

Abstract
Genome maintenance in germ cells is critical for fertility and the stable propagation of species. While mechanisms of meiotic DNA repair and chromosome behavior are well-characterized, the same is not true for primordial germ cells (PGCs), which arise and propagate during very early stages of mammalian development. Fanconi anemia (FA), a genomic instability syndrome that includes hypogonadism and testicular failure phenotypes, is caused by mutations in genes encoding a complex of proteins involved in repair of DNA lesions associated with DNA replication. The signaling mechanisms underlying hypogonadism and testicular failure in FA patients or mouse models are unknown. We conducted genetic studies to show that hypogonadism of Fancm mutant mice is a result of reduced proliferation, but not apoptosis, of PGCs, resulting in reduced germ cells in neonates of both sexes. Progressive loss of germ cells in adult males also occurs, overlaid with an elevated level of meiotic DNA damage. Genetic studies indicated that ATM-p53-p21 signaling is partially responsible for the germ cell deficiency.
Author Summary
The precursors to sperm and eggs begin are a group of <100 cells in the embryo, called primordial germ cells (PGCs). They migrate in the primitive embryo to the location of the future gonads, then undergo a rapid proliferation over the next few days to a population of many thousands. Because these cells contain the precious genetic information for our offspring, and the DNA replication associated with rapid PGC proliferation is subject to spontaneous errors, mechanisms exist to avoid propagation of mutations. A manifestation of this is the high sensitivity of PGCs to genetic perturbations affecting DNA repair. We studied mice defective for a gene called Fanconi anemia M (Fancm) that is important for repair of DNA damage that occurs during replication. Although it is expressed in all tissues, only the PGCs are affected in mutants, and are reduced in number. We find that PGCs lacking Fancm respond by slowing cell division, and identified the genetic pathway responsible for this protective response.
Introduction
Fanconi anemia (FA) is a genomic instability (GIN) syndrome characterized by developmental abnormalities affecting the renal, gastrointestinal and reproductive systems, the skeleton, skin pigmentation, and heart. It also causes progressive bone marrow failure and increased incidence of cancer [1], [2]. It can be caused by germline mutations in any of at least 17 genes (FANCA, FANCB, FANCC, FANCD1(BRCA2), FANCD2, FANCE, FANCF, FANCG, FANCI, FANCJ, FANCL, FANCM, FANCN (PALB2), FANCO(RAD51C), FANCP(SLX4), FANCQ(ERCC1 or 4)) [3], [4]). The products of these genes coordinately function in the repair of DNA interstrand crosslinks (ICL) during DNA replication [5]. A key event in FA pathway activation is the monoubiquitination of FANCI-FANCD2 (ID) heterodimers by the FA “core complex” (FANCA/B/C/E/F/G/L/M) [6]–[8]. The monoubiquitinated ID complex is recruited to DNA ICLs, and coordinates ICL repair together with downstream FA proteins (D1/J/N/O/P) and other (BRCA1, ATR) DNA repair proteins [1], [9], [10]. FANCM complexed with FAAP24 initiates FA pathway activity by recognizing DNA damage and loading the FA core complex. FAAP24 is particularly important in activating ATR in response to ICLs [11]. FANCM also has translocase activity that promotes branch migration of Holliday junctions and replication forks independent of FAAP24 [12].
FA deficient cells are hypersensitive to agents that induce ICLs, such as mitomycin C [MMC] or cisplatin. Most FA patients manifest anemia and bone marrow failure during childhood and are predisposed to cancer. Reduced fertility, hypogonadism and testicular failure, which is a consequence of impaired gametogenesis, are also common [13], [14], and this is reflected in most mouse models for FA, including knockouts for Fanca, Fancc, Fancd2, Fancf, Fancg, Fancl, Fancm, and Fancp, though Fancd1 is an exception [15]–[22]. While the severity varies amongst mutants, males generally present a partial Sertoli Cell Only-like phenotype whereby a subset of seminiferous tubule sections are depleted of germ cells. In mutant females, the number of ovarian follicles is typically reduced. Although most of these mutants have been characterized only as adults, the germ cell defects in three have been investigated perinatally or earlier. Germ cell depletion in Fancd2 −/− is evident in newborn mice [22], and defects in the proliferation of PGCs were reported in Fancc and Fancl mutants [15], [23]. While defects in DNA repair presumably underlie these germ cell phenotypes, the downstream DNA damage signaling pathway(s) that respond to these defects, ultimately leading to germ cell depletion, have not been identified.
The FA pathway appears to function in all cell types, including germ cells. However, experimental difficulties in studying the mammalian germline – particularly those stages occurring during embryonic development – have limited investigations into the roles of the FA and other DNA damage response (DDR) pathways in these cells. Importantly, the germline mutation rate is significant lower than that in somatic cells [24], [25], indicating a fundamental difference in genome maintenance that appears to reflect the biological importance of minimizing the germline mutation rate. While specific DDRs in the C. elegans germline have been identified [26], the DDRs operative in mammalian PGCs have not.
Here we investigate a Fancm mouse model (FancmChaos4) that was recovered in a forward genetic screen for GIN mutants. Mutant mice exhibit GIN and PGC depletion during embryogenesis. Using a genetic approach, we found that the ATM-p53-p21 axis contributes to the PGC depletion in this model, underscoring the critical importance of genome maintenance in these cells that undergo rapid cellular proliferation during a short period of time during development.
Results
Isolation of a new Fancm allele, FancmChaos4, from a forward genetic screen for GIN mutations in mice
We previously conducted an N-ethyl-N-nitrosourea (ENU) mutagenesis screen in mice for mutants showing chromosome instability, as assessed by micronucleus levels in erythrocytes [27]. Chaos4 (chromosome aberrations occurring spontaneously 4) was one mutation identified in this screen. Homozygous mutants show a mildly elevated (3 fold) frequency of erythrocytes with micronuclei (Figure 1A). Using combined SNP- [28] and microsatellite-based mapping, Chaos4 was genetically localized to a 9-Mb region between RS13481482 and D12Mit71 containing 9 RefSeq genes, including Fancm (Figure 1B). Sequencing of Fancm cDNA from mutants and controls identified a de novo T to C transition at nucleotide 524 of the coding region (Figure 1C). This point mutation changes a highly conserved cysteine residue to arginine (C142A) that is located within the DEXDc domain of this DEAD-like helicase superfamily region of FANCM (Figure 1D).
Figure 1. The Chaos4 allele is a point mutation in Fancm.

(A) Flow cytometric analysis of erythrocytes to quantify red blood cells (RBC) with micronuclei. (B) Genetic mapping of Chaos4 to a 9-Mb region of chromosome 12 containing Fancm between rs13481482 and D12Mit71. (C). Sequence traces showing the T524C transversion (arrows) identified in the Chaos4 allele of Fancm. (D) The Chaos4 point mutation is in the first exon, and the XH297 gene-trap is in the 14th exon. DEXDc, DEAD-like helicase domain; HELICc, Helicase superfamily c-terminal domain; ERCC4, ERCC4 endonuclease domain; HhH, Helix-hairpin-helix domain which interacts with FAAP24. Sequence alignment surrounding C142 is highly conserved from human to budding yeast. (E) SCE rates are significantly increased in FancmC4/C4 MEFs (p<0.05).
To confirm that the point mutation in Chaos4 underlies the GIN phenotype, we performed complementation analysis with a Fancm gene-trap allele, FancmGt(XH297)Byg, abbreviated hereafter as FancmXH. The gene-trap vector resides in exon 14, between the helicase and endonuclease domains (Figure 1D). FancmXH homozygotes also had elevated erythrocyte micronuclei (Figure 1A) as did FancmC4/XH mice, providing strong evidence that the FancmChaos4 allele (hereafter abbreviated FancmC4) is responsible for the GIN phenotype. We further assessed the chromosomal instability phenotype of our alleles via the sister chromatid exchange (SCE) assay. Consistent with results from a FancmΔ2 knockout mouse model [18], untreated FancmC4/C4 and FancmXH/XH MEFs both had elevated DNA breaks and radial chromosomes (Figure 1E; Figure S1), further confirming that the Chaos4 phenotype is attributable to the mutation in Fancm. Both FancmC4/C4 and FancmXH/XH mice were born at a Mendelian ratios, indicating that the mutations do not compromise embryonic viability (Table S1).
FancmC4/C4 primary MEFs undergo premature immortalization and mutant mice are cancer prone
The proliferation of untreated FancmC4/C4 primary MEFs during early passages was diminished compared to wild-type (Figure 2A, B). However, they recovered from senescent crisis and became immortalized much earlier (by passage 7) than wild-type (passage 10 or later) (Figure 2A, B).
Figure 2. FancmC4/C4 MEFs undergo premature senescence, but are not sensitive to interstrand crosslinks.
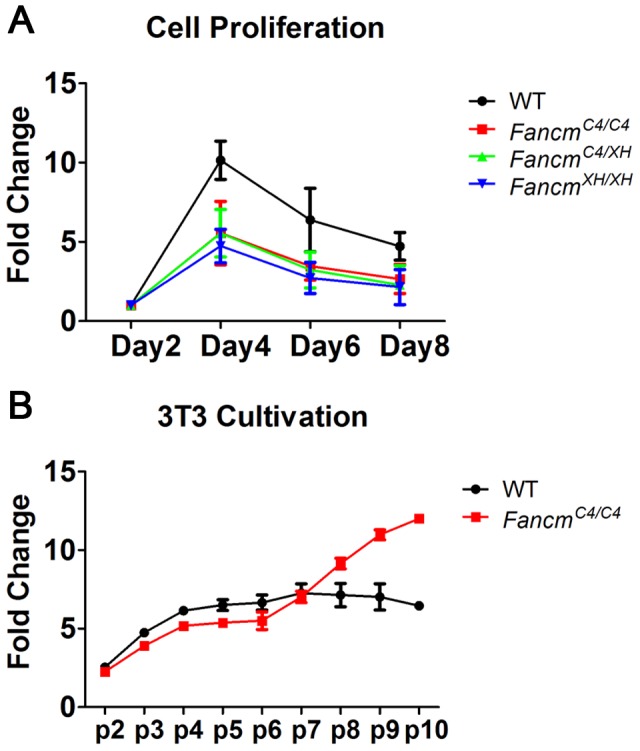
(A) MEF growth assays. (B) Immortalization timeline of primary MEFs using a 3T3 growth protocol. Cultures were passaged every 3 days.
Cancer predisposition is a defining feature of Fanconi Anemia. To determine if the early immortalization was an indicator of cancer susceptibility, FancmC4 mutants were aged for up to 1.5 years. Fancm+/C4 and FancmC4/C4 females congenic in the C3HeB/FeJ background had significantly elevated cancer/neoplasia susceptibility (Table S2), developing multiple tumor types (Table S3). Thirty-three percent (33%) of heterozygotes (9/27) and 58% of homozygous females (15/26) developed tumors by ∼1 year of age, compared to none of the 28 WT controls (p = 0.004 and p = 0.0002, respectively). The most common tumor types were ovarian, mammary and uterine. Heterozygous and homozygous FancmC4 males also were significantly tumor prone (42%, p = 0.001 and 47%, p = 0.002, respectively, vs. 9% of WT males; Tables S2, S3). Fancm null mice were reported to have a similar degree of tumor susceptibility [18].
FANCM deficiency compromises primordial germ cell proliferation and causes meiotic defects
In a limited gross and histological study, adult Fancm null mice were reported to have smaller gonads, germ cell loss in a subset of seminiferous tubule sections, and a reduced number of ovarian follicles [18]. Similar to those findings, we found that although FancmC4/C4 males appear grossly normal and were fertile, they had markedly smaller testes and about 60% the amount of sperm as wild-type littermates at 12 weeks of age (Figure 3A,B). Testis histology of young mice (≤16 weeks of age) revealed subtle seminiferous tubule abnormalities, namely the presence of occasional giant multinucleated cells that are not present in WT (Figure 3C, D). Prior to inbreeding onto strain C3HeB/FeJ, young FancmC4/C4 also exhibited germ-cell depleted individual tubules (not shown). Spermatogenesis defects in FancmC4/C4 mice (but not WT controls) became more severe over time, such that most seminiferous tubules in mice over 1 year of age were highly disrupted (Figure 3E, F). Gonadal defects in FancmC4/C4 mutants were sex independent; females manifested a significant depletion of primordial follicles compared to WT animals (Figure 3G).
Figure 3. Hypogonadism and spermatogenesis defects in Fancm mutant males.

(A,B) FancmC4/C4 mice (12 weeks old) have smaller testes and lower epididymal sperm counts. (C–F) H&E-staining of testis sections of the indicated genotypes. Samples in panels C–D are from 16 week old males. Arrowhead points to a giant multinucleated cell. (*) indicates an example of a seminiferous tubule with abnormal spermatogenesis. Panels E and F are from 80-wk-old males. Most tubules in 80 week FancmC4/C4 testes have only Sertoli cells. (G) Primordial follicles quantification in mutants. * p<0.05, ** p<0.01 n>10 for each genotype.
The presence of multinucleate cells in younger animals was suggestive of abnormal meiotic or premeiotic cell divisions. To investigate potential meiosis defects, we immunolabeled meiotic chromosomes from 12-week FancmC4/C4 males with markers of DSB signaling (γH2AX, the phosphorylated form of H2AX), DSB repair (RAD51), and meiotic chromosome structure (SYCP3, which detects axial elements of the synaptonemal complex). H2AX phosphorylation is also a marker of, and is involved in, transcriptional Meiotic Silencing of Unsynapsed Chromatin (MSUC) during meiosis [29]. As in WT (Figure 4A, E), most mutant pachytene spermatocytes had a normal XY body (marked by an intense γH2AX domain) and no RAD51 foci or autosomal γH2AX staining (Figure 4B, F), indicative of proper chromosome synapsis and recombinational repair of programmed (SPO11-induced) meiotic DSBs. However, 42% of the pachytene nuclei showed abnormal γH2AX staining, either spreading as a cloud into autosomes (Figure 4C) or as punctate foci on chromosome axes (Figure 4D), reflective of unsynapsed chromosomes and unrepaired DSBs, respectively. Consistent with the γH2AX results, twenty-seven percent of the spreads showed persistent RAD51 foci (Figure 4G, H). The data suggest that FancmC4/C4 spermatocytes have a defect in meiotic DSB repair, which in turn may affect synapsis of chromosomes in a subset of spermatocytes.
Figure 4. Meiotic defects in Chaos4 mutant spermatocytes.

(A–H) Shown are surface spread meiotic chromosomes from the indicated genotypes of males. Antibodies used for immunolabeling are as indicated with color coding. SYCP3 is a synaptonemal complex protein marking chromosome cores. The large domains of γH2AX staining in A,B and D correspond to the XY body. In panel C, there is an extended region of XY body-like staining over autosomes, a pattern typically called a “pseudo sex body” and usually marks asynapsed autosomes. All nuclei are in the pachytene stage of meiosis.
The incomplete, sex-independent germ cell depletion in young adults, characterized by primordial follicle reduction, reduced testis size, and germ cell losses in some seminiferous tubules was suggestive of premeiotic germ cell defects. To explore this, newborn gonads were serially sectioned and probed with the germ cell-specific marker MVH (mouse vasa homolog) to quantify the number of germ cells at birth. In FancmC4/C4 males and females, there were markedly fewer germ cells (55% and 30%, respectively) compared to wild-type littermates (Figure 5). This indicates that the germ cell depletion is initiated during embryogenesis.
Figure 5. Germ cell depletion in FancmC4/C4 occurs before birth.

Immunofluorescence of 1(A and B) and ovaries (C and D) of the indicated genotypes. MVH (green) stains germ cells and DAPI stains nuclei (blue). (E and F) Germ-cell counts at 1 dpp. Germ cell number is averaged on a per seminiferous tubule cross-section basis for males. Female counts correspond to the total from three medial sections. **, p<0.01; ***, p<0.001; Error bars indicate SD.
To identify the stage at which germ cell depletion starts, we examined the PGC population at various times of gestation. PGCs are first specified extra-embryonically at embryonic day 7.5 (E7.5). Between E8.5 and E10.5, this pool of alkaline phosphatase-positive PGCs then migrates along the epithelia of the hindgut towards the urogenital ridge, undergoing a modest degree of proliferation along the way. From there, they traverse the dorsal mesentery and populate the primitive gonad. They then undergo a dramatic proliferation after which male PGCs enter mitotic arrest until 3–4 dpp, while female PGCs enter meiosis at ∼E13.5 and arrest in meiotic prophase I until puberty (reviewed in [30]). We quantified PGCs at E11.5, E12.5 and E13.5. The numbers were not significantly decreased in either male or female FancmC4/C4 embryos at E11.5 (Figure 6). However, a significant reduction was evident by E12.5 and E13.5 (Figure 6).
Figure 6. PGC depletion in FancmC4/C4 mice is associated with reduced proliferation, not apoptosis.

(A) Male and female embryonic gonads from E11.5 and E12.5 stained for alkaline phosphatase activity. A decrease in PGCs is becomes evident only at the latter time point. The graphs of germ cells in (B) represent quantification of germ cells by immunolabelling fetal gonads with either Stella (E11.5 and E12.5) or MVH (E13.5). Representative images for E13.5 gonads are shown in Figure S2. The graphs of BrdU+ cells represent data from BrdU incorporation assays shown in (C and Figure S2). BrdU and Stella double-positive cells, which represent PGCs in S phase, were quantified as percentage of total Stella-positive PGCs.
These combined data suggest that FANCM deficiency does not significantly impair PGC specification or migration, but rather that mutant PGCs either proliferate more slowly or undergo elevated apoptosis. To distinguish between these possibilities, we assessed PGC proliferation and apoptosis using BrdU incorporation and TUNEL assays, respectively. The BrdU incorporation assays indicated that PGC proliferation is reduced in both male and female FancmC4/C4 gonads at E12.5 and E13.5 (Figure 6B; Figure S2). Furthermore, apoptosis was not evident in either wild type or FancmC4/C4 gonads at E12.5 (Figure S3).
Previous studies estimated the number and the doubling time of PGCs between E11.5 and E13.5 [31], [32]. The doubling time of wild type PGCs is 15.8 h in males, and 16.1 h in females (see Methods). Based on our PGC quantification, the doubling time of FancmC4/C4 PGC increased to 17.9±0.2 h in males, and 18.9±0.3 h in females.
DNA damage response pathways involved in PGC depletion
Although hypogonadism and testicular failure is characteristic of FA, a possible link between this and FA-related GIN has not been established. We hypothesized that if activation of a particular DDR pathway triggers PGC growth arrest or attenuation, then genetic disruption of that pathway would relieve the PGC depletion. Accordingly, we crossed FancmC4/C4 with various checkpoint mutants, including alleles of Atm, Chk2 (Chek2), p53 (Trp53), p21 (Cdkn1a), and Hus1 to obtain double mutants. All mutations were congenic or near congenic (at least 7 backcross generations) on the C3H strain background. The numbers of MVH-positive germ cells in newborn gonads were then quantified.
We first analyzed the role of p53 and its downstream effector p21 [33], [34]. Deletion of one or both p53 alleles partially but significantly rescued germ cell loss in FancmC4/C4 male newborns (Figure 7A). This partial rescue implies that some but not all germ cell depletion is due to p53 activation. Similar partial rescue was observed in FancmC4/C4 p21−/− males (Figure 7B). The involvement of p21, a CDK inhibitor and downstream effector of p53 [35], [36], is consistent with our previous finding that PGC depletion in FancmC4/C4 is a result of reduced proliferation. Surprisingly, the partial rescue was sexually dimorphic; neither p53 nor p21 knockout ameliorated the germ cell deficiency in newborn FancmC4/C4 females.
Figure 7. Genetic analysis of checkpoint signaling in FancmC4/C4 germ cells.

(A–E) Compound mutant gonads with indicated genotypes were collected at 1 dpp. Germ-cell counts were performed following MVH labeling. Male germ cell number was averaged on a per tubule cross-section basis for males. Values for females equal the total of germ cells detected in three medial sections. *, p<0.05; **, p<0.01; ***, p<0.001; Error bars indicate SD.
Next, we focused on the upstream kinases of two major DDR pathways, ATM and ATR [37]. These two proteins primarily respond to DSBs and sites of replication errors (RPA-coated ssDNA), respectively. Intercrosses of FancmC4/C4 Atm+/− mice produced 49 pups, none of which were homozygous for both mutations (p<0.001; expected = 12.25). Whereas doubly deficient mice were not born, FancmC4/C4 mice heterozygous for Atm were viable, and the genetic reduction of ATM partially rescued the germ cell loss in males but not females (Figure 7C). Therefore, Atm may respond to increased DNA damage in FancmC4/C4 PGCs, ultimately activating p53-p21 signaling to protect the fidelity of genetic information in the PGC pool. In contrast, a hypomorphic viable allele (Hus1neo) of the ATR-pathway gene Hus1 [38] had no apparent impact on the depletion of FancmC4/C4 PGCs (Figure 7D).
Given the partial phenotypic rescue of FancmC4/C4 PGCs by Atm haploinsufficiency and p53 nullizygosity, we hypothesized that the ATM target CHK2 served as the intermediate transducer kinase. However, Chk2 deficiency did not rescue germ cells loss in FancmC4/C4 males, but significantly rescued the germ cell population in FancmC4/C4 females (Figure 7E). Interestingly, Chk2−/− newborn females had more germ cells than WT controls (Figure 7E). Therefore, the rescue effect of Chk2 mutation is probably independent of FancmC4 mutation. As previously reported [39], we observed that Chk2−/− adults had histologically normal gonads. Chk2−/− males did not have more gonocytes at birth than WT siblings (Figure 7E). Since female but not male PGCs enter meiosis before birth, and Chk2 was recently found to play a crucial DNA damage checkpoint role in female meiosis [40], this may account for the elevated number of oocytes in double mutants.
Discussion
FANCM is a key component of the FA signaling pathway. Numerous in vitro studies have suggested that FANCM is a sensor of DNA damage at replication forks and helps anchor the FA core complex to chromatin [8], [41]–[44]. Fancm was also reported to have the non-canonical function of regulating meiotic crossovers in Arabidopsis thaliana and Saccharomyces pombe, specifically by catalyzing interference-independent recombination intermediates to undergo noncrossover rather than crossover resolution [45]–[47]. It was recently shown that FANCM, via its translocase activity, interacts with MHF to allow replication to “traverse” ICLs without repair, and that this activity is independent of other FA members [48]. Despite the substantial biochemical and mechanistic information on Fancm function, the physiological roles of Fancm in vertebrates are incompletely characterized.
A previous study found that Fancm null mice not only phenocopied other FA mouse models in causing hypogonadism and hypersensitivity to cross-linking agents (in MEFs), but also had decreased longevity and tumor-free survival [18]. As with the null mutant, FancmC4/C4 mice had elevated SCE and tumor susceptibility, and FancmC4/C4 MEFs underwent senescence prematurely. The general similarity in phenotypes between the null and FancmC4 alleles indicates that the single amino acid change in the DEAH helicase domain disrupts the crucial function of this protein in mice. This domain has no detectable helicase activity, but does encode the translocase activity of FANCM that is important for promoting the recovery of stalled replication forks [49], [50]. Given that mutating the translocase function of FANCM alone disrupts replication traverse of ICLs in the same manner as null cells [48], we speculate that the FancmC4 mutation disrupts translocase function to yield phenotypes that are essentially indistinguishable from nulls. Future studies to test this and other possibilities, such as protein stability, would be of interest.
We traced the cause of germ cell depletion in newborn FANCM-deficient mice to defects in PGC proliferation, which was not reported for the knockout, but which has been noted for knockouts of other FA genes (discussed earlier). Specifically, we found that the ATM-p53-p21 DDR pathway is operative in regulating PGC proliferation in males. Mutations of each partially restored germ cell numbers in newborns. However, the results with compound Atm mutants suggest a complex relationship with FANCM in PGCs. It has been reported that FANCM is actually regulated in part by ATR and ATM in response to damaged DNA in a Xenopus extract system [51], but the synthetic lethality between Fancm and complete ATM deficiency (Atm−/−) suggests that ATM and FANCM also have parallel, non-epistatic roles in DDRs during development. The Fancg−/− Atm−/− genotype also causes embryonic lethality [52], and inhibition of the FA pathway selectively kills ATM-deficient cells [53], [54], supporting the idea that the DNA damage to which the ATM and the FA pathway responds overlap. The viability of, and partial rescue of PGC loss in, FancmC4/C4 Atm+/− males suggests that the parallel DNA repair role of reduced ATM is sufficient to overcome the lack of functional FA pathway repair, but compromises checkpoint-mediated cell cycle delay in PGCs, presumably via reduced signaling to p53.
p53 is a key transcription factor that regulates several signaling pathways involved in the response to cellular stress, DNA damage, oncogene activation and other physiological signals [55]. Genetic experiments in mice have shown that p53 plays a role in FA signaling. p53 deficiency partially rescues the embryonic lethality in Fancn (Palb2) and Fanco (Rad51c) mutants [56], [57] and bone marrow failure in Fancd2 mutants [58]. Our studies provide the first evidence that p53 is involved in genome surveillance of PGCs during their expansion phase in development, at least in males. In the context of Fancm deficiency and the presumed increase of DNA lesions this causes, p53 appears to slow cell cycle progression rather than causing apoptosis (see model in Figure 8). Mutations in Fancl and Fancc also cause germ cell reduction traced to reduced PGC proliferation and not apoptosis [15], [23], suggesting that the level of endogenous DNA damage induced by FA pathway defects is not sufficient to stimulate p53-mediated apoptotic signaling. In contrast, p53 was reported to mediate germ cell apoptosis in Zebrafish fancl mutants [59], implying either that germ cells in this organism are more sensitive to DNA replication defects, the p53 pathway is more active in zebrafish germ cells, and/or zebrafish lack a redundant repair pathway(s).
Figure 8. Model of checkpoint responses to replication stress in primordial germ cells.

The activity of p53 alone doesn't fully account for germ cell depletion in Fancm mutants. Aside from only partial rescue in FancmC4/C4 males by p53 deletion, which suggests that an additional or parallel DDR pathway might still be operative such as one involving paralogs p63 and p73, p53 deficiency did not rescue loss of oocytes in newborn females. One possible explanation for this sexual dimorphism may relate to the direct entry of female PGCs into meiosis at ∼E13.5, unlike the mitotic arrest that male PGCs undergo. Since quantification of germ cell number in compound mutants was conducted in newborns, the number of oocytes at birth reflects events that occur both during PGC proliferation and during meiotic prophase I. Considering that male FancmC4/C4 meiocytes had substantially elevated DSBs, and mouse oocytes have a stringent meiotic DNA damage checkpoint that causes apoptotic elimination perinatally [60], it is possible that any rescue of PGC proliferation in FancmC4/C4 p53−/− females was counteracted by subsequent meiotic losses of those oocytes derived from damage-bearing “rescued” PGCs. Importantly, the oocyte DNA damage checkpoint involves signaling of CHK2 to both p53 and TAp63, and that in the absence of p53, DSB-bearing oocytes are still efficiently eliminated by CHK2-activated TAp63 [40]. As mentioned earlier, our observation that perinatal FancmC4/C4 germ cell numbers were rescued in CHK2-deficient females but not males likely reflects this oocyte-specific meiotic DNA damage pathway, not a PGC DDR.
Few DNA repair gene mutations are known to impact PGC growth or maintenance. Beyond FA mutants, Pin1, Mcm9, Rev7 and Helq are four other genes that have been correlated with both a function in genome maintenance and a PGC depletion phenotype [61]–[66]. Pin1 is a prolyl isomerase which directly regulates cell cycle genes. Pin1 deletion depletes PGCs by delaying their proliferation [64]. Mcm9 and Helq appear to be involved in homologous recombination repair (HRR) of ICLs. HELQ interacts with the RAD51 paralog complex, but appears to function in a pathway in parallel to FA [61], [62], [67]–[70]. MCM9 is required for normal homologous recombination, promoting recruitment of RAD51 to DNA damage sites and repair of ICLs [68]–[70] It also appears to act downstream of the FA pathway [70]. Interestingly, FANCM was reported to be required for HR-independent ICL repair [11]. Despite these indications of multiple pathways for DNA repair in PGCs, that these cells remain highly sensitive to perturbations of any of them.
FancmC4/C4 males also exhibited progressive germ cell depletion with age. The reason for this is unclear, since histological analysis revealed only subtle seminiferous tubule abnormalities in young mice. The progression to a near Sertoli Cell Only-like phenotype in many tubules suggests a defect in spermatogonial proliferation or renewal. The lack of more dramatic testicular pathology in young mice is also curious in light of evidence for DNA repair and XY-body defects in a substantial fraction of spermatocytes. Aside from the occasional appearance of abnormal multinucleated cells near the lumen of seminiferous tubules, coordinated arrest of pachytene stage spermatocytes was not observed as is typical for mutants that are recombination-defective and which disrupt XY silencing, an event proposed to underlie meiotic arrest [71]. One possible explanation is that the level of defects is below the threshold that would trigger a checkpoint, or that the unrepaired DNA damage is eventually repaired before checkpoint-mediated elimination. It may be relevant in this regard that we have not noticed visual abnormalities in offspring of Fancm mutants. Another possibility is that the DNA damage in FancmC4/C4 spermatocytes, inferred as such by the presence of γH2AX and RAD51 foci, may be of a nature that does not trigger elimination. For example, it is possible that these foci correspond to sites of damage incurred during premeiotic DNA replication, as opposed to SPO11-induced DSBs. Another example of apparently tolerated meiotic damage is the case of Rad54−/− spermatocytes, which are not eliminated despite bearing extensive RAD51 foci in late pachynema [72]. Finally, it is possible that Fancm has a hitherto unknown role in meiotic checkpoint activation in addition to DNA repair.
This study contributes to an emerging picture that the FA pathway is particularly important in stem cell biology [2]. Reprogramming of fibroblasts into induced pluripotent stem cells requires FA pathway function [73], [74]. Furthermore, not only is bone marrow failure a hallmark of FA, but this failure depends upon p53/p21 signaling [58]. The involvement of p53/p21 activation in both hematopoietic and germline stem cells bearing FA mutations, and the particular sensitivity of these lineage, emphasizes the importance of expanding studies of the FA pathway into diverse cell types including additional stem cell lineages.
Materials and Methods
Micronucleus assays
These were performed as described [75].
Positional cloning
The Chaos4 mutation was ENU-induced on the C57BL/6J (“B6”) background [27]. To identify the causative mutation, the mutation was outcrossed to strain C3HeB/FeJ (“C3H”), then intercrossed to produce potential homozygotes. F2 offspring were screened for micronucleus levels and a genome scan with a collection of microsatellite markers polymorphic between C3H and the parental strain B6 was performed [28]. This localized Chaos4 to a 44-Mb interval on chromosome 12, between D12Mit285 and D12Mit71. Subsequently, we conducted an inter-subspecific mapping cross with Mus castaneus (CAST/Ei). The F1s were either intercrossed or backcrossed to CAST/Ei and scored for micronuclei. A total of 956 informative meioses were examined, defining a 9-Mb critical region (Figure 1B).
Embryonic stem cell culture and microinjection for chimera production
The XH297 ES cell line (derived from the 129/Ola strain; BayGenomics) [76] bearing a gene trap insertion of Fancm (abbreviated FancmXH) were cultured in DMEM (Gibco) supplemented with 15% FBS (HyClone), 0.1 mM MEM nonessential amino acids, 1 mM sodium pyruvate, penicillin-streptomycin (100 units/ml), 100 µM beta-mercaptoethanol (Sigma) and recombinant leukemia inhibitory factor (produced in-house). Cells were microinjected into C57BL/6J blastocysts by standard methods. Fancm+/XH mice were then backcrossed into the C3HeB/FeJ background.
Mice and genotyping
Genotyping of FancmC4 mice was performed by PCR amplification of a 240 bp mutated segment with two primers: Chaos4L (CTTCTGGCAAGGTGGTTTTC) and Chaos4R (TTTGCTACCCACAGACGATG). PCR products were then digested by restriction enzyme AciI, which is present in the Chaos4 allele only. The Chaos4 allele is cut into 180 bp and 60 bp fragments. Genotyping of FancmXH mice was performed indirectly using microsatellite markers D12Mit69 and D12Mit71 that flank Fancm, and which are polymorphic between strain C3H and B6 (B6 alleles at D12Mit69 and D12Mit71 are indicative of the Chaos4 allele). The use of mice in this study was approved by Cornell's Institutional Animal Care and Use Committee. Mice bearing alleles of other mutations were: Atm (Atmtm1Led, abbreviated as Atm−), Chk2 (Chek2tm1Mak, abbreviated as Chk2−), p53 (Trp53tm1Tyj, abbreviated as p53−), p21 (Cdkn1atm1Tyj, abbreviated as p21−), and Hus1 (Hus1tm2Rsw, abbreviated as Hus1neo) [39], [77]–[80]. The stocks of mice bearing the p53, p21 and Hus1 alleles were all congenic in the C3H background (N10 or greater). The Atm, Chk2 stocks were at the N7 backcross generation. Euthanasia was performed by CO2 administration.
Mouse embryonic fibroblast (MEF) growth analyses
MEFs were generated from 12.5- to 14.5-dpc embryos. Cells were cultured in DMEM supplemented with 15% FBS (fetal bovine serum), 0.1 mM MEM nonessential amino acids, 1 mM sodium pyruvate, penicillin-streptomycin (100 units/ml), and beta-mercaptoethanol. For cell proliferation assays, 0.5×106 cells were seeded per 100-mm plate and then cultured and harvested to count cell numbers at various time points. For the cell senescence assay, 0.5×106 cells were seeded per 100-mm plate and then cultured and passaged every 3 days until they became immortalized. MEF metaphase spreads and the sister chromatid exchange assay were performed as previously described [18], [81].
Histology and immunohistochemistry
For basic histology, tissues were fixed in 4% paraformaldehyde (PFA) overnight, paraffin-embedded, sectioned at 5 µm, and stained with H&E (hematoxylin and eosin). Statistical differences in tumor types were assessed via Fisher's exact test. For germ-cell counts on embryonic or newborn gonads, 5 µm sections were immunostained as previously described [82]. Germ cells in postnatal gonads were counted in three sections from the midportion of each gonad and averaged. Antibodies: Rabbit anti-DDX4/MVH (Abcam ab13840; 1∶250); rabbit anti-Stella (Abcam ab19878; 1∶250); goat anti-mouse Alexa 594 conjugate (Molecular Probes A11005; 1∶1,000); goat anti-rabbit Alexa 488 conjugate (Molecular Probes A11008; 1∶1,000). The data were analyzed using one-way ANOVA with Bonferroni correction (Prism software package). The resulting P values were used to determine significance (P<0.05).
BrdU incorporation assay
Pregnant females received a single BrdU intraperitoneal injection (50 mg/kg) at 11, 12, or 13 days after vaginal plug detection (their corresponding embryos were E11.5, E12.5 and E13.5). Injected mice were sacrificed two hours later, and embryos were collected. Embryonic gonads together with mesonephric tubules (for E12.5 and E13.5 embryos) or the dorsal part of the trunk without other internal organs (for E11.5 embryos) were fixed in 4% PFA. Tissues were embedded in paraffin and sectioned. BrdU was detected by the Invitrogen BrdU Staining Kit (Cat. No. 93-3944), and PGCs were detected with rabbit anti-Stella (Abcam ab19878; 1∶250). At least three sagittal sections across the central part of the gonads were used for PGC quantification and BrdU scoring.
Since no cell apoptosis was obvious and no cell migration occurs between E11.5 and E13.5, PGC doubling time was calculated based on an exponential growth model:
NE13.5 and NE11.5 are the absolute number of PGCs in the whole gonad, which was estimated based on the previous studies and the relative ratio between wild type and mutants.
Alkaline phosphatase staining
Embryonic gonads were stained as described [83]. Briefly, fixed gonads were washed with dH2O and stained with freshly made staining solution (0.1 mg/ml Sodium α-naphthyl phosphate, 5 mg/ml Borax, 0.6 mg/ml MgCl2, and 0.5 mg/ml Fast Red TR salt) for 15–30 min. Tissues were then washed in dH2O and cleared with 70% glycerol.
TUNEL staining
Five µm paraffin sections of embryonic gonads were TUNEL stained using the In Situ Cell Death Detection Kit (Roche 11684817910). Atm−/− adult testes were used as a positive control [84].
Meiotic chromosome analysis
This was performed as described [75]. Primary antibodies used in this study: rabbit anti-SYCP3 (1∶500, Abcam); mouse anti-γH2AX (1∶500, JBW301 Upstate Biotechnology); rabbit anti-RAD51 (1∶250, this polyclonal antibody recognizes both RAD51 and DMC1; Oncogene Research Products).
Ethics statement regarding vertebrate animal use
The use of mice in this study was approved by Cornell's Institutional Animal Care and Use Committee, under the approved protocol of JCS (2004-0038). Euthanasia was performed by CO2 administration.
Supporting Information
Chromosomal instability in Fancm mutant MEFs. (A–F) Metaphase chromosomes from the indicated genotypes of MEFs. Chromosomal breaks (black arrowhead in B), sister chromatid exchanges (white arrowheads in C and D), and radial chromosomes (arrow in E and F) are observed in Fancm mutant MEFs, but not wild type MEFs (A).
(TIF)
Representative images for PGC quantification and proliferation in E13.5 embryonic gonads. Wild type (A, C) and FancmC4/C4 (B, D) male (A, B) and female (C, D) gonads are immunolabeled for Stella, a PGC marker, in red and BrdU in green.
(PDF)
TUNEL assay of PGCs in E12.5 gonads. Wild type (A, C) and FancmC4/C4 (B, D) female (A, B) and male (C, D) gonads are immunolabeled for Stella, a PGC marker, in red and TUNEL in green. (E) Atm−/− testis was used as a positive control for TUNEL signal (green).
(TIF)
Viability of Fancm mutant mice.
(DOCX)
Tumor Frequency of Fancm mutants.
(DOCX)
Histopathology of Fancm mutant mice.
(XLS)
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. All data are within the submission.
Funding Statement
This work was supported by 1) Empire State Stem Cell Fund (NYSTEM; stemcell.ny.gov) C026442, C024174 (JCS); 2) National Institutes of Health (T32 HD052471; R01GM45415, JCS). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Kim H, D'Andrea AD (2012) Regulation of DNA cross-link repair by the Fanconi anemia/BRCA pathway. Genes Dev 26: 1393–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kottemann MC, Smogorzewska A (2013) Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature 493: 356–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bogliolo M, Schuster B, Stoepker C, Derkunt B, Su Y, et al. (2013) Mutations in ERCC4, encoding the DNA-repair endonuclease XPF, cause Fanconi anemia. Am J Hum Genet 92: 800–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kashiyama K, Nakazawa Y, Pilz DT, Guo C, Shimada M, et al. (2013) Malfunction of nuclease ERCC1-XPF results in diverse clinical manifestations and causes Cockayne syndrome, xeroderma pigmentosum, and Fanconi anemia. Am J Hum Genet 92: 807–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Knipscheer P, Raschle M, Smogorzewska A, Enoiu M, Ho TV, et al. (2009) The Fanconi anemia pathway promotes replication-dependent DNA interstrand cross-link repair. Science 326: 1698–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Garcia-Higuera I, Taniguchi T, Ganesan S, Meyn MS, Timmers C, et al. (2001) Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol Cell 7: 249–262. [DOI] [PubMed] [Google Scholar]
- 7. Singh TR, Bakker ST, Agarwal S, Jansen M, Grassman E, et al. (2009) Impaired FANCD2 monoubiquitination and hypersensitivity to camptothecin uniquely characterize Fanconi anemia complementation group M. Blood 114: 174–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kim JM, Kee Y, Gurtan A, D'Andrea AD (2008) Cell cycle-dependent chromatin loading of the Fanconi anemia core complex by FANCM/FAAP24. Blood 111: 5215–5222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Andreassen PR, D'Andrea AD, Taniguchi T (2004) ATR couples FANCD2 monoubiquitination to the DNA-damage response. Genes Dev 18: 1958–1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang X, Andreassen PR, D'Andrea AD (2004) Functional interaction of monoubiquitinated FANCD2 and BRCA2/FANCD1 in chromatin. Mol Cell Biol 24: 5850–5862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang Y, Leung JW, Jiang Y, Lowery MG, Do H, et al. (2013) FANCM and FAAP24 maintain genome stability via cooperative as well as unique functions. Mol Cell 49: 997–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gari K, Decaillet C, Stasiak AZ, Stasiak A, Constantinou A (2008) The Fanconi anemia protein FANCM can promote branch migration of Holliday junctions and replication forks. Mol Cell 29: 141–148. [DOI] [PubMed] [Google Scholar]
- 13. Alter BP, Frissora CL, Halperin DS, Freedman MH, Chitkara U, et al. (1991) Fanconi's anaemia and pregnancy. Br J Haematol 77: 410–418. [DOI] [PubMed] [Google Scholar]
- 14. Auerbach AD (2009) Fanconi anemia and its diagnosis. Mutat Res 668: 4–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Agoulnik AI, Lu B, Zhu Q, Truong C, Ty MT, et al. (2002) A novel gene, Pog, is necessary for primordial germ cell proliferation in the mouse and underlies the germ cell deficient mutation, gcd. Hum Mol Genet 11: 3047–3053. [DOI] [PubMed] [Google Scholar]
- 16. Bakker ST, van de Vrugt HJ, Visser JA, Delzenne-Goette E, van der Wal A, et al. (2012) Fancf-deficient mice are prone to develop ovarian tumours. J Pathol 226: 28–39. [DOI] [PubMed] [Google Scholar]
- 17. Parmar K, D'Andrea A, Niedernhofer LJ (2009) Mouse models of Fanconi anemia. Mutat Res 668: 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bakker ST, van de Vrugt HJ, Rooimans MA, Oostra AB, Steltenpool J, et al. (2009) Fancm-deficient mice reveal unique features of Fanconi anemia complementation group M. Hum Mol Genet 18: 3484–3495. [DOI] [PubMed] [Google Scholar]
- 19. Crossan GP, van der Weyden L, Rosado IV, Langevin F, Gaillard PH, et al. (2011) Disruption of mouse Slx4, a regulator of structure-specific nucleases, phenocopies Fanconi anemia. Nat Genet 43: 147–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Whitney MA, Royle G, Low MJ, Kelly MA, Axthelm MK, et al. (1996) Germ cell defects and hematopoietic hypersensitivity to gamma-interferon in mice with a targeted disruption of the Fanconi anemia C gene. Blood 88: 49–58. [PubMed] [Google Scholar]
- 21. Wong JC, Alon N, McKerlie C, Huang JR, Meyn MS, et al. (2003) Targeted disruption of exons 1 to 6 of the Fanconi Anemia group A gene leads to growth retardation, strain-specific microphthalmia, meiotic defects and primordial germ cell hypoplasia. Hum Mol Genet 12: 2063–2076. [DOI] [PubMed] [Google Scholar]
- 22. Houghtaling S, Timmers C, Noll M, Finegold MJ, Jones SN, et al. (2003) Epithelial cancer in Fanconi anemia complementation group D2 (Fancd2) knockout mice. Genes Dev 17: 2021–2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nadler JJ, Braun RE (2000) Fanconi anemia complementation group C is required for proliferation of murine primordial germ cells. Genesis 27: 117–123. [DOI] [PubMed] [Google Scholar]
- 24. Conrad DF, Keebler JE, DePristo MA, Lindsay SJ, Zhang Y, et al. (2011) Variation in genome-wide mutation rates within and between human families. Nat Genet 43: 712–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Simpson AJ (1997) The natural somatic mutation frequency and human carcinogenesis. Adv Cancer Res 71: 209–240. [DOI] [PubMed] [Google Scholar]
- 26. Gartner A, Milstein S, Ahmed S, Hodgkin J, Hengartner MO (2000) A conserved checkpoint pathway mediates DNA damage–induced apoptosis and cell cycle arrest in C. elegans. Mol Cell 5: 435–443. [DOI] [PubMed] [Google Scholar]
- 27. Shima N, Hartford SA, Duffy T, Wilson LA, Schimenti KJ, et al. (2003) Phenotype-based identification of mouse chromosome instability mutants. Genetics 163: 1031–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Moran JL, Bolton AD, Tran PV, Brown A, Dwyer ND, et al. (2006) Utilization of a whole genome SNP panel for efficient genetic mapping in the mouse. Genome Res 16: 436–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schimenti J (2005) Synapsis or silence. Nat Genet 37: 11–13. [DOI] [PubMed] [Google Scholar]
- 30. Durcova-Hills G, Capel B (2008) Development of germ cells in the mouse. Curr Top Dev Biol 83: 185–212. [DOI] [PubMed] [Google Scholar]
- 31. Tam PP, Snow MH (1981) Proliferation and migration of primordial germ cells during compensatory growth in mouse embryos. J Embryol Exp Morphol 64: 133–147. [PubMed] [Google Scholar]
- 32. Kim B, Kim Y, Sakuma R, Hui CC, Ruther U, et al. (2011) Primordial germ cell proliferation is impaired in Fused Toes mutant embryos. Dev Biol 349: 417–426. [DOI] [PubMed] [Google Scholar]
- 33. Waga S, Li R, Stillman B (1997) p53-induced p21 controls DNA replication. Leukemia 11 Suppl 3: 321–323. [PubMed] [Google Scholar]
- 34. el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, et al. (1993) WAF1, a potential mediator of p53 tumor suppression. Cell 75: 817–825. [DOI] [PubMed] [Google Scholar]
- 35. Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ (1993) The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell 75: 805–816. [DOI] [PubMed] [Google Scholar]
- 36. Xiong Y, Hannon GJ, Zhang H, Casso D, Kobayashi R, et al. (1993) p21 is a universal inhibitor of cyclin kinases. Nature 366: 701–704. [DOI] [PubMed] [Google Scholar]
- 37. Sirbu BM, Cortez D (2013) DNA damage response: three levels of DNA repair regulation. Cold Spring Harb Perspect Biol 5: a012724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Levitt PS, Zhu M, Cassano A, Yazinski SA, Liu H, et al. (2007) Genome maintenance defects in cultured cells and mice following partial inactivation of the essential cell cycle checkpoint gene Hus1. Mol Cell Biol 27: 2189–2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hirao A, Cheung A, Duncan G, Girard PM, Elia AJ, et al. (2002) Chk2 is a tumor suppressor that regulates apoptosis in both an ataxia telangiectasia mutated (ATM)-dependent and an ATM-independent manner. Mol Cell Biol 22: 6521–6532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bolcun-Filas E, Rinaldi VD, White ME, Schimenti JC (2014) Reversal of female infertility by Chk2 ablation reveals the oocyte DNA damage checkpoint pathway. Science 343: 533–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tao Y, Jin C, Li X, Qi S, Chu L, et al. (2012) The structure of the FANCM-MHF complex reveals physical features for functional assembly. Nat Commun 3: 782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Huang M, Kennedy R, Ali AM, Moreau LA, Meetei AR, et al. (2011) Human MutS and FANCM complexes function as redundant DNA damage sensors in the Fanconi Anemia pathway. DNA Repair (Amst) 10: 1203–1212. [DOI] [PubMed] [Google Scholar]
- 43. Mosedale G, Niedzwiedz W, Alpi A, Perrina F, Pereira-Leal JB, et al. (2005) The vertebrate Hef ortholog is a component of the Fanconi anemia tumor-suppressor pathway. Nat Struct Mol Biol 12: 763–771. [DOI] [PubMed] [Google Scholar]
- 44. Meetei AR, Medhurst AL, Ling C, Xue Y, Singh TR, et al. (2005) A human ortholog of archaeal DNA repair protein Hef is defective in Fanconi anemia complementation group M. Nat Genet 37: 958–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Crismani W, Girard C, Froger N, Pradillo M, Santos JL, et al. (2012) FANCM limits meiotic crossovers. Science 336: 1588–1590. [DOI] [PubMed] [Google Scholar]
- 46. Knoll A, Higgins JD, Seeliger K, Reha SJ, Dangel NJ, et al. (2012) The Fanconi anemia ortholog FANCM ensures ordered homologous recombination in both somatic and meiotic cells in Arabidopsis. Plant Cell 24: 1448–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lorenz A, Osman F, Sun W, Nandi S, Steinacher R, et al. (2012) The fission yeast FANCM ortholog directs non-crossover recombination during meiosis. Science 336: 1585–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Huang J, Liu S, Bellani MA, Thazhathveetil AK, Ling C, et al. (2013) The DNA translocase FANCM/MHF promotes replication traverse of DNA interstrand crosslinks. Mol Cell 52: 434–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Meetei AR, Medhurst AL, Ling C, Xue Y, Singh TR, et al. (2005) A human ortholog of archaeal DNA repair protein Hef is defective in Fanconi anemia complementation group M. Nature genetics 37: 958–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Blackford AN, Schwab RA, Nieminuszczy J, Deans AJ, West SC, et al. (2012) The DNA translocase activity of FANCM protects stalled replication forks. Human molecular genetics 21: 2005–2016. [DOI] [PubMed] [Google Scholar]
- 51. Sobeck A, Stone S, Landais I, de Graaf B, Hoatlin ME (2009) The Fanconi anemia protein FANCM is controlled by FANCD2 and the ATR/ATM pathways. J Biol Chem 284: 25560–25568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kennedy RD, Chen CC, Stuckert P, Archila EM, De la Vega MA, et al. (2007) Fanconi anemia pathway-deficient tumor cells are hypersensitive to inhibition of ataxia telangiectasia mutated. J Clin Invest 117: 1440–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jenkins C, Kan J, Hoatlin ME (2012) Targeting the fanconi anemia pathway to identify tailored anticancer therapeutics. Anemia 2012: 481583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Landais I, Hiddingh S, McCarroll M, Yang C, Sun A, et al. (2009) Monoketone analogs of curcumin, a new class of Fanconi anemia pathway inhibitors. Mol Cancer 8: 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Vousden KH, Prives C (2009) Blinded by the Light: The Growing Complexity of p53. Cell 137: 413–431. [DOI] [PubMed] [Google Scholar]
- 56. Bouwman P, Drost R, Klijn C, Pieterse M, van der Gulden H, et al. (2011) Loss of p53 partially rescues embryonic development of Palb2 knockout mice but does not foster haploinsufficiency of Palb2 in tumour suppression. J Pathol 224: 10–21. [DOI] [PubMed] [Google Scholar]
- 57. Kuznetsov S, Pellegrini M, Shuda K, Fernandez-Capetillo O, Liu Y, et al. (2007) RAD51C deficiency in mice results in early prophase I arrest in males and sister chromatid separation at metaphase II in females. J Cell Biol 176: 581–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ceccaldi R, Parmar K, Mouly E, Delord M, Kim JM, et al. (2012) Bone marrow failure in Fanconi anemia is triggered by an exacerbated p53/p21 DNA damage response that impairs hematopoietic stem and progenitor cells. Cell Stem Cell 11: 36–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Rodriguez-Mari A, Canestro C, Bremiller RA, Nguyen-Johnson A, Asakawa K, et al. (2010) Sex reversal in zebrafish fancl mutants is caused by Tp53-mediated germ cell apoptosis. PLoS Genet 6: e1001034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Di Giacomo M, Barchi M, Baudat F, Edelmann W, Keeney S, et al. (2005) Distinct DNA-damage-dependent and -independent responses drive the loss of oocytes in recombination-defective mouse mutants. PNAS 102: 737–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Adelman CA, Lolo RL, Birkbak NJ, Murina O, Matsuzaki K, et al. (2013) HELQ promotes RAD51 paralogue-dependent repair to avert germ cell loss and tumorigenesis. Nature 502: 381–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Luebben SW, Kawabata T, Akre MK, Lee WL, Johnson CS, et al. (2013) Helq acts in parallel to Fancc to suppress replication-associated genome instability. Nucleic Acids Res 41: 10283–10297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hartford SA, Luo Y, Southard TL, Min IM, Lis JT, et al. (2011) Minichromosome maintenance helicase paralog MCM9 is dispensible for DNA replication but functions in germ-line stem cells and tumor suppression. Proc Natl Acad Sci U S A 108: 17702–17707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Atchison FW, Capel B, Means AR (2003) Pin1 regulates the timing of mammalian primordial germ cell proliferation. Development 130: 3579–3586. [DOI] [PubMed] [Google Scholar]
- 65. Watanabe N, Mii S, Asai N, Asai M, Niimi K, et al. (2013) The REV7 subunit of DNA polymerase zeta is essential for primordial germ cell maintenance in the mouse. J Biol Chem 288: 10459–10471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Khalaj M, Abbasi A, Yamanishi H, Akiyama K, Wakitani S, et al. (2014) A Missense Mutation in Rev7 Disrupts Formation of Polzeta, Impairing Mouse Development and Repair of Genotoxic Agent-induced DNA Lesions. J Biol Chem 289: 3811–3824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Takata K, Reh S, Tomida J, Person MD, Wood RD (2013) Human DNA helicase HELQ participates in DNA interstrand crosslink tolerance with ATR and RAD51 paralogs. Nat Commun 4: 2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Park J, Long DT, Lee KY, Abbas T, Shibata E, et al. (2013) The MCM8-MCM9 complex promotes RAD51 recruitment at DNA damage sites to facilitate homologous recombination. Mol Cell Biol 33: 1632–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Lutzmann M, Grey C, Traver S, Ganier O, Maya-Mendoza A, et al. (2012) MCM8- and MCM9-deficient mice reveal gametogenesis defects and genome instability due to impaired homologous recombination. Mol Cell 47: 523–534. [DOI] [PubMed] [Google Scholar]
- 70. Nishimura K, Ishiai M, Horikawa K, Fukagawa T, Takata M, et al. (2012) Mcm8 and Mcm9 form a complex that functions in homologous recombination repair induced by DNA interstrand crosslinks. Mol Cell 47: 511–522. [DOI] [PubMed] [Google Scholar]
- 71. Royo H, Polikiewicz G, Mahadevaiah SK, Prosser H, Mitchell M, et al. (2010) Evidence that meiotic sex chromosome inactivation is essential for male fertility. Current Biol 20: 2117–2123. [DOI] [PubMed] [Google Scholar]
- 72. Wesoly J, Agarwal S, Sigurdsson S, Bussen W, Van Komen S, et al. (2006) Differential contributions of mammalian Rad54 paralogs to recombination, DNA damage repair, and meiosis. Mol Cell Biol 26: 976–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Raya A, Rodriguez-Piza I, Guenechea G, Vassena R, Navarro S, et al. (2009) Disease-corrected haematopoietic progenitors from Fanconi anaemia induced pluripotent stem cells. Nature 460: 53–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Muller LUW, Milsom MD, Harris CE, Vyas R, Brumme KM, et al. (2012) Overcoming reprogramming resistance of Fanconi anemia cells. Blood 119: 5449–5457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Reinholdt L, Ashley T, Schimenti J, Shima N (2004) Forward genetic screens for meiotic and mitotic recombination-defective mutants in mice. Methods Mol Biol 262: 87–107. [DOI] [PubMed] [Google Scholar]
- 76. Stryke D, Kawamoto M, Huang CC, Johns SJ, King LA, et al. (2003) BayGenomics: a resource of insertional mutations in mouse embryonic stem cells. Nucleic Acids Res 31: 278–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Levitt PS, Liu H, Manning C, Weiss RS (2005) Conditional inactivation of the mouse Hus1 cell cycle checkpoint gene. Genomics 86: 212–224. [DOI] [PubMed] [Google Scholar]
- 78. Brugarolas J, Chandrasekaran C, Gordon JI, Beach D, Jacks T, et al. (1995) Radiation-induced cell cycle arrest compromised by p21 deficiency. Nature 377: 552–557. [DOI] [PubMed] [Google Scholar]
- 79. Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, et al. (1994) Tumor spectrum analysis in p53-mutant mice. Curr Biol 4: 1–7. [DOI] [PubMed] [Google Scholar]
- 80. Elson A, Wang Y, Daugherty CJ, Morton CC, Zhou F, et al. (1996) Pleiotropic defects in ataxia-telangiectasia protein-deficient mice. Proc Natl Acad Sci U S A 93: 13084–13089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Shima N, Munroe RJ, Schimenti JC (2004) The mouse genomic instability mutation chaos1 is an allele of Polq that exhibits genetic interaction with Atm. Mol Cell Biol 24: 10381–10389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Reinholdt LG, Munroe RJ, Kamdar S, Schimenti JC (2006) The mouse gcd2 mutation causes primordial germ cell depletion. Mech Dev 123: 559–569. [DOI] [PubMed] [Google Scholar]
- 83. Ginsburg M, Snow MH, McLaren A (1990) Primordial germ cells in the mouse embryo during gastrulation. Development 110: 521–528. [DOI] [PubMed] [Google Scholar]
- 84. Takubo K, Hirao A, Ohmura M, Azuma M, Arai F, et al. (2006) Premeiotic germ cell defect in seminiferous tubules of Atm-null testis. Biochem Biophys Res Commun 351: 993–998. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Chromosomal instability in Fancm mutant MEFs. (A–F) Metaphase chromosomes from the indicated genotypes of MEFs. Chromosomal breaks (black arrowhead in B), sister chromatid exchanges (white arrowheads in C and D), and radial chromosomes (arrow in E and F) are observed in Fancm mutant MEFs, but not wild type MEFs (A).
(TIF)
Representative images for PGC quantification and proliferation in E13.5 embryonic gonads. Wild type (A, C) and FancmC4/C4 (B, D) male (A, B) and female (C, D) gonads are immunolabeled for Stella, a PGC marker, in red and BrdU in green.
(PDF)
TUNEL assay of PGCs in E12.5 gonads. Wild type (A, C) and FancmC4/C4 (B, D) female (A, B) and male (C, D) gonads are immunolabeled for Stella, a PGC marker, in red and TUNEL in green. (E) Atm−/− testis was used as a positive control for TUNEL signal (green).
(TIF)
Viability of Fancm mutant mice.
(DOCX)
Tumor Frequency of Fancm mutants.
(DOCX)
Histopathology of Fancm mutant mice.
(XLS)
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. All data are within the submission.