivas NR. Curr Clin Pharmacol. 2009;4:67. doi: 10.2174/157488409787236065. [DOI] [PubMed] [Google Scholar]
- 24.Shukla S, Gupta S. Pharm Res. 2010;27:962. doi: 10.1007/s11095-010-0089-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Patel D, Shukla S, Gupta S. Int J Oncol. 2007;30:233. [PubMed] [Google Scholar]
- 26.Keller TH, Pichota A, Yin Z. Curr Opin Chem Biol. 2006;10:357. doi: 10.1016/j.cbpa.2006.06.014. [DOI] [PubMed] [Google Scholar]
- 27.Di L, Kerns EH, Carter GT. Curr Pharm Des. 2009;15:2184. doi: 10.2174/138161209788682479. [DOI] [PubMed] [Google Scholar]
- 28.Manthey JA, Guthrie N. J Agric Food Chem. 2002;50:5837. doi: 10.1021/jf020121d. [DOI] [PubMed] [Google Scholar]
- 29.Chan KF, Zhao Y, Burkett BA, Wong IL, Chow LM, Chan TH. J Med Chem. 2006;49:6742. doi: 10.1021/jm060593+. [DOI] [PubMed] [Google Scholar]
- 30.Lin AS, Nakagawa-Goto K, Chang FR, Yu D, Morris-Natschke SL, Wu CC, Chen SL, Wu YC, Lee KH. J Med Chem. 2007;50:3921. doi: 10.1021/jm070363a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wong IL, Chan KF, Tsang KH, Lam CY, Zhao Y, Chan TH, Chow LM. J Med Chem. 2009;52:5311. doi: 10.1021/jm900194w. [DOI] [PubMed] [Google Scholar]
- 32.Walle T. Mol Pharm. 2007;4:826. doi: 10.1021/mp700071d. [DOI] [PubMed] [Google Scholar]
- 33.Gradolatto A, Canivenc-Lavier MC, Basly JP, Siess MH, Teyssier C. Drug Metab Dispos. 2004;32:58. doi: 10.1124/dmd.32.1.58. [DOI] [PubMed] [Google Scholar]
- 34.Gradolatto A, Basly JP, Berges R, Teyssier C, Chagnon MC, Siess MH, Canivenc-Lavier MC. Drug Metab Dispos. 2005;33:49. doi: 10.1124/dmd.104.000893. [DOI] [PubMed] [Google Scholar]
- 35.Wen X, Walle T. Drug Metab Dispos. 2006;34:1786. doi: 10.1124/dmd.106.011122. [DOI] [PubMed] [Google Scholar]
- 36.Chan KF, Zhao Y, Chow TW, Yan CS, Ma DL, Burkett BA, Wong IL, Chow LM, Chan TH. ChemMedChem. 2009;4:594. doi: 10.1002/cmdc.200800413. [DOI] [PubMed] [Google Scholar]
- 37.Chen H, Yang Z, Ding C, Chu L, Zhang Y, Terry K, Liu H, Shen Q, Zhou J. ACS Med Chem Lett. 2013;4:180. doi: 10.1021/ml3003082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Apte M, Pirola R, Wilson J. Antioxid Redox Signal. 2011;15:2711. doi: 10.1089/ars.2011.4079. [DOI] [PubMed] [Google Scholar]
- 39.Lerch MM, Gorelick FS. Gastroenterology. 2013;144:1180. doi: 10.1053/j.gastro.2012.12.043. [DOI] [PubMed] [Google Scholar]
- 40.Aghdassi AA, Mayerle J, Christochowitz S, Weiss FU, Sendler M, Lerch MM. Fibrogenesis Tissue Repair. 2011;4:26. doi: 10.1186/1755-1536-4-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chao C, Tallman ML, Ives KL, Townsend CM, Jr, Hellmich MR. J Surg Res. 2005;129:313. doi: 10.1016/j.jss.2005.04.038. [DOI] [PubMed] [Google Scholar]
- 42.Apte MV, Haber PS, Applegate TL, Norton ID, McCaughan GW, Korsten MA, Pirola RC, Wilson JS. Gut. 1998;43:128. doi: 10.1136/gut.43.1.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bachem MG, Schneider E, Gross H, Weidenbach H, Schmid RM, Menke A, Siech M, Beger H, Grunert A, Adler G. Gastroenterology. 1998;115:421. doi: 10.1016/s0016-5085(98)70209-4. [DOI] [PubMed] [Google Scholar]
- 44.Ruifrok AC, Johnston DA. Anal Quant Cytol Histol. 2001;23:291. [PubMed] [Google Scholar]
- 45.Schneider CA, Rasband WS, Eliceiri KW. Nat Methods. 2012;9:671. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]