

Abstract
Variation in plasma levels of cortisol, an essential hormone in the stress response, is associated in population-based studies with cardio-metabolic, inflammatory and neuro-cognitive traits and diseases. Heritability of plasma cortisol is estimated at 30–60% but no common genetic contribution has been identified. The CORtisol NETwork (CORNET) consortium undertook genome wide association meta-analysis for plasma cortisol in 12,597 Caucasian participants, replicated in 2,795 participants. The results indicate that <1% of variance in plasma cortisol is accounted for by genetic variation in a single region of chromosome 14. This locus spans SERPINA6, encoding corticosteroid binding globulin (CBG, the major cortisol-binding protein in plasma), and SERPINA1, encoding α1-antitrypsin (which inhibits cleavage of the reactive centre loop that releases cortisol from CBG). Three partially independent signals were identified within the region, represented by common SNPs; detailed biochemical investigation in a nested sub-cohort showed all these SNPs were associated with variation in total cortisol binding activity in plasma, but some variants influenced total CBG concentrations while the top hit (rs12589136) influenced the immunoreactivity of the reactive centre loop of CBG. Exome chip and 1000 Genomes imputation analysis of this locus in the CROATIA-Korcula cohort identified missense mutations in SERPINA6 and SERPINA1 that did not account for the effects of common variants. These findings reveal a novel common genetic source of variation in binding of cortisol by CBG, and reinforce the key role of CBG in determining plasma cortisol levels. In turn this genetic variation may contribute to cortisol-associated degenerative diseases.
Author Summary
Cortisol is a steroid hormone from the adrenal glands that is essential in the response to stress. Most cortisol in blood is bound to corticosteroid binding globulin (CBG). Diseases causing cortisol deficiency (Addison's disease) or excess (Cushing's syndrome) are life-threatening. Variations in plasma cortisol have been associated with cardiovascular and psychiatric diseases and their risk factors. To dissect the genetic contribution to variation in plasma cortisol, we formed the CORtisol NETwork (CORNET) consortium and recruited collaborators with suitable samples from more than 15,000 people. The results reveal that the major genetic influence on plasma cortisol is mediated by variations in the binding capacity of CBG. This is determined by differences in the circulating concentrations of CBG and also in the immunoreactivity of its ‘reactive centre loop’, potentially influencing not only binding affinity for cortisol but also the stability of CBG and hence the tissue delivery of cortisol. These findings provide the first evidence for a common genetic effect on levels of this clinically important hormone, suggest that differences in CBG between individuals are biologically important, and pave the way for further research to dissect causality in the associations of plasma cortisol with common diseases.
Introduction
The adrenal steroid hormone cortisol plays a vital role in adaptation to environmental stress. In response to stressors such as starvation, infection or injury, cortisol secretion is elevated by activation of the hypothalamic-pituitary-adrenal (HPA) axis. Cortisol acts predominantly through glucocorticoid receptors to induce a wide range of physiological responses, including liberating fuel (by facilitating gluconeogenesis and lipolysis), maintaining cardiovascular homeostasis (by inducing sodium retention and vasoconstriction), altering mood and memory (in favour of focusing on ‘fight or flight’ responses), and acting as a ‘brake’ on the innate immune response (preventing bystander damage from unrestrained inflammation) [1]. Chronic elevations in cortisol, however, may be maladaptive, as exemplified in patients with tumours of the pituitary or adrenal gland causing Cushing's syndrome; here, elevated plasma cortisol is responsible for obesity, type 2 diabetes, hypertension, dyslipidaemia, depression, memory loss, impaired wound healing, osteoporosis, myopathy, and many other features.
Epidemiological data suggest that subtle activation of the HPA axis associates with many of these traits within the population, in people who do not harbour the tumours which cause overt Cushing's syndrome. In these studies higher plasma cortisol concentration, measured in the morning, provided a robust marker of the activation of the HPA axis which accompanies high blood pressure, hyperglycaemia and dyslipidaemia [2]–[5], age-associated cognitive dysfunction [6], and low mood [7]. Conversely, lower cortisol associates with immunological abnormalities [8], post-traumatic stress disorder (PTSD) [9], and obesity [1] (the inverse association with obesity is likely due to increased metabolic clearance of cortisol and confounds the positive association of cortisol with other cardiovascular risk factors, explaining some inconsistencies in the associations of cortisol with ‘metabolic syndrome’ [1]). Mechanisms underlying these associations remain uncertain, with most investigators suggesting abnormal central control of the HPA axis [1], [10], [11]. A high proportion of cortisol in plasma is protein bound, mostly to corticosteroid binding globulin (CBG). Although variations in total CBG concentrations have been associated with features of metabolic syndrome [12], [13], this does not account entirely for associations of total plasma cortisol with other quantitative traits [5], [14], [15].
Morning plasma cortisol has a heritability of 30–60% [16]–[18]. Identifying genetic variants which contribute to variation in morning cortisol values could provide key insights into the mechanism of HPA axis activation associated with common quantitative traits, and an opportunity to dissect causality using Mendelian randomisation [19]. Attempts to identify these genetic variants to date have been limited to small candidate gene studies [18]. We therefore established the CORtisol NETwork (CORNET) consortium with the initial aim of identifying genetic determinants of inter-individual variation in HPA axis function.
Results
Genome-wide association meta-analysis
We conducted a discovery meta-analysis of genome-wide association studies (GWAMA) of morning plasma cortisol levels, investigating ∼2.5 M SNPs in 12,597 men and women, aged 14–102 years, of European origin (Table S1 for participant characteristics). There was very little inflation of test statistics (λGC = 1.005, Table S2). The −log10 P values by chromosome for age- and sex-adjusted cortisol z-scores are shown in Figure 1a. A quantile–quantile plot (Figure 1b) showed marked departure from the null for SNPs with low P values, listed in Table S3. Analysis of data for men and women separately showed no sex-specific effects (data not shown). The results were similar between all multivariable adjusted models, and whether or not time of sampling was included as a covariate. The results reported are therefore adjusted only for age and sex.
Figure 1. Meta-analysis of genome wide association studies for morning plasma cortisol.
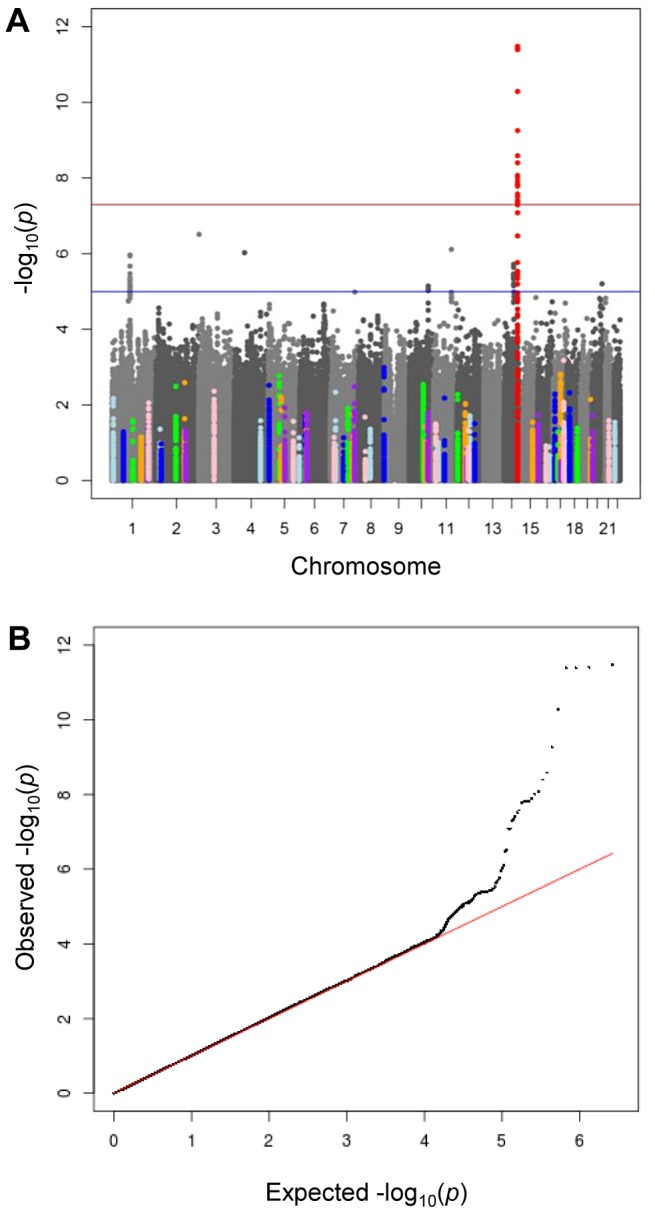
A) Manhattan plot of −lop10 P values by chromosome. The red horizontal line indicates genome-wide significance (P<5×10−8) and the blue horizontal line indicates moderate significance (P<5×10−5). The lead SNP rs12589136 (chr14:94,793,686; b37) in red is genome-wide significant. SNPs within ±50 kb of cortisol-related candidate genes (listed in Table S6) are highlighted in colours. B) Quantile-quantile plot of −log10 P, comparing the distribution of observed −log10 P-values and that expected by chance.
There was strong evidence for associations between plasma cortisol and genetic variation found at chromosome 14q32. In an additive genetic model, the lead SNP rs12589136 reported a per minor allele effect of 0.10 cortisol z-score (95%CI 0.07,0.13; P = 4.0×10−12 after genomic control). The effect allele frequency was 0.22 and this variation explained 0.13% of the morning plasma cortisol variance. A forest plot showed consistent directional effects in all studies, with the T allele at rs12589136 associated with higher morning plasma cortisol (Figure 2). Only minimal heterogeneity was observed between studies (I2 = 0.18).
Figure 2. Forest plot of association of morning plasma cortisol with rs12589136.
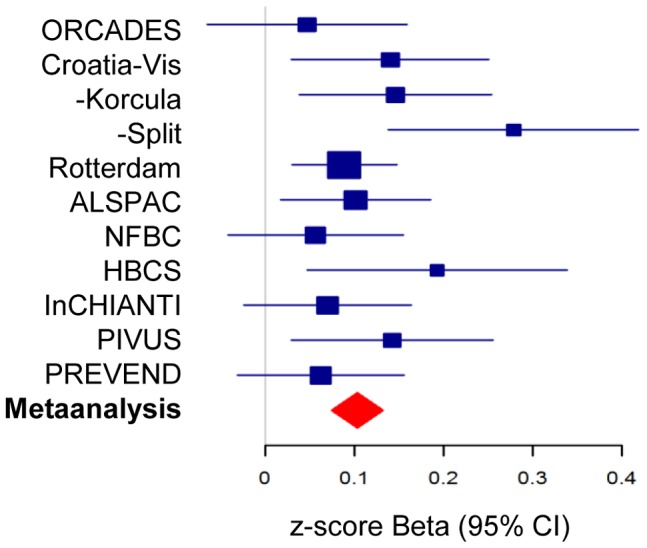
Plot shows association as beta values with 95%CI for morning plasma cortisol z-scores for rs12589136 (T allele) in discovery cohorts (blue) and meta-analysis (red).
A recombination boundary containing SERPINA6 and SERPINA1 was found to contain all variants at this locus contributing to association with plasma cortisol (Figure 3). A clumping procedure [20] identified rs12589136 (4 kb upstream of SERPINA6), rs11621961 (1 kb downstream of SERPINA6) and rs2749527 (30 kb upstream of SERPINA6) as markers representing genome-wide significant signals in this region. Individually, the beta (for effect on cortisol z score, 95%CI) for minor (all T) alleles at rs12589136, rs2749527, and rs11621961 were 0.10 (0.07,0.13; P = 3.3×10−12), −0.08 (−0.11,−0.06; P = 5.2×10−11), and −0.08 (−0.10,−0.05; P = 4.0×10−8); joint analysis showed these SNPs have partially independent effects, with beta (95%CI) 0.07 (0.04,0.10; P = 3.1×10−5), −0.04 (−0.07,−0.01; P = 0.012), and −0.03 (−0.07,0.00; P = 0.037), respectively.
Figure 3. Regional associations surrounding lead SNP rs12589136 in genome-wide meta-analysis of morning plasma cortisol.

Regional plot shows −log10 P values of all SNPs, and degree of correlation between all SNPs and lead SNP rs12589136. SNPs with lower P values span SERPINA6 and SERPINA1 genes within a recombination boundary.
The SERPINA6 gene encodes corticosteroid binding globulin (CBG). The neighbouring (upstream) gene, SERPINA1, encodes α1-antitrypsin, the inhibitor of neutrophil elastase which cleaves and inactivates CBG [21].
Conditional analysis
A quantile-quantile plot after removal of the SERPINA6/SERPINA1 region (chr14: 94,768,859–94,843,565; Genome Reference Consortium build 37) showed no evident inflation of test statistics (not shown). Conditional analyses adjusting for each of the partially independent genome-wide significant variants (rs12589136, rs11621961, rs2749527) in a subset of the meta-analysis population reduced the significance of all other SNPs to P>1×10−5 for an association with plasma cortisol.
Gene-centric associations
We used a gene-centric approach to analyse the combined effect of all SNPs within a gene, rather than individual SNP associations, using VEGAS [22]. This produced a gene-based test statistic from meta-analysis results, allowing identification of genes containing multiple SNPs that individually did not reach genome-wide significance (Table S4). Only SERPINA6 and SERPINA1 were identified as gene-wide significant (P<3×10−6); both included rs12589136 in the gene boundary.
Candidate gene analysis
A list of 61 candidate genes thought likely to influence plasma cortisol was collated by a panel of experts. A Manhattan plot of the −log10 P values highlighted for SNPs in these candidate genes showed that only SERPINA6 reached genome-wide significance (Figure 1a). Using gene-based p-values from VEGAS, after adjusting for multiple testing, only SERPINA6 was associated with plasma cortisol (Table S5). SERPINA1 was not included in the candidate gene list.
Replication
In 2,795 participants in additional cohort studies, the association with plasma cortisol was replicated for the lead SNP rs12589136 (P = 0.0002), rs11621961 (P = 0.003) and rs2749529 (used as proxy for rs2749527, P = 0.019)(Table 1; Table S1 for participant characteristics; Table S6 for results in each cohort).
Table 1. Association with morning plasma cortisol of SNPs representing signals in the SERPINA6/SERPINA1 region from meta-analyses of discovery genome-wide association studies and of replication studies.
| GWAMA (n = 12,597) | REPLICATION (n = 2,795) | |||||||||||
| SNP ID | Chr | Number of supporting SNPs | Position (b37) | Alleles effect/other | EAF | Effects | Beta (95%CI) | Pa | EAF | Effects | Beta (95%CI) | Pa |
| rs12589136 | 14 | 30 | 94,793,686 | T/G | 0.22 | +++++++++++ | 0.10 (0.07,0.13) | 3.3×10−12 | 0.21 | +++ | 0.12 (0.06,0.18) | 0.00024 |
| rs2749527 | 14 | 17 | 94,827,068 | T/C | 0.49 | ----------- | −0.08 (−0.11,−0.06) | 5.2×10−11 | n/a | |||
| rs2749529b | 14 | n/a | 94,820,459 | T/A | 0.47 | +++++++++++ | 0.07 (0.05,0.10) | 2.6×10−9 | 0.45 | +++ | 0.06 (0.01,0.11) | 0.019 |
| rs11621961 | 14 | 0 | 94,769,476 | T/C | 0.36 | --------?— | −0.08 (−0.10,−0.05) | 4.0×10−8 | 0.37 | --- | −0.08 (−0.14,−0.03) | 0.003 |
adjusted for age and sex.
rs2749527 was replaced with rs2749529 (r2 = 0.905, D' = 1.0) as rs2749527 failed manufacture for replication. LD patterns (from SNAP HapMap CEU build 22): rs11621961-rs12589136 (r2 = 0.131), rs11621961-rs2749529 (r2 = 0.260), rs12589136-rs2749529 (r2 = 0.291), rs11621961-rs2749527 (r2 = 0.255). Independent SNPs were defined by PLINK using the clumping function (within 500kb, LD r2 >0.2, P-value <5x10−5)
Functional consequences of genetic variation in the SERPINA6/SERPINA1 region
We explored the associations between rs12589136, rs11621961, rs2749527 with cortisol and CBG phenotypes in more detail in 316 subjects from the CROATIA-Korcula cohort (Table 2). Together these three SNPs explained 0.54% of the variance in total plasma cortisol in CROATIA-Korcula. However, there were distinct patterns of association of ‘high cortisol’ alleles with CBG. After adjusting for age and sex, although all three variants were associated with differences in total cortisol binding activity, measured by the binding of [3H]-cortisol, there were different associations with CBG immunoreactivity. The T allele at rs2749527 was associated with higher ‘total’ CBG concentration by radioimmunoassay, and there were similar, but weaker associations of total CBG immunoreactivity with variation at rs11621961. Differences in calculated free plasma cortisol reflected these differences in total CBG immunoreactivity, which is used in the calculation of free cortisol. In contrast, however, the minor (T) allele at rs12589136 was not associated with ‘total’ CBG immunoreactivity but was strongly associated with the proportion of CBG bound by a monoclonal antibody against an epitope in the reactive centre loop of CBG [23]. None of these SNPs representing signals in the SERPINA6/SERPINA1 region was associated with α1-antitrypsin concentrations in blood. However, α1-antitrypsin levels were negatively correlated with plasma total cortisol (beta −0.17 (95%CI −0.28, −0.06); P = 0.002) and calculated free cortisol (beta −0.13 (95%CI −0.24, −0.02); P = 0.021), although they did not correlate with ratio of intact/total CBG 0.02 (95%CI −0.09, 0.13; P = 0.715).
Table 2. Functional consequences of variants in the SERPINA6/A1 locus significantly associated with morning plasma cortisol in GWAMA, and of the Leuven variant, in CROATIA-Korcula.
| rs11621961 | Beta (95%CI) | P c | |||
| TT | CT | CC | |||
| Total cortisola | 655 (618,694) [129] | 664 (644,685)[411] | 675 (654,696)[357] | −0.05 (−0.14,0.05) | 0.305 |
| Calculated free cortisol | 42 (35.0,51)[56] | 47 (42,52)[126] | 50 (46,55)[136] | −0.14 (−0.28,0.00) | 0.049 |
| Measured free cortisolb | 14 (11,17)[27] | 14 (11,16)[70] | 13 (10,16)[67] | 0.01 (−0.2,0.22) | 0.933 |
| Total CBG | 0.90 (0.82,0.99)[56] | 0.90 (0.85,0.94)[126] | 0.96 (0.92,1.01)[136] | −0.12 (−0.26,0.02) | 0.103 |
| RCL/total CBG | 0.76 (0.71,0.80)[56] | 0.77 (0.74,0.79)[125] | 0.76 (0.73,0.78)[136] | 0.01 (−0.14,0.15) | 0.940 |
| Cortisol binding activity | 503 (473,532)[56] | 492 (474,511)[125] | 531 (512,550)[136] | −0.17 (−0.32,−0.03) | 0.016 |
| α1-antitrypsin | 2.74 (2.37,3.16)[56] | 2.74 (2.51,2.99)[124] | 2.66 (2.43,2.91)[134] | 0.02 (−0.14,0.17) | 0.840 |
Data for total cortisol is from the whole Croatia-Korcula sample, n = 898.
Samples selected for measured free cortisol assay were age and sex matched homozygotes at rs12589136.
adjusted for age, sex, and first three principal components, using kinship matrix derived from GWAS data.
Data are geometric mean (95% CI)[n]. Cortisol values are nmol/L, CBG µmol/L, cortisol binding activity nmol/L, and α1-antitrypsin g/L. RCL = reactive centre loop.
We investigated exome chip data for this locus in all CROATIA-Korcula participants to identify non-synonymous variants in the SERPINA6/SERPINA1 region associated with plasma cortisol. From 34 variants on the exome chip in this region (chr14:94,770,585–94,857,029, build 37), 9 were polymorphic in this sample (Table S7) but only two were associated with plasma cortisol: rs113418909 in SERPINA6 (Leu115His in CBG, previously reported as the Leuven mutation associated with low total cortisol [24]); and rs28931570 in SERPINA1 (Arg63Cys in α1-antitrypsin, not recognised as a disease-causing variant [25]). We also analysed 735 additional SNPs in the same region imputed from 1000 Genomes data in all CROATIA-Korcula participants but did not find any additional SNPs associated with plasma cortisol, using a P threshold of 0.05/735 = 7×10−5. The two rare variants rs113418909 and rs28931570 were in perfect linkage disequilibrium amongst participants with detailed biochemical phenotyping performed, so results are reported for rs113418909 only (Table 2). Prevalence of the Leuven variant in CROATIA-Korcula was higher than expected (MAF = 0.017, compared with MAF = 0.0046 in dbSNP). After adjusting for age, sex, and accounting for kinship, participants who were heterozygote for the Leuven variant had lower total cortisol and markedly lower total cortisol binding activity, but normal CBG immunoreactivity (Table 2).
After removing subjects with the rare Leuven variant (ie. heterozygotes), rs12589136 remained associated with total cortisol (0.15, 95%CI 0.04,0.26; P = 0.009), calculated free cortisol (0.17, 95%CI 0.02,0.32; P = 0.031), and the proportion of CBG bound by the reactive centre loop antibody (−0.45, 95%CI −0.60,−0.29; P = 2.8×10−8).
Discussion
These results clearly attribute inter-individual differences in morning plasma cortisol amongst Europeans to genetic variation within a region on chromosome 14 containing the SERPINA6 and SERPINA1 genes. The association of this region with plasma cortisol was consistent across multiple cohorts and was observed not only in genome-wide meta-analysis of individual SNPs, but also in gene-based hypothesis-free analysis, and in a candidate gene analysis. Investigation of the functional consequences of genetic variation in this region in a genetic isolate population in Croatia indicates that the effects of variation at SERPINA6 and SERPINA1 on plasma cortisol are likely to be mediated through alterations in total cortisol binding by corticosteroid binding globulin (CBG). In part, this is determined by differences in total CBG concentrations, and in part in association with a previously unrecognised variability in the immunoreactivity of the reactive centre loop of CBG. Since the process of CBG cleavage by neutrophil elastase and resultant reconfiguration of the reactive centre loop is considered important in the release of bioavailable cortisol within target tissues [21], [26], this finding provides a novel insight into a biological pathway controlling cortisol action.
The diverse actions of cortisol, and the striking clinical consequences of glucocorticoid excess or deficiency, have led many investigators to propose a central role for variations in cortisol levels in determining common quantitative traits. However, cortisol has not been measured as widely in epidemiological cohort studies as many other phenotypes. This may reflect the perceived difficulty of obtaining samples at a fixed time of day and in un-stressed conditions, to avoid confounding effects. The CORNET consortium had to decline samples from many cohorts in which time of sampling was inadequately controlled, and even then there was high variability in plasma cortisol. Thus, although the variants we identified in the SERPINA6/SERPINA1 region of chromosome 14 accounted for <1% of the variance in plasma cortisol, this signal may be obscured by substantial unmeasured confounding and measurement error and may comprise a considerable component of the estimated 30–60% heritability of plasma cortisol [16]–[18]. We identified 3 SNPs with partially independent effects on plasma cortisol. There may be a small degree of linkage disequilibrium between these SNPs, but they also show different associations with CBG biochemistry, suggesting that they represent independent effects. None of these 3 SNPs appears directly to affect CBG function; although rs12589136 is close to a consensus estrogen response element, there was no gender difference in its association with plasma cortisol.
Previous investigations of the genetic determinants of plasma cortisol (reviewed in [18]) have been underpowered candidate gene studies, including some which included a tandem repeat in intron 1 of SERPINA6 [27], [28]. Interestingly, many of the genetic variants previously associated with cortisol, eg for glucocorticoid [1] and mineralocorticoid [29], [30] receptors, showed no signal whatsoever in the adequately powered GWAMA and candidate gene analysis conducted here.
Rare mutations in SERPINA6 have been described which cause absent CBG protein or, more often, reduced affinity of CBG for cortisol [31]–[35]. Affected individuals have low total plasma cortisol but normal free plasma cortisol. However, they also have abnormal pulsatility of plasma cortisol, and non-specific symptoms including fatigue which are unresponsive to cortisol supplementation; features which have been attributed to abnormal function of CBG in delivering cortisol to target tissues, including in brain regions involved in negative feedback regulation of the HPA axis [21]. Although one of these mutations, A51V, has been found to be surprisingly prevalent (MAF>3%) amongst Chinese subjects [34], it has not been found in non-Asian populations and we did not find Caucasians carrying this mutation when tested by exome chip analysis. In cohort studies, plasma CBG concentrations have been associated with features of the metabolic syndrome [12], [13], [15] and one previous candidate gene study with >900 participants showed that SNPs in SERPINA6, including some identified as being associated with plasma cortisol in this GWAMA, were predictive of somatic symptoms [36]. We found evidence that genetic variation in the SERPINA6/SERPINA1 region influences total plasma cortisol not only through changes in total CBG concentrations, but also in association with alterations in the immunoreactivity of the reactive centre loop of the CBG protein.
Cleavage of the reactive centre loop (RCL) of CBG by neutrophil elastase and inhibition of elastase activity by α1-antitrypsin has been recognised for more than 20 years [26]. However, the recent development of monoclonal antibodies which recognise the intact RCL of CBG has allowed this process to be studied in vivo for the first time [23]. Using these tools in samples from Croatia-Korcula has provided the novel insight that immunoreactivity of the RCL of CBG is variable in the population, and further that this is explained in part by genetic variations in the SERPINA6/SERPINA1 region. It remains to be determined whether this difference in immunoreactivity of the RCL represents altered susceptibility to CBG cleavage. We show that a common variant (rs12589136) associated with impaired RCL antibody binding was associated with higher total plasma cortisol and higher cortisol binding activity. These observations are inconsistent with the interpretation that impaired RCL antibody binding represents enhanced RCL cleavage [23], given that cleaved CBG has a lower affinity than intact CBG for cortisol binding [37]. Alternatively, the altered immunoreactivity of the RCL epitope may represent resistance to cleavage and hence enhanced cortisol binding. It is possible that the genetically determined difference in the RCL epitope of CBG is associated with impaired negative feedback of the HPA axis due to reduced tissue delivery of cortisol by CBG, analogous with findings in CBG knockout mice [38]. Although we could not confirm associated elevation in free plasma cortisol concentrations, these measurements are notoriously unreliable, for example being similarly unhelpful in dissecting the consequences of CBG deficiency described above.
We found further evidence for the importance of CBG using exome chips in the genetic isolate population of Korcula in Croatia, where we discovered an unusually high prevalence of heterozygotes for the Leuven mutation in SERPINA6 [24]. These individuals have lower total plasma cortisol despite normal total CBG concentrations, and we confirmed substantial reductions in total cortisol binding activity, without any difference in RCL antibody binding. The presence of the Leuven variant, however, did not account for the association of the top hit SNPs identified by GWAMA with plasma cortisol or CBG RCL antibody binding.
It is possible that a combination of alterations in CBG substrate as well as in neutrophil elastase level and/or activity may determine cleavage of CBG and tissue delivery of cortisol, especially in local sites of inflammation [21]. Intriguingly, we found inverse associations between levels of α1-antitrypsin, the inhibitor of neutrophil elastase, and plasma cortisol concentrations, consistent with instability of CBG resulting in HPA axis activation as proposed above; however, we could not identify a genetic influence on this relationship, or confirm its association with CBG RCL immunoreactivity. Specifically, we did not identify independent signals for SERPINA6 and SERPINA1 in conditional analysis, and the rare variant Leuven mutation was in linkage disequilibrium with the only rare variant we identified in SERPINA1. Recent studies have identified variants in SERPINA1 that are associated with coronary artery calcification [39] and serum lipid profile [40], the latter represented by rs1303 which is in linkage disequilibrium with the top hit rs12589136 identified by GWAMA (r2 = 0.35). These findings are consistent with variation in the SERPINA6/SERPINA1 locus affecting downstream actions of cortisol, but it remains unclear if an interaction exists between the variants at these two genes. Mutations in SERPINA1 cause the syndrome of α1-antitrypsin deficiency, but we are not aware of any investigations of CBG or cortisol in these patients, and their HPA axis may be disturbed anyway by un-restrained neutrophil-mediated tissue damage. Rare variants in SERPINA1 (notably rs112635299) have been associated with α1-globulin plasma protein levels, of which α1-antitrypsin is a major constituent, using GWAS with 1000 Genomes imputation [41]. However, in the CROATIA-Korcula cohort neither rs1303 (Table S7) nor SNPs imputed from 1000 Genomes (including rs112635299) were associated with plasma cortisol. More detailed phenotyping amongst participants with contrasting genotypes at the SERPINA6/SERPINA1 region will be required to clarify the basis for altered interaction between the two gene products.
These findings emphasise the biological importance of plasma protein binding for steroid hormones, and are analogous to recent findings that a common variant in sex hormone binding globulin contributes to variation in total testosterone levels [42]. Given the consequences of altered binding protein function for steroid volume of distribution and clearance, and documented effects on HPA axis function [21], this is an important finding of itself. However, potentially of greater importance is the novel observation that a key protein domain of CBG, the reactive centre loop, is subject to inter-individual differences which are influenced by genetic variation and may constitute a novel influence on tissue steroid action.
Materials and Methods
Gene discovery
We performed a meta-analysis of genome-wide association studies of morning plasma cortisol in 12,597 subjects from 11 western European population-based cohorts: CROATIA-Vis (n = 885), CROATIA-Korcula (n = 898), CROATIA-Split (n = 493), ORCADES (n = 886), Rotterdam Study (n = 2945), NFBC1966 (n = 1195), Helsinki Birth Cohort Study 1934–44 (n = 451), ALSPAC (n = 1567), InChianti (n = 1207), PREVEND (n = 1151), and PIVUS (n = 919). Replication was tested in 2,795 subjects from three independent cohorts: Raine Study (n = 797), ET2DS (n = 1,069), and MrOS-Sweden (n = 929). Cortisol was measured by immunoassay in blood samples collected from study participants between 0700 and 1100 h. Inclusion criteria were adults aged 17 years or older from Caucasian populations; exclusion criteria were current glucocorticoid use, pregnant or breast feeding women, and twins (exclusion of one). Characteristics of the study populations are presented in Table S1 and details of each cohort are provided in Text S1. All participants provided written informed consent and studies were approved by local Research Ethics Committees and/or Institutional Review Boards.
Association analysis with morning plasma cortisol
Each study performed single marker association tests, and study-specific linear regression models which used z-scores of log-transformed cortisol, additive SNP effects, and were adjusted for age and sex (model 1); age, sex, and smoking (model 2); or age, sex, smoking and body mass index (model 3). Imputation of the gene-chip results used the HapMap CEU population, build 36. In cohorts with consanguineous populations (ORCADES and Croatia), adjustments for principal components in kinship matrices were performed using ProbABEL; for other cohorts, Identity-By-Descent coefficients were calculated using PLINK and related participants excluded. In the majority of cohorts, participants were only included if blood samples had been obtained within a 60 minute time interval, when variations in time of sampling were ignored. In a subset of cohorts, samples were obtained over a wider time interval (but always in the morning before 1100 h) and time of blood sampling recorded; for these cohorts, three further models were run as above but also including time of sampling, calculated as minutes from first sampling time, as an additional covariate [43].
Quality control was carried out on the imputed genome-wide data for all 11 studies prior to meta-analysis; this excluded all samples with a minor allele frequency (MAF) <2%, call rate <95%, Hardy-Weinberg equilibrium (HWE)<1×10−8 and poor imputation quality (MACH R2_HAT<0.30, IMPUTE PROPER_INFO<0.60, BEAGLE INFO<0.30, as appropriate). Quantile-quantile (QQ) plots and genomic control (lambda) were used to confirm quality control. Sex chromosomes were not analysed.
Meta-analysis of association results
We performed fixed effects meta-analysis, which used combined allelic effects weighted by the inverse of their variance for each of the models using the GWAMA program [44]. This aligned all studies to the same reference allele at each SNP, thus avoiding strand errors, and excluded SNPs with obvious input errors (eg. discrepancies in effect allele frequencies). The results from analysis with or without genomic control were nearly identical, as expected with λGC = 1.005. The genome-wide significance threshold for the meta-analysis was P<5×10−8. Percentage variation of cortisol was calculated from meta-analysis results as (2*effect allele frequency)*(1-effect allele frequency)*(beta2/sd2). A regional plot was generated using LocusZoom [45], and heat map using snp.plotter [46] in R version 2.15.2. Joint analysis of meta-analysis was performed with the GCTA program [47].
Clumping analysis
To detect independent top SNPs on the basis of empirical estimates of linkage disequilibrium between the SNPs, we used the clumping function as implemented in PLINK [20]. All the SNPs with a P-value<5×10−5 in meta-analysis were used for clumping. We grouped the SNPs within 500 kb of the index SNP that have r2 >0.2 with the index SNP.
Replication genotyping and analysis
Genes identified in the meta-analysis were evaluated in the Raine Study, MrOS-Sweden, and Edinburgh type 2 Diabetes Study (ET2DS). Raine Study and MrOS-Sweden had GWAS data so we extracted the replication SNP results, and ET2DS was genotyped at the Wellcome Trust Clinical Research Facility Genetics Core Laboratory in Edinburgh using the OpenArray genotyping platform. rs2749527 failed manufacture for the OpenArray, and was replaced with rs2749529 (r2 = 0.905, D′ = 1.0) in all replication cohorts. Genotypic association analysis in these studies followed the same methods as those described above for the discovery meta-analysis, adjusting for age and sex.
Conditional & sex-specific analysis
We performed meta-analyses of sex-specific GWAS and conditional GWAS in a subset of populations, using single marker association tests of z-scores of log-transformed cortisol. For the sex-specific analysis, each study adjusted for age in both men (n = 3,546) and women (n = 5,956). For the conditional analysis, each study adjusted for age, sex, and each of the SERPINA6 loci SNPs rs12589136 (n = 9,308), rs11621961 (n = 7,687), and rs2749527 (n = 9,307) individually. We then did fixed effects meta-analysis using GWAMA program.
Gene-based analysis
We used the Versatile Gene-based Association Study (VEGAS) program [22] to perform gene-centric analysis. This used individual SNP p-values derived from the meta-analysis results to compute a gene-based p-value. We used two methods: all SNPs within a gene, or a subset of the 10% most significant SNPs in each gene boundary. VEGAS accounted for linkage disequilibrium between SNPs using the HapMap phase 2 population (CEU). SNPs were assigned to ∼18,000 genes based on positions in build 36 (hg18), with gene boundaries of ±50 kb of the UTR. Bonferroni corrected threshold for gene-wide significance was 3×10−6. The overlap of SNPs included in the gene boundaries in our results indicates this is likely to be an overly conservative correction factor [22].
A list of 61 candidate genes with known biological function in the regulation of cortisol was compiled by a panel of experts in the field. All SNPs within and ±50 kb of these genes were examined in the GWAMA results, and gene-based p values were inspected in VEGAS results.
Exome chip and 1000 Genomes imputed data analysis
Genotypes for the SERPINA6/SERPINA1 gene region (chr14:94,770,585–94,857,029, build 37) in the CROATIA-Korcula samples (n = 898) were extracted from an Illumina Exome Chip v1 analysis. Genotypes were called in GenomeStudio (Illumina) using the CHARGE Consortium joint calling cluster file (http://www.chargeconsortium.com/main/exomechip) [48]. 1000 Genomes imputation was performed using genotypes from Illumina HumanHap370CNV after quality control (Individual Call Rate 97%, SNP Call Rate 98%, MAF 0.01, HWE 1×10−6); prephasing was performed using ShapeIt v2 [49] and imputation using IMPUTE2 [50] and the ALL (Phase 1 integrated release v3, April 2012) reference panel. Associations with plasma cortisol were analysed in GenABEL [51].
Detailed biochemical studies
More detailed phenotyping was undertaken in samples from 316 participants in the CROATIA-Korcula cohort, comprising 158 age- and sex-matched homozygotes at the top hit variant rs12589136 (53 T/T, 106 G/G; however, one T/T sample had insufficient sample for CBG measurement resulting in 52 T/T and 106 G/G). An additional 160 randomly selected samples had CBG measured, however two samples failed genotyping resulting in an additional 47 T/G and 111 G/G.
Total plasma cortisol was measured with a commercial radioimmunoassay (MP Biomedicals, UK). Total CBG was also measured in CROATIA-Korcula samples using a commercial radioimmunoassay (DiaSource, Louvain-la-Neuve, Belgium). Total cortisol binding capacity was measured using a ligand-saturation assay, with [3H]-cortisol (PerkinElmer Life Sciences, Waltham, MA) as the labelled ligand and dextran-coated charcoal to separate the CBG-bound [3H]cortisol, as previously described [52]. α1-Antitrypsin was measured with a commercial ELISA (Genway Biotech, San Diego, USA). Unbound free plasma cortisol was calculated from measured total plasma cortisol and total CBG values using Coolens' equation [53]. Free cortisol was also measured by ELISA following equilibrium dialysis. Briefly, dialysis tubing (12–14 kD, Medicell, London, UK) was heated to 80C for 30 min in 2% Na bicarbonate and 1 mM EDTA before overnight dialysis of plasma into phosphate buffered saline containing 1% gelatin at 37C and measurement of dialysed free cortisol by commercial ELISA (Salimetrics Europe Ltd, Newmarket, UK). CBG were also measured, as previously described [23], by ELISAs using monoclonal antibodies 12G2 and 9G12. Antibody 9G12 binds to an epitope in the reactive centre loop (RCL), the elastase cleavage site on CBG, and has been used to infer intact (uncleaved) CBG, whereas antibody 12G2 binds to a distant epitope and measures total (cleaved and uncleaved) CBG.
As CROATIA-Korcula is a population isolate, we used the polygenic and mmscore functions in GenABEL [51]. All regression equations included the first three principal components and kinship matrix derived from GWAS data in this population and used z-scores of the traits. All variables were normalised using log transformation (cortisol, calculated free cortisol, measured free cortisol, CBG, α1-antitrypsin), and reported means are geometric means, the ratio of intact/cleaved was normally distributed.
Supporting Information
Characteristics of participants in cohorts included in genome-wide association meta-analysis and replication for morning plasma cortisol.
(DOCX)
Genome-wide data characteristics for discovery cohorts used in meta-analysis.
(DOCX)
All SNPs with p-values <5×10−5 in discovery genome wide association meta-analysis for morning plasma cortisol.
(DOCX)
Top 10 genes identified in VEGAS as associated with morning plasma cortisol from genome wide association meta-analysis with adjustment for age and sex.
(DOCX)
VEGAS results for candidate genes based on genome wide association meta-analysis results adjusted for age and sex.
(DOCX)
Replication results in individual cohorts and by meta-analysis for association with morning plasma cortisol of SNPs representing independent signals in the SERPINA6/SERPINA1 region discovered in genome-wide association meta-analysis.
(DOCX)
Associations with plasma cortisol of variants in SERPINA6/A1 locus on chromosome 14 identified by exome chip in n = 808 subjects from CROATIA-Korcula.
(DOCX)
Supporting methods.
(DOCX)
Acknowledgments
We thank Veronique Vitart for her critical review of the manuscript.
CORNET Consortium
We are grateful to Jill Harrison, Lynne Ramage and Carolynn Cairns for excellent technical assistance. We thank Karen Chapman, Megan Holmes and Jonathan Seckl for assistance in compiling a list of candidate genes.
Croatia studies
We acknowledge the staff of several institutions in Croatia that supported the field work, including but not limited to The University of Split and Zagreb Medical Schools, the Institute for Anthropological Research in Zagreb, the Croatian Institute for Public Health and the recruitment teams in Korcula and Split, the administrative teams in Croatia and Edinburgh and the people of Vis, Korcula and Split. The SNP genotyping for the CROATIA-Vis cohort was performed in the core genotyping laboratory of the Wellcome Trust Clinical Research Facility, Edinburgh, UK. The SNP genotyping for the CROATIA-Korcula cohort was performed in Helmholtz Zentrum München, Neuherberg, Germany. The SNP genotyping for the CROATIA-Split cohort was performed by AROS Applied Biotechnology, Aarhus, Denmark.
ORCADES study
ORCADES was supported by the staff of the MRC Human Genetics Unit. DNA extractions were performed at the Wellcome Trust Clinical Research Facility in Edinburgh. We acknowledge the invaluable contributions of Lorraine Anderson and the research nurses in Orkney, the administrative team in Edinburgh and the people of Orkney.
Rotterdam Study
We are grateful to the study participants, the staff of the Rotterdam Study, and the participating general practitioners and pharmacists. We thank Pascal Arp, Mila Jhamai, Marijn Verkerk, Lizbeth Herrera, and Marjolein Peters for their help in creating the genome-wide association study database, and Dr. Karol Estrada and Maksim V. Struchalin for their support in creation and analysis of imputed data. We thank Dr. Karol Estrada, Dr. Fernando Rivadeneira, Dr. Tobias A. Knoch, Anis Abuseiris, Luc V. de Zeeuw, and Rob de Graaf (Erasmus MC Rotterdam, The Netherlands) for their help in creating GRIMP and BigGRID, MediGRID, and Services@MediGRID/D-Grid (funded by the German Bundesministerium für Forschung und Technology; Grants 01 AK 803 A-H, 01 IG 07015 G) for access to their grid computing resources.
HBCS
We thank all study participants and everybody involved in the Helsinki Birth Cohort Study.
Northern Finland Birth Cohort 1966 (NFBC1966)
We are grateful to all investigators and participants involved in the NFBCS1966.
ALSPAC
We are grateful to all the families who took part, the midwives for their help in recruiting them, and the whole ALSPAC team, which includes interviewers, computer and laboratory technicians, clerical workers, research scientists, volunteers, managers, receptionists and nurses.
InCHIANTI
We are grateful to all investigators and participants involved in the InCHIANTI study.
PIVUS
We are grateful to all investigators and participants involved in the PIVUS study.
PREVEND
We are grateful to all investigators and participants involved in the PREVEND study.
Edinburgh Type 2 Diabetes Study
Data collection and SNP genotyping was performed in the Wellcome Trust Clinical Research Facility, Edinburgh, UK. We are grateful to all ET2DS participants, staff, investigators and collaborators.
Raine Study
We gratefully acknowledge the Raine Study participants and their families and the Raine Study team for cohort co-ordination and data collection.
MrOS-Sweden Study
We are grateful to all investigators and participants involved in the MrOS-Sweden study.
Funding Statement
The CORtisol NETwork Consortium is funded by the Chief Scientist Office of the Scottish Government (grant CZB-4-733) and the British Heart Foundation (grant RG11/4/28734). The CROATIA-Vis, CROATIA-Korcula and CROATIA-Split studies were funded by grants from the Medical Research Council (UK), the Republic of Croatia Ministry of Science, Education and Sports research grants to IR (108-1080315-0302), and European Commission Framework 6 project EUROSPAN (Contract No. LSHG-CT-2006-018947). ORCADES was supported by the Chief Scientist Office of the Scottish Government, the Royal Society, the MRC Human Genetics Unit, Arthritis Research UK and the European Union framework program 6 EUROSPAN project (contract no. LSHG-CT-2006-018947). In the Rotterdam Study, the generation and management of genome-wide association study genotype data are supported by the Netherlands Organisation of Scientific Research Investments (number 175.010.2005.011, 911-03-012). This study is funded by the Research Institute for Diseases in the Elderly (014-93-015) and the Netherlands Genomics Initiative/Netherlands Organisation for Scientific Research project number 050-060-810. The Rotterdam Study is funded by Erasmus MC and Erasmus University, Rotterdam, the Netherlands Organization for the Health Research and Development, the Ministry of Education, Culture and Science, the Ministry for Health, Welfare and Sports, the European Commission (Directorate-General XII), and the Municipality of Rotterdam. HT was supported by the Vidi Grant of Netherlands Organization for the Health Research and Development (2009-017.106.370). ND was supported by the Netherlands Consortium for Healthy Ageing. The Helsinki Birth Cohort Study has been supported by grants from the Academy of Finland, the Finnish Diabetes Research Society, Folkhälsan Research Foundation, Novo Nordisk Foundation, Finska Läkaresällskapet, Signe and Ane Gyllenberg Foundation, University of Helsinki, European Science Foundation (EUROSTRESS), Ministry of Education, Ahokas Foundation, Emil Aaltonen Foundation, Juho Vainio Foundation, and Wellcome Trust (grant number WT089062). The Northern Finland Birth Cohort 1966 study is supported by the Academy of Finland [project grants 104781, 120315, 129418, Center of Excellence in Complex Disease Genetics and Public Health Challenges Research Program (SALVE)], University Hospital Oulu, Biocenter, University of Oulu, Finland (75617), the European Commission [EUROBLCS, Framework 5 award QLG1-CT-2000-01643], The National Heart, Lung and Blood Institute [5R01HL087679-02] through the SNP Typing for Association with Multiple Phenotypes from Existing Epidemiologic Data (STAMPEED) program [1RL1MH083268-01], The National Institute of Health/The National Institute of Mental Health [5R01MH63706:02], European Network of Genomic and Genetic Epidemiology (ENGAGE) project and grant agreement [HEALTH-F4-2007-201413], and the Medical Research Council, UK [G0500539, G0600705, PrevMetSyn/Public Health Challenges Research Program (SALVE)]. ALSPAC was funded by the UK Medical Research Council and the Wellcome Trust (Grant ref: 092731) and the University of Bristol. The InCHIANTI study has been supported by the Italian Ministry of Health and the U.S. National Institute on Aging. The PIVUS study was supported by Wellcome Trust Grants WT098017, WT064890, WT090532, Uppsala University, Uppsala University Hospital, the Swedish Research Council and the Swedish Heart-Lung Foundation. The PREVEND Study has been supported by the Dutch Kidney Foundation (Grant E.033), the Groningen University Medical Center (Beleidsruimte), Bristol Myers Squibb, Dade Behring, Ausam, Roche, Abbott, The Netherlands Organization of Scientific Research, The Dutch Heart Foundation, and the De Cock Foundation. The Edinburgh Type 2 Diabetes Study was funded by the UK Medical Research Council (G0500877). For the Raine Study, this work was supported by funding for the 17 year follow-up and genotyping provided by the National Health and Medical Research Council of Australia (353514, 572613, 403981) and the Canadian Institutes of Health Research (MOP82893). Core funding for the Raine Study is provided by the University of Western Australia (UWA), Raine Medical Research Foundation, the Telethon Institute for Child Health Research, UWA Faculty of Medicine, Dentistry and Health Sciences, the Women and Infants Research Foundation and Curtin University. The MrOS-Sweden study was supported by the Swedish Research Council, the Swedish Foundation for Strategic Research, European Commission Grant QLK4-CT-2002-02528, the ALF/LUA research grant in Gothenburg, the Lundberg Foundation, the Torsten and Ragnar Söderber's Foundation, Petrus and Augusta Hedlunds Foundation, and the Novo Nordisk Foundation. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Walker BR (2007) Glucocorticoids and cardiovascular disease. Eur J Endocrinol 157: 545–559. [DOI] [PubMed] [Google Scholar]
- 2. Phillips DIW, Barker DJP, Fall CHD, Whorwood CB, Seckl JR, et al. (1998) Elevated plasma cortisol concentrations: an explanation for the relationship between low birthweight and adult cardiovascular risk factors. J Clin Endocrinol Metab 83: 757–760. [DOI] [PubMed] [Google Scholar]
- 3. Filipovsky J, Ducimetiere P, Eschwege E, Richard JL, Rosselin G, et al. (1996) The relationship of blood pressure with glucose, insulin, heart rate, free fatty acids and plasma cortisol levels according to degree of obesity in middle-aged men. J Hypertens 14: 229–235. [DOI] [PubMed] [Google Scholar]
- 4. Fraser R, Ingram MC, Anderson NH, Morrison C, Davies E, et al. (1999) Cortisol effects on body mass, blood pressure, and cholesterol in the general population. Hypertension 33: 1364–1368. [DOI] [PubMed] [Google Scholar]
- 5. Reynolds RM, Walker BR, Phillips DIW, Sydall HE, Andrew R, et al. (2001) Altered control of cortisol secretion in adult men with low birthweight and cardiovascular risk factors. J Clin Endocrinol Metab 86: 245–250. [DOI] [PubMed] [Google Scholar]
- 6. Lupien SJ, de Leon M, de Santi S, Convit A, Tarshish C, et al. (1998) Cortisol levels during human aging predict hippocampal atrophy and memory deficits. Nature Neuroscience 1: 69–73. [DOI] [PubMed] [Google Scholar]
- 7. Holsboer F (2001) Stress, hypercortisolism and corticosteroid receptors in depression: implications for therapy. J Affect Disord 62: 77–91. [DOI] [PubMed] [Google Scholar]
- 8. Ball TM (2006) Cortisol circadian rhythms and stress responses in infants at risk of allergic disease. Neuroimmunomodulation 13: 294–300. [DOI] [PubMed] [Google Scholar]
- 9. Seckl JR, Meaney MJ (2006) Glucocorticoid “programming” and PTSD risk. Ann N Y Acad Sci 1071: 351–378. [DOI] [PubMed] [Google Scholar]
- 10. Bjorntorp P, Holm G, Rosmond R (1999) Hypothalamic arousal, insulin resistance and type 2 diabetes mellitus. Diabetic Med 16: 373–381. [DOI] [PubMed] [Google Scholar]
- 11. Mattsson C, Reynolds RM, Simonyte K, Olsson T, Walker BR (2009) Combined receptor antagonist stimulation of the hypothalamic-pituitary-adrenal axis test identifies impaired negative feedback sensitivity to cortisol in obese men. J Clin Endocrinol Metab 94: 1347–1352. [DOI] [PubMed] [Google Scholar]
- 12. Fernandez-Real JM, Grasa M, Casamitjana R, Ricart W (2000) The insulin resistance syndrome and the binding capacity of cortisol binding globulin (CBG) in men and women. Clin Endocrinol (Oxf) 52: 93–99. [DOI] [PubMed] [Google Scholar]
- 13. Fernandez-Real JM, Pugeat M, Grasa M, Broch M, Vendrell J, et al. (2002) Serum corticosteroid-binding globulin concentration and insulin resistance syndrome: a population study. J Clin Endocrinol Metab 87: 4686–4690. [DOI] [PubMed] [Google Scholar]
- 14. Reynolds RM, Walker BR, Syddall HE, Andrew R, Wood PJ, et al. (2005) Is there a gender differences in the associations of low birthweight with adult hypothalamic-pituitary-adrenal axis activity? European Journal of Endocrinology 152: 249–253. [DOI] [PubMed] [Google Scholar]
- 15. Lewis JG, Borowski KK, Shand BI, George PM, Scott RS (2010) Plasma sex hormone-binding globulin, corticosteroid-binding globulin, cortisol, and free cortisol levels in outpatients attending a lipid disorders clinic: a cross-sectional study of 1137 subjects. Horm Metab Res 42: 274–279. [DOI] [PubMed] [Google Scholar]
- 16. Inglis GC, Ingram MC, Holloway CD, Swan L, Birnie D, et al. (1999) Familial pattern of corticosteroids and their metabolism in adult human subjects - The Scottish adult twin study. J Clin Endocrinol Metab 84: 4132–4137. [DOI] [PubMed] [Google Scholar]
- 17. Bartels M, de Geus EJC, Kirschbaum C, Sluyter F, Boomsma DI (2003) Heritability of daytime cortisol levels in children. Behavior Genetics 33: 421–433. [DOI] [PubMed] [Google Scholar]
- 18. Mormede P, Foury A, Barat P, Corcuff JB, Terenina E, et al. (2011) Molecular genetics of hypothalamic-pituitary-adrenal axis activity and function. Ann N Y Acad Sci 1220: 127–136. [DOI] [PubMed] [Google Scholar]
- 19. Smith GD, Ebrahim S (2003) ‘Mendelian randomization’: can genetic epidemiology contribute to understanding environmental determinants of disease? Int J Epidemiol 32: 1–22. [DOI] [PubMed] [Google Scholar]
- 20. Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, et al. (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81: 559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Henley DE, Lightman SL (2011) New insights into corticosteroid-binding globulin and glucocorticoid delivery. Neuroscience 180: 1–8. [DOI] [PubMed] [Google Scholar]
- 22. Liu JZ, McRae AF, Nyholt DR, Medland SE, Wray NR, et al. (2010) A versatile gene-based test for genome-wide association studies. Am J Hum Genet 87: 139–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lewis JG, Elder PA (2011) Corticosteroid-binding globulin reactive centre loop antibodies recognise only the intact natured protein: elastase cleaved and uncleaved CBG may coexist in circulation. J Steroid Biochem Mol Biol 127: 289–294. [DOI] [PubMed] [Google Scholar]
- 24. Van Baelen H, Brepoels R, De Moor P (1982) Transcortin Leuven: a variant of human corticosteroid-binding globulin with decreased cortisol-binding affinity. J Biol Chem 257: 3397–3400. [PubMed] [Google Scholar]
- 25. Ekeowa UI, Gooptu B, Belorgey D, Hagglof P, Karlsson-Li S, et al. (2009) alpha1-Antitrypsin deficiency, chronic obstructive pulmonary disease and the serpinopathies. Clin Sci (Lond) 116: 837–850. [DOI] [PubMed] [Google Scholar]
- 26. Hammond GL, Smith CL, Paterson NAM, Sibbald WJ (1990) A role for corticosteroid-binding globulin in delivery of cortisol to activated neutrophils. J Clin Endocrinol Metab 71: 34–39. [DOI] [PubMed] [Google Scholar]
- 27. Barat P, Corcuff JB, Tauber M, Moisan MP (2012) Associations of glucocorticoid receptor and corticosteroid-binding globulin gene polymorphisms on fat mass and fat mass distribution in prepubertal obese children. J Physiol Biochem [DOI] [PubMed] [Google Scholar]
- 28. Barat P, Duclos M, Gatta B, Roger P, Mormede P, et al. (2005) Corticosteroid binding globulin gene polymorphism influences cortisol driven fat distribution in obese women. Obes Res 13: 1485–1490. [DOI] [PubMed] [Google Scholar]
- 29. van Leeuwen N, Kumsta R, Entringer S, de Kloet ER, Zitman FG, et al. (2010) Functional mineralocorticoid receptor (MR) gene variation influences the cortisol awakening response after dexamethasone. Psychoneuroendocrinology 35: 339–349. [DOI] [PubMed] [Google Scholar]
- 30. DeRijk RH, Wust S, Meijer OC, Zennaro MC, Federenko IS, et al. (2006) A common polymorphism in the mineralocorticoid receptor modulates stress responsiveness. J Clin Endocrinol Metab 91: 5083–5089. [DOI] [PubMed] [Google Scholar]
- 31. Cizza G, Rother KI (2012) Cortisol binding globulin: more than just a carrier? J Clin Endocrinol Metab 97: 77–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hill LA, Vassiliadi DA, Simard M, Pavlaki A, Perogamvros I, et al. (2012) Two different corticosteroid-binding globulin variants that lack cortisol-binding activity in a greek woman. J Clin Endocrinol Metab 97: 4260–4267. [DOI] [PubMed] [Google Scholar]
- 33. Perogamvros I, Underhill C, Henley DE, Hadfield KD, Newman WG, et al. (2010) Novel corticosteroid-binding globulin variant that lacks steroid binding activity. J Clin Endocrinol Metab 95: E142–E150. [DOI] [PubMed] [Google Scholar]
- 34. Lin HY, Underhill C, Lei JH, Helander-Claesson A, Lee HY, et al. (2012) High frequency of SERPINA6 polymorphisms that reduce plasma corticosteroid-binding globulin activity in Chinese subjects. J Clin Endocrinol Metab 97: E678–E686. [DOI] [PubMed] [Google Scholar]
- 35. Hill LA, Vassiliadi DA, Simard M, Pavlaki A, Perogamvros I, et al. (2012) Two different corticosteroid-binding globulin variants that lack cortisol-binding activity in a greek woman. J Clin Endocrinol Metab 97: 4260–4267. [DOI] [PubMed] [Google Scholar]
- 36. Holliday KL, Macfarlane GJ, Nicholl BI, Creed F, Thomson W, et al. (2010) Genetic variation in neuroendocrine genes associates with somatic symptoms in the general population: results from the EPIFUND study. J Psychosom Res 68: 469–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lin HY, Underhill C, Gardill BR, Muller YA, Hammond GL (2009) Residues in the human corticosteroid-binding globulin reactive center loop that influence steroid binding before and after elastase cleavage. J Biol Chem 284: 884–896. [DOI] [PubMed] [Google Scholar]
- 38. Petersen HH, Andreassen TK, Breiderhoff T, Brasen JH, Schulz H, et al. (2006) Hyporesponsiveness to glucocorticoids in mice genetically deficient for the corticosteroid binding globulin. Mol Cell Biol 26: 7236–7245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Miller VM, Petterson TM, Jeavons EN, Lnu AS, Rider DN, et al. (2013) Genetic polymorphisms associated with carotid artery intima-media thickness and coronary artery calcification in women of the Kronos Early Estrogen Prevention Study. Physiol Genomics 45: 79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Inouye M, Ripatti S, Kettunen J, Lyytikainen LP, Oksala N, et al. (2012) Novel Loci for metabolic networks and multi-tissue expression studies reveal genes for atherosclerosis. PLoS Genet 8: e1002907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wood AR, Perry JR, Tanaka T, Hernandez DG, Zheng HF, et al. (2013) Imputation of variants from the 1000 Genomes Project modestly improves known associations and can identify low-frequency variant-phenotype associations undetected by HapMap based imputation. PLoS One 8: e64343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ohlsson C, Wallaschofski H, Lunetta KL, Stolk L, Perry JR, et al. (2011) Genetic determinants of serum testosterone concentrations in men. PLoS Genet 7: e1002313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Reynolds RM, Fischbacher C, Bhopal R, Byrne CD, White M, et al. (2006) Differences in cortisol concentrations in South Asian and European men living in the United Kingdom. Clin Endocrinol (Oxf) 64: 530–534. [DOI] [PubMed] [Google Scholar]
- 44. Magi R, Morris AP (2010) GWAMA: software for genome-wide association meta-analysis. BMC Bioinformatics 11: 288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pruim RJ, Welch RP, Sanna S, Teslovich TM, Chines PS, et al. (2010) LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics 26: 2336–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Luna A, Nicodemus KK (2007) snp.plotter: an R-based SNP/haplotype association and linkage disequilibrium plotting package. Bioinformatics 23: 774–776. [DOI] [PubMed] [Google Scholar]
- 47. Yang J, Lee SH, Goddard ME, Visscher PM (2011) GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet 88: 76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Grove ML, Yu B, Cochran BJ, Haritunians T, Bis JC, et al. (2013) Best practices and joint calling of the HumanExome BeadChip: the CHARGE Consortium. PLoS One 8: e68095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Delaneau O, Zagury JF, Marchini J (2013) Improved whole-chromosome phasing for disease and population genetic studies. Nat Methods 10: 5–6. [DOI] [PubMed] [Google Scholar]
- 50. Howie BN, Donnelly P, Marchini J (2009) A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet 5: e1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Aulchenko YS, Ripke S, Isaacs A, van Duijn CM (2007) GenABEL: an R library for genome-wide association analysis. Bioinformatics 23: 1294–1296. [DOI] [PubMed] [Google Scholar]
- 52. Hammond GL, Lahteenmaki PL (1983) A versatile method for the determination of serum cortisol binding globulin and sex hormone binding globulin binding capacities. Clin Chim Acta 132: 101–110. [DOI] [PubMed] [Google Scholar]
- 53. Coolens JL, Van BH, Heyns W (1987) Clinical use of unbound plasma cortisol as calculated from total cortisol and corticosteroid-binding globulin. J Steroid Biochem 26: 197–202. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Characteristics of participants in cohorts included in genome-wide association meta-analysis and replication for morning plasma cortisol.
(DOCX)
Genome-wide data characteristics for discovery cohorts used in meta-analysis.
(DOCX)
All SNPs with p-values <5×10−5 in discovery genome wide association meta-analysis for morning plasma cortisol.
(DOCX)
Top 10 genes identified in VEGAS as associated with morning plasma cortisol from genome wide association meta-analysis with adjustment for age and sex.
(DOCX)
VEGAS results for candidate genes based on genome wide association meta-analysis results adjusted for age and sex.
(DOCX)
Replication results in individual cohorts and by meta-analysis for association with morning plasma cortisol of SNPs representing independent signals in the SERPINA6/SERPINA1 region discovered in genome-wide association meta-analysis.
(DOCX)
Associations with plasma cortisol of variants in SERPINA6/A1 locus on chromosome 14 identified by exome chip in n = 808 subjects from CROATIA-Korcula.
(DOCX)
Supporting methods.
(DOCX)