

Abstract
Our understanding of cancer has grown considerably with recent advances in high-throughput genome and transcriptome sequencing, but techniques to comprehensively analyze protein activity are still in development. Methods to quantitatively measure the activation of signaling pathways within tumors at baseline and following therapeutic intervention will prove critical to the design of proper treatment regimens. Focusing on breast cancer, we present such a method to understand kinase signaling using multiplexed kinase inhibitor beads coupled with mass spectrometry (MIB/MS).
The National Cancer Institute Cancer Genome Atlas Project has brought us a deep understanding of the genetic landscape of human cancer. In samples from breast cancer patients, mutations in TP53 (37%) and PIK3CA (36%) account for the majority of somatic mutations observed across all subtypes,1 and although certain genetic aberrations are enriched in specific subtypes, the basis of this correlation is not yet clear. This massive data set importantly presents a snapshot of the genetic heterogeneity of breast cancer, but an understanding of novel missense and nonsense mutations and the attendant genomic and epigenetic changes will require careful functional analysis; which alterations significantly influence the oncogenic phenotype and which of these are low-impact passenger mutations? The high occurrence of amplifications and deletions will probably also influence tumorigenic signaling. The most recent compilation of the National Cancer Institute Cancer Genome Atlas data demonstrates that among basal-like breast cancer tumors, nearly 80% have potential MEK/ERK pathway activation (23% EGFR amplification, 32% KRAS amplification, and 30% BRAF amplification) and ∼90% contain a possible PIK3CA pathway activation (49% PIK3CA amplified, 9% PIK3CA mutant, 68% AKT3 amplified, 35% PTEN mutated or lost, and 28% deleted for a related phosphatase, INPP4B), illustrating the significance of kinase signaling pathways.1 Combining this genetic information with recent advances in high-throughput whole-transcriptome sequencing, we now have the ability to quantitatively measure the gene expression, mRNA splicing, and noncoding RNA levels present in human tumors. Deep RNA sequencing offers the promise of identifying outliers and predicting targeted therapies2 but also has its limitations because the expression level of a given protein does not necessarily correlate with its enzymatic activity level or commitment to the oncogenic phenotype. Another crucial level to our understanding of cancer biology will come from quantitative measurements of signaling network activity within tumors. To complement whole-genome and whole-transcriptome sequencing, it would be ideal to observe tumor signaling on a global scale.
MIB/MS as a Method for Whole-Kinome Activation Profiling
Kinases play essential roles in driving cancer cell proliferation, survival, and metastasis. Nearly half of all molecularly targeted cancer therapeutics are kinase inhibitors, making our knowledge of the kinome and its activity critical to cancer treatment. To better understand oncogenic signaling, our laboratory has optimized methods of determining the global activation state and behavior of the cancer kinome.3 Using an affinity column composed of multiple kinase inhibitors conjugated to Sepharose beads, we have the capability to capture >85% of known protein kinases. Kinase capture is a function of the expression and the activity level of the kinase and its affinity for the different kinase inhibitors. Kinases eluted from the Multiplexed Inhibitor Bead (MIB) matrix are identified and quantitated by mass spectrometry (MS), a process known as MIB/MS, providing a global snapshot of the signaling within tumors. Using reagents such as isobaric tags for relative and absolute quantitation (iTRAQ) and tandem mass tags (TMT), multiple samples can be compared in a single MS run. For instance, kinome activation profiles of multiple tumors can be compared or global changes in kinase activation can be determined after drug treatment (Figure 1). MIB/MS exhibits multiple important advances relative to traditional antibody techniques: (i) the ability to quantitatively measure activation changes in the majority of kinases simultaneously, (ii) the opportunity to characterize the behavior of kinases that are understudied due to lack of biological knowledge or reagent availability, and (iii) the ability to distinguish between closely related family members—for example, activity differences between MEK1 and MEK2 and between ERK1 and ERK2 cannot be resolved by current phospho-antibodies.
Figure 1.
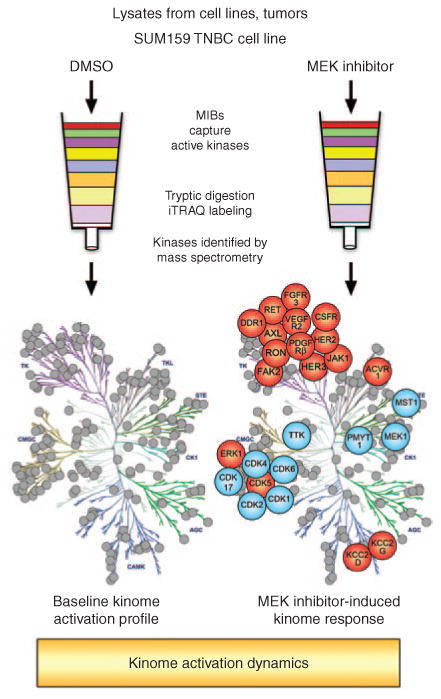
Multiplexed kinase inhibitor beads coupled with mass spectrometry (MIB/MS) as a technique for whole-kinome activation profiling. Total protein lysates from cell lines, tumors, or other tissues are flowed over affinity columns composed of MIBs: custom-synthesized, linker-adapted small-molecule kinase inhibitors covalently attached to Sepharose beads. Multiple type I kinase inhibitors are used to enrich for active kinases from every branch of the kinome tree. Kinases are eluted from the column and digested to generate a complex peptide mixture. Peptides are separated by liquid chromatography and identified by mass spectrometry. Labeling with isobaric tags for relative and absolute quantitation (iTRAQ) allows multiple samples to be compared in a single mass spectrometry run. In this manner, a 24-hour treatment of the SUM159 triple-negative breast cancer (TNBC) cell line with a MEK inhibitor was compared with dimethyl sulfoxide (DMSO) control–treated cells. MEK inhibition generates global changes in kinase activation. In particular, multiple tyrosine kinases demonstrate increased activity (in red) following MEK inhibitor treatment (blue indicates loss of activity).
Using MIB/MS, we identified elevated activity of the MAP kinase pathway in triple-negative breast cancer (TNBC) tumors, as compared with adjacent uninvolved breast tissue, and revealed the surprising resiliency of the kinome toward node-directed kinase inhibition.4 Treatment of TNBC cell lines with a highly selective inhibitor of MEK1/2 (AZD6244, selumetinib) induced a rapid rewiring of kinase signaling networks, effectively reprogramming the kinome within 24 hours of drug treatment to signal through a series of receptor tyrosine kinases. This adaptive response overcomes the activity of selumetinib and reactivates MEK/ERK oncogenic signaling in a matter of days. Similar results were seen in genetically engineered mouse models of breast cancer. Using MIB/MS to assess reprogramming, we were able to rationally predict a dual combination therapy to inhibit those kinases responsible for compensating for MEK inhibition. The combination of sorafenib and selumetinib induced apoptosis and tumor regression in the C3Tag mouse model of TNBC.
Mechanisms of Kinome Adaptation Imply the Need for Initial Combination Therapies
Kinome reprogramming in response to MEK inhibition in claudin-low TNBC cell lines is dependent on powerful transcriptional feedback loops downstream of c-Myc activity. Loss of MEK/ERK signaling causes a rapid degradation of c-Myc protein, and a derepression of PDGFRB, DDR1, and VEGFR2 transcription, among others. A similar transcriptional upregulation of the ligands PDGFB and PDGFD is observed, forming an autocrine feed-forward loop to perpetuate receptor tyrosine kinase signaling and reinitiate MEK/ERK signaling. MIB/MS allowed us to further determine that MEK inhibitor–induced reprogramming occurs specifically through reactivation of MEK2 and ERK1 (Figure 2). Our unpublished work demonstrates that kinome reprogramming occurs in every subtype of breast cancer, and although some commonalities exist, the overall response to MEK inhibition is unique for each subtype. Furthermore, inhibition of other signaling nodes such as AKT and mTOR elicits different kinome responses, although similarly based on the disruption of transcriptional feedback loops controlling receptor tyrosine kinase expression.5 Thus, cancer cells that adopt these kinase signaling nodes for growth and survival also gain powerful homeostatic mechanisms that reactivate growth signaling when therapeutics interdict these pathways. Together, these findings argue that the best initial treatment for such cancers would be a combination therapy: one drug to inhibit the oncogenic signaling node, and another to inhibit the adaptive kinome reprogramming response. However, variability in the response exists even between cell lines and genetically engineered mouse models of the same subtype. This is probably a function of inherent heterogeneity in the mutational and epigenetic landscape of cancer. Determining the kinome response on a case-by-case basis may be necessary to determine the optimal initial combination therapy. Continuing research on the adaptive responses of cell lines, patient-derived xenografts, and genetically engineered mouse models to evaluate clinical therapeutics will help to characterize the behavior of the cancer kinome and will further our understanding of kinase signaling as a series of overlapping and integrated networks. In these studies, special consideration needs to be given to cataloging and correlating the impact that mutations, amplifications, and deletions have on kinome reprogramming.
Figure 2.
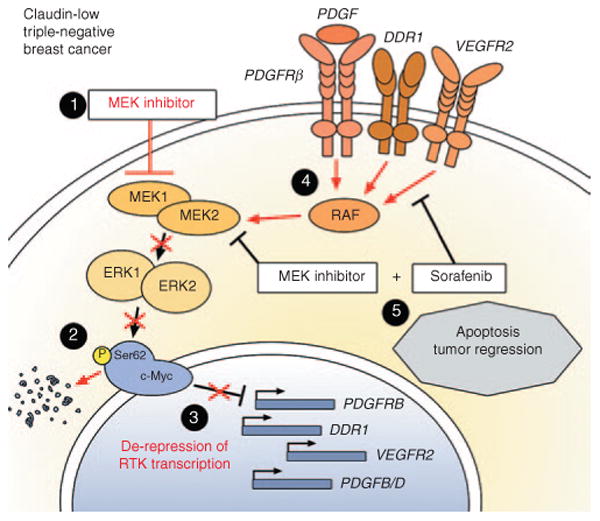
MEK inhibition induces receptor tyrosine kinase (RTK)-based kinome reprogramming in claudin-low triple-negative breast cancer. Responses to MEK inhibition are shown in red. (1) Application of small-molecule MEK inhibitor causes initial inhibition of MEK1/2 and ERK1/2 activity. (2) ERK1/2 can no longer phosphorylate c-Myc on serine 62, leading to degradation of c-Myc protein. (3) Loss of c-Myc protein relieves transcriptional repression of c-Myc target genes, including PDGFRB, DDR1, and VEGFR2. (4) Transcriptional upregulation and increased signaling by these RTKs reactivates an RAF/MEK2/ERK1 signaling cascade, leading to increased oncogenic signaling and resistance to MEK inhibitor. (5) Rational combination therapy of MEK inhibitor + sorafenib (a broad-spectrum RTK inhibitor) prevents reactivation of MEK/ERK signaling, leading to apoptosis and tumor regression.
Kinome Profiling in Patient Tumors
Our work (currently under review) extends MIB/MS to a clinical window trial of TNBC patients. Individuals enrolled in this trial had a punch biopsy taken before being placed on a 7-day treatment of trametinib (a MEK1/2 inhibitor) followed by resection of the tumor. Advances in MS and optimization of MIB purification of kinases have allowed us to successfully pursue analysis of small clinical samples. In this study, we profiled biopsies and tumors with MIB/MS and gene arrays to define the adaptive kinome response. Interestingly, changes in the kinome activity and gene expression of patient tumors resembled those of preclinical models, including a level of heterogeneity. Importantly, these new data validate the use of MIB/MS with clinical samples and demonstrate that actual tumors within patients undergo kinome reprogramming in response to cancer therapeutics as early as at 7 days of treatment. A second window trial combines targeted exome sequencing, deep RNA sequencing, and MIB/MS to define 7-day kinome responses to trastuzumab, pertuzumab, and lapatinib in HER2-positive cancer. MIB/MS provides a quantitative measure of the activation state of the kinome, and coupling this technology with a comprehensive background of genetic mutations and RNA expression, this trial has the potential to provide an unprecedented understanding of cancer biology and kinome dynamics on an individual-tumor basis. One goal is to discover kinome-based biomarkers that could be used as predictors of response to specific therapies. Such biomarkers could be assayed by developing appropriate immunohistochemistry reagents (or sequencing analysis applicable to formalin-fixed, paraffin-embedded tissue) with which to study prognostic/predictive value of the signature in large clinical formalin-fixed, paraffin-embedded specimen sets.
Combining our knowledge of driver mutations with the basal activity of the kinome in tumor biopsies may in fact predict the best initial cancer therapy. Continuing studies will provide a growing number of biomarkers to be monitored in the first days and weeks of treatment, identifying the adaptive responses of tumors and correlating how genetic alterations affect these responses. In the near future, patient tumors could first be screened for kinome activation and the presence of key mutations, followed by an analysis of the molecular response of the tumor. A major challenge confronting the cancer field is variation within individual tumors; adjacent regions of the same tumor can contain different mutations and probably display differing basal kinome states. Tumor heterogeneity creates a dilemma for both next-generation deep sequencing and adoption of baseline and adaptive kinome responses as predictors for therapy. We believe the combination of next-generation sequencing and measurement of kinome response to targeted inhibitors will provide the knowledge needed to predict combination therapies to overcome adaptive kinome reprogramming and resistance.
Footnotes
Conflict of Interest: G.L.J. is a cofounder of KinoDyn, Inc. The other authors declared no conflict of interest.
References
- 1.Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumors. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kothari V, et al. Outlier kinase expression by RNA sequencing as targets for precision therapy. Cancer Discov. 2013;3:280–293. doi: 10.1158/2159-8290.CD-12-0336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bantscheff M, et al. Quantitative chemical proteomics reveals mechanisms of action of clinical ABL kinase inhibitors. Nat Biotechnol. 2007;25:1035–1044. doi: 10.1038/nbt1328. [DOI] [PubMed] [Google Scholar]
- 4.Duncan JS, et al. Dynamic reprogramming of the kinome in response to targeted MEK inhibition in triple-negative breast cancer. Cell. 2012;149:307–321. doi: 10.1016/j.cell.2012.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chandarlapaty S, et al. AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell. 2011;19:58–71. doi: 10.1016/j.ccr.2010.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]