

Abstract
The interplay between Clostridium difficile and the host's metabolome is believed to influence the severity of infection. However, the mechanism for this phenomenon remains unclear. In this study we model one of these metabolic pathways by focussing on tryptophan metabolism in the host. We found that inhibition of tryptophan catabolism in indoleamine 2,3-dioxygenase (IDO1) knockout mice led to increased mucosal destruction and cecal haemorrhage, increased production of IFNγ in response to C. difficile infection, but no significant change in mucosal effector or regulatory T cell numbers or IL-10 mRNA expression. The increased immunopathology in infected IDO1 knockout mice was associated with a lower Clostridium difficile burden and an increased percentage of IFNγ-expressing neutrophils. We further demonstrated the ability of kynurenine to induce apoptosis in bone marrow-derived neutrophils while the presence of tryptophan reversed this effect, providing a possible mechanism for the increased neutrophil accumulation in IDO1-/- mice. We conclude that Clostridium difficile induces tryptophan catabolism in cecal lamina propria cells, which restricts Clostridium difficile-associated immunopathology and the accumulation of IFNγ-expressing neutrophils. This might represent a self-regulatory mechanism for neutrophils, via the IFNγ-IDO1 pathway, to restrict their own accumulation during infection. These findings have important clinical implications as IDO inhibitors are currently used to treat cancer in clinical trials (in patients particularly susceptible to getting Clostridium difficile infection) and treatment with IDO1 inhibitors may exacerbate the severity of Clostridium difficile colitis.
Keywords: Metabolomics, Kynurenine, IDO1, inflammation, myeloid
Introduction
The interplay between Clostridium difficile (C. difficile) and the intestinal metabolome is believed to influence the severity of infection (1, 2). For example, the host's bile acid metabolites deoxycholate and taurocholate potently stimulate germination of C. difficile spores (3), while chenodeoxycholate inhibits it (3). Bile salts can also regulate the activity of Rho proteins in host cells thereby providing protection against C. difficile (4). Moreover, antibiotics, which can predispose to C. difficile infection, alter the microbiome and metabolome profiles in the mouse gut (5) and feces (6), for example by decreasing short chain fatty acids (6). These metabolic alterations increase the susceptibility to C. difficile infection (5). In addition, C. difficile induces dysbiosis of the intestinal microbiota (7), which is associated with changes in their metabolomic profile (6). Therefore, while there is considerable evidence to support the conclusion that intestinal metabolites regulate the severity of C. difficile infection, few studies to date have demonstrated a clear mechanism for the function of these metabolites during infection. Determining these mechanisms is particularly useful for the development of therapeutic drugs that inhibit the metabolic pathways contributing to disease.
In this study we model one of the C. difficile-regulated metabolic pathways by focussing on tryptophan metabolism in the host. Recent studies have reported an induction of this pathway during Citrobacter rodentium infection and TNBS-induced colitis of the intestine (8, 9). Tryptophan metabolism is mediated by an enzyme called indoleamine 2,3-dioxygenase-1 (IDO1) (10). IDO1 induces the conversion of tryptophan into kynurenine and downstream metabolites (11). This conversion mainly occurs in myeloid cells and restricts T cell proliferation by diminishing the tryptophan pool due to conversion to kynurenine (12-14). Moreover, this conversion also promotes T regulatory cell (Treg) generation (15, 16), which is partly mediated by IDO1-generated ligands for the aryl hydrocarbon receptor (AhR) (17, 18). Based on these observations, we hypothesize that tryptophan catabolism by IDO1 may down-regulate the immunopathology of Clostridium difficile.
To test our hypothesis, we infected wild-type (WT) versus IDO1 knockout (IDO1-/-) mice with C. difficile following the administration of cefoperazone according to a previously established protocol (19). The outcome of this study would therefore determine the specific role of tryptophan-kynurenine conversion during C. difficile infection.
Materials and Methods
Animals
IDO1-/- mice on a C57BL/6 background, which were previously generated by Baban et al. (20), were obtained from Jackson Labs (stock #005867). All animals used were 8 week old males. Animals were housed in groups (3-5 animals per cage; mixed genotypes) in microisolator cages under specific pathogen-free conditions. Prior to C. difficile infection, cage bedding was replaced between cages repetitively for 7 days to normalize the microflora between cages. All studies were approved by the University of Michigan Institutional Animal Care and Use Committee.
Antibiotic Administration and Infection with C. difficile
C. difficile strain VPI 10463 (ATCC 43255) was kindly provided by G. Huffnagle. Spores were prepared as described previously (19). WT and IDO1-/- mice were given cefoperazone (0.5 mg / ml) (MP Bioworks, Cat# 199695) in sterile drinking water for 5 days. Antibiotic water was refreshed every other day to prevent antibiotic breakdown. After 5 days, mice were switched to regular water to recover for 2 days before being orally gavaged with 6 × 106 C. difficile spores. Mice were euthanized after 2 days of infection.
Tissue Collection
Cecal tissue was divided intro different groups: 1) For mass spectrometric analysis of kynurenine, cecal tissue was snap frozen in liquid nitrogen; 2) for RNA tissue samples were homogenized in TRIzol (Invitrogen, Carlsbad, CA) and cleaned up using the RNEasy Microkit (Qiagen, Valencia, CA) as previously described (21); 3) for histology, the cecum was cut longitudinally and the section incubated in formalin for 2 days followed by 70% ethanol, processing and paraffin embedding; 4) for frozen blocks, longitudinal cecal tissue was frozen in OCT compound (Fisher Scientific, Houston, TX) on dry ice (22); 5) for DNA samples to measure toxin DNA, cecal tissue was snap frozen in liquid nitrogen and extracted using the DNEasy Blood and Tissue Kit (Qiagen). All tissue segments were collected from equivalent positions within the ceca of different mice.
Mass Spectrometry
Mass spectrometry was performed by the Michigan Regional Comprehensive Metabolomics Resource Core. Metabolites were extracted from snap-frozen cecal tissue using an extraction solvent made of an 8:1:1 mixture of methanol, chloroform and water. 250μl of the extraction solvent were added to the tissue samples in Eppendorf tubes, and tissue samples were then sonicated. All the samples were incubated on ice for 10 min, vortexed, and then centrifuged at 16,000 g for 10 min. Supernatants containing metabolites were dried and reconstituted in 100ml of mobile phase A (water with 0.1% formic acid) for LC-MS analysis. Samples were analysed by liquid chromatography-mass spectrometry (LC-MS) using a Waters BEH C18 column (Waters Corporation, Milford, MA) for chromatographic separation on an Agilent 1200 chromatography system (Agilent Technologies, Santa Clara, CA), followed by analysis on an Agilent 6410 series triple quadruple mass spectrometer with electrospray ionization source (ESI) (Agilent Technologies), operated in a positive mode. The following transition was used to identify and quantify kynurenine: m/z 209.1 → m/z 192.1. Data was processed using MassHunter workstation software, version B.04 (Agilent Technologies).
Immunofluorescence and Antibodies
Immunofluorescence on frozen sections was performed as previously described (21). The following antibodies were used: polyclonal goat anti-mouse IDO1 (I-17 Cat # sc-25121, or M-20 Cat # sc-25123, Santa Cruz Biotechnology, Santa Cruz, CA); FITC conjugated monoclonal hamster anti CD11c (clone N418, Cat # 117306, BioLegend, San Diego, CA); monoclonal rabbit anti E-Cadherin (clone 24E10, Cat #3195, Cell Signaling, Boston, MA);FITC conjugated monoclonal mouse anti MPO (clone 2D4, Cat #ab90812, Abcam, Cambridge, MA); Alexa Fluor 647 conjugated (far red) monoclonal rat anti IFNγ (clone XMG1.2, Cat #505816, BioLegend). Staining was visualized using an Olympus Fluoview scanning confocal microscope (Olympus, Center Valley, PA).
Flow Cytometry
Cecal cells were digested using a modified version of the protocol by Geem et al.(23). Briefly ceca were cut into 1 cm pieces and digested twice, 20 min each, in 2mM EDTA in calcium- and magnesium-free HBSS containing 5% FBS. Each digestion was performed in an orbital shaker at 37 °C and 200 rpm. The tissue was passed through a mesh wire strainer after each digestion and the solutions collected. The remaining tissue was digested in 17.9 μg/ml Liberase TM (Cat #05401119001, Roche Diagnostics Corporation, Indianapolis, IN) for 11 min and passed through a 100 μm cell strainer. The digested cells from the EDTA and Liberase steps were pooled together for flow cytometric staining. The cells were stained with the following: 1) LIVE/DEAD Fixable Aqua Dead Cell Stain Kit (Cat #L34957, Life Technologies, Green Island, NY); 2) IFNγ-APC (clone XMG1.2, Cat #505809, BioLegend); 3) CD11b-eFluor 450 (clone M1/70, Cat #48-0112-82, eBioscience, San Diego, CA); 4) Ly6G-PE (clone 1A8, Cat #127607, BioLegend); 5) CD4-FITC (clone GK1.5, Cat #11-0041-85, eBioscience); 6) CD25-PE-Cy7 (clone PC61.5, Cat #25-0251-81, eBioscience). Foxp3 staining was performed separately using Foxp3-PE (clone FJK-16s, Cat #12-5773-82, eBioscience) and combined with the LIVE/DEAD-Aqua, CD4-FITC and CD25-PE antibodies described above. Flow cytometric analysis was performed using a BD FACSAria III (BD Biosciences, San Jose, CA).
Isolation of Bone Marrow-Derived Neutrophils and Measurement of Apoptosis
Isolation of bone marrow-derived neutrophils was performed according to a previously described protocol (24). Briefly, bone marrow cells were flushed with tryptophan-free DMEM F-12 (Cat #D9807-04, United States Biological, Salem, MA), and separated on a gradient prepared by overlaying 3 ml Histopaque 1119 (bottom layer, Cat #11191, Sigma-Aldrich, St. Louis, MO), 3 ml Histopaque 1077 (middle layer, Cat #10771, Sigma-Aldrich), and 1 ml cell-containing PBS (upper layer). The gradient was centrifuged for 30 min at 2,000 rpm without brakes. The neutrophils were collected from the interface of Histopaque 1119 and Histopaque 1077. The cells were plated in tryptophan-free media, treated with 100μM kynurenine (Cat #K8625, Sigma-Aldrich) with or without 100μM L-tryptophan (Cat #T0254, Sigma-Aldrich), or vehicle control, and assayed for apoptosis using the Annexin V staining kit (Cat #88-8007-72, eBioscience).
Quantitative PCR and real time quantitative PCR (RT-qPCR)
For DNA quantification of C. difficile toxins A (TcdA) and B (TcdB), quantitative PCR was performed on a CFX96 real-time PCR detection system using the following primers:
TcdA Forward 5′-GGT AAT AAT TCA AAA GCG GCT-3′
TcdA Reverse 5′-AGC ATC CGT ATT AGC AGG TG-3′
TcdB Forward 5′-GAA AGT CCA AGT TTA CGC TCA-AT-3′
TcdB Reverse 5′-GCT GCA CCT AAA CTT ACA CCA-3′
The DNA signal was normalized to TNF-α using the following primers:
TNF-α Forward 5′-GGC TTT CCG AAT TCA CTG GAG-3′
TNF-α Reverse 5′-CCC CGG CCT TCC AAA TAA A-3′
The method used for qPCR quantification and the RT-qPCR primers for IFNγ, IL-4, IL-17A, TNF-α, IL-1β, and HPRT was described previously (21). The RT-qPCR primers used for IL-10 and Foxp3 were as follows:
IL-10 Forward 5′ AGT GGA GCA GGT GAA GAG TG 3′
IL-10 Reverse 5′ TTC GGA GAG AGG TAC AAA CG 3′
Foxp3 Forward 5′ TCT CCA GGT TGC TCA AAG TC 3′
Foxp3 Reverse 5′ GCA GAA GTT GCT GCT TTA GG 3′
Pathological Scoring and Morphometric Analysis
The criteria for pathological scoring of C. difficile-infected ceca were previously described (25). Ceca were scored blindly and assigned pathological scores 1-4 depending on severity. For morphometric analysis, positive cells were counted in 20× fields and then divided by the total number of fields examined. The data was compared in 3-4 mice per group.
Statistical Analysis
Data were tested for normality using the Shapiro-Wilk W test (Prism, GraphPad Software, La Jolla, CA). One-way ANOVA followed by Dunnett's (parametric) or Dunn's (non-parametric) multiple comparisons test was performed using GraphPad Prism (Version 6.00 for Windows, GraphPad Software, La Jolla, CA, www.graphpad.com). P values less than 0.05 were considered significant.
Results
C. difficile induces tryptophan catabolism via IDO1-expressing lamina propria cells
We have postulated that different components of the metabolome regulate the severity of C. difficile infection. This would suggest that tryptophan metabolism, being one of these components, might affect the immunopathogenicity of C. difficile. To begin to address this hypothesis, we measured the host's cecal kynurenine levels in response C. difficile. C. difficile infection (Figure 1A) induced kynurenine levels in the cecal tissue from a baseline level of 345 pg/mg to 2753 pg/mg (p < 0.05) (Figure 1B). Next, we investigated the role of IDO1 during C. difficile infection using an IDO1-/- mouse model (20) (Figure 1C) to study the requirement of IDO1 in mediating tryptophan to kynurenine conversion. We observed that C. difficile infection induced IDO1 4.7-fold in the ceca of wild-type mice, but not IDO1-/- mice (Figure 1D). To determine whether IDO1 was required for the conversion of tryptophan to kynurenine, we measured kynurenine levels in IDO1-/- ceca of C. difficile-infected mice. IDO1 deficiency prevented the production of kynurenine in response to C. difficile infection (Figure 1B). To investigate which host cells were responsible for the conversion of tryptophan into kynurenine in response to C. difficile infection, we used immunofluorescent microscopy to further characterize the cellular distribution of IDO1 during C. difficile infection. IDO1 was not readily detectable in uninfected wild-type ceca (Figure 1E, upper right panel). However, in response to C. difficile, IDO1 (red) was induced in cells within the E-cadherin-negative lamina propria compartment (Figure 1E, lower right panel). Some of these IDO1+ cells (red) coincided with CD11c expression (orange) (Figure 1E, lower panels, white arrows). Indeed, IDO1 (red) was not expressed in CD11c+ cells of the uninfected (Figure 1F, upper panel), but was induced in these cells during C. difficile infection (Figure 1F, lower panel). Hence IDO1 is induced in CD11c+ myeloid cells, amongst other stromal cells, during C. difficile infection. Our data indicate that C. difficile infection induces conversion of tryptophan into kynurenine via intestinal lamina propria cellular expression of IDO1. This is consistent with prior reports that IDO1 is mainly expressed in myeloid cell (12-14).
Figure 1. C. difficile induces tryptophan catabolism via lamina propria IDO1 expression.
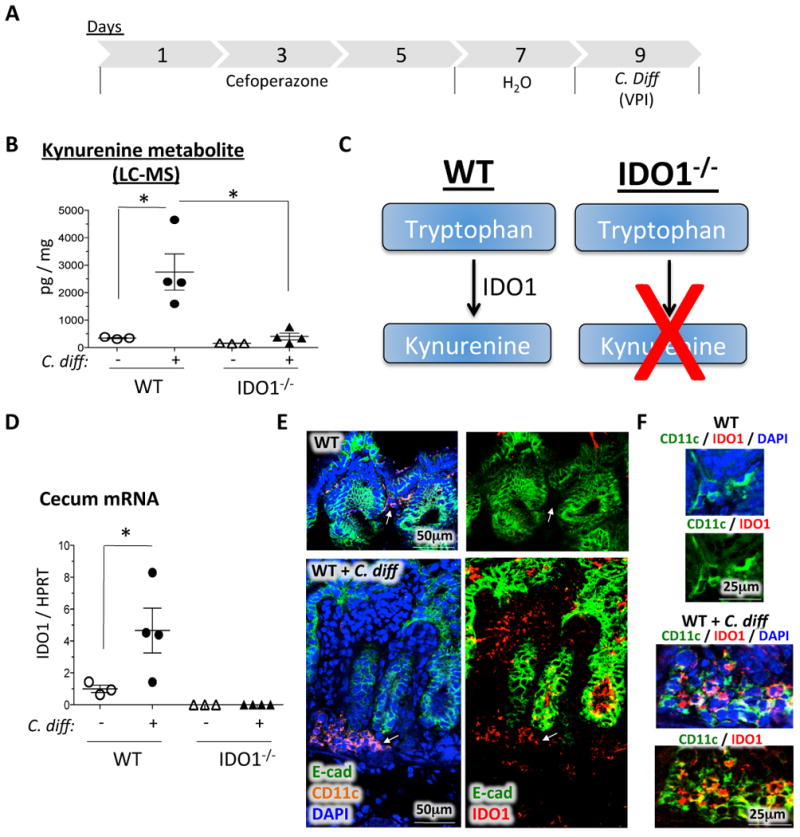
A) Protocol for C. difficile infection. Mice were treated with cefoperazone for 5 days, switched to regular water for 2 days, and then infected with C. difficile for 2 days. B) Liquid chromatography-mass spectrometry (LC-MS) measurements of cecal tissue kynurenine levels in uninfected and C. difficile-infected WT versus IDO1-/- mice. Values are normalized to weight of cecal tissue used in the experiment. C) Diagram illustrating the difference in tryptophan conversion to kynurenine between WT and IDO1-/- mice. D) RT-qPCR of IDO1 in C. difficile-infected versus uninfected WT and IDO1-/- cecal tissue. E) Confocal immunofluorescent analysis of IDO1 (red), CD11c (orange), E-cadherin (green), and DAPI (blue) in uninfected versus C. difficile-infected ceca. The left panels demonstrate the E-cadherin (green), CD11c (orange) and DAPI (blue) staining patterns. The right panels demonstrate the IDO1 (red) and E-cadherin (green) staining patterns. Arrows indicate the location of CD11c+ cells (orange, left panels) and the status of IDO1 expression (red, right panels) in these cells. F) Confocal immunofluorescent analysis of IDO1 (red) and CD11c (green) in uninfected (upper panels) versus C. difficile-infected (lower panels) ceca. Images are presented with and without DAPI (blue). The areas presented are high power images of those highlighted by arrows in (E), but distinctly pseudocolored using the confocal microscope to highlight the co-localization. Uninfected controls were cefoperazone-treated similar to the infected group. Each data point represents one mouse. Error bars represent the standard errors of the mean. P-values are indicated such that * P < 0.05.
Tryptophan-kynurenine metabolism restricts C. difficile immunopathology
To examine the role of tryptophan-kynurenine metabolism during C. difficile infection, we assessed the macroscopic pathology of C. difficile-infected WT versus IDO1-/- mice. We observed that infected IDO1-/- ceca were haemorrhagic compared to infected WT mice (Figure 2A), despite no significant difference in the overall body weight of the mice in either group (Figure 2B). There was, however, increased immunopathology characterized by more severe polymorphonuclear infiltration (inflammation), and epithelial damage in the C. difficile-infected IDO1-/- mice compared to WT mice (Figure 2C and 2D). The worsened pathology did not arise from higher bacterial loads of C. difficile as the infected IDO1-/- mice exhibited lower toxin A (TcdA) and toxin B (TcdB) DNA than WT (Figure 3). Our data indicate that the loss of tryptophan conversion by IDO1 into kynurenine exacerbates C. difficile-induced immunopathology.
Figure 2. Tryptophan-kynurenine metabolism restricts C. difficile immunopathology.

A) Representative macroscopic imaging of the cecum and colon from C. difficile-infected WT versus IDO1-/- mice. The arrows indicate the areas of haemorrhage that were observed reproducibly in C. difficile-infected IDO1-/- ceca. B) Graphical representation of mouse weight fluctuations in uninfected and C. difficile-infected WT and IDO1-/- mice. Mouse weights are expressed as a percentage of original mouse weight at day 1. Uninfected controls were cefoperazone-treated similar to the infected group. Error bars represent the standard error of the mean. C) H&E staining of cecal sections from uninfected and C. difficile-infected WT versus IDO1-/- mice. D) Pathological scoring of edema, inflammation, and epithelial damage in uninfected and C. difficile-infected WT versus IDO1-/- mice. Bars represent the mean values. Uninfected controls were cefoperazone-treated similar to the infected group. Each data point represents one mouse. * P < 0.05. ** P < 0.01.
Figure 3. IDO deletion reduces C. difficile toxin levels in cecal tissue.

qPCR of TcdA (A) and TcdB (B) in uninfected and C. difficile-infected WT versus IDO1-/- cecal tissue. Each data point represents one mouse. Error bars represent the standard errors of the mean. * P < 0.05. ** P < 0.01.
IDO1 deletion exacerbates pro-inflammatory cytokine production
To investigate further the mechanism by which IDO1 regulates C. difficile immunopathology, we measured the helper T (Th) cell-derived cytokine expression in infected WT versus IDO1-/- ceca by qRT-PCR. We observed a significant increase in IFNγ expression and moderate increase in IL-4 and IL-17A in IDO1-/- ceca compared to WT ceca (Figure 4A-C). We did not detect a difference in myeloid cell-associated cytokines, TNF-α and IL-1β between WT and IDO1-/- mice (Figure 4D-E). The increase in effector helper T cell-derived cytokines suggested that IDO1 might have altered the activity of CD4 helper T cells. We therefore studied the cecal Th1/Treg cell populations by flow cytometry. Following live cell gating (Supplementary Figure 1), we did not observe an appreciable difference in the abundance of CD4+CD25- effector T cells or CD4+CD25+Foxp3+ Tregs between infected WT and IDO1-/- ceca (Figure 5A-D). Because IDO1 was reported to be a source of ligands for AhR-dependent Treg generation (17, 18), we measured further the mRNA levels of cecal Foxp3 and IL-10 and found similar levels between WT vs IDO1-/- mice (Figure 5E and F). Hence, the lack of association between T cell frequency and IFNγ induction prompted us to investigate the source of IFNγ in the infected cecal tissue.
Figure 4. IDO1 deletion exacerbates pro-inflammatory but not myeloid cell-associated cytokine production.

A-E) RT-qPCR quantification of IFN-γ, IL-4, IL-17A, TNF-α, and IL-1β in uninfected and C. difficile-infected WT versus IDO1-/- mice. Each data point represents one mouse. Uninfected controls were cefoperazone-treated similar to the infected group. Error bars represent the standard errors of the mean. * P < 0.05.
Figure 5. IDO1 deletion does not alter effector T cell or Treg populations.
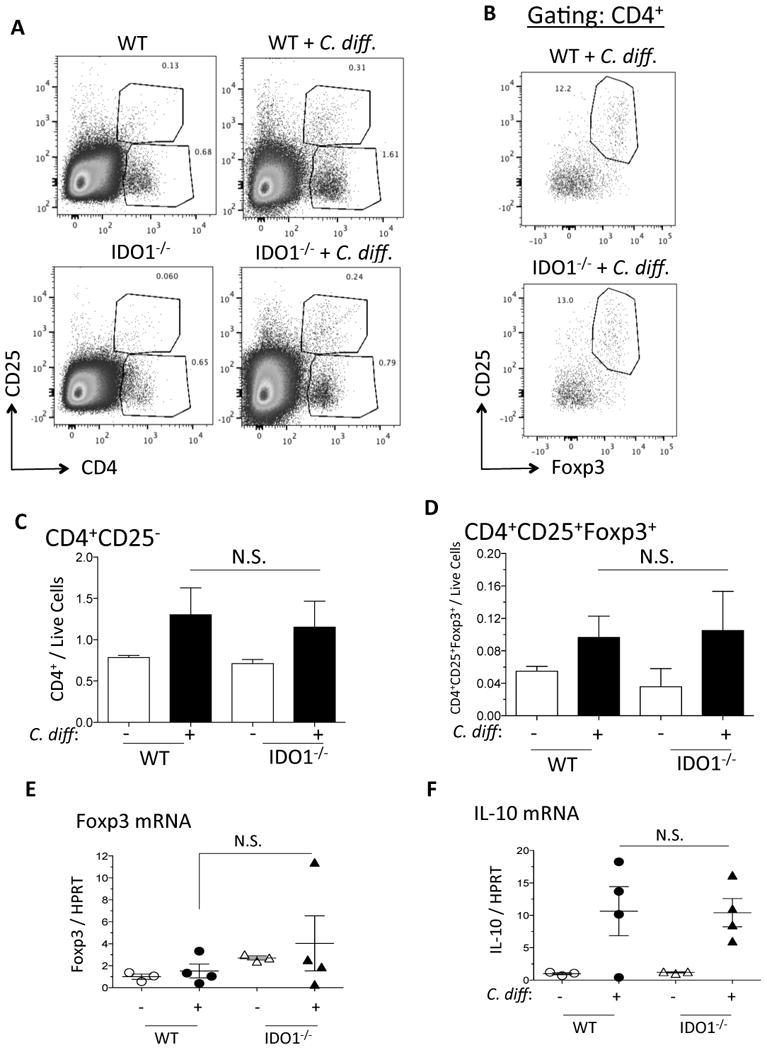
A) Representative flow plots of CD4+CD25- effector T cells and CD4+CD25+ Tregs in uninfected and C. difficile-infected WT and IDO1-/- ceca. B) Representative flow plots of CD4+CD25+Foxp3+ cells in infected WT versus IDO1-/- ceca. Flow plots in (B) represent cell populations that had been previously gated to CD4+. C-D) Flow cytometric quantification of CD4+CD25- T cells and CD4+CD25+Foxp3+ Tregs from 3 mice per group of uninfected and C. difficile-infected WT versus IDO1-/- ceca. E-F) RT-qPCR quantification of Foxp3 and IL-10 in uninfected and C. difficile-infected WT versus IDO1-/- ceca. Error bars represent the standard errors of the mean. N.S. = not significant.
IFNγ is produced by neutrophils, not T cells, during C. difficile infection
We sought to determine the cellular sources of IFNγ because it was significantly increased in IDO1-/- mice and IFNγ had been shown to mediate C. difficile toxin A-induced enteritis (26). We first analysed IFNγ-expressing populations in the cecum of uninfected vs infected WT and IDO1-/- mice. In WT mice, we measured an increase in the percentage of IFNγ-expressing CD4-negative population following C. difficile infection and this increase was even higher in IDO1-/- mice (Figure 6A). Because a previous study indicated that C. difficile toxin A might induce IFNγ production by neutrophils (26), we examined the neutrophil population as a potential source of IFNγ. We observed that the majority of IFNγ-expressing cells were found in the CD11b+Ly6G+ myeloid population, which includes neutrophils (Figure 6B). In contrast, the IFNγ-negative population contained both populations of CD11b+Ly6G- and CD11b+Ly6G+ myeloid cells (Figure 6B). To determine whether these IFNγ-expressing CD11b+Ly6G+ cells represent neutrophils, we measured the co-localization of myeloperoxidase (MPO) and IFNγ in mouse cecal tissue and the result indicated that neutrophils were the major source of IFNγ in the cecum of C. difficile infected mice (Figure 6C and Supplementary Figure 2).
Figure 6. C. difficile infection induces IFNγ production by neutrophils, but not T cells.

A) Flow cytometric dot plots of IFNγ versus CD4 in uninfected and infected WT versus IDO1-/- ceca. B) Upper panel, flow cytometric gating of IFNγ-negative (P1) versus IFNγ-positive (P2) cecal populations following C. difficile infection. Lower panels, flow cytometric analysis of CD11b and Ly6G in IFNγ-negative (P1) versus IFNγ-positive (P2) cecal populations. C) Confocal immunofluorescent analysis of MPO (green) and IFNγ (red) in infected WT and IDO1-/- ceca. High power fields (right panels) represent the numbered areas from the low power field image.
IDO1 deficiency increases neutrophil abundance
In the previous sections, we showed that IDO1 deficiency increases IFNγ production during C. difficile infection, and that neutrophils are the major source of IFNγ. We therefore investigated the possibility that the induction of IFNγ was attributed to an increase in neutrophils in the IDO1-/- infected ceca. First, we showed increased CD11b+Ly6G+ neutrophil accumulation in the cecum during C. difficile infection (Figure 7A, upper panels). The percentage of neutrophils was significantly higher in infected IDO1-/- than WT ceca (Figure 7A and 7B). We did not observe any changes in CD11b+Ly6G- myeloid cells between infected WT and IDO1-/- ceca. Therefore, our data revealed the role of tryptophan metabolism in restricting neutrophil accumulation during C. difficile infection.
Figure 7. Tryptophan catabolism restricts neutrophil accumulation.

A) Representative flow plots of CD11b+Ly6G+ neutrophils and CD11b+Ly6G- myeloid cells in uninfected and C. difficile-infected WT versus IDO1-/- ceca. B) Flow cytometric quantification of CD11b+Ly6G+ neutrophils (upper panel) and CD11b+Ly6G- myeloid cells (lower panel) from 3 mice per group of uninfected and C. difficile-infected WT versus IDO1-/- ceca. C) Representative flow plot of Annexin-V staining in bone marrow-derived neutrophils treated with or without 100μm kynurenine overnight, in tryptophan-free media. D) Bar graph representation of apoptotic fold increase from 3 experiments of bone marrow-derived neutrophils treated with or without 100μm kynurenine in the presence/absence of 100μm tryptophan, in tryptophan-free media. Error bars represent the standard errors of the mean. N.S. = not significant. * P < 0.05.
Kynurenine promotes bone marrow-derived neutrophil apoptosis in vitro
Based on a preliminary report by van der Sluijs et al. that kynurenine induces human neutrophil apoptosis (27), we studied the effect of kynurenine on bone marrow-derived neutrophils (Supplementary Figure 3) and showed that kynurenine promoted neutrophil apoptosis in vitro (Figure 7C and 7D). This effect was reversed by tryptophan (Figure 7D) indicating that the increase in IDO1 activity and the build up of kynurenine in the intestinal mucosa during C. difficile infection serves to restrict an over-exuberant inflammatory response, which may be harmful to the host fitness. Deficiency in IDO1 therefore leads to worsening disease outcome during C. difficile infection as observed in our IDO1-deficient mice.
Discussion
This study focuses on a specific mechanism in which one of the host's metabolic pathways, i.e. tryptophan catabolism, regulates the severity of C. difficile infection. The major conclusion is that tryptophan catabolism in cecal lamina propria cells limits neutrophil accumulation leading to less severe C. difficile immunopathology (Figure 8). These findings are based on the following experimental evidence. First, we established a mouse model of IDO1 deficiency in which the conversion of tryptophan to kynurenine is absent during C. difficile infection. Second, we showed that IDO1 deficiency exacerbates C. difficile immunopathology and increases pro-inflammatory cytokine production, e.g., IFNγ. Third, we demonstrated that the more severe immunopathology in IDO1-/- mice is associated with an increase in IFNγ– expressing neutrophils. Fourth, we showed that the neutrophil population is sensitive to kynurenine-triggered apoptosis. Collectively, our study describes a previously unknown mechanism of tryptophan catabolism in C. difficile infection that limits neutrophil accumulation and immunopathology by the activity of kynurenine.
Figure 8. A proposed model of the tryptophan-kynurenine pathway in C. difficile infection.

Upper panel, in WT mice C. difficile induces IDO1 expression in dendritic cells of the cecum. This converts tryptophan into kynurenine, which diminishes the neutrophil population. One mechanism for this process is mediated via the induction of neutrophil apoptosis by kynurenine. The reduction of neutrophils reduces IFNγ production and C. difficile immunopathology. Hence, the production of IFNγ by neutrophils initiates a self-regulatory mechanism, mediated by IDO1, to limit their own accumulation. Lower panel, in IDO1-/- mice C. difficile is unable to induce the conversion of tryptophan into kynurenine in dendritic cells. This leads to a reduction in kynurenine levels, reduction in apoptosis leading to an increase in neutrophils, an increase in IFNγ production, and an increase in C. difficile immunopathology.
The interplay between C. difficile and the host's metabolites has been previously described (1-5), but the precise mechanisms of this interplay remain unclear. However, the roles of tryptophan catabolism and IDO1 have been described in other systems. For example, IDO1 inhibition, which prevents the conversion of tryptophan to kynurenine (10), exacerbates the immunopathology in the TNBS colitis model (9, 28). Moreover, induction of IDO1 by immunostimulatory DNA limits the severity of TNBS colitis in mice (29). Indeed, the immunoregulatory function of IDO1 has been previously established, including its ability to prevent T-regulatory cell reprogramming into Th17-like cells (30) or to restrict T cell proliferation (12-14). These previous reports suggest that tryptophan catabolism plays a robust and versatile role in intestinal inflammation. However, no studies to date have described the mechanism in which IDO1 regulates acute bacteria-associated inflammation, which does not require T cell immunity. In this study we determined that tryptophan catabolism regulates neutrophils, which is a mechanism of the innate immune response. In addition, little is known regarding the extent to which IDO1 contributes to kynurenine production versus other tryptophan converting enzymes (IDO2 and TDO) (31), or tryptophan converting genes (Kyn1 and Kyn2) in the intestinal flora (32). By measuring the kynurenine levels in cecal tissue by mass spectrometry, we showed that the conversion of tryptophan to kynurenine is dependent on IDO1 of the host. However, since the microbiota has previously been shown to produce kynurenine and regulate AhR (33), we suggest that this mechanism cannot be observed in our model due to cefoperazone-induced dysbiosis. We therefore propose that the increased susceptibility to C. difficile is likely due to a combination of reduced AhR ligands from both the IDO1-deficient host and the dysbiotic microflora.
In contrast to the above reports, several studies have reached disparate conclusions about the role of tryptophan catabolism in the GI tract. A recent report demonstrates that IDO1 deletion did not affect the inflammatory response but rather reduced epithelial proliferation and tumor burden in a colon carcinogenesis model of azoxymethane (AOM) plus dextran sodium sulphate (DSS) (34). However, it is important to note that this phenotype is associated with a chronic model of tumor progression, which might trigger considerable epithelial expression of IDO1 during this process. In our study of short-term infection, IDO1 expression was predominantly stromal and strictly played an anti-inflammatory role. Hence, the contexts of a short-term infection versus a long-term tumorigenesis model are clearly not suitable for comparison. In addition, another study by Harrington et al. demonstrated that the deficiency of IDO1 attenuated the intestinal response to Citrobacter rodentium-induced colitis (8). However, the authors explained this disparity by proposing that the IDO1-/- mice exhibited reduced colonization by Citrobacter rodentium due to higher IgA secretions (8). Their observation that Citrobacter rodentium colonization is lower in IDO1-/- mice is consistent with our observation that C. difficile toxins are also reduced in our mouse model. However, in our model, the immunopathology was exacerbated despite lower colonization, which contrasts with their study. One potential explanation could be due to the contextual differences in neutrophil activation – and their ability to produce IFNγ – in response to different bacterial pathogens, e.g., C. difficile versus C. rodentium.
One conundrum in this study is while neutralization of neutrophils in mice has been shown to increase C. difficile-associated mortality (35) and intestinal bacterial translocation (36), our data suggest an exaggerated neutrophil response during C. difficile infection may lead to increased severity of the disease. Thus regulation of neutrophil activity during acute C. difficile infection may be critical to optimize host defense against pathogens while limit immunopathology. In our study, we uncovered the role of tryptophan catabolism through IDO1 activity in restraining neutrophil proinflammatory activity.
The findings from our study are significant because they provide a mechanism for one of the host's metabolic pathways that can influence the severity of C. difficile infection. This finding may pave the way for other similar investigations of other metabolic pathway mechanisms. Moreover, a therapeutic drug has already been developed to block the function of the IDO1 pathway and is currently being tested in clinical trials (37). It is therefore critical to understand the function of this pathway during C. difficile infection to determine the potential usefulness of this drug in this context. It appears from our results that this drug would exacerbate the C. difficile-associated colitis, which is more common in this patient population (38). Furthermore, this study highlights an important novel observation in which neutrophils mediate C. difficile immunopathology by producing cytokines that are usually believed to be associated with an effector T cell response. In addition, it appears that neutrophils regulate their own accumulation during infection by producing IFNγ, which is known to induce IDO1 in dendritic cells (39) (Figure 8). This will prompt further investigation about the activation status of these neutrophils to produce these cytokines and their exact functions. Hence new avenues are now open for investigating the regulation of neutrophils by myeloid cells during the innate immune response, and in a manner independent of adaptive immunity.
In conclusion, our study demonstrates a specific mechanism by which the host's metabolites regulate C. difficile infection, and identifies a novel role for tryptophan metabolism in regulating neutrophil accumulation. The study provides new avenues to investigate the mechanisms of neutrophil activation to produce IFNγ, and provides insights about the potential side effects of the IDO1 inhibitor in cancer patients. These findings have important clinical implications as IDO inhibitors are currently used to treat cancer in clinical trials (in patients particularly susceptible to getting C. difficile infection) and treatment with IDO inhibitors may exacerbate the severity of C. difficile colitis.
Supplementary Material
Acknowledgments
Grant support: Research reported in this publication was supported by NIDDK and NIAID of the National Institutes of Health under award number 5P30DK034933 (MEZ), R01DK087708-01 (JYK), U24DK097153 (CFB), U19 090871 (VBY, GH), and T32DK094775 (YMC, AS). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
- 1.Britton RA, Young VB. Interaction between the intestinal microbiota and host in Clostridium difficile colonization resistance. Trends Microbiol. 2012;20:313–319. doi: 10.1016/j.tim.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Peniche AG, Savidge TC, Dann SM. Recent insights into Clostridium difficile pathogenesis. Curr Opin Infect Dis. 2013;26:447–453. doi: 10.1097/01.qco.0000433318.82618.c6. [DOI] [PubMed] [Google Scholar]
- 3.Sorg JA, Sonenshein AL. Chenodeoxycholate is an inhibitor of Clostridium difficile spore germination. J Bacteriol. 2009;191:1115–1117. doi: 10.1128/JB.01260-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brandes V, Schelle I, Brinkmann S, Schulz F, Schwarz J, Gerhard R, Genth H. Protection from Clostridium difficile toxin B-catalysed Rac1/Cdc42 glucosylation by tauroursodeoxycholic acid-induced Rac1/Cdc42 phosphorylation. Biol Chem. 2012;393:77–84. doi: 10.1515/BC-2011-198. [DOI] [PubMed] [Google Scholar]
- 5.Theriot CM, Koenigsknecht MJ, Carlson PE, Jr, Hatton GE, Nelson AM, Li B, Huffnagle GB, L ZJ, Young VB. Antibiotic-induced shifts in the mouse gut microbiome and metabolome increase susceptibility to Clostridium difficile infection. Nature communications. 2014;5:3114. doi: 10.1038/ncomms4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhao Y, Wu J, Li JV, Zhou NY, Tang H, Wang Y. Gut microbiota composition modifies fecal metabolic profiles in mice. J Proteome Res. 2013;12:2987–2999. doi: 10.1021/pr400263n. [DOI] [PubMed] [Google Scholar]
- 7.Lawley TD, Clare S, Walker AW, Stares MD, Connor TR, Raisen C, Goulding D, Rad R, Schreiber F, Brandt C, Deakin LJ, Pickard DJ, Duncan SH, Flint HJ, Clark TG, Parkhill J, Dougan G. Targeted restoration of the intestinal microbiota with a simple, defined bacteriotherapy resolves relapsing Clostridium difficile disease in mice. PLoS Pathog. 2012;8:e1002995. doi: 10.1371/journal.ppat.1002995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harrington L, Srikanth CV, Antony R, Rhee SJ, Mellor AL, Shi HN, Cherayil BJ. Deficiency of indoleamine 2,3-dioxygenase enhances commensal-induced antibody responses and protects against Citrobacter rodentium-induced colitis. Infect Immun. 2008;76:3045–3053. doi: 10.1128/IAI.00193-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Takamatsu M, Hirata A, Ohtaki H, Hoshi M, Hatano Y, Tomita H, Kuno T, Saito K, Hara A. IDO1 Plays an Immunosuppressive Role in 2,4,6-Trinitrobenzene Sulfate-Induced Colitis in Mice. J Immunol. 2013;191:3057–3064. doi: 10.4049/jimmunol.1203306. [DOI] [PubMed] [Google Scholar]
- 10.Shimizu T, Nomiyama S, Hirata F, Hayaishi O. Indoleamine 2,3-dioxygenase. Purification and some properties. J Biol Chem. 1978;253:4700–4706. [PubMed] [Google Scholar]
- 11.Takikawa O, Yoshida R, Kido R, Hayaishi O. Tryptophan degradation in mice initiated by indoleamine 2,3-dioxygenase. J Biol Chem. 1986;261:3648–3653. [PubMed] [Google Scholar]
- 12.Munn DH, Zhou M, Attwood JT, Bondarev I, Conway SJ, Marshall B, Brown C, Mellor AL. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science. 1998;281:1191–1193. doi: 10.1126/science.281.5380.1191. [DOI] [PubMed] [Google Scholar]
- 13.Mellor AL, Sivakumar J, Chandler P, Smith K, Molina H, Mao D, Munn DH. Prevention of T cell-driven complement activation and inflammation by tryptophan catabolism during pregnancy. Nat Immunol. 2001;2:64–68. doi: 10.1038/83183. [DOI] [PubMed] [Google Scholar]
- 14.Munn DH, Shafizadeh E, Attwood JT, Bondarev I, Pashine A, Mellor AL. Inhibition of T cell proliferation by macrophage tryptophan catabolism. J Exp Med. 1999;189:1363–1372. doi: 10.1084/jem.189.9.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sharma MD, Baban B, Chandler P, Hou DY, Singh N, Yagita H, Azuma M, Blazar BR, Mellor AL, Munn DH. Plasmacytoid dendritic cells from mouse tumor-draining lymph nodes directly activate mature Tregs via indoleamine 2,3-dioxygenase. J Clin Invest. 2007;117:2570–2582. doi: 10.1172/JCI31911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen W, Liang X, Peterson AJ, Munn DH, Blazar BR. The indoleamine 2,3-dioxygenase pathway is essential for human plasmacytoid dendritic cell-induced adaptive T regulatory cell generation. J Immunol. 2008;181:5396–5404. doi: 10.4049/jimmunol.181.8.5396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mezrich JD, Fechner JH, Zhang X, Johnson BP, Burlingham WJ, Bradfield CA. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J Immunol. 2010;185:3190–3198. doi: 10.4049/jimmunol.0903670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nguyen NT, Kimura A, Nakahama T, Chinen I, Masuda K, Nohara K, Fujii-Kuriyama Y, Kishimoto T. Aryl hydrocarbon receptor negatively regulates dendritic cell immunogenicity via a kynurenine-dependent mechanism. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:19961–19966. doi: 10.1073/pnas.1014465107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Theriot CM, Koumpouras CC, Carlson PE, Bergin, Aronoff DM, Young VB. Cefoperazone-treated mice as an experimental platform to assess differential virulence of Clostridium difficile strains. Gut Microbes. 2011;2:326–334. doi: 10.4161/gmic.19142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baban B, Chandler P, McCool D, Marshall B, Munn DH, Mellor AL. Indoleamine 2,3-dioxygenase expression is restricted to fetal trophoblast giant cells during murine gestation and is maternal genome specific. J Reprod Immunol. 2004;61:67–77. doi: 10.1016/j.jri.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 21.El-Zaatari M, Kao JY, Tessier A, Bai L, Hayes MM, Fontaine C, Eaton KA, Merchant JL. Gli1 deletion prevents Helicobacter-induced gastric metaplasia and expansion of myeloid cell subsets. PLoS One. 2013;8:e58935. doi: 10.1371/journal.pone.0058935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun Y, Zhang M, Chen CC, Gillilland M, 3rd, Sun X, El-Zaatari M, Huffnagle GB, Young VB, Zhang J, Hong SC, Chang YM, Gumucio DL, Owyang C, Kao JY. Stress-induced corticotropin-releasing hormone-mediated NLRP6 inflammasome inhibition and transmissible enteritis in mice. Gastroenterology. 2013;144:1478–1487. 1487 e1471–1478. doi: 10.1053/j.gastro.2013.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Geem D, Medina-Contreras O, Kim W, Huang CS, Denning TL. Isolation and characterization of dendritic cells and macrophages from the mouse intestine. J Vis Exp. 2012:e4040. doi: 10.3791/4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Swamydas M, Lionakis MS. Isolation, purification and labeling of mouse bone marrow neutrophils for functional studies and adoptive transfer experiments. J Vis Exp. 2013:e50586. doi: 10.3791/50586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen X, Katchar K, Goldsmith JD, Nanthakumar N, Cheknis A, Gerding DN, Kelly CP. A mouse model of Clostridium difficile-associated disease. Gastroenterology. 2008;135:1984–1992. doi: 10.1053/j.gastro.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 26.Ishida Y, Maegawa T, Kondo T, Kimura A, Iwakura Y, Nakamura S, Mukaida N. Essential involvement of IFN-gamma in Clostridium difficile toxin A-induced enteritis. J Immunol. 2004;172:3018–3025. doi: 10.4049/jimmunol.172.5.3018. [DOI] [PubMed] [Google Scholar]
- 27.van der Sluijs K, Singh R, Dijkhuis A, Snoek M, Lutter R. Indoleamine-2,3-dioxygenase activity induces neutrophil apoptosis. Critical care. 2011;15:P208. [Google Scholar]
- 28.Gurtner GJ, Newberry RD, Schloemann SR, McDonald KG, Stenson WF. Inhibition of indoleamine 2,3-dioxygenase augments trinitrobenzene sulfonic acid colitis in mice. Gastroenterology. 2003;125:1762–1773. doi: 10.1053/j.gastro.2003.08.031. [DOI] [PubMed] [Google Scholar]
- 29.Ciorba MA, Bettonville EE, McDonald KG, Metz R, Prendergast GC, Newberry RD, Stenson WF. Induction of IDO-1 by immunostimulatory DNA limits severity of experimental colitis. J Immunol. 2010;184:3907–3916. doi: 10.4049/jimmunol.0900291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sharma MD, Hou DY, Liu Y, Koni PA, Metz R, Chandler P, Mellor AL, He Y, Munn DH. Indoleamine 2,3-dioxygenase controls conversion of Foxp3+ Tregs to TH17-like cells in tumor-draining lymph nodes. Blood. 2009;113:6102–6111. doi: 10.1182/blood-2008-12-195354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Platten M, Wick W, Van den Eynde BJ. Tryptophan catabolism in cancer: beyond IDO and tryptophan depletion. Cancer Res. 2012;72:5435–5440. doi: 10.1158/0008-5472.CAN-12-0569. [DOI] [PubMed] [Google Scholar]
- 32.Lima WC, Varani AM, Menck CF. NAD biosynthesis evolution in bacteria: lateral gene transfer of kynurenine pathway in Xanthomonadales and Flavobacteriales. Mol Biol Evol. 2009;26:399–406. doi: 10.1093/molbev/msn261. [DOI] [PubMed] [Google Scholar]
- 33.Zelante T, Iannitti RG, Cunha C, De Luca A, Giovannini G, Pieraccini G, Zecchi R, D'Angelo C, Massi-Benedetti C, Fallarino F, Carvalho A, Puccetti P, Romani L. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity. 2013;39:372–385. doi: 10.1016/j.immuni.2013.08.003. [DOI] [PubMed] [Google Scholar]
- 34.Thaker AI, Rao MS, Bishnupuri KS, Kerr TA, Foster L, Marinshaw JM, Newberry RD, Stenson WF, Ciorba MA. IDO1 metabolites activate beta-catenin signaling to promote cancer cell proliferation and colon tumorigenesis in mice. Gastroenterology. 2013;145:416–425 e411. 414. doi: 10.1053/j.gastro.2013.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jarchum I, Liu M, Shi C, Equinda M, Pamer EG. Critical role for MyD88-mediated neutrophil recruitment during Clostridium difficile colitis. Infect Immun. 2012;80:2989–2996. doi: 10.1128/IAI.00448-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hasegawa M, Yamazaki T, Kamada N, Tawaratsumida K, Kim YG, Nunez G, Inohara N. Nucleotide-binding oligomerization domain 1 mediates recognition of Clostridium difficile and induces neutrophil recruitment and protection against the pathogen. J Immunol. 2011;186:4872–4880. doi: 10.4049/jimmunol.1003761. [DOI] [PubMed] [Google Scholar]
- 37.Liu X, Shin N, Koblish HK, Yang G, Wang Q, Wang K, Leffet L, Hansbury MJ, Thomas B, Rupar M, Waeltz P, Bowman KJ, Polam P, Sparks RB, Yue EW, Li Y, Wynn R, Fridman JS, Burn TC, Combs AP, Newton RC, Scherle PA. Selective inhibition of IDO1 effectively regulates mediators of antitumor immunity. Blood. 2010;115:3520–3530. doi: 10.1182/blood-2009-09-246124. [DOI] [PubMed] [Google Scholar]
- 38.Chopra T, Alangaden GJ, Chandrasekar P. Clostridium difficile infection in cancer patients and hematopoietic stem cell transplant recipients. Expert Rev Anti Infect Ther. 2010;8:1113–1119. doi: 10.1586/eri.10.95. [DOI] [PubMed] [Google Scholar]
- 39.Chen W. IDO: more than an enzyme. Nat Immunol. 2011;12:809–811. doi: 10.1038/ni.2088. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.