

Abstract
Athero-prone flow promotes inflammation in endothelial cells, and this process is critical for pathogenesis of many chronic inflammatory conditions such as coronary and carotid artery atherosclerosis, as well as abdominal aortic aneurysm. Signal mediators activated by athero-prone (disturbed) flow that have been described include NF-κB and protein kinase C, which is very different from athero-protective (steady laminar) flow1. In this issue a publication from Shyy’s lab shows the critical role of sterol regulatory element binding protein 2 (SREBP2) on athero-prone flow-mediated NLRP3 inflammasome activation2. In particular, they showed that athero-prone flow induced both mature form of SREBP2 (SREBP2-N) and SREBP2 mRNA induction, which transcriptionally increase NADPH oxidase 2 (Nox2) and NLRP3 expression, thereby leading to IL-1β expression and endothelial inflammation (Figure 1). In this editorial, we will briefly review the NLRP3 inflammasome and SREBP activation system, which play a key role in modulating athero-prone flow-mediated EC inflammation. We will also discuss the following important questions for the future; the role of local NLRP3 and IL-1β expression, mechanisms for two different types of flow (athero-prone flow vs. athero-protective flow) on SREBP2 activation, and other NLRP3 activators including thioredoxin-interacting protein (TXNIP).
Keywords: SREBP, NLRP3 inflammasome, Editorial, inflammation, flow shear stress, signal transduction
The inflammasome is a protein complex that serves as a platform for the maturation of caspase-1 subsequent activation, leading to proteolytic maturation and secretion of interleukin 1β (IL-1β) and IL-18 (Figure 8 in 2). Three essential components of inflammasome are a sensor protein, the adapter protein ASC, and the inflammatory protease caspase-1. As a sensor protein, Nod-like receptor (NLR) family (NLRP1, NLRP3, and NLRP4) and AIM2 function as pathogen sensors and form an inflammasome complex. NLRP3 is unique in that it responds to numerous diverse physical and chemical triggers, unlike other NLRP members. NLRP3 also integrates concomitant stresses of cell damage present during inflammation such as extracellular adenosine triphosphate (ATP), reactive oxygen species (ROS), uric acid crystals, cholesterol crystals, and IAPP (islet amyloid polypeptide)3, 4. In the current study, Xiao et al have shown that athero-prone flow (oscillatory flow) increased NLRP3 expression and subsequent cleaved caspase-1 and IL-1β expression compared with pulsatile shear flow in vitro. The authors also found that the levels of the cleaved caspase-1, IL-1β, and NLRP3 were higher in the aortic arch (predominantly exposed to athero-prone flow) than thoracic aorta in vivo, supporting the crucial role of athero-prone flow-induced NLRP3 inflammasome activation on the development of atherosclerosis. The induction of NLRP3 and Nox2 by SREBP2 transactivation is proposed as a novel mechanism of athero-prone flow effects.
However, the evidence for the role of NLRP3 activation during the process of atherosclerosis is controversial5–7 (Figure 1). Cholesterol crystal in the plaque itself can activate NLRP3 inflammasome and release IL-1β from mouse and human macrophages6. Cholesterol crystals are recognized as a hallmark of atherosclerotic lesions. While crystals are prevalent in advanced atherosclerotic lesions, Duewell et al have reported that cholesterol crystals were not only detected in necrotic cores but also in subendothelial areas that are rich in immune cells, even in an early stage of atherosclerosis6. Depletion of NLRP3 or ACS in macrophages inhibits cholesterol crystals-mediated caspase-1 cleavage and IL-1β release and subsequent atherosclerosis formation in the background of low-density lipoprotein receptor knock out mice (LDLR−/−). These data support the crucial role of NLRP3 inflammasome complex in cholesterol crystals-mediated inflammation and atherosclerosis6. However, these results have been challenged by the recent publication from Menu et al7, which showed no differences of atherosclerosis between double knock mice of Nlrp3−/− crossed with ApoE−/− mice and control ApoE−/− mice7. Two notable differences of those studies are 1) LDLR−/− vs. ApoE−/− mice, and 2) knock out from bone marrow-derived cells vs. whole body. In this current issue, Xiao et al. nicely showed that endothelial specific SREBP2 deficiency inhibited NLRP3 induction and atherosclerosis in the ApoE−/− background2. But since the function of NLRP3 inflammasome in vessels wall including endothelial cells, especially in ApoE−/− background, remains unclear, it will be crucial to determine the role of endothelial NLRP3 inflammasome in atherosclerosis.
Figure 1.
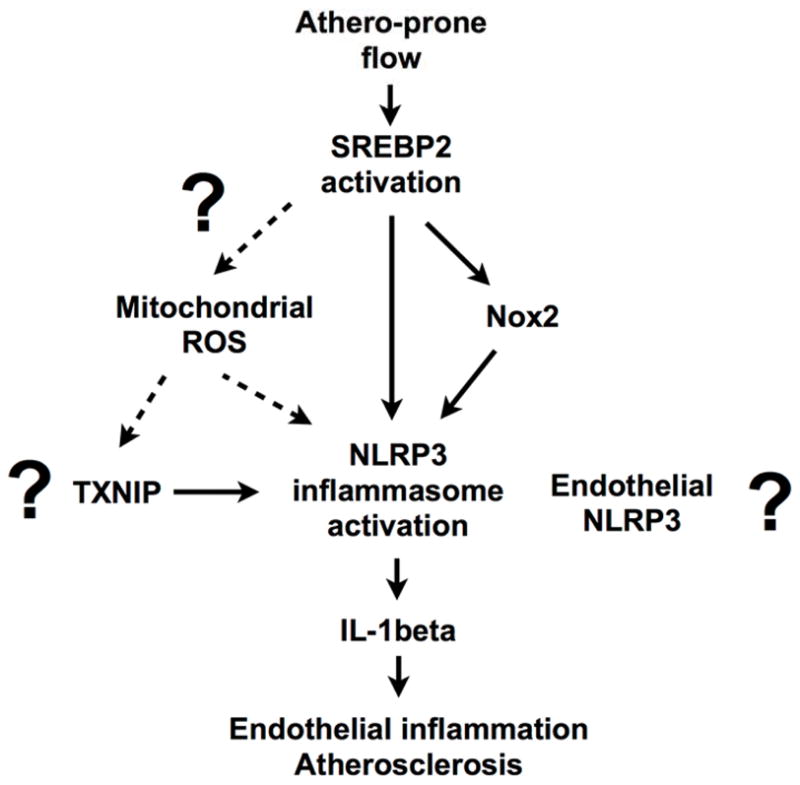
Scheme for SREBP2-mediated NLRP3 inflammasome activation.
The importance of NLRP3 inflammasome in the process of atherosclerosis has been supported by several studies that focus on IL-1β. In IL-1β deficient mice crossed with atherogenic apolipoprotein E knock out mice (ApoE−/−) exhibited a significant decrease in atherosclerosis with stabilization of the atherosclerosis plaque. Importantly, treatment of ApoE−/− mice with IL-1 receptor antagonists also significantly inhibit atherosclerosis 5. To evaluate the clinical efficacy of this concept, the CANTOS (Canakinumab Anti-inflammatory Thrombosis Outcomes Study) has been ongoing8. The purpose of this study is to evaluate the effect of IL-1β inhibition by canakinumab (a human monoclonal antibody that selectively neutralize IL-1β) as compared with placebo on recurrent myocardial infarction, stroke, and cardiovascular death among stable patients with coronary artery disease who show persistent elevation of high sensitivity C-reactive protein despite contemporary prevention strategies8.
SREBPs, including SREBP-1a, -1c, and 2, regulate the transcription of a number of genes involved in the cellular sterol and lipid homeostasis. Especially, SREBP2 is ubiquitouslly expressed, and relatively selective activator of cholesterol synthesis, as opposed to fatty acid synthesis, and regulates HMG-CoA reductase and low-density lipoprotein receptor (LDLR) genes9, 10. Since low levels of cholesterol stimulates SREBPs, the crucial role of SREBPs on liver steatosis and hyperlipedemia has been reported9, 11. However, the other roles of SREBPs in innate immunity and autophagy, which are not directly related with lipid homeostatis, have also been reported12, 13. The functions of SREBP in endothelial cells unrelated to lipid homeostatis remains unclear. To our knowledge, this is the first report to show SREBP2 regulates endothelial inflammation via increasing NLRP3 and Nox2 expression, which is not directly related to the regulation of lipid homeostatis2.
Importance of shear stress in vascular biology and pathophysiology is highlighted by the fact that steady laminar flow is protective against atherosclerosis while disturbed flow is a strong risk factor of the disease1. Previously, the authors have reported that athero-prone flow can cause sustained SREBP1 activation with nuclear localization. In contrast, athero-protective flow also temporarily increased nuclear transport, but could not be sustained14. SREBPs are bHLH-LZ family transcription factors, and inactive precursors (pre-SREBPs) of newly synthesized SREBPs are cleaved subsequently by proteases, then release the SREBPs NH2-terminal portion of SREBPs (SREBP-N) (Figure 2). This mature form of SREBP-N enters the nucleus and binds the sterol regulatory element 1 (SRE-1), and increases target genes expression9 (Figure 2). Therefore, there are two possible steps, which flow can regulate SREBPs activation. The first one is the process of pre-SREBPs cleavage in the extra-nucleus, and the second is nuclear SREBP-N transcriptional activity regulation. In the current issue Xiao et al showed the acceleration of the process of pre-SREBPs cleavage, which is the first step of SREBPs activation, by showing the increased level of SREBP-N induced by athero-prone flow. In the discussion the possible involvement of Akt induced by athero-prone flow was suggested by the authors, but since Akt directly regulates nuclear SREBP-N, which is the second step of SREBPs activation as we will explain below (Figure 2), further evaluation will be necessary.
Figure 2.

Scheme for SREBPs activation regulated by two different types of flow (athero-prone flow vs. athero-protective flow).
Recently, the role of posttranslational modification on the regulation of SREBPs activation has been discussed12. Insulin increases SREBP-N transcriptional activity via activating phosphatidylinositol 3-kinase (PI3K)/Akt/mammalian target of rapamycin complex 1 (mTORC1) kinase pathway12. Furthermore, Akt activation inhibits GSK3 kinase activation, which leads to ubiquitination and proteosomal degradation of the active transcription factor of SREBP-N by direct phosphorylation. In contrast to Akt, the inhibitory effect of AMP-activated protein kinase (AMPK) and Sirtulin1 (SIRT1) on SREBPs activation has been reported12. SREBP can directly be phosphorylated by AMPK, leading to suppress protein processing and nuclear translocation, and repress target gene expression, and SIRT1 directly deacetylate SREBP-1c and increases its degradation12. Of note, Dr. Shyy’s group has previously reported the specific activation of AMPK and induction of SIRT1 by athero-protective flow15 (Figure 2). However, it is not clear whether athero-prone flow-mediated Akt activation can explain sustained activation and nuclear localization of SREBP, and athero-protective flow-induced AMPK and SIRT1 induction inhibit these events. These issues including the specificity of each flows pattern differences on SREBP activation need to be clarified in the future studies. As mentioned above, since cholesterol crystal formation was observed in subendothelial areas in an early stage of atherosclerosis, athero-prone flow-induced SREBP2 activation may accelerate cholesterol crystal formation, which may indirectly activate NLRP3 inflammasome.
Our group has reported that athero-prone flow increased TXNIP expression16. TXNIP can promote atherosclerosis by multiple mechanisms17. These data are relevant because it has been reported that ROS induced TXNIP-NLRP3 association, and TXNIP deficiency inhibited ROS-mediated NLRP3 inflammasome activation18. Therefore, it is possible that athero-prone flow-induced TXNIP expression may be involved to this NLRP3 inflammasome activation. However, since other reports were unable to reproduce some of TXNIP function on NLRP34, 19, further investigation is necessary (Figure 1). Furthermore, although the crucial role of ROS on NLRP3 inflammasome activation has been well established, the source and type of inflammasome-activating ROS is under controversy (Figure 1). It is very intriguing that Xiao et al. showed the crucial role of NADPH oxidase 2 (Nox2) on caspase-1 activation and IL-1β expression in endothelial cells in the current paper. However, the idea of the involvement of Noxs on NLRP3 inflammasome activation was not supported in macrophages4, 20. The deletion of Nox1, Nox2, and Nox4 in macrophages do not show any defect in inflammasome activation. The macrophage deficient Nox2 showed robust decrease of ROS production, but only a minimum reduction in inflammasome activation. Instead ROS generated by mitochondria having reduced mitochondria membrane potential leads to NLRP3 inflammasome activation4, 20. Of note, mitochondria is also very sensitive to ROS, which disrupts the electron respiratory chain, and further increases ROS production. The contribution of Noxs family and mitochondrial ROS production on endothelial NLRP3 inflammasome activation needs further investigation.
In conclusion, Xiao et al. have shown that athero-prone flow-mediated SREBP2 activation increases NLRP3 inflammasome activation via increasing NLRP3 and Nox2 expression, leading to endothelial inflammation and atherosclerosis formation. Previously, the authors’ group showed the unique sustained SREBP nuclear localization induced by athero-prone flow but not by athero-protective flow, supporting the critical role of SREBP2 activation under athero-prone flow, but the mechanisms by which the athero-prone flow but not athero-protective flow activates SREBP activation needs more clarification. In addition, the contribution of endothelial NLRP3 inflammasome on atherosclerosis and source of ROS, which activates endothelial NLRP3, will be important to understand. It is clear that athero-prone flow-mediated endothelial inflammation is a highly regulated process that is increasingly as attractive a target for therapeutic intervention as steady laminar flow mediated athero-protective pathways.
Acknowledgments
Funding Sources: This study was supported by a grant from National Institutes of Health to Drs Bradford C. Berk (HL-064839, HL 106158), Jun-ichi Abe (HL-064839, HL-108551, HL-102746).
Footnotes
Conflict of Interest Disclosures: None.
References
- 1.Heo KS, Fujiwara K, Abe J. Disturbed-flow-mediated vascular reactive oxygen species induce endothelial dysfunction. Circ J. 2011;75:2722–2730. doi: 10.1253/circj.cj-11-1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xiao H, Lu M, Lin TY, Chen Z, Chen G, Wang WC, Marin T, TPS, Wen L, Gongol B, Sun W, Liang X, Chen J, Huang HD, Pedra JHF, Johnson DA, Shyy JYJ. Srebp2 activation of nlrp3 inflammasome in endothelium mediates hemodynamic-induced atherosclerosis susceptibility. Circulation. 2013;128:XX–XXX. doi: 10.1161/CIRCULATIONAHA.113.002714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kanneganti TD. Central roles of nlrs and inflammasomes in viral infection. Nat Rev Immunol. 2010;10:688–698. doi: 10.1038/nri2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gross O, Thomas CJ, Guarda G, Tschopp J. The inflammasome: An integrated view. Immunol Rev. 2011;243:136–151. doi: 10.1111/j.1600-065X.2011.01046.x. [DOI] [PubMed] [Google Scholar]
- 5.Wen H, Ting JP, O’Neill LA. A role for the nlrp3 inflammasome in metabolic diseases--did warburg miss inflammation? Nat Immunol. 2012;13:352–357. doi: 10.1038/ni.2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, Abela GS, Franchi L, Nunez G, Schnurr M, Espevik T, Lien E, Fitzgerald KA, Rock KL, Moore KJ, Wright SD, Hornung V, Latz E. Nlrp3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357–1361. doi: 10.1038/nature08938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Menu P, Pellegrin M, Aubert JF, Bouzourene K, Tardivel A, Mazzolai L, Tschopp J. Atherosclerosis in apoe-deficient mice progresses independently of the nlrp3 inflammasome. Cell Death Dis. 2011;2:e137. doi: 10.1038/cddis.2011.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ridker PM, Thuren T, Zalewski A, Libby P. Interleukin-1beta inhibition and the prevention of recurrent cardiovascular events: Rationale and design of the canakinumab anti-inflammatory thrombosis outcomes study (cantos) Am Heart J. 2011;162:597–605. doi: 10.1016/j.ahj.2011.06.012. [DOI] [PubMed] [Google Scholar]
- 9.Xiao X, Song BL. Srebp: A novel therapeutic target. Acta Biochim Biophys Sin (Shanghai) 2013;45:2–10. doi: 10.1093/abbs/gms112. [DOI] [PubMed] [Google Scholar]
- 10.Daemen S, Kutmon M, Evelo CT. A pathway approach to investigate the function and regulation of srebps. Genes Nutr. 2013;8:289–300. doi: 10.1007/s12263-013-0342-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tang JJ, Li JG, Qi W, Qiu WW, Li PS, Li BL, Song BL. Inhibition of srebp by a small molecule, betulin, improves hyperlipidemia and insulin resistance and reduces atherosclerotic plaques. Cell Metab. 2011;13:44–56. doi: 10.1016/j.cmet.2010.12.004. [DOI] [PubMed] [Google Scholar]
- 12.Shao W, Espenshade PJ. Expanding roles for srebp in metabolism. Cell Metab. 2012;16:414–419. doi: 10.1016/j.cmet.2012.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Im SS, Yousef L, Blaschitz C, Liu JZ, Edwards RA, Young SG, Raffatellu M, Osborne TF. Linking lipid metabolism to the innate immune response in macrophages through sterol regulatory element binding protein-1a. Cell Metab. 2011;13:540–549. doi: 10.1016/j.cmet.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu Y, Chen BP, Lu M, Zhu Y, Stemerman MB, Chien S, Shyy JY. Shear stress activation of srebp1 in endothelial cells is mediated by integrins. Arterioscler Thromb Vasc Biol. 2002;22:76–81. doi: 10.1161/hq0102.101822. [DOI] [PubMed] [Google Scholar]
- 15.Chen Z, Peng IC, Cui X, Li YS, Chien S, Shyy JY. Shear stress, sirt1, and vascular homeostasis. Proc Natl Acad Sci U S A. 2010;107:10268–10273. doi: 10.1073/pnas.1003833107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang XQ, Nigro P, World C, Fujiwara K, Yan C, Berk BC. Thioredoxin interacting protein promotes endothelial cell inflammation in response to disturbed flow by increasing leukocyte adhesion and repressing kruppel-like factor 2. Circ Res. 2012;110:560–568. doi: 10.1161/CIRCRESAHA.111.256362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Spindel ON, World C, Berk BC. Thioredoxin interacting protein: Redox dependent and independent regulatory mechanisms. Antioxid Redox Signal. 2012;16:587–596. doi: 10.1089/ars.2011.4137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol. 2010;11:136–140. doi: 10.1038/ni.1831. [DOI] [PubMed] [Google Scholar]
- 19.Masters SL, Dunne A, Subramanian SL, Hull RL, Tannahill GM, Sharp FA, Becker C, Franchi L, Yoshihara E, Chen Z, Mullooly N, Mielke LA, Harris J, Coll RC, Mills KH, Mok KH, Newsholme P, Nunez G, Yodoi J, Kahn SE, Lavelle EC, O’Neill LA. Activation of the nlrp3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced il-1beta in type 2 diabetes. Nat Immunol. 2010;11:897–904. doi: 10.1038/ni.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in nlrp3 inflammasome activation. Nature. 2011;469:221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]