

Abstract
In this study, we synthesized a series of trans-indole-3-acrylamide derivatives (3a–k) and investigated their activity for inhibition of cell proliferation against five human cancer cell lines (HeLa, MCF7, MDA-MB-231, Raji and HL-60) by MTT assay. Compound 3e showed significant antiproliferative activity against both the Raji and HL-60 cell lines with IC50 values of 9.5 and 5.1 µM, respectively. Compound 3e also exhibited moderate inhibitory activity on tubulin polymerization (IC50 = 17 µM). Flow cytometric analysis of cultured cells treated with 3e also demonstrated that the compound caused cell cycle arrest at the G2/M phase in HL-60 and HeLa cells. Moreover, 3e, the most active compound, caused an apoptotic cell death through the activation of caspase-3. Docking simulations suggested that 3e binds to the colchicine site of tubulin.
Keywords: Synthesis, Anticancer activity, Indole, Tubulin polymerization, Colchicine binding, Apoptosis, Cell cycle arrest, Molecular docking
1. Introduction
Cancer remains one of the leading causes of death worldwide and requires a pressing need for the development of novel and more effective treatments. Although the chemotherapy is the major method for treatment for various cancer types, the narrow dosing window of current drugs with regard to their efficacy and safety and significant drug resistance resulting a failure of antitumor drugs to exert their effects in certain cancer types limit the use of contemporary cancer chemotherapy. Therefore, the design and discovery of more effective and safer anticancer drug candidates are of interest in contemporary medicinal chemistry.1, 2
Microtubules are important in mitosis and have been recognized as an important target for the development of novel anticancer drugs.3 Agents targeting tubulin such as the vinca alkaloids and taxoids are potent chemotherapeutics currently used in the clinic. Among them, colchicine was the first tubulin-binding agent to have antivascular effects causing hemorrhagic necrosis in human tumors.4 Combretastatins which are isolated from the South African tree Combretum caffrum are also a group of antimitotic compounds and combretastatin A-4 (CA-4, Fig. 1) is one of the well-known natural tubulin-binding molecule affecting microtubule dynamics.5 CA-4 has provided researchers a simple structural template for the design of related compounds with potent activity and a large number of combretastatin analogues have been prepared as potential anticancer agents including chalcones, some of which have recently been reviewed.6–10 Chalcones (1,3-diaryl-2-propen-1-ones) with an ionone system between two aromatic rings (Fig. 1) serve as precursors for the preparation of various flavonoids and exhibit interesting pharmacological activities11, 12 such as anticancer13– 15 and antiproliferative activities.16–21 Their broad biological properties are reported to be due to the α,β-unsaturated ketone moiety.22 Chalcones in which both the 1,3-diaryl rings are separated by α,β-unsaturated carbonyl system with three-carbon lengths are structurally similar to indolyl heterocycles (Fig. 1). There are many indole-based compounds found to be effective as tubulin assembly inhibitors such as the recently reported 3-arylthioindoles, which induced significant apoptotic cell death.23 However, indole-based chalcones remain largely unexplored for their anticancer potential.24–26 Kumar et al. showed that indolyl chalcones inhibited the growth of A549, PaCa-2 and PC-3 cancer cell lines at micromolar concentrations.27 Cinnamon amides having an α,β-unsaturated ketone moiety (phenylcinnamides in Fig. 1) are shown to bind to tubulin, thereby causing an inhibition of its polymerization and alteration in the tubulin-microtubule equilibrium.28–31
Figure 1.

Chemical structures of known tubulin inhibitors and synthesized compounds
Encouraged with these results and to discover novel anticancer agents, we have synthesized a series of novel indolylacrylamide derivatives and evaluated their anticancer activities. The newly synthesized compounds structurally resemble the indolyl chalcone structure (Fig. 1).
2. Results and Discussion
2.1. Chemistry
We synthesized a series of amide derivatives of trans-indole-3-acrylic acid as illustrated in Scheme 1. First, trans-indole-3-acrylic acid 2 was generated by Knoevenagel condensation of indole-3-carbaldehyde with malonic acid in the presence of piperidine as reported previously.32 Treatment of 2 with appropriate amines in the presence of triethylamine and ethyl chloroformate, which was used as the carboxylate activator, produced trans-indole-3-acrylamide derivatives 3a–k in moderate to good yields (40%–69%). Compounds were purified by automated flash chromatography and checked for purity with UPLC before being tested in biological assays (purity was >97%). The structures of these compounds were confirmed by high resolution mass spectrometry (HRMS), IR and 1H- and 13C-NMR spectral data. Final acryl amide derivatives exhibited a characteristic strong absorption peaks in the area of 1638–1733 cm−1, which was attributable to the C=O of the amide moiety. In the 1HNMR spectra of compounds 3a–k, one of the olefinic protons (CH=CH-CO) was observed as a doublet at about 7.61–7.83 ppm, while the other (CH=CH-CO) was observed as a doublet at about 6.64–7.15 ppm, with coupling constants of 15.6 or 16.0 Hz indicating the presence of the (E) isomer.
Scheme 1.

Reagents and conditions; a) Malonic acid, pyridine, piperidine, 5 h, 40 °C; b) 2, ethyl chloroformate, NEt3, amine derivative, CH2Cl2, overnight, rt.
2.2. Biological Evaluations
2.2.1. Effects of the compounds on the viability of cancer cells
trans-Indolyl-3-acrylamide derivatives 3a–k were screened against five human cancer cell lines (HeLa, MCF7, MDA-MB-231, Raji and HL-60) using the 3-(4,5-dimethyldiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. The MTT cell proliferation assay has been widely accepted as a reliable way to measure the cell proliferation rate when metabolic events lead to apoptosis or necrosis.33–36 After incubation with compounds at different concentrations for 48 h, the cells were treated with MTT to measure their growth/viability (% of the untreated control) using a spectrophotometer as described previously.37, 38 Experiments were performed in quadruplicate. The IC50 values (Table 1) were calculated from concentration– response curves by means of the PRISM 5, GraphPad Software.39
Table 1.
In vitro cytotoxic activity of compounds 3a–k
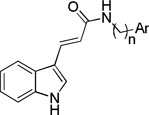 | |||||||
|---|---|---|---|---|---|---|---|
| IC50 (µM)a | |||||||
| Compnd. | n | Ar | HeLa | MDA-MB-231 | MCF7 | Raji | HL-60 |
| 3a | 0 | 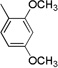 |
>100 | >100 | >100 | 22.6±1.45 | 17.8±1.54 |
| 3b | 0 | 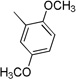 |
>100 | >100 | >100 | 22.9±1.76 | 10.1±0.87 |
| 3c | 0 | 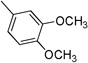 |
31.4± 9.01 | >100 | >100 | 10.3±2.43 | 6.2±0.44 |
| 3d | 0 | 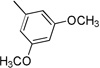 |
>100 | >100 | >100 | 9.3±1.93 | 7.1±0.31 |
| 3e | 0 | 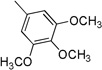 |
23.7±2.46 | >100 | 36.3±3.07 | 9.5±1.28 | 5.1±0.32 |
| 3f | 0 | 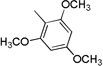 |
>100 | >100 | >100 | >100 | >100 |
| 3g | 1 |  |
>100 | >100 | >100 | 33.1±1.40 | >100 |
| 3h | 1 | 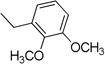 |
36.5±6.26 | >100 | >100 | >100 | >100 |
| 3i | 1 | 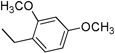 |
>100 | >100 | >100 | 32.9±0.94 | >100 |
| 3j | 1 | 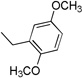 |
>100 | >100 | >100 | >100 | >100 |
| 3k | 1 | >100 | >100 | >100 | >100 | >100 | |
Each experiment was independently performed four times, and data are shown as means ± SD.
In general, compounds having a phenylamidic moiety (3a–e) displayed greater antiproliferative potency than the molecules possessing a benzylamidic moiety (3g–k). Compounds having methoxy substituent at positions 3 and 4 or at 3 and 5 on the phenyl amidic moiety of the molecules (3c, 3d) exerted potency on the Raji and HL-60 cell lines (IC50 values of 6.2–10.3 µM). Compound 3e showed appreciable antiproliferative activity against both Raji and HL-60 cell lines, with IC50 values of 9.5 and 5.1 µM, respectively. Breast cancer cell lines MDA-MB-231 and MCF7 were not sensitive towards the newly synthesized compounds with the exception of the 3,4,5-trimethoxy-substituted phenyl amide derivative 3e, which was the most effective compound found in the phenyl amide series.
In the series of benzylamide derivatives, compound 3h, with 2,3-dimethoxy substitutions on the amide moiety, showed weak activity against HeLa cells. Derivatives 3g (3-methoxy-4-hydroxy-substituted benzylamide derivative) and 3i (2,5-dimethoxy-substituted benzylamide derivative) had weak activity against Raji cells.
2.2.2. Inhibition of tubulin polymerization and colchicine binding
Based on their structural resemblance to phenylcinnamides, we considered tubulin as a potential target31 for our active compounds. To investigate whether the antiproliferative activity of these compounds is due to an interaction with tubulin, we evaluated the effects of 3a–k on the polymerization of purified tubulin, using the highly potent CA-4 as reference (Table 2).40
Table 2.
Inhibition of tubulin polymerization and colchicine binding by synthesized compounds and CA-4.
| Compound | Inhibition of tubulin assemblya IC50 (µM)± SD |
Inhibition of colchicine bindingb % Inhibition ± SD |
||
|---|---|---|---|---|
| 50 µM | 5 µM | 1 µM | ||
| 3a | > 40 | 13 ± 5 | - | - |
| 3b | > 40 | 15 ± 5 | - | - |
| 3c | > 40 | 11 ± 2 | - | - |
| 3d | > 40 | 26 ± 5 | - | - |
| 3e | 17 ± 1 | 50 ± 4 | 11 ± 4 | - |
| 3f | > 40 | 11 ± 1 | - | - |
| 3g | > 40 | 14 ± 5 | - | - |
| 3h | > 40 | 12 ± 4 | - | - |
| 3i | > 40 | 13 ± 4 | - | - |
| 3j | > 40 | 23 ± 2 | - | - |
| 3k | > 40 | 13 ± 0.2 | - | - |
| CA-4 | 1.3 ± 0.07 | 98 ± 0.8 | 88 ± 0.3 | |
Tubulin was at 10 µM.
Tubulin and colchicine were at 1 and 5 µM concentrations, respectively.
Only 3e inhibited tubulin polymerization with IC50 values below 20 µM. Compounds 3e with 3,4,5-trimethoxy substituents on the amide moiety had IC50 value of 17 µM. This was in agreement with 3e being the compound with the greatest antiproliferative activity. Three compounds (3b, 3c, 3d) of the five best antiproliferative agents were inactive as antitubulin agents. Therefore, the results suggest that there may be another, more dominant molecular mechanism of action for these three compounds for their antiproliferative activity.
All synthesized compounds were also examined for their inhibitory effects at 5 and/or 50 µM on the binding of 5 µM [3H]colchicine to 1 µM tubulin (Table 2).41 Significant inhibition was only observed with 3e at 50 µM, in accord with their effects on the assembly reaction.
2.2.3. Analysis of cell cycle effects
Considering the cytotoxic activity of 3e and its antitubulin properties, we further analyzed its effects on cell cycle distribution of cultured HL-60 and HeLa cells, as determined by flow cytometry (Fig. 2). After treatment of cells with 3e for 24 h, we observed a concentration dependent increase of G2/M-phase cells with a concomitant reduction in the proportion of cells in G1 and S phases with both cell lines. The increase in the G2/M phase cells occurred at a concentration as low as 10 µM, while more than 80% of the HeLa and 60% of the HL-60 cells were blocked in G2/M at higher concentrations (15–30 µM). These results were in good agreement with the inhibitory effect of 3e on tubulin polymerization and also on the proliferation of both cell lines.
Figure 2.

Percentage of cells in each phase of the cell cycle in HL-60 (Panel A) and HeLa cells (Panel B) treated with 3e at the indicated concentrations for 24 h. Cells were fixed and labeled with PI and analyzed by flow cytometry as described in the experimental section. Data are represented as means ± SEM of three independent experiments
2.2.4. Compound 3e induces apoptosis
To characterize the mode of cell death by 3e, the annexin-V/propidium iodide (PI) biparametric cytofluorimetric assay was performed on both the HeLa and HL-60 cell lines. Since PI, which stains DNA and only enters the dead cells, and annexin-V [conjugated to fluorescein isothiocyanate (FITC)], which binds to phosphatidylserine (PS) located only on the outer membrane of apoptotic cells, the assay permits quantitation of live cells (annexin-V−/PI−), early apoptotic cells (annexin-V+/PI−), late apoptotic cells (annexin-V+/PI+) and necrotic cells (annexin-V−/PI+). As shown in Fig. 3, treatment of both cell lines with compound 3e induced a concentration- and time-dependent increase in annexin-V+/PI− and annexin-V+/PI+ fractions, indicating activation of the apoptotic cell death machinery.
Figure 3.

Flow cytometric analysis of apoptotic cells after treatment of HL-60 (panels A and B) and HeLa (panels C and D) cells with 3e at the indicated concentrations after incubation for 24 (panels A and C) or 48 h (panels B and D). The cells were harvested and labeled with annexin-V-FITC and PI and analyzed by flow cytometry. Data are represented as means ± SEM of three independent experiments
2.2.5. Effects of 3e on caspase activation
To further evaluate the apoptotic process induced by 3e, we performed immunoblot analysis on HL-60 protein extracts. During apoptosis, the endogenous 113 kDa Poly-ADP-ribosyl-polymerase (PARP) protein is cleaved into 89 kDa and 24 kDa fragments, which can be shown by Western blot analysis. As shown in Fig. 4, 3e induced the activation of caspase-3 with the appearance of the cleaved fragments and the subsequent cleavage of its substrate PARP, after 24 h and 48 h treatments. This observation indicated that the 3e-induced apoptosis was caspase-dependent.
Figure 4.

Western blot analysis of caspase-3 and PARP after treatment of HL-60 cells with 3e at the indicated concentrations and for the indicated times. To confirm equal protein loading, each membrane was stripped and reprobed with anti-β-actin antibody
2.2.6. Molecular docking studies
To determine the possible binding modes of the most active compound 3e to tubulin, docking studies were carried out using the high-resolution crystal structure of the tubulin/DAMA-colchicine complex (PDB ID: 1SA0).42
By following the reported docking technique43, 44 for 1SA042, a Glide 5.845 run in single precision mode (GlideScore SP) was used to collect the best ranked pose of each ligand as shown in Fig. 5. Ligand 3e assumed a V-shaped conformation in the same binding region of DAMA-colchicine (X-Ray ligand, Fig. 5-A). The trimethoxyphenyl group of 3e was positioned in a similar orientation of the corresponding ring in the co-crystallized DAMA-colchicine. In both structures, the middle methoxy group formed a hydrogen bond with Cys241.
Figure 5.

3D visualization of (A) DAMA-colchicine (X-Ray Ligand)42 and docking results of the best ranked docking pose of 3e (B). The main interacting residues are shown and labeled. Hydrogen bonds are indicated by black dashed lines
3. Conclusions
We synthesized a series of indolylacrylamides as potential anticancer agents and determined their cytotoxic activity against five human cancer cell lines. Compound 3e, the most potent derivative in this series, displayed antiproliferative activity with IC50 values in the range of 5.1 to 36.3 µM against the tested cancer cell lines. Compound 3e was also the most active inhibitor of tubulin polymerization and inhibited the binding of [3H]colchicine to tubulin. Compound 3e caused HeLa and HL-60 cells to accumulate in the G2/M phase of the cell cycle as was the case with most antitubulin agents. 3e also induced apoptotic cell death by activation of caspase-3 activity. Moreover, molecular docking studies demonstrated a potential binding mode for compound 3e in the colchicine site of tubulin resembling the co-crystallized DAMA-colchicine binding mode.
4. Experimental
4.1. Chemistry
Chemicals purchased from commercial vendors were used without purification. Thin-layer chromatography (TLC) was performed on Merck 60F254 plates. Reactions were monitored by TLC on silica gel, with detection by UV light (254 nm) or charring Dragendorff reagent.46 Melting points were determined with an SMP-II Digital Melting Point Apparatus and are uncorrected (Schorpp Geaetetechnik, Germany). IR spectra were obtained using a Perkin Elmer Spectrum 400 FTIR/FTNIR spectrometer equipped with a Universal ATR Sampling Accessory. 1H-NMR spectra were recorded in CDCl3 or DMSO-d6 on a Varian Mercury 400 MHz High Performance Digital FT-NMR spectrometer using tetramethylsilane as the internal standard at the NMR facility of the Faculty of Pharmacy, Ankara University. 13C-NMR spectra were recorded in CDCl3 on a Varian Mercury 300 MHz FT-NMR spectrometer using tetramethylsilane as the internal standard at the NMR facility of FARGEM (Pharmaceutical Research and Development Center) Inc. All chemical shifts were recorded as δ (ppm). High resolution mass spectra data (HRMS) were collected using a Waters LCT Premier XE Mass Spectrometer (high sensitivity orthogonal acceleration time-of-flight instrument) using ESI (+) method. The instrument was coupled to an AQUITY Ultra Performance Liquid Chromatography system (Waters Corporation, Milford, MA, USA). Flash chromatography was performed with a Combiflash®Rf automated flash chromatography system with RediSep columns (Teledyne-Isco, Lincoln, NE, USA) using CH2Cl2-methanol (0%–5%) solvent gradients.
4.2. Synthesis of trans-indole-3-acrylic acid 2
trans-Indole-3-acrylic acid was synthesized by Knoevenagel condensation between indole-3-carbaldehyde and malonic acid as previously reported.32 (Yield 91%, mp 195 °C)
4.3. General procedure for the preparation of amide derivatives of trans-indole-3-acrylic acid
To a solution of the acid derivative (1 mmol) in CH2Cl2 were added triethylamine (2 mmol) and ethyl chloroformate (1 mmol), followed by stirring at 0 °C for 30 min. After addition of the appropriate amine derivative (1.2 mmol), the mixture was stirred for an additional 1 h at 0 °C. Then, the reaction mixture was warmed to room temperature and stirred overnight. After the solvent was evaporated under reduced pressure, acetone was added, filtered, and evaporated. The residue was dissolved in CH2Cl2, and the organic phase was washed with a 1% NaHCO3 solution and brine, dried over Na2SO4, and evaporated under vacuum. The final residue was purified by flash column chromatography (Combiflash® Rf) using CH2Cl2-MeOH (0%–5%) as eluents.
4.3.1. (E)-N-(2,4-Dimethoxyphenyl)-3-(1H-indol-3-yl)acrylamide 3a (CAS Registry Number: 953135-26-7)
Yield 40%, mp 165–167°C; IR (FTIR/FTNIR-ATR): 1644 cm−1 (C=O), 3172 cm−1 (N-H). 1H-NMR (DMSO-d6) δ: 11.60 (1H, s), 9.15 (1H, s), 8.11 (1H, d, J=6.8 Hz), 8.04 (1H, d, J=9.2 Hz), 7.97 (1H, s), 7.72 (1H, d, J=16 Hz), 7.47 (1H, d, J=7.2 Hz), 7.20 (2H, m), 7.07 (1H, d, J=15.6 Hz), 6.64 (1H, d, J=2.8 Hz), 6.52 (1H, m), 3.87 (3H, s), 3.75 (3H, s). 13C-NMR (DMSO-d6) δ: 165.6, 156.7, 151.2, 138.1, 134.8, 131.6, 125.5, 123.1, 122.9, 121.9, 121.2, 121.0, 117.1, 113.1, 112.9, 104.6, 99.3, 56.4, 55.9; HRMS C19H19N2O3 [M+H]+ Calc. 323.1396, Found m/z 323.1397.
4.3.2. (E)-N-(2,5-Dimethoxyphenyl)-3-(1H-indol-3-yl)acrylamide 3b (CAS Registry Number: 953194-76-8)
Yield 63%, mp 158–160 °C; IR (FTIR/FTNIR-ATR): 1655 cm−1 (C=O), 3403 cm−1 (N-H). 1H-NMR (DMSO-d6) δ: 11.64 (1H, s), 9.29 (1H, s), 8.15 (1H, d, J=7.2 Hz), 8.02 (1H, m), 7.81 (1H, s), 7.75 (1H, d, J=15.6 Hz), 7.47 (1H, d, J=7.6 Hz), 7.20 (2H, m), 7.15 (1H, d, J=15.6 Hz), 6.97 (1H, d, J=9.2 Hz), 6.60 (1H, m), 3.84 (3H, s), 3.70 (3H, s). 13C-NMR (DMSO-d6) δ: 166.0, 153.6, 143.7, 138.1, 135.6, 132.0, 129.6, 125.4, 123.0, 121.3, 121.0, 116.8, 113.1, 112.9, 112.1, 108.3, 107.7, 56.9, 55.9; HRMS C19H19N2O3 [M+H]+ Calc. 323.1396, Found m/z 323.1393.
4.3.3. (E)-N-(3,4-Dimethoxyphenyl)-3-(1H-indol-3-yl)acrylamide 3c (CAS Registry Number: 953243-47-5)
Yield 68%, mp 244–246 °C; IR (FTIR/FTNIR-ATR): 1649 cm−1 (C=O), 3328 cm−1 (N-H). 1H-NMR (DMSO-d6) δ: 11.63 (1H, s), 9.86 (1H, s), 7.94 (1H, m), 7.81 (1H, s), 7.73 (1H, d, J=15.6 Hz), 7.48 (1H, m), 7.43 (1H, s), 7.21 (3H, m), 6.92 (1H, d, J=8.8 Hz), 6.78 (1H, d, J=15.6 Hz), 3.76 (3H, s), 3.73 (3H, s). 13C-NMR (DMSO-d6) δ: 165.3, 149.2, 145.1, 138.1, 134.9, 134.1, 131.6, 125.5, 123.0, 121.1, 120.6, 116.8, 113.0, 112.8, 112.8, 111.2, 104.5, 56.3, 55.9; HRMS C19H19N2O3 [M+H]+ Calc. 323.1396, Found m/z 323.1393.
4.3.4. (E)-N-(3,5-Dimethoxyphenyl)-3-(1H-indol-3-yl)acrylamide 3d
Yield 45%, mp 218–221 °C; IR (FTIR/FTNIR-ATR): 1649 cm−1 (C=O), 3206 cm−1 (N-H). 1H-NMR (DMSO-d6) δ: 11.68 (1H, s), 9.96 (1H, s), 7.95 (1H, m), 7.84 (1H, s), 7.75 (1H, d, J=15.6 Hz), 7.49 (1H, m), 7.23 (2H, m), 6.96 (2H, s), 6.79 (1H, d, J=15.6 Hz), 6.21 (1H, m), 3.74 (6H, s). 13C-NMR (DMSO-d6) δ: 165.7, 161.2, 142.1, 138.1, 135.6, 131.9, 125.5, 123.1, 121.2, 120.7, 116.5, 113.1, 112.8, 97.8, 95.5, 55.7. HRMS C19H19N2O3 [M+H]+ Calc. 323.1396, Found m/z 323.1391.
4.3.5. (E)-3-(1H-Indol-3-yl)-N-(3,4,5-trimethoxyphenyl)acrylamide 3e
Yield 53%, mp 246–248 °C; IR (FTIR/FTNIR-ATR): 1733 cm−1 (C=O), 3304 cm−1 (N-H). 1H-NMR (DMSO-d6) δ: 11.68 (1H, s), 9.96 (1H, s), 7.95 (1H, m), 7.83 (1H, s), 7.74 (1H, d, J=15.6 Hz), 7.48 (1H, m), 7.23 (2H, m), 7.10 (2H, s), 6.79 (1H, d, J=15.6 Hz), 3.77 (6H, s), 3.63 (3H, s). 13C-NMR (DMSO-d6) δ: 165.5, 153.4, 138.1, 136.6, 135.2, 133.7, 131.8, 125.5, 123.0, 121.1, 120.6, 116.7, 113.1, 112.8, 97.1, 60.8, 56.3; HRMS C20H21N2O4 [M+H]+ Calc. 353.1501, Found m/z 353.1503.
4.3.6. (E)-3-(1H-Indol-3-yl)-N-(2,4,6-trimethoxyphenyl)acrylamide 3f
Yield 41%, mp 221–223 °C; IR (FTIR/FTNIR-ATR): 1651 cm−1 (C=O), 3324 cm− (N-H). 1H-NMR (DMSO-d6) δ: 11.57 (1H, s), 8.68 (1H, s), 7.96 (1H, d, J=7.2 Hz), 7.77 (1H, s), 7.61 (1H, d, J=16 Hz), 7.47 (1H, d, J=7.2 Hz), 7.20 (2H, m), 6.83 (1H, d, J=15.6 Hz), 6.27 (2H, s), 3.79 (3H, s), 3.73 (6H, s). 13C-NMR (DMSO-d6) δ: 165.9, 159.7, 157.2, 138.0, 134.0, 131.1, 125.5, 122.8, 121.0, 120.6, 117.0, 112.9, 112.8, 108.7, 91.6, 56.3, 56.0; HRMS C20H21N2O4 [M+H]+ Calc. 353.1501, Found m/z 353.1490.
4.3.7. (E)-N-(4-Hydroxy-3-methoxybenzyl)-3-(1H-indol-3-yl)acrylamide 3g
Yield 55%, mp 213–215 °C; IR (FTIR/FTNIR-ATR): 1702 cm−1 (C=O), 3266 cm−1 (N-H). 1HNMR (DMSO-d6) δ: 11.55 (1H, s), 8.87 (1H, s), 8.23 (1H, t, J=5.6 Hz), 7.89 (1H, d, J=7.2 Hz), 7.57 (1H, m), 7.63 (1H, d, J=15.6 Hz), 7.45 (1H, d, J=7.6 Hz), 7.18 (2H, m), 6.88 (1H, s), 6.72 (1H, m), 6.67 (1H, d, J=15.6 Hz), 4.29 (2H, d, J=5.2 Hz), 3.75 (3H, s). The OH-signal was not observed in the spectrum, due to solvent exchange. 13C-NMR (DMSO-d6) δ: 166.8, 148.1, 146.1, 138.0, 133.8, 131.0, 130.9, 125.5, 125.2, 122.8, 120.9, 120.7, 116.9, 115.9, 112.9, 112.8, 112.6, 56.2, 42.9; HRMS C19H19N2O3 [M+H]+ Calc. 323.1396, Found m/z 323.1397.
4.3.8. (E)-N-(2,3-Dimethoxybenzyl)-3-(1H-indol-3-yl)acrylamide 3h
Yield 69%, mp 211–213 °C; IR (FTIR/FTNIR-ATR): 1639 cm−1 (C=O), 3206 cm−1 (N-H). 1H-NMR (DMSO-d6) δ: 11.56 (1H, s), 8.21 (1H, t, J=5.6 Hz), 7.91 (1H, d, J=7.6 Hz), 7.75 (1H, s), 7.63 (1H, d, J=15.6 Hz), 7.45 (1H, d, J=7.6 Hz), 7.18 (2H, m), 6.88 (3H, m), 6.71 (1H, d, J=16 Hz), 4.40 (2H, d, J=6 Hz), 3.80 (3H, s), 3.76 (3H, s). 13C-NMR (DMSO-d6) δ: 166.9, 152.9, 146.9, 138.0, 133.8, 133.5, 131.0, 125.5, 124.5, 122.8, 121.1, 120.9, 120.6, 116.8, 112.9, 112.8, 112.3, 60.8, 56.3, 37.7; HRMS C20H21N2O3 [M+H]+ Calc. 337.1552, Found m/z 337.1544.
4.3.9. (E)-N-(2,4-Dimethoxybenzyl)-3-(1H-indol-3-yl)acrylamide 3i
Yield 48%, mp 164–166 °C; IR (FTIR/FTNIR-ATR): 1644 cm−1 (C=O), 3241 cm−1 (N-H). 1H-NMR (DMSO-d6) δ: 11.54 (1H, s), 8.07 (1H, t, J=6 Hz), 7.92 (1H, d, J=8 Hz), 7.74 (1H, s), 7.61 (1H, d, J=15.6 Hz), 7.45 (1H, d, J=7.6 Hz), 7.18 (3H, m), 6.71 (1H, d, J=16 Hz), 6.57 (1H, s), 6.50 (1H, d, J=8 Hz), 4.28 (2H, d, J=5.6 Hz), 3.81 (3H, s), 3.74 (3H, s). 13C-NMR (DMSO-d6) δ: 166.9, 160.4, 158.4, 138.0, 133.6, 131.0, 129.8, 125.5, 122.8, 120.9, 120.7, 119.8, 116.9, 112.9, 112.8, 104.9, 98.8, 56.0, 55.8, 37.7; HRMS C20H21N2O3 [M+H]+ Calc. 337.1552, Found m/z 337.1539.
4.3.10. (E)-N-(2,5-Dimethoxybenzyl)-3-(1H-indol-3-yl)acrylamide 3j
Yield 57%, mp 190–193 °C; IR (FTIR/FTNIR-ATR): 1638 cm−1 (C=O), 3299 cm−1 (N-H). 1H-NMR (DMSO-d6) δ: 11.56 (1H, s), 8.18 (1H, t, J=5.6 Hz), 7.93 (1H, d, J=8 Hz), 7.76 (1H, s), 7.63 (1H, d, J=15.6 Hz), 7.46 (1H, d, J=8 Hz), 7.18 (2H, m), 6.93 (1H, d, J=8.8 Hz), 6.79 (2H, m), 6.73 (1H, d, J=15.6 Hz), 4.34 (2H, d, J=5.6 Hz), 3.77 (3H, s), 3.68 (3H, s). 13C-NMR (DMSO-d6) δ: 172.2, 168.4, 168.3, 164.1, 155.2, 155.0, 136.7, 133.2, 132.7, 125.6, 121.2, 121.0, 119.8, 115.1, 115.0, 112.5, 111.6, 68.4, 24.4; HRMS C20H21N2O3 [M+H]+ Calc. 337.1552, Found m/z 337.1548.
4.3.11. (E)-N-(3,4-Dimethoxybenzyl)-3-(1H-indol-3-yl)acrylamide 3k47
Yield 68%, mp 176–178 °C; IR (FTIR/FTNIR-ATR): 1657 cm−1 (C=O), 3292 cm−1 (N-H). 1H-NMR (DMSO-d6) δ: 11.52 (1H, s), 8.24 (1H, t, J=5.6 Hz), 7.86 (1H, d, J=7.6 Hz), 7.72 (1H, s), 7.61 (1H, d, J=16 Hz), 7.42 (1H, d, J=7.6 Hz), 7.13 (2H, m), 6.89 (2H, m), 6.80 (1H, m), 6.64 (1H, d, J=16 Hz), 4.30 (2H, d, J=5.2 Hz), 3.71 (3H, s), 3.69 (3H, s). 13C-NMR (DMSOd6) δ: 166.8, 149.2, 148.4, 138.0, 133.9, 132.7, 131.0, 125.5, 122.8, 120.9, 120.6, 120.2, 116.8, 112.9, 112.7, 112.3, 112.1, 56.2, 56.0, 42.8; HRMS C20H21N2O3 [M+H]+ Calc. 337.1552, Found m/z 337.1544.
4.4. Anticancer activity
4.4.1. Cell lines and cell culture
The human cancer cell lines, cervical carcinoma (HeLa), estrogen receptor positive breast carcinoma (MCF7), estrogen receptor negative breast carcinoma (MDA-MB-231), Burkitt’s lymphoma (Raji) and human promyelocytic leukemia (HL-60), were obtained from ATCC.
4.4.2. Cytotoxicity assay
MDA-MB-231 and MCF7 cells were cultured in DMEM whereas HeLa, HL-60 and Raji cells were grown in RPMI-1640 medium in a humidified atmosphere containing 5% CO2 at 37 °C. Both DMEM and RPMI-1640 medium were supplemented with 10% fetal bovine serum (FBS), 200 mM L-glutamine, 100 IU/mL penicillin and 100 µg/mL streptomycin (all from Hyclone Laboratories, Logan, UT, USA). Cell viability was determined using the MTT assay (Cell Proliferation Kit I, Roche, Germany). Briefly, cells were seeded in a 96-well plate at 10,000 cells per well and cultured overnight in growth medium containing 1% FBS. Then cells were treated with test compounds at different concentrations (from 0.1 to 125 µM) for 48 h. As a solvent control, cells were also treated with dimethyl sulfoxide (DMSO) at a final concentration of 0.1%. At the end of the incubation time, MTT (final concentration, 0.5 mg/mL) was added to each well, and the plate was incubated for an additional 4 h. After formation of blue formazan crystals, medium containing MTT was discarded, and DMSO was added to the wells to dissolve the MTT crystals. The absorbance of samples was measured with a Spectra Max M3 microplate reader (Molecular Devices, Sunnyvale, CA, USA) at 570 nm. Average absorbance values from quadruplicate replicates per test compound and solvent control (DMSO) were calculated. Mean solvent control values were set to 100% viability, and then the effects of test compounds on cell viability were calculated by comparing mean values obtained from compound treated culture wells with those of the solvent controls. IC50 values were calculated from concentration–response curves by means of PRISM 5, Graph Pad Software.39
4.4.3. Tubulin polymerization assays
Purified bovine brain tubulin48 was used in turbidimetric polymerization studies and in the colchicine binding assay. Tubulin and the desired concentrations of compound were preincubated at 30 °C for 15 min in a 0.24 mL reaction volume. Reaction mixtures were then placed on ice, and 10 µL of 10 mM GTP was added. All concentrations are in terms of the final reaction volume of 0.25 mL: tubulin at 1.0 mg/mL (10 µM), 0.8 M monosodium glutamate (adjusted to pH 6.6 in a 2 M stock solution with HCl), 4% (v/v) DMSO, and 0.4 mM GTP. The ice-cold reaction mixtures were transferred to cuvettes held at 0 °C in recording spectrophotometers (Beckman models DU7400 and DU7500) equipped with electronic temperature controllers. After baselines were established at 350 nm, the temperature was jumped to 30 °C over about 30 s, and the IC50 is defined as the concentration of compound that inhibits turbidity development by 50% at 20 min. Detailed method description has been published previously.40 The method is generally most reliable at compound concentrations up to 20 µM. At higher concentrations, compound precipitation and/or absorbance often leads to interference with the turbidity readings caused by tubulin assembly.
4.4.4. Inhibition of colchicine binding assays
The colchicine binding assay was performed in 0.1 mL reaction volumes. Each assay tube contained 0.1 mg/mL (1.0 µM) tubulin, 5% (v/v) DMSO, 5.0 µM [3H]colchicine (Perkin Elmer), varying concentrations of potential inhibitors, as indicated, and additional components, including 1.0 M glutamate, shown to stabilize the colchicine binding activity of tubulin41, 49 for prolonged times at 37 °C. Samples were incubated for 10 min at 37 °C, at which time the reaction in control samples is 40–60% complete. The reactions were stopped with water at 0 °C, and the diluted samples (total volume, about 2 mL, with several 2 mL rinses) were filtered through a stack of two DEAE-cellulose filters (Whatman) in a 12-place manifold (Millipore) under a weak vacuum (filtration time about 10 min). The filters were then rapidly washed three times (2 mL each) with ice-cold water, using a stronger vacuum. Radiolabel bound to the filters was measured in a liquid scintillation counter.
4.4.5. Flow cytometric analysis of cell cycle distribution
For flow cytometric analysis of DNA content, 5×105 HeLa and HL-60 cells were treated with different concentrations of the test compounds for 24 h. After the incubation period, the cells were collected, centrifuged and fixed with ice-cold ethanol (70%). The cells were then treated with lysis buffer containing RNAse A and 0.1% Triton X-100 and then stained with PI. Samples were analyzed on a Cytomic FC500 flow cytometer (Beckman Coulter). DNA histograms were analyzed using MultiCycle for Windows (Phoenix Flow Systems).
4.4.6. Annexin-V assay
Surface exposure of PS on apoptotic cells was measured by flow cytometry with a Coulter Cytomics FC500 (Beckman Coulter) by adding annexin-V-FITC to cells according to the manufacturer’s instructions (Annexin-V Fluos, Roche Diagnostic). Simultaneously, the cells were stained with PI. Excitation was set at 488 nm, and the emission filters were at 525 and 585 nm, respectively, for FITC and PI.
4.4.7. Western blot analysis
HL-60 cells were incubated in the presence of test compounds and, after different times, were collected, centrifuged and washed twice with ice-cold phosphate-buffered saline. The pellet was then resuspended in lysis buffer. After the cells were lysed on ice for 30 min, lysates were centrifuged at 15000 × g at 4 °C for 10 min. The protein concentration in the supernatant was determined using the BCA protein assay reagents (Pierce, Italy). Equal amounts of protein (20 µg) were resolved using sodium dodecyl sulfate polyacrylamide gel electrophoresis (7.5–15 % acrylamide gels) and transferred to a PVDF Hybond-p membrane (GE Healthcare). Membranes were blocked with 5% bovine serum albumin for 2 h. Membranes were then incubated with primary antibodies against caspase-3 (Alexis) and PARP (Cell Signalling) overnight at 4 °C. Membranes were next incubated with peroxidase-labeled secondary antibodies for 60 min. All membranes were visualized using ECL Select (GE Healthcare) and exposed to Hyperfilm MP (GE Healthcare). To ensure equal protein loading, each membrane was stripped and reprobed with anti-β-actin antibody (Sigma-Aldrich).
4.5. Docking and Modeling Studies
A structure based procedure was applied to discover and study the putative binding mode of the most potent compounds in our compound series. The PDB used (PDB ID: 1SA042) in this work was first submitted to the Protein Preparation Wizard (only Chain A and B) protocol of the Schrödinger Suite45 by following similar procedures described previously.43, 44 Later, ligand preparation was applied by the LigPrep 2.545 run to correctly assign the protonation states and atom types of the molecule. An additional conformational search by using the Mixed torsional/Low-mode sampling method present in MacroModel 9.945 and setting 50 conformations as output for each compound was needed to reproduce correctly the binding pose of the X-Ray ligand (RMSD 0.877 Ǻ on heavy atoms) and was then applied to all the other chemical entities. The grid generation and all docking runs were performed by leaving all the variables at default values in the Glide 5.845 program used in single precision docking mode (SP) and saving up to the 10 best poses per conformation. The best ranking pose was visualized with PyMOL 1.6.45
Supplementary Material
Acknowledgment
This research was supported by Scientific Research Grant 02/2011-09, awarded by Gazi University BAP.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cozzi P. Farmaco. 2003;58:213. doi: 10.1016/S0014-827X(03)00014-4. [DOI] [PubMed] [Google Scholar]
- 2.Johnston SRD, Ford H, Ross P. In: The Royal Marsden Hospital Hand Book of Cancer Chemotherapy. Brighton D, Wood M, editors. London, New York, Oxford: Elsevier Churchill Livingstone; 2005. pp. 1–17. [Google Scholar]
- 3.Jordan MA, Wilson L. Nat. Rev. Cancer. 2004;4:253. doi: 10.1038/nrc1317. [DOI] [PubMed] [Google Scholar]
- 4.Seed L, Slaughter DP, Limarzi LR. Surgery. 1940;7:696. [Google Scholar]
- 5.Pettit GR, Singh SB, Hamel E, Lin CM, Alberts DS, Garia-Kendall D. Experientia. 1989;45:205. doi: 10.1007/BF01954881. [DOI] [PubMed] [Google Scholar]
- 6.Simoni D, Romagnoli R, Baruchello R, Rondanin R, Grisolia G, Eleopra M, Rizzi M, Tolomeo M, Giannini G, Alloatti D, Castorina M, Marcellini M, Pisano C. J. Med. Chem. 2008;51:6211. doi: 10.1021/jm8005004. [DOI] [PubMed] [Google Scholar]
- 7.Simoni D, Romagnoli R, Baruchello R, Rondanin R, Rizzi M, Pavani MG, Alloatti D, Giannini G, Marcellini M, Riccioni T, Castorina M, Guglielmi MB, Bucci F, Carminati P, Pisano C. J. Med. Chem. 2006;49:3143. doi: 10.1021/jm0510732. [DOI] [PubMed] [Google Scholar]
- 8.Odlo K, Hentzen J, dit Chabert JF, Ducki S, Gani O, Sylte I, Skrede M, Florenes VA, Hansen TV. Bioorg. Med. Chem. 2008;16:4829. doi: 10.1016/j.bmc.2008.03.049. [DOI] [PubMed] [Google Scholar]
- 9.Tron GC, Pirali T, Sorba G, Pagliai F, Busacca S, Genazzani AA. J. Med. Chem. 2006;49:3033. doi: 10.1021/jm0512903. [DOI] [PubMed] [Google Scholar]
- 10.Lawrence NJ, McGown AT. Curr. Pharm. Des. 2005;11:1679. doi: 10.2174/1381612053764733. [DOI] [PubMed] [Google Scholar]
- 11.Dhar DN. The Chemistry of Chalcones and Related Compounds. New York: John Wiley & Sons; 1981. [Google Scholar]
- 12.Stu AW, Marby TJ. Phytochemistry. 1971;10:2812. [Google Scholar]
- 13.Park EJ, Park HR, Lee JS, Kim J. Planta Med. 1998;64:464. doi: 10.1055/s-2006-957485. [DOI] [PubMed] [Google Scholar]
- 14.Claude AC, Jean CL, Patric T, Christelle P, Gerard H, Albert JC, Jean LD. Anticancer Res. 2001;21:3949. [Google Scholar]
- 15.Kumar SK, Erin H, Catherine P, Halluru G, Davidson NE, Khan SR. J. Med. Chem. 2003;46:2813. doi: 10.1021/jm030213+. [DOI] [PubMed] [Google Scholar]
- 16.Aponte J, Verastegui M, Malaga E, Zimic M, Quiliano M, Vaisberg AJ, Gilman RH, Hammond GB. J. Med. Chem. 2008;51:6230. doi: 10.1021/jm800812k. [DOI] [PubMed] [Google Scholar]
- 17.Lahtchev KL, Batovska DI, Parushev PSt, Ubiyvovk VM, Sibirny AA. Eur. J. Med. Chem. 2008;43:2220. doi: 10.1016/j.ejmech.2007.12.027. [DOI] [PubMed] [Google Scholar]
- 18.Boumendjel A, Boccard J, Carrupt P-A, Nicolle E, Blanc M, Geze A, Choisnard L, Wouessidjewe D, Matera E-L, Dumontet C. J. Med. Chem. 2008;51:2307. doi: 10.1021/jm0708331. [DOI] [PubMed] [Google Scholar]
- 19.Cabrera M, Simoens M, Falchi G, Lavaggi ML, Piro OE, Castellano EE, Vidal A, Azqueta A, Monge A, Lopez de Cerain A, Sagrera G, Seoane G, Cerecetto H, Gonzalez M. Bioorg. Med. Chem. 2007;15:3356. doi: 10.1016/j.bmc.2007.03.031. [DOI] [PubMed] [Google Scholar]
- 20.Kim DY, Kim KH, Kim ND, Lee KY, Han CK, Yoon JH, Moon SK, Lee SS, Seong BL. J. Med. Chem. 2006;49:5664. doi: 10.1021/jm050761i. [DOI] [PubMed] [Google Scholar]
- 21.Go ML, Wu X, Liu XL. Curr. Med. Chem. 2005;12:483. doi: 10.2174/0929867053363153. [DOI] [PubMed] [Google Scholar]
- 22.Dimmock JR, Elias DW, Beazely MA, Kandepu NM. Curr. Med. Chem. 1999;6:1125. [PubMed] [Google Scholar]
- 23.Kumar D, Swapna S, Johnson EO, Shah K. Bioorg. Med. Chem. Lett. 2009;19:4492. doi: 10.1016/j.bmcl.2009.03.172. [DOI] [PubMed] [Google Scholar]
- 24.Rani P, Srivastava VK, Kumar A. Eur. J. Med. Chem. 2004;39:449. doi: 10.1016/j.ejmech.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 25.Agarwal A, Srivastava K, Puri SK, Chauhan PMS. Bioorg. Med. Chem. Lett. 2005;15:3133. doi: 10.1016/j.bmcl.2005.04.011. [DOI] [PubMed] [Google Scholar]
- 26.Manna F, Chimenti F, Bolasco A, Bizzarri B, Filippelli W, Filippelli A, Gagliardi L. Eur. J. Med. Chem. 1999;34:245. [Google Scholar]
- 27.Kumar D, Kumar NM, Akamatsu K, Kusaka E, Harada H, Ito T. Bioorg. Med. Chem. Lett. 2010;20:3916. doi: 10.1016/j.bmcl.2010.05.016. [DOI] [PubMed] [Google Scholar]
- 28.Luo Y, Qiu KM, Lu X, Liu K, Fu J, Zhu HL. Bioorg. Med. Chem. 2011;19:4730. doi: 10.1016/j.bmc.2011.06.088. [DOI] [PubMed] [Google Scholar]
- 29.Raffa D, Maggio B, Plescia F, Cascioferro S, Plescia S, Raimondi MV, Daidone G, Tolomeo M, Grimaudo S, Cristina AD, Pipitone RM, Bai R, Hamel E. Eur. J. Med. Chem. 2011;46:2786. doi: 10.1016/j.ejmech.2011.03.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang X-H, Wen Q, Zhao T-T, Sun J, Li X, Xing M, Lu X, Zhu H-L. Bioorg. Med. Chem. 2012;20:1181. doi: 10.1016/j.bmc.2011.12.057. [DOI] [PubMed] [Google Scholar]
- 31.Leslie BJ, Holaday CR, Nguyen T, Hergenrother PJ. J. Med. Chem. 2010;53:3964. doi: 10.1021/jm901805m. [DOI] [PubMed] [Google Scholar]
- 32.Shaw KNF, McMillan A, Gudmundson AG, Armstrong MD. J. Org.Chem. 1958;23:1171. [Google Scholar]
- 33.Holst-Hansen C, Brünner N. In: Cell Biology. A Laboratory Handbook. MTT cell proliferation assay. Celis JE, editor. San Diego: Academic press; 1998. pp. 16–18. [Google Scholar]
- 34.Reile H, Birnbock H, Bernhardt G, Spruss T, Schonenberger H. Anal. Biochem. 1990;187:262. doi: 10.1016/0003-2697(90)90454-h. [DOI] [PubMed] [Google Scholar]
- 35.Kueng W, Silber E, Eppenberger U. Anal. Biochem. 1989;182:16. doi: 10.1016/0003-2697(89)90710-0. [DOI] [PubMed] [Google Scholar]
- 36.Senaratne SG, Pirianov G, Mansi JL, Arnett TR, Colston KW. Br. J. Cancer. 2000;82:1459. doi: 10.1054/bjoc.1999.1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mosmann T. J. Immunol. Methods. 1983;65:55. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 38.Denizot F, Lang R. J. Immunol. Methods. 1986;89:271. doi: 10.1016/0022-1759(86)90368-6. [DOI] [PubMed] [Google Scholar]
- 39.GraphPad Prism Version 5.00 for Windows, G.S. San Diego, CA: U. ' www.graphpad.com’. [Google Scholar]
- 40.Hamel E. Cell Biochem. Biophys. 2003;38:1. doi: 10.1385/CBB:38:1:1. [DOI] [PubMed] [Google Scholar]
- 41.Verdier-Pinard P, Lai J-Y, Yoo H-D, Yu J, Márquez B, Nagle DG, Nambu M, White JD, Falck JR, Gerwick WH, Day BW, Hamel E. Mol. Pharmacol. 1998;53:62. doi: 10.1124/mol.53.1.62. [DOI] [PubMed] [Google Scholar]
- 42.Ravelli RB, Gigant B, Curmi PA, Jourdain I, Lachkar S, Sobel A, Knossow M. Nature. 2004;428:198. doi: 10.1038/nature02393. [DOI] [PubMed] [Google Scholar]
- 43.Ty N, Pontikis R, Chabot GG, Devillers E, Quentin L, Bourg S, Florent J-C. Bioorg. Med. Chem. 2013;21:1357. doi: 10.1016/j.bmc.2012.11.056. [DOI] [PubMed] [Google Scholar]
- 44.Marinozzia M, Carottia A, Sardellaa R, Buonerbaa F, Iannia F, Natalinia B, Passerib D, Rizzoc G, Pellicciaria R. Bioorg. Med. Chem. 2013;21:3780. doi: 10.1016/j.bmc.2013.04.038. [DOI] [PubMed] [Google Scholar]
- 45.Schrödinger L. New York, NY: 2012. [Google Scholar]
- 46.Stahl E. Thin-layer Chromatography. New York: Springer; 1969. [Google Scholar]
- 47.Lee K, Lee CH. WO 2013187696 A1 20131219 PCT Int. Appl. 2013
- 48.Hamel E, Lin CM. Biochemistry. 1984;23:4173. doi: 10.1021/bi00313a026. [DOI] [PubMed] [Google Scholar]
- 49.Hamel E, Lin CM. Biochim. Biophys. Acta. 1981;675:226. doi: 10.1016/0304-4165(81)90231-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.