

Abstract
Seeds of flowering plants can be formed sexually or asexually through apomixis. Apomixis occurs in about 400 species and is of great interest for agriculture as it produces clonal offspring. It differs from sexual reproduction in three major aspects: (1) While the sexual megaspore mother cell (MMC) undergoes meiosis, the apomictic initial cell (AIC) omits or aborts meiosis (apomeiosis); (2) the unreduced egg cell of apomicts forms an embryo without fertilization (parthenogenesis); and (3) the formation of functional endosperm requires specific developmental adaptations. Currently, our knowledge about the gene regulatory programs underlying apomixis is scarce. We used the apomict Boechera gunnisoniana, a close relative of Arabidopsis thaliana, to investigate the transcriptional basis underlying apomeiosis and parthenogenesis. Here, we present the first comprehensive reference transcriptome for reproductive development in an apomict. To compare sexual and apomictic development at the cellular level, we used laser-assisted microdissection combined with microarray and RNA-Seq analyses. Conservation of enriched gene ontologies between the AIC and the MMC likely reflects functions of importance to germline initiation, illustrating the close developmental relationship of sexuality and apomixis. However, several regulatory pathways differ between sexual and apomictic germlines, including cell cycle control, hormonal pathways, epigenetic and transcriptional regulation. Enrichment of specific signal transduction pathways are a feature of the apomictic germline, as is spermidine metabolism, which is associated with somatic embryogenesis in various plants. Our study provides a comprehensive reference dataset for apomictic development and yields important new insights into the transcriptional basis underlying apomixis in relation to sexual reproduction.
Author Summary
In flowering plants, asexual reproduction through seeds (apomixis) likely evolved from sexual ancestors several times independently. Only three key developmental steps differ between sexual reproduction and apomixis. In contrast to sexual reproduction, in apomicts the first cell of the female reproductive lineage omits or aborts meiosis (apomeiosis) to initiate gamete formation. Subsequently, the egg cell develops into an embryo without fertilization (parthenogenesis), and endosperm formation can either be autonomous or depend on fertilization. Consequently, the offspring of apomicts is genetically identical to the mother plant. The production of clonal seeds bears great promise for agricultural applications. However, the targeted manipulation of reproductive pathways for seed production has proven difficult as knowledge about the underlying gene regulatory processes is limited. We performed cell type-specific transcriptome analyses to study apomictic germline development in Boechera gunnisoniana, an apomictic species closely related to Arabidopsis thaliana. To facilitate these analyses, we first characterized a floral reference transcriptome. In comparison, we identified several regulatory pathways, including core cell cycle regulation, protein degradation, transcription factor activity, and hormonal pathways to be differentially regulated between sexual and apomictic plants. Apart from new insights into the underlying transcriptional networks, our dataset provides a valuable starting point for functional investigations.
Introduction
In flowering plants, both sexual and asexual reproduction through seeds (apomixis) is common. Apomixis occurs in more than 400 plant species belonging to over 40 families, but it is poorly represented in crop species. Apomixis leads to clonal offspring by conservation of the maternal genotype through the absence of meiosis and fertilization [1]–[4]. Engineering of apomixis in crop species is perceived as one of the greatest challenges faced by modern agriculture [5]. However, achieving this goal proved to be difficult, particularly as the knowledge about the genetic basis and regulatory programs underlying apomictic reproduction is very limited.
Sexual reproduction and apomixis only differ in a number of key developmental steps [6], [7]. During sexual reproduction, the female and male reproductive lineages are initiated by spore formation from a spore mother cell during megasporogenesis and microsporogenesis, respectively. The megaspore mother cell (MMC) is the first cell of the female germline. It is specified by selection of one subepidermal, somatic (sporophytic) cell within an ovule, the precursor of the seed. The MMC undergoes meiosis and gives rise to a tetrad of reduced megaspores. Typically, only one of those - the functional megaspore (FMS) - survives to form the female gametophyte (embryo sac). The FMS divides mitotically and subsequently cellularizes to form the mature female gametophyte harbouring the gametes (egg cell and central cell) and several accessory cells, including the synergids that play an important role in fertilization [8]. Double fertilization of the egg and central cell with one sperm each initiates the development of embryo and endosperm, respectively. In contrast, in gametophytic apomixis an unreduced sporophytic cell of the ovule in proximity to the MMC (apospory), or the MMC itself becoming an apomictic initial cell (AIC) that omits or aborts meiosis (diplospory), gives rise to an unreduced embryo sac (apomeiosis) [9]. The egg cell subsequently develops into an embryo without fertilization (parthenogenesis). Endosperm development can either be autonomous or require fertilization (pseudogamy). It is likely that signals from sporophytic ovule tissues are important for the development of the sexual and apomictic germline [6], [9]. During meiosis the MMC is shielded by incorporation of callose into its cell wall [10], which may temporarily reduce or block such signaling. However, to our knowledge such signaling events have so far not been investigated in detail.
While recent studies uncovered the transcriptional basis of key steps of female germline development in the sexual model species Arabidopsis thaliana [11]–[13], relatively little is known about the genetic and transcriptional basis governing apomictic reproduction. Gametophytic apomixis is genetically controlled by usually two or more loci - or potentially clusters of linked loci - in different aposporous and diplosporous species [14]–[24]. In the Boechera genus, there is evidence for a complex genetic control of apomixis [25]. At the transcriptional level it has been hypothesized that apomixis is derived from a deregulation of the sexual pathway [6], [7], [26]. Indeed, evidence for differential regulation of many genes between apomictic and sexual accessions comes from comparative gene expression analyses. These studies mostly use ovule or flower tissues from a variety of species, including Boechera spp. [27], [28], Brachiaria spp. [29], [30], Hieracium perforatum [31], Pennisetum spp. [32], [33], Paspalum spp. [34]–[36], apomeiotic mutants of Medicago falcata [37], Panicum maximum [38], and Poa pratensis [39], [40]. In addition, recent findings indicate spatial and temporal shifts in the expression of genes important for reproductive development between sexual and apomictic plants [27]–[29], [41].
To coordinate such complex transcriptional deregulation, the involvement of epigenetic regulatory pathways has been proposed [3], [6], [7]. Epigenetic pathways play important roles in regulating developmental and cell-fate decisions through the modification of gene activity by histone modifications, DNA methylation or gene silencing by small RNAs. Interestingly, features of apospory or diplospory have recently been observed in Arabidopsis and maize carrying mutant alleles of genes involved in DNA methylation and small RNA pathways [42]–[44]. In Arabidopsis plants carrying mutations in ARGONAUTE9 (AGO9), or genes encoding additional members of a small RNA pathway (RNA-DEPENDENT RNA POLYMERASE 6 (RDR6), SUPPRESSOR OF GENE SILENCING 3 (SGS3)), additional MMC-like cells in the ovule gave rise to developing female gametophytes in a process resembling apospory [42]. Maize plants with mutations in homologues of the Arabidopsis DNA methyltransferases DOMAINS REARRANGED METHYLASE1 (DRM1)/DRM2 and CHROMOMETHYLTRANSFERASE3 (CMT3) show also features of apospory [43]. However, in maize plants carrying mutations in AGO104, a homologue of Arabidopsis AGO9, formation of unreduced viable gametes occurs by a diplospory-like mechanism [44].
In addition, features of apospory have been observed in Arabidopsis plants carrying mutations in the RNA helicase gene MNEME (MEM), which restricts germline fate to one cell per ovule [12]. As in ago9 mutants, the additional MMC-like cells initiate development of unreduced female gametophytes [12]. Apomeiosis has also been achieved by mutating important meiotic genes in Arabidopsis, such as DYAD/SWITCH1 (SWI1), a regulator of meiotic chromosome organisation, or a combination of three mutations in the MiMe triple mutant (sporulation11-1 (spo11-1); omission of second division1 (osd1); recombination8 (rec8)) [45], [46]. However, to date the potential role of these genes in naturally occurring apomixis has not been elucidated.
To study the transcriptional basis of key steps of apomictic reproduction we used the triploid, diplosporous species Boechera gunnisoniana as an apomictic model. The genus Boechera is closely related to the sexual model species Arabidopsis thaliana, facilitating comparative studies. We demonstrate the obligate apomictic behaviour of B. gunnisoniana by analysing the ploidy of embryo and endosperm in single seeds by means of a flow cytometric seed screen [47]. As no annotated, genome-wide sequence information is available for this species, we used RNA-Seq (Illumina HiSeq2000) to generate a reference transcriptome based on ovule tissues isolated by microdissection at the developmental stages of interest. We annotated the reference transcriptome, including the identification of homologous genes in Arabidopsis. Using a combination of laser-assisted microdissection (LAM), Affymetrix GeneChip profiling (ATH1), and RNA-Seq (SOLiD), we studied the transcriptome of isolated AICs, as well as egg, central and synergid cells from B. gunnisoniana. Statistical data analysis revealed the significant enrichment of polyamine and spermidine metabolism in the AIC as compared to the cells of the mature female gametophyte in Boechera. In addition, we compared the gene expression profiles of the AIC and the MMC, egg cells and central cells between apomictic Boechera and sexual Arabidopsis. This uncovered differential expression of genes in important regulatory pathways, including protein degradation, hormonal pathways, cell cycle control, signal transduction, transcriptional regulation, and epigenetic pathways.
Results
Boechera gunnisoniana seeds are derived from unreduced female gametes
B. gunnisoniana has previously been described as diplosporous apomict [48], [49]. While the embryo develops parthenogenetically, the endosperm requires fertilization (pseudogamy) [48], [49]. Based on flow cytometric studies of single seeds, a high variability of the reproductive mode - ranging from obligate sexual to obligate apomictic - has been reported among 71 Boechera accessions analysed [50]. We applied this technique to test the frequency of apomictic reproduction in B. gunnisoniana. From 84 individual seeds tested, ∼98% showed a 3C∶9C (embryo∶endosperm) ploidy ratio in the seed, as expected for a triploid, pseudogamous apomict (Figure 1A). In two seeds (∼2%) a 6C embryo resulted from fertilization of an unreduced egg cell (Figure 1B). In conclusion, B. gunnisoniana reproduces obligatory by pseudogamous apomixis. In all seeds analysed an unreduced egg cell gave rise to the embryo, and embryos developed parthenogenetically at very high frequency.
Figure 1. Flow cytometric seed screen on single seeds of Boechera gunnisoniana to analyse ploidy.

Fluorescence intensity was measured on individual green seeds of Boechera to determine the ploidy level of embryo and endosperm. 98% of the seeds measured (N = 84) showed a 3C∶9C ratio of embryo∶endosperm, indicating diplosporous apomixis (A). The remaining 2% showed a 6C∶9C embryo∶endosperm ratio indicative of a BIII hybrid where the embryo is derived from fertilization of an unreduced egg cell (B).
Nevertheless, the possibility of developmental variations during germline formation cannot be excluded based on a flow cytometric analysis alone. We used ovule and seed clearings for cytological analyses to address the question whether there is potential variation of reproductive development. In young ovules typically a single enlarged subepidermal cell specified to an AIC (Figure S1A, B), while in 3.6% of all ovules (N = 551) an additional enlarged, subepidermal cell was observed (Figure S1A). As previously reported, the AICs give rise to the formation of dyads [48], [49], [51]. Dyad formation was seen at a frequency of 85% (N = 224; Figure S1E, Q). In an additional 10% of all ovules, either dyads accompanied by large parietal cells and or triads were formed (Figure S1F, Q). These two possibilities could not clearly be discriminated based on morphology. Unexpected numbers of nuclei during AIC division or the formation tetrads were observed in ∼2% of all cases (Figure S1G, Q). In the remaining 3% of ovules the AICs apparently failed to divide (Figure S1C, Q), likely leading to developmental arrest (Figure S1D). Formation of a mature gametophyte was observed in 92% of all ovules (N = 353) in agreement with previously published results [49], the majority showing a delay or defect in the fusion of the polar nuclei (Figure S1I, J, R). In 7.4% of the ovules development was arrested early (at AIC or FMS stage), was delayed, or resulted in an unexpected number of nuclei (Figure S1R). At a very low frequency (0.6%) more than one gametophyte developed in a single ovule (Figure S1K, R). In agreement with previous reports, in the absence of pseudogamous fertilization no evidence for the initiation of embryo development was observed [48], [51]. After fertilization, 62% of the seeds developed normally (N = 477; Figure S1L, M). In the remainder, ovules harbouring mature gametophytes or enlarging seeds due to seed coat growth without embryo or endosperm development were observed, or only embryo or endosperm development initiated (Figure S1N–P), suggesting a problem in fertilization. In summary, in B. gunnisoniana the large majority of mature gametophytes are formed by diplospory and 98% of the seeds are derived parthenogenetically under our growing conditions. Thus B. gunnisoniana is well suited as a model species for molecular studies of apomixis.
Sequencing, assembly, and annotation of a Boechera gunnisoniana reference transcriptome based on ovule tissues
The close relation of the apomict B. gunnisoniana with the sexual model species A. thaliana provides an excellent basis for comparative analyses. However, while genome sequencing projects for Boechera species are currently ongoing (http://www.jgi.doe.gov/), this initiative does not include B. gunnisoniana, which is fast cycling and obligatory diplosporous. Thus, as a tool for transcriptomic studies, we generated a reference transcriptome for this species. We isolated ovules at the two developmental stages of interest, megasporogenesis (i.e. ovule stages from the initiation of integument development until the integuments start to overgrow the nucellus; Figure S1A, B) and mature gametophyte stage (Figure S1I–K). The highly enriched ovule samples included some pistil tissue, particularly for the early developmental stage. We prepared two libraries that were sequenced with the Illumina HiSeq2000. Both libraries were assembled together using trinity [52]. Following removal of reads with low average quality scores (Q<30) or adaptor sequences, and trimming of low quality (Q<20) ends, around 697 million reads were assembled into 112'232 sequences corresponding to 30'298 distinct genes with 50% having a sequence length of ≥2'153 bp. The reference transcriptome was annotated using Blast2GO [53] and BLAT [54] (Table S1). Using Blast2GO, 51% of all hits matched best to A. thaliana and an additional 25% to A. lyrata sequences. Gene ontology (GO) terms could be successfully assigned to 62% of all hits. In addition, we aligned the sequences to cDNA (TAIR10) using BLAT and identified 19'617 close A. thaliana homologues of B. gunnisoniana genes (hereafter denoted as Arabidopsis homologues, Table S2). In summary, the length of assembled sequences and annotation results indicate a good quality of our apomictic reference transcriptome.
Transcriptional profiling of cells involved in key steps of gametophytic apomixis
For the sexual model plant Arabidopsis, transcriptomes of the cell types of the mature gametophyte (egg, central, and synergid cells) and the MMC have been described [11]–[13]. From these studies, important new insights into the transcriptional basis of sexual germline development could be gained. We applied LAM to isolate the AIC and the surrounding sporophytic nucellus tissue, as well as the egg, central, and synergid cells from B. gunnisoniana (Figure 2A, B; Figure S2A). For the AIC, small contamination with surrounding nucellus tissue cannot be completely avoided (Figure 2A, B). Due to the dense structure of the mature embryo sac, samples for egg, central, and synergid cells are highly enriched in these cell types, but contain some contamination from neighbouring gametophytic cells (Figure S2A). For transcriptional profiling, 300–650 cell- or tissue-specific sections were pooled per sample. Transcriptional profiling was done using two alternative strategies: heterologous hybridization of amplified and labelled Boechera RNA to the Affymetrix ATH1 GeneChip designed for Arabidopsis and SOLiD V4 sequencing (Table 1, Figure 2C, D). For GeneChip analysis, the extracted RNA was subjected to linear amplification, labelled and hybridized to the microarray as described [12]. Cross-species hybridization of microarrays with RNA from a species other than the original target species is largely influenced by the degree of sequence similarities between the probes on the array and the mRNA sequence of the species under investigation [55]. To account for this effect we used an adapted BgPANP algorithm for the generation of presence/absence p values, similar to the AtPANP previously shown to outperform the default algorithm [11]. These algorithms use probes that do not match to the reference genome or transcriptome of the target species as “negative probes” to estimate the true background of each array. For our BgPANP algorithm probes not aligning to the reference transcriptome (allowing for three mismatches) were defined as negative. In this way, several thousand genes were detected significantly above background (hereafter referred to as present/“P”) in each cell type-specific sample (Table 1, Figure 2C, Figure S2B, C, Table S3). For RNA-Seq, the isolated RNA was subjected to linear amplification following an established protocol [13], [56]. Each library was sequenced on one eights of a slide, resulting in 53'701'313 (AIC, apo_initial3), 50'453'327 (egg cell, egg_cell2), 49'331'759 (central cell, central_cell2), and 46'240'916 (synergid cells, synergid_cell2) reads. Reads were processed and aligned to the assembled reference transcriptome as described [13]. Under the applied criteria, between 30% and 37% of the reads had at least one valid alignment, corresponding to 16'371'464 (apo_initial3), 18'783'550 (egg_cell2), 17'348'718 (central_cell2), and 15'353'384 (synergid_cell2) weighted alignments. Gene expression values were calculated as the sum of expression of individual variants (Table S4). We identified 16'385, 17'828, 19'091, and 10'409 B. gunnisoniana genes to be expressed (i.e. to have at least 5 mapped reads) in the AIC, egg, central, and synergid cells, respectively (Table 1). This corresponds to 13'047, 13'811, 14'893, and 9'390 expressed (≥5 read counts) Arabidopsis homologues in the AIC, egg, central, and synergid cells, respectively (Table 1, Table S2, Table S4). Between ∼2'000 and 6'000 genes were consistently identified in at least two independent cell type-specific samples (Table 1), in agreement with previous observations on the comparability of microarray and RNA-Seq data and the higher sensitivity and genome-wide coverage reached by RNA-Seq [13].
Figure 2. Laser-assisted microdissection (LAM) and transcriptome analysis to study the Boechera apomictic initial cell (AIC).

(A, B) LAM of the AIC from a 6 µm dry section (scale bar = 40 µm). (A) An ovule harbouring the AIC before LAM. Arrows point to the AICs. (B) The ovule after the AIC has been dissected and collected. (C, D) Venn diagrams showing the overlaps of prediction of expression (P calls; apo_initial1, apo_initial2, MMC_M/P) as determined with the BgPANP algorithm or the AtPANP algorithm described previously [12], and the genes with ≥5 read counts on Boechera homologues (apo_initial3) to the Arabidopsis genes as determined by mapping to the Boechera reference transcriptome.
Table 1. Transcriptome analysis of 11 samples from apomictic Boechera isolated by LAM.
| Sample | Genes present on microarray with BgPANP (p value≤0.02) | Number of B. gunnisoniana genes with ≥5 read counts | Number of Arabidopsis homologues with ≥5 read counts | Expressed in at least 2 samples (column 1 and column 3) |
| apo_initial1 | 5'595 | 6'006 | ||
| apo_initial2 | 5'686 | |||
| apo_initial3 | 16'385 | 13'047 | ||
| sporo_nucellus1 | 5'706 | 4'345 | ||
| sporo_nucellus2 | 5'236 | |||
| egg_cell1 | 5'835 | 4'490 | ||
| egg_cell2 | 17'828 | 13'811 | ||
| central_cell1 | 2'973 | 2'192 | ||
| central_cell2 | 19'091 | 14'893 | ||
| synergid_cell1 | 4'149 | 2'472 | ||
| synergid_cell2 | 10'409 | 9'390 |
Summary of gene expression from Boechera germline samples. Samples apo_initial1 and 2, sporo_nucellus1 and 2, egg_cell1, central_cell1, and synergid_cell1 were hybridized on ATH1 microarrays, apo_initial3, egg_cell2, central_cell2, and synergid_cell2 were analysed using RNA-Seq (SOLiD V4) by mapping of reads to the B. gunnisoniana reference transcriptome.
Independent data confirmation shows apomictic initial cell-enriched expression
Apomixis and sexual reproduction are interrelated developmental processes. Therefore, it is likely that the cell type-specific transcriptome profiles are largely overlapping between the sexual and apomictic mode of reproduction. Nevertheless, differences in expression of a subset of genes are expected due to the differences in reproductive mode and species. To compare the cell type-specific transcriptome profiles between Boechera and Arabidopsis, we used genes designated as P in two (for AIC) or one (for egg and central cell) microarray sample(s), or were identified as an expressed Arabidopsis homologue using RNA-Seq (Table 1). For Arabidopsis we used the 9'115 genes with evidence of expression in the MMC [12], 12'769 genes expressed in the egg cell (as described in [11], [12] and SOLiD reads aligned to the reference genome of Arabidopsis thaliana (TAIR10)), and 14'661 genes expressed in the central cell ([11], [12] and both samples from [13]). Comparing the genes with evidence of expression from Arabidopsis and Boechera for MMC/AIC, egg and central cells, we found overlapping expression of 7'606, 9'883, and 10'772 genes, respectively (Figure 2C, D; Figure S2B, C).
In addition, we selected several genes for independent data confirmation by in situ hybridization. Based on our analyses, these genes were expressed in the Boechera AIC but not in the Arabidopsis MMC (Table S5). Probes for in situ hybridization on B. gunnisoniana ovule sections were designed based on the Arabidopsis Col-0 cDNA for three transcription factors (Figure 3, (A–D) AT1G06170, basic helix-loop-helix (bHLH) DNA-binding superfamily protein; (E–G) AT1G28050, B-BOX DOMAIN PROTEIN 13; (H–J) AT1G76580, Squamosa promoter-binding protein-like (SBP domain) transcription factor family protein), an oligopeptide transporter (AT1G59740, Figure 3 K,L), and a HIGH MOBILITY GROUP A protein (HMGA, AT1G14900, Figure 3 M–O). The probes were designed to have significant sequence homologies only to the respective Boechera homologue (Figure S3, Supporting Information S1). For all selected genes we could confirm enriched expression in the AIC. Taken together, our analyses confirm the accuracy of the B. gunnisoniana transcriptome dataset.
Figure 3. Independent data validation for selected genes by in situ hybridization on B. gunnisoniana ovules.
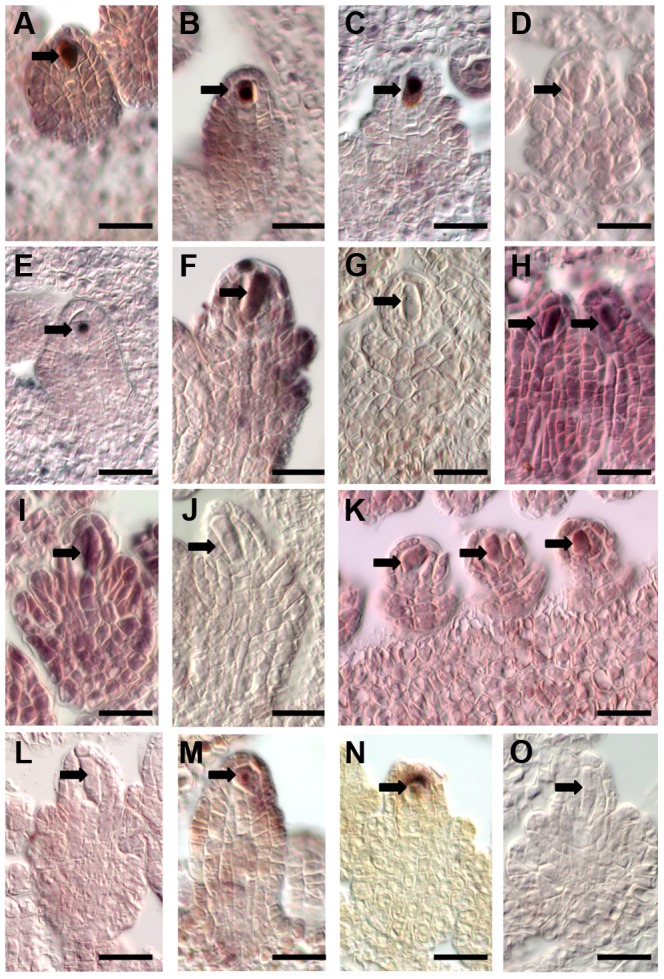
Data validation for selected genes found expressed in the Boechera AIC but not the Arabidopsis MMC. Scale bars are 20 µm, arrows point to the AICs. In situ hybridizations on B. gunnisoniana ovule sections were performed with antisense probes (A–C, E, F, H, I, K, M, N) or sense probes as controls (D, G, J, L, O) for the transcription factors AT1G06170, a basic helix-loop-helix (bHLH) DNA-binding superfamily protein (A–D), AT1G28050, a B-BOX DOMAIN PROTEIN 13 (E–G), and AT1G76580 a Squamosa promoter-binding protein-like (SBP domain) transcription factor family protein (H–J), an oligopeptide transporter, AT1G59740 (K,L), and AT1G14900, encoding the HIGH MOBILITY GROUP A protein.
Gene expression and gene ontology enrichment analysis uncovers upregulation of spermidine metabolism in the apomictic initial cell
Between sexual and apomictic reproduction, there are important differences in cell specification and cell fate decisions. Heterochronic shifts in expression patterns have been reported previously using isolated Boechera ovules from sexual and apomictic accessions [27], [28]. However, gene expression has not yet been profiled in a germline-specific manner without the confounding effects of the surrounding sporophytic tissue in Boechera. Based on genes significantly enriched in the MMC as compared to the cell types of the mature gametophyte, we previously identified translational control pathways and the activity of RNA-helicases as crucial for the acquisition of germline fate and MMC specification in Arabidopsis [12]. To see if similar or different functions are prominent in the Boechera AIC as compared to the mature gametophyte, we used read counts obtained by mapping to the Boechera reference transcriptome. To identify genes significantly enriched we used NOIseq-sim, a non-parametric approach for differential expression analysis based on simulated replicate samples [57]. We identified 1'487 genes to be significantly enriched in the AIC as compared to the cell types of the mature gametophyte (Figure 4A). In addition, 3'509, 1'466, and 1'806 genes were significantly enriched in the egg, central, and synergid cells, respectively, as compared to the other three cell types of the germline under investigation.
Figure 4. Heatmap of log2 transformed normalized read counts.

Heatmap of 1'487 genes enriched in the Boechera AIC as compared to all cell types of the mature female gametophyte as identified using NOISeq-sim (A). Heatmap of 3'792 genes enriched in the AIC as compared to all cell types of the mature gametophyte or differentially expressed between any cell type of the mature gametophyte identified using EdgeR (B). The hierarchical clustering of samples and genes was based on euclidean distance and hierarchical agglomerative clustering. Colours are scaled per row and red denotes high expression and black low expression.
In a gene ontology (GO) analysis, we identified functions important for pollen germination and sperm cell and pollen maturation as significantly enriched in the AIC (p<0.01, Table 2). In addition, different metabolic and transport processes were upregulated, in addition to spermidine metabolism and polyamine biosynthesis (p<0.01, Table 2). Functions related to plant cell wall modification and epigenetic regulatory pathways (histone H3K4 demethylation and maintenance of DNA methylation) were also amongst the enriched functions (p<0.01, Table 2). Furthermore, cytokinin catabolism was among the near-significantly enriched processes (p = 0.012, Table 2).
Table 2. Gene ontology analysis.
| GO.ID | Term | Annotated | Significant | Expected | p value |
| GO:0010584 | pollen exine formation | 92 | 44 | 4.73 | <1e-30 |
| GO:0009827 | plant-type cell wall modification | 336 | 46 | 17.26 | 1.20E-09 |
| GO:0008216 | spermidine metabolic process | 25 | 10 | 1.28 | 2.00E-07 |
| GO:0009860 | pollen tube growth | 393 | 43 | 20.19 | 2.50E-06 |
| GO:0006527 | arginine catabolic process | 8 | 5 | 0.41 | 1.70E-05 |
| GO:0006817 | phosphate ion transport | 32 | 9 | 1.64 | 2.30E-05 |
| GO:0008643 | carbohydrate transport | 97 | 18 | 4.98 | 2.30E-05 |
| GO:0046467 | membrane lipid biosynthetic process | 210 | 25 | 10.79 | 8.00E-05 |
| GO:0006596 | polyamine biosynthetic process | 24 | 7 | 1.23 | 0.00015 |
| GO:0016036 | cellular response to phosphate starvation | 194 | 22 | 9.97 | 0.00042 |
| GO:0055085 | transmembrane transport | 1020 | 80 | 52.4 | 0.00048 |
| GO:0034720 | histone H3-K4 demethylation | 5 | 3 | 0.26 | 0.00125 |
| GO:0009396 | folic acid-containing compound biosynthetic process | 18 | 5 | 0.92 | 0.00173 |
| GO:0030162 | regulation of proteolysis | 6 | 3 | 0.31 | 0.0024 |
| GO:0048235 | pollen sperm cell differentiation | 47 | 8 | 2.41 | 0.00248 |
| GO:0046938 | phytochelatin biosynthetic process | 7 | 3 | 0.36 | 0.00405 |
| GO:0006665 | sphingolipid metabolic process | 88 | 12 | 4.52 | 0.00582 |
| GO:0030036 | actin cytoskeleton organization | 254 | 14 | 13.05 | 0.00587 |
| GO:0010216 | maintenance of DNA methylation | 15 | 4 | 0.77 | 0.00599 |
| GO:0009395 | phospholipid catabolic process | 15 | 4 | 0.77 | 0.00599 |
| GO:0010951 | negative regulation of endopeptidase activity | 8 | 3 | 0.41 | 0.00623 |
| GO:0010199 | organ boundary specification between lateral organs and meristems | 8 | 3 | 0.41 | 0.00623 |
| GO:0015800 | acidic amino acid transport | 7 | 3 | 0.36 | 0.00763 |
| GO:0010084 | specification of organ axis polarity | 3 | 2 | 0.15 | 0.00764 |
| GO:0090408 | phloem nitrate loading | 3 | 2 | 0.15 | 0.00764 |
| GO:0042398 | cellular modified amino acid biosynthetic process | 84 | 10 | 4.32 | 0.01076 |
| GO:0010205 | photoinhibition | 18 | 4 | 0.92 | 0.01188 |
| GO:0009823 | cytokinin catabolic process | 10 | 3 | 0.51 | 0.01236 |
Biological Processes identified to be up-regulated based on 1'487 genes identified to be up-regulated in the AIC as compared to the cell types composing the mature gametophyte (egg cell, central cell, synergid cells).
In the egg cell of Boechera, cytokinin metabolism is a dominant molecular function as discovered by analysis of GO enrichment based on the 3'509 significantly upregulated genes, in addition to transcription factor activity (Table S6A). The central cell transcriptome is dominated by different epigenetic regulatory pathways, cell cycle regulation, and regulation of cell fate decisions (p<0.01, Table S6B).
At higher stringency, using EdgeR with an estimated biological variation coefficient of 0.8, we identified 142 genes to be significantly enriched in all pairwise comparisons of the AIC with the transcriptomes of cells of the mature gametophyte (adjusted p value (FDR)<0.05, Benjamini-Hochberg adjustment; Figure 4B) [58]. Based in these genes, GO enrichment analysis confirmed spermidine metabolism, cytokinin catabolism, and functions related to pollen development and germination as significantly enriched in the AIC (p<0.01; Table S7). Notably, also the term “sexual reproduction” was an enriched function based on upregulated genes. In addition, 3'792 genes were differentially expressed in any pairwise comparison between the cell types of the mature gametophyte (FDR≤0.05 for comparisons between synergid cells and egg- or central cell, or an unadjusted p value≤0.001 for comparisons between egg cell and central cell (Figure 4B)).
In summary this indicates interesting differences in the functions underlying the specification of the germline lineage and the female gametes in the apomict B. gunnisoniana as compared to the sexual pathway in Arabidopsis. Consistently, spermidine metabolism was identified as enriched in the AIC. Our analysis also indicated a distinct regulation of cytokinin metabolism and degradation in the apomictic germline lineage.
Evidence for different regulation of important regulatory pathways in apomictic and sexual germline cells
To analyse differences in gene activity between the sexual and apomictic germline in more detail, we identified Arabidopsis genes and their homologues only expressed in a certain cell type in Arabidopsis or Boechera. Boechera genes were designated as expressed when having at least 5 read counts by mapping against the reference transcriptome, or a P call on one or both microarrays. For a conservative estimate of genes only expressed in Arabidopsis, we also aligned the SOLiD reads to the reference genome of A. thaliana (TAIR10) and only considered genes with at least 5 read counts. We included the latter method as annotation of the closest Arabidopsis homologue is not always unambiguous. Sometimes sequence variants for one Boechera gene have their highest sequence similarity to different Arabidopsis genes (see below), complicating a direct comparison. Of the 9'115 MMC-expressed genes, no evidence of expression has been found for 852 genes in the AIC. GO analysis on this set of genes identified a significant enrichment of different molecular functions, including metabolism, regulation of physiological responses, auxin turnover, translation initiation, and functions related to cell wall structure and cell cycle control (p<0.01, Table 3A). Also the “core cell cycle genes” were found to be significantly enriched (Fisher's exact test, p = 0.006), in agreement with the meiotic fate of the MMC. In addition, 14 protein family (PFAM) domains were identified as enriched (Fisher's exact test, p value<0.01, Table S8) including F-box domain and F-box related domains, as well as the cyclin C- and N-terminal domains. This suggests that protein ubiquitinylation and degradation, as well as cell cycle control, may be differentially regulated between MMCs and AICs. Using a similar approach, out of 12'679 genes expressed in the Arabidopsis egg cell (Figure S2B) we identified 1'731 for which no homologues were expressed in the Boechera egg cell. GO analysis in this set of genes identified biological processes related to RNA modification and splicing, transport and metabolism, and methylation-dependent chromatin silencing as significantly enriched, and also functions related to double fertilization and endosperm formation (p<0.01, Table 4A). In addition, two transcription factor families, the “AtRKD Transcription Factor Family” and the “MYB Transcription Factor Family” were identified as significantly enriched gene families (Fisher's exact test, p = 0.0087 and p = 0.0038, respectively). For the Arabidopsis central cell, out of 14'661 expressed genes (Figure S2C) no evidence for expression of homologues in the Boechera central cell was found for 2'146 genes. As in the Arabidopsis egg cell, biological processes related to RNA modification and splicing (GO:0000154, rRNA modification, p = 5.1e-17; GO:0045292, nuclear mRNA cis-splicing, via splicosome, p = 5.4e-5) and “endosperm development” (p = 0.0078) were significantly enriched. In addition, out of the 12 PFAM domains identified as enriched, three were related to F-box domains (Fisher's exact test, p<0.01, Table S9).
Table 3. Gene ontology analysis on MMC and AIC.
| A) Arabidopsis thaliana MMC | |||||
| GO.ID | Term | Annotated | Significant | Expected | p value |
| GO:0016538 | cyclin-dependent protein kinase regulator activity | 29 | 6 | 1.14 | 0.00081 |
| GO:0005199 | structural constituent of cell wall | 30 | 6 | 1.18 | 0.00098 |
| GO:0016844 | strictosidine synthase activity | 14 | 4 | 0.55 | 0.00176 |
| GO:0004129 | cytochrome-c oxidase activity | 15 | 4 | 0.59 | 0.00232 |
| GO:0010178 | IAA-amino acid conjugate hydrolase activity | 3 | 2 | 0.12 | 0.00455 |
| GO:0003743 | translation initiation factor activity | 82 | 9 | 3.24 | 0.00496 |
| GO:0005034 | osmosensor activity | 4 | 2 | 0.16 | 0.00885 |
Significant upregulation of molecular functions based on 852 genes expressed in the Arabidopsis MMC but not in the Boechera AIC (A). Significant upregulation of molecular functions based on 901 genes expressed in the apomictic initial cell but not in the MMC (B). A p value<0.01 was considered significant.
Table 4. Gene ontology analysis on sexual and parthenogenetic egg cells of Arabidopsis and Boechera, respectively.
| (A) Arabidopsis thaliana egg cell | |||||
| GO.ID | Term | Annotated | Significant | Expected | p value |
| GO:0000154 | rRNA modification | 74 | 43 | 4.02 | <1e-30 |
| GO:0045292 | nuclear mRNA cis splicing, via spliceosome | 8 | 4 | 0.43 | 0.00051 |
| GO:0045490 | pectin catabolic process | 2 | 2 | 0.11 | 0.00295 |
| GO:0015986 | ATP synthesis coupled proton transport | 38 | 7 | 2.07 | 0.00395 |
| GO:0043086 | negative regulation of catalytic activity | 82 | 11 | 4.46 | 0.00474 |
| GO:0019432 | triglyceride biosynthetic process | 7 | 3 | 0.38 | 0.00475 |
| GO:0080155 | regulation of double fertilization forming a zygote and endosperm | 3 | 2 | 0.16 | 0.00854 |
| GO:2000014 | regulation of endosperm development | 3 | 2 | 0.16 | 0.00854 |
| GO:0090309 | positive regulation of methylation-dependent chromatin silencing | 3 | 2 | 0.16 | 0.00854 |
Significant enrichment of biological processes based on 1'731 genes with evidence of expression only in the sexual Arabidopsis egg cell (A) and 5'273 Boechera homologues with evidence of expression only in the parthenogenetic egg cell (B). A p value<0.01 was considered significant.
For the identification of genes only expressed in the apomictic Boechera germline and not in Arabidopsis, we used the Arabidopsis homologues identified and mapping to the Boechera reference transcriptome, combined with the microarray data. We identified 5'273 and 4'902 genes expressed in the apomictic egg and central cell, respectively, that were absent in the corresponding Arabidopsis cell type. We used more restrictive criteria to identify the 901 genes expressed in the AIC but not in the MMC: we considered only Arabidopsis homologues with ≥5 reads in the SOLiD dataset and detected as P in at least one microarray dataset of the AIC. Interestingly, for all three cell types of the apomictic Boechera germline, GO and/or PFAM analyses revealed a significant enrichment of signal transduction processes and protein kinases (Table 3B, Table 4B, Table 5, Table S10). For instance, we identified the significant enrichment of “MAP kinase kinase activity” in the AIC (p<0.01, Table 3B). In addition, transport and metabolic processes were enriched, and spermidine metabolism was confirmed as an important feature (p<0.01, Table 3B). Analysis of gene families revealed the Squamosa promoter Binding Proteins as enriched (Fisher's exact test, p value<0.01, SBP transcription factor family). Analysis of gene families and PFAM domains also identified a significant enrichment of the AMINO ACID/AUXIN PERMEASE (AAAP) family, the ARF transcription factor family, and the protein domain of the AUX/IAA family during apomictic germline specification (Fisher's exact test, p<0.01, Table 6). Also the family of B3 transcription factors (B3_TFs), including the ARF transcription factor family, was identified as significantly enriched (Fisher's exact test, p<0.01, Table 6). In the parthenogenetic Boechera egg cell, GO analysis suggests the importance of signal transduction pathways, cell cycle regulation, and transcription factor activity (p<0.01). In general, in the female gametes analysis of gene families identified the enriched expression of several transcription factor families, particularly the basic Helix-Loop-Helix transcription factors both in the egg and central cell (Table S11).
Table 5. Analysis of PFAM domains significantly enriched in the Boechera germline.
| AIC | ||||
| ID | Significant | Expected | p value | description |
| PF01535 | 217 | 124.07 | 2.30E-10 | Pentatricopeptide repeat |
| PF00069 | 268 | 200.47 | 0.00012525 | Protein kinase domain |
| PF00225 | 38 | 18.94 | 0.00123647 | Kinesin motor domain |
| PF00612 | 32 | 16.10 | 0.00295969 | IQ calmodulin-binding motif |
| PF07714 | 130 | 96.92 | 0.00671958 | Protein typrosine kinase |
PFAM domains significantly enriched in 901, 5'273 and 4'902 genes expressed only in the AIC, egg cell and central cell of Boechera but without evidence of expression in the Arabidopsis MMC, egg cell, and central cell, respectively (p value<0.01).
Table 6. Gene family enrichment.
| Gene family | Significant | Expected | p value |
| AAAP family | 8 | 1.55 | 0.0004025 |
| Acyl Lipid Metabolism Family | 35 | 20.65 | 0.0050592 |
| ARF Transcription Factor Family | 5 | 0.71 | 0.0015891 |
| B3_TFs | 8 | 2.77 | 0.0099967 |
| Glycoside Hydrolase Gene Families | 24 | 13.30 | 0.0084674 |
| Monolignol Biosynthesis | 8 | 2.31 | 0.0037947 |
| Organic Solute Cotransporters | 28 | 10.20 | 7.53E-06 |
| SBP Transcription Factor Family | 4 | 0.59 | 0.0051551 |
| Superfamily of zinc-coordinating DNA-binding proteins | 4 | 0.59 | 0.0051551 |
Enrichment of gene families in 901 genes with evidence of expression in the AIC but not in the sexual MMC as analysed by Fisher's exact test. A p value<0.01 was considered significant.
In summary, our analysis reveals interesting differences in the regulatory programs underlying the acquisition of germline fate and between the female gametes. While the subset of genes only expressed in the sexual germline is significantly enriched in protein degradation pathways, the apomictic Boechera germline is marked by the activity of signal transduction processes. In addition, indications for a role of auxin signalling and metabolism were observed in both germlines. Among the genes identified as active in the apomictic germline lineage only, we found the enrichment of different transcription factors families, particularly basic helix-loop-helix transcription factors in the female Boechera gametes. The comparison between the sexual and apomictic germlines further revealed differential regulation of genes involved in cell cycle control and posttranscriptional regulatory processes, including mRNA splicing, and epigenetic regulatory pathways related to methylation-dependent chromatin modifications.
Expression analysis of selected candidate genes and pathways related to apomixis
For a number of genes, enriched expression in the Arabidopsis MMC or the aposporic initial cell of Hieracium praealtum have previously been described [12], [31]. In addition, for sexual or apomictic germline development, evidence for the importance of different genes including core cell cycle genes, meiotic genes, and genes involved in epigenetic regulatory pathways has previously been reported based on mutant analyses or expression patterns [42], [43], [59]. Thus, we compared the expression of selected genes of interest upon sexual and diplosporic germline initiation.
From a list of 89 core cell cycle genes as defined before [60], [61], 75 are represented on the ATH1 array and for 66 Arabidopsis genes, homologues were identified in the Boechera reference transcriptome. From these, 41 genes are expressed in the Arabidopsis MMC and 49 homologues are present in the Boechera AIC. 16 and 24 cell cycle regulators have only been detected in the MMC or the AIC, respectively (Table S12). In particular, the genes only detected in the apomict upon germline specification include genes involved in different cell cycle transitions, e.g. G1/S phase, including a number of genes involved in the cyclin D/retinoblastoma/E2F pathway (Table S12). The observed differences in cell cycle regulation are in agreement with the different mechanisms of cell division in the meiotic MMC versus the diplosporous AIC.
Interestingly, for 14 selected meiotic genes and genes expressed in the sexual MMC no evidence for expression was found in the H. praealtum aposporous initial cell [31]. However, although the aposporous and the diplosporous initial cell both give rise to unreduced embryo sacs, cell lineage and developmental fate are markedly different. So far it is unknown whether common regulators underlie apomeiosis in these distinct types of apomixis. Interestingly, for all 14 genes except for SWITCH/DYAD and SPO11-2 evidence for expression was found in the Boechera AIC; although at very low levels for most genes (Figure 5). The Arabidopsis male meiocytes cluster separately from the expression data of different Arabidopsis cell- and tissue-types publicly available [13], [62]–[66]. Furthermore, the RNA helicase MEM previously identified as predominantly expressed in the Arabidopsis MMC is only expressed at low levels in the AIC but higher in the apomictic egg cell (Figure 5). Interestingly, this indicates differences in the expression of genes previously identified to have important functions for MMC specification and meiosis in Arabidopsis. In agreement with the differences in developmental fate, the data also suggest differences in cell specification of aposporous and diplosporous initial cells. Nevertheless, the majority of 35 genes described as enriched in the H. praealtum aposporous initial cell or the early apomictic embryo sac as compared to sporophytic ovule tissues [31] was also expressed in the Boechera AIC, except for HISTONE ACETYLTRANSFERASE OF THE CBP FAMILY1, LIKE HETEROCHROMATIN PROTEIN1, BEL1-LIKE HOMEODOMAIN1, CONSTITUTIVE DISEASE RESISTANCE1, genes involved in lipid localization (AT1G03103, AT5G38170, AT3G18280, AT1G43666), and a pathogenesis-related lipid-transfer protein gene (AT2G18370).
Figure 5. Heatmap of log2 transformed normalized read counts for 14 selected meiotic or MMC-expressed genes.

Hierachical clustering of read counts from different Arabidopsis and Boechera cell- and tissue types [13], [62]–[66]. The hierarchical clustering of samples and genes was based on euclidean distance and hierarchical agglomerative clustering. Colours are scaled per row. Red denotes high expression and black low expression.
Increasing evidence suggests the involvement of epigenetic regulatory pathways in the discrimination between sexual reproduction and apomixis. Therefore, we were interested in a closer investigation of the expression of 69 genes involved in DNA methylation and small RNA pathways (as used in [12]). 58 of these genes have annotated homologues in Boechera (Table S1). 40 genes are consistently present both in the AIC and in the MMC, supporting the important role of epigenetic regulatory pathways for the initiation of germline development [12]. Heatmap clustering suggests the closest relation between the AIC dataset and the datasets of the Boechera female gametes (Figure S4). Together, these datasets cluster with the Arabidopsis egg and synergid cells, but distantly from male meiocytes or the central cell of the sexual germline lineage (Figure S4). Nevertheless, a number of genes were only detected in the MMC or the AIC, respectively (Supporting Information S2). Genes only detected in the AIC included ENHANCED SILENCING PHENOTYPE3 (ESP3). Also AGO9 and RDR6, mutations in which cause an apospory-like behaviour in Arabidopsis [42], were both detected at low levels in the Boechera AIC (Figure S4, Figure S5). In summary, for a subset of genes involved in DNA methylation and small RNA pathways, we observed distinct expression patterns during germline specification in sexual Arabidopsis MMCs versus apomictic Boechera AICs, which may be of importance to distinguish fate decisions between these alternative reproductive modes.
Influence of sequence similarities between Arabidopsis and Boechera homologues on the distribution of count data
Particularly within a gene family, the assignment of the closest Boechera homologues to Arabidopsis genes is not always unambiguous. For selected gene families of interest we aimed to test the influence of sequence divergence and annotation criteria on the expression estimates for B. gunnisoniana homologues of A. thaliana genes. Identification of the closest homologues in the Boechera reference transcriptome was based on the highest bit score sum with BLAT, using only the best mappings per Arabidopsis gene. For this analysis we selected the AtRKD gene family (Figure 6, 7). In addition, similar analysis of the ARIADNE (ARI) gene family, and the AGO gene family are shown in “Supporting Information S2” (Figure S5, Figure S6, Figure S7, Supporting Information S2).
Figure 6. Analysis of sequence divergence of members of the AtRKD gene family.

Analysis of sequence divergence of members of the AtRKD gene family and close Boechera homologues as analysed with ClustalX based on protein sequences and read counts assigned. Boechera gene model variants are indicated with compxxx_cY_seqZ.
Figure 7. Heatmap clustering of members of the AtRKD gene family.

Heatmap of normalized log2 transformed read counts from different Arabidopsis and Boechera cell- and tissue-types [13], [62]–[66]. The hierarchical clustering of samples and genes was based on euclidean distance and hierarchical agglomerative clustering. No row scaling of colours was applied. Red denotes high expression and black low expression.
The RKD gene family has been identified in our analysis to be enriched among the genes expressed only in the Arabidopsis but not the Boechera egg cell. Instead of the five members of the Arabidopsis RKD family, two gene models of homologues with one variant each have been identified in the Boechera reference transcriptome (Figure 6). This suggests either that the gene family is smaller in Boechera as compared to Arabidopsis, or that additional members of this family are not expressed in Boechera ovules at the developmental stages used to generate the reference transcriptome. Analysis of sequence similarities indicates the closest similarity between comp76373_c0_seq1 and AtRKD2. In agreement, counts for reads mapped to comp76373_c0_seq1 are assigned to AtRKD2. However, while clustering of comp83606_c0_seq1 indicates higher sequence divergence from all AtRKDs, the reads are assigned to AtRKD5. The expression and role of members of the RKD family in Arabidopsis, where they play a role in egg cell specification, has been described previously [67]. As the two Boechera gene models homologous to the AtRKD genes are expressed in the egg apparatus (egg and synergid cells; Figure 6, 7), the Arabidopsis family as a whole is predominantly expressed in the Arabidopsis egg apparatus (Figure 7), in agreement with our gene set enrichment analysis.
Discussion
Boechera gunnisoniana as a model species to study apomixis
To investigate apomictic reproduction, the female germline is in particular of interest, as in apomicts clonal offspring genetically identical to the mother plant is generated. In B. gunnisoniana, based on a flow cytometric seed screen using single seeds, we observed exclusively apomeiotic behaviour and only a very low percentage of fertilized, unreduced egg cells. In agreement, the formation of dyads and mature Polygonum type embryo sacs were observed at high frequencies. At low frequencies, developmental variations during germline development were observed, including the formation of more than one female gametophyte per ovule. This could either be due to a failure of degradation of the second megaspore resulting from diplospory, or indicate the rare occurrence of apospory. Interestingly, parthenogenesis remains repressed in the absence of pseudogamous fertilization. In maturing siliques, likely due to a lack of successful fertilization, not all female gametes give rise to an embryo or endosperm. As a consequence of deviations from apomictic germline development and fertilization, reproductive development seems to arrests, so that the vast majority of mature seeds are derived apomictically. This obligate apomictic behaviour, together with its fast cycling (about 4 months from seed to seed) and the close relation to the sexual model species A. thaliana, make B. gunnisoniana an ideal system to study apomixis. We generated the first comprehensive, annotated reference transcriptome for reproductive development in B. gunnisoniana, including the identification of Arabidopsis homologues, as an essential tool for further studies.
Spermidine and polyamine metabolism are enriched in the apomictic initial cell
Previously, similarities of germline development were reported even across kingdoms, between the plant and animal germline. These are likely of general importance for the acquisition of germline fate [12]. Nevertheless, cell type specification and developmental fate is markedly different during germline specification in sexual, aposporous, and diplosporous species. Consistently, a number of differences in gene expression profiles have been observed between the apomictic and the sexual germline. In the B. gunnisoniana AIC, a number of functions related to pollen development and germination were enriched, consistent with gene activities observed during germline development in apomeiotic, non-parthenogenetic hybrids of Pennisetum glaucum [33].
Polyamine biosynthesis and spermidine metabolism were also identified as features of the Boechera AIC. Interestingly, spermidine synthesis is essential for embryo development in Arabidopsis [68]. In addition, a possible role of polyamines in promoting somatic plant embryogenesis was reported [69]–[71]. This indicates the importance of spermidine for plant reproduction and provides an interesting link between polyamines and somatic embryogenesis, a form of asexual reproduction different from gametophytic apomixis. Interestingly, spermidine is involved in the protection of DNA from oxidative stress by quenching free radicals mostly arising from reactive oxygen species (ROS) [72]. In line with the high activity of spermidine metabolism in the apomeiotic AIC, it has been hypothesized that repair of DNA damage after oxidative stress has been a major driving force for the evolution of meiosis [73]. Apart from being cytotoxic, the role of ROS in signalling and for plant reproductive development has recently been demonstrated [74]. Notably, a spermine/spermidine synthase has previously been identified to be present in the apospory-specific region of P. squamulatum and hypothesized to be expressed [75], supporting a potential role of these substances for the specification of the apomictic germline. However, further studies will be required to conclude which, if any, role polyamine and spermidine metabolism plays during germline development or the determination of the asexual reproductive fate.
Differentially regulated genes and pathways during sexual and apomictic reproduction include hormonal and protein degradation pathways and transcription factor activity
In addition to polyamine and spermidine metabolism, the activities of important hormonal pathways were also observed in the AIC. Upregulation of cytokinin degradation was detected upon apomictic germline specification as compared to the mature gametophyte, while the egg cell is marked by gene activities leading to cytokinin modifications. In addition, genes involved in auxin signalling were enriched in the set of genes expressed in the AIC but not in the sexual MMC, in line with the identification of genes involved in auxin signal transduction in the H. praealtum apospory initial cell [31]. In the Boechera AIC, we detected an enriched activity of the AUX/IAA and the ARF transcription factor gene families. These play crucial roles in auxin-regulated gene expression, for example to control cell type-specific auxin responses during Arabidopsis embryo development [76], [77]. Evidence for differential expression of ARF genes has previously been reported during early stages of reproductive development in a comparative cDNA-AFLP analysis targeting sexual and apomictic Paspalum simplex flowers [35].
In contrast, genes active only during sexual reproduction and MMC specification are marked by an enrichment of F-box proteins. F-box proteins play important roles in ubiquitin-dependent protein degradation involved in signal transduction pathways, cell cycle control, and a variety of other processes [78], [79]. The expression of miRNAs targeting genes encoding F-box proteins and ARF transcription factors in Boechera floral tissues supports the importance of these pathways in plant reproductive development [80]. This is in line with the identification of a truncated ARI allele with homology to Arabidopsis ARI7 as a candidate apospory locus in Hypericum perforatum [81]. ARI7 encodes a ring finger protein predicted to be involved in ubiquitin-dependent protein degradation [82]. Interestingly, we found evidence for higher activity of ARI family members in the sexual MMC compared to the AIC.
In addition to miRNAs targeting F-box proteins and ARF transcription factors, miRNAs involved in regulation of SPL and MYB transcription factors have been identified in Boechera spp. [80]. Together with the enrichment observed for SPL transcription factors in the B. gunnisoniana AIC and of MYB transcription factors in the sexual Arabidopsis MMC and egg cell, respectively, this suggests that these transcription factors play important roles in plant reproduction. Differences in activity were also observed for additional transcription factor families in agreement with previously identified differences in transcriptional regulation at later developmental stages in sexual and apomictic P. simplex flowers [35]. In the sexual Arabidopsis egg cell as compared to the apomictic Boechera egg cell, we observed the enriched expression of the RKD transcription factor family, which are important regulators of egg cell gene expression programs in Arabidopsis and wheat [67]. This suggests that RKD transcription factors might be specifically involved in the determination of the developmental fate of the sexual egg cell. Taken together, our findings indicate differences in the activity of important regulatory pathways during sexual and apomictic germline specification and development.
Germline specification during sexual reproduction and apomixis
Development of an unreduced embryo sac from an AIC is common to both diplospory and apospory. However, the founder cell of the female germline differs in position and cell fate between these two types of gametophytic apomixis. It is unknown whether a common regulator or a set of regulatory genes determines apomeiosis, or whether apomeiosis is mediated by unrelated developmental programs during apospory and diplospory. Interestingly, a number of important differences in gene expression have been observed in the aposporous initial cell in H. praealtum and the AIC of diplosporous B. gunnisoniana. This is consistent with the differences in cellular fate and identity between these apomicts. While the aposporous initial cell acquires a FMS-like fate without intervening cell division, the transcriptome of the AIC in a diplosporous apomict is expected to be more similar to the sexual MMC. This is in agreement with the lack of expression of several meiotic genes and other genes expressed in the sexual MMC in the aposporous initial cell in H. praealtum [31], differing from the transcriptome of the AIC in Boechera. Interestingly, in the Boechera AIC we did not observe evidence of expression of DYAD/SWITCH. In Arabidopsis, mutations in this gene have previously been shown to cause a diplospory-like phenotype with rare seed formation by the fertilization of unreduced egg cells [45]. The manipulation of cell cycle progression or meiotic genes has also been shown to lead to the formation of unreduced gametophytes [46], [83], [ reviewed in 84]. The comparison between the Arabidopsis MMC and the Boechera AIC identified a number of core cell cycle genes to be differentially regulated. While a small number of genes important for meiotic cell cycle progression in Arabidopsis has already been described [46], [ reviewed in 84], detailed functional studies of candidate genes showing differential expression in the MMC and AIC will be required to elucidate their putative role in the discrimination between meiosis and apomeiosis. Interestingly, the Arabidopsis gene encoding WEE1 is only detected in the Arabidopsis MMC. The WEE1 protein is specifically removed to allow progression of mitosis [85]. In addition, homologues of three members of the Arabidopsis E2F transcription factor family have only been detected in the Boechera AIC but not in the Arabidopsis MMC. Members of this family are involved in the regulation of the centromer-specific histone 3 variant CENH3 in Arabidopsis [86]. Manipulation of CENH3 can induce genome elimination, a capacity that has already been successfully applied for the generation of synthetic clonal seeds from Arabidopsis in combination with dyad or MiMe mutants [83]. Based on our transcriptome analysis, different levels of CENH3 expression have been observed in the Boechera germline as compared to Arabidopsis. In contrast to very low expression or absence in Arabidopsis gametes, higher expression levels of the CENH3 homologue have been observed in Boechera gametes. It is thus possible that the absence of DYAD/SWITCH expression in the AIC combined with elevated expression levels of CENH3 in apomictic Boechera as compared to sexual gametes might play a role in naturally occurring diplospory. In addition to unknown parthenogenesis factors, the regulation of CENH3 activity might provide an additional control mechanism to secure the absence of a paternal contribution in the offspring.
While mutations in the gene encoding for DYAD lead to features of diplospory, mutations in MEM, AGO9 and additional genes involved in a small RNA pathway have recently been reported to cause phenotypes reminiscent of apospory [12], [42], [43]. We identified additional genes involved in gene silencing and small RNA pathways to be differentially expressed in the MMC and the AIC. The expression of ESP3 in the AIC is reminiscent of the previous identification of ESP4 among the transcripts form the apospory-specific region in P. squamulatum [87]. This supports the importance of epigenetic regulatory pathways for sexual and apomictic reproduction.
Taken together, upon specification of the apomictic and sexual germline a number of differences involving regulatory processes such as hormone signalling, cell cycle control, and protein turnover have been observed. In addition, increased activity of signal transduction processes was identified as a typical feature of the apomictic germline. The potential role of positioning of the MMC or AIC and the signalling from the surrounding sporophytic tissues has previously been discussed [88], [89], and our study has shown that signalling pathways are indeed modulated in the two modes of reproduction.
In conclusion, our study provides the first comprehensive transcriptional analysis of germline cells at key steps of apomictic reproduction in B. gunnisoniana. The generation and annotation of an apomictic reference transcriptome forms an essential basis for further analyses and allows the comparison of gene expression to Arabidopsis as sexual model species. Important differences in the development of the apomictic as compared to the sexual germline have been observed. While translational regulation is a feature conserved in both types of germline, polyamine and spermine/spermidine metabolism is only enriched upon initiation of the apomictic germline. In addition, key regulatory mechanisms are differentially regulated, involving hormone pathways, cell cycle control, signal transduction, and epigenetic regulatory processes. Thus, our analysis provides important new insights into gene regulation during apomictic germline development.
Methods
Plant material
A. thaliana Col-0 plants were used to isolate RNA for cloning of in situ probes. Plants were grown as described previously [12]. Seeds of B. gunnisoniana were kindly provided by Bitty Roy (University of Oregon, previously ETH Zürich) [48]. Seeds were surface sterilized and grown on MS plates for 10–14 days before transfer to a mixture of soil (ED73, Universalerde, Germany) and sand (5∶1), fertilized with Plantomaag (Syngenta, Basel, Switzerland) and Osmocote (Scotts, Marysville, USA). Plants were grown in a greenhouse chamber with 60% humidity and 16 h light/ 8 h darkness at 20°C and 16°C, respectively.
Flow cytometry
Matured green seeds were harvested from B. gunnisoniana plants and individually analysed in a Quanta SC MPL flow cytometer (Beckman-Coulter, Nyon, Switzerland). Seeds were individually transferred to 1.2 ml cluster tubes (Thermo Scientific, Wohlen, Switzerland) containing 80 µl 0.1 M citric acid and 0.1% Triton X-100. A 3 mm stainless steel bead (Schieritz & Hauenstein AG, Zwingen, Switzerland) was added to each tube prior to shaking for 4 minutes at 30 Hz on a mixer mill (MM300, Retsch GmbH, Germany). Afterwards, 80 µl of 0.1 M citric acid containing 1% Triton X-100 was added and each tube was inverted 40 times. The solution containing the nuclei was filtered though fritted deep well plates (Nunc, Thermo Scientific, Wohlen, Switzerland) into 96-well V-bottom plates (Sarstedt, Numbrecht, Germany). Nuclei were collected by filtering in a centrifuge for 5 minutes at 150 g (Centrifuge 5810R, Eppendorf, Schönebuch, Switzerland). The nuclei were resuspended in 30 µl 0.1 M citric acid containing 1% Triton X-100. The samples were either analysed directly by flow cytometer robotics (Quanta SC MPL, Beckman-Coulter, Nyon, Switzerland) or stored at 4°C overnight prior to analysis. 120 µl of staining solution (0.4 M Na2HPO4, 2.6 ml H2O, 27.4 µl DAPI (5.5 µg/µl), 0.2 µl β-mercaptoethanol) were added 2 min prior to analysis. The protocol was set to count nuclei for six minutes or until a maximum of 10'000 counts was reached. The Photo Multiplier Tube and the gain were set to have the embryo peak at around 200 on the linear fluorescent scale. B. stricta nuclei were used as external standard.
Cytological characterization
To quantitatively characterize developmental stages during germline development in B. gunnisoniana, material of 5 plants was used and averaged. Tissues were fixed in an ice-cold solution of ethanol∶acetic acid (3∶1; v/v), vacuum infiltrated on ice two times for 15 min, and left in fixative on ice over night before replacing the fixative with 70% ethanol. Tissues were cleared in chloral hydrate/glycerol/water (8∶1∶2; w/v/v), and microdissected with dissecting needles. Pictures were taken as previously described [12].
In situ hybridization
Genes for data confirmation by in situ hybridization were selected based on the following criteria: (1) expression in the B. gunnisoniana AIC and no evidence of expression in the A. thaliana MMC, (2) representing different expression levels (Table S5), (3) high homology only to the respective homologue in B. gunnisoniana (82–96% identity between A. thaliana and B. gunnisoniana nucleotide sequences; Figure S3; Supporting Information S1), and (4) gene specificity in A. thaliana. Total RNA was isolated from Arabidopsis Col-0 inflorescences and from B. gunnisoniana buds and opened flowers using the RNeasy Plant Mini Kit (QIAGEN, Hilden, Germany). During the isolation procedure, RNA was treated with DNAseI on column. Reverse transcription was done as previously described ([12]; see Table S13 for a summary of primers and cDNA templates used). Fragment cloning and in situ hybridizations were done as previously described with modifications [12], [51], [90]: in situ hybridizations were performed on 8 µm thick sections of fixed and embedded Boechera buds or flowers. Pictures were taken and processed as previously described [12].
Laser-assisted microdissection
To prepare samples for LAM, buds with ovules harbouring the AIC were chosen as previously described for selection of buds with ovules harbouring the MMC in Arabidopsis [12] with modifications: for Boechera individual buds were harvested instead of inflorescences. To obtain ovules harbouring mature gametophytes, flowers were emasculated ∼7 hours prior to fixation. The buds and flowers were fixed on ice in farmer's fixative (ethanol∶acetic acid 3∶1; v/v), vacuum infiltrated on ice two times for 15 min, and stored on ice over night before replacing the fixative with 70% ethanol. Embedding, microdissection, and LAM were done as previously described [12]. On average ∼60 sections of AICs were collected per day, or ∼25 sections for each cell-type of the mature female gametophyte. Egg and synergid cells from Arabidopsis were isolated as described previously for the central cell of Arabidopsis [13].
RNA isolation and quality control
LAM samples were stored dry at −80°C before RNA isolation. RNA isolation and quality control was done as previously described [12], [13].
Array hybridization
RNA amplification and labelling was done with the MessageAmpII Kit (Ambion, Foster City, USA) as described previously. ∼15 mg labeled aaRNA was fragmented and hybridized onto the Arabidopsis ATH1 GeneChip (Affymetrix) for 16 h at 45°C as described in the technical manual. The hybridization, staining, washing, and subsequent array scanning were performed as described previously [12]. Original data files are deposited under the Gene Expression Omnibus at NCBI (Accession Number GSE51996).
SOLiD sequencing
RNA isolation, amplification, library preparation, and SOLiD Sequencing were performed as described previously [12], except that SOLiD V4 was used for paired-end sequencing. Original data files are deposited in the NCBI database (Accession Number: SRP032961).
Reference transcriptome
As a tool for our data analysis we generated a reference transcriptome from female reproductive tissues of B. gunnisoniana at the two developmental stages of interest: (I) at megasporogenesis and (II) at the mature gametophyte stage. After isolation of mRNA and library preparation, sequencing was performed on an Illumina HiSeq 2000 instrument (see Supporting Methods S1 for details). Original data files are deposited in the NCBI SRA database (Accession Number SRP032960). The Transcriptome Shotgun Assembly project has been deposited at DDBJ/EMBL/GenBank under the accession GBAD00000000. The version described in this paper is the first version, GBAD01000000.
Blast2GO annotation of the B. gunnisoniana reference transcriptome
After quality filtering, pre-processed reads were assembled using Trinity (version r2012-06-08) with default parameter settings, except that min_kmer_cov was set to 2. For annotation with Blast2GO, trinity assembled transcripts were compared to the NCBI non-redundant protein database (nr) using blastx (in blastall version 2.2.21). E-value cutoff was set to 0.00001. Top five hits were recorded. BLASTX results in XML format were analysed using b2g4pipe (version 2.5, [53]) to assign GO terms to the query transcript sequences.
BLAT comparison of the B. gunnisoniana reference transcriptome to TAIR10 cDNA
The BLAT (version 34) comparison of the Boechera reference transcriptome and the TAIR10 cDNA sequences (updated 12/14/2010) was done with default parameters for cross species DNA mapping (-q = dnax -t = dnax). The top hits were selected using the blat utility script pslCDnafilter (globalNearBest, globalNearBest plus minCov of 80%). TAIR10 cDNA annotation of the top hits was then transferred to the query transcripts.
Mapping of SOLiD reads
To obtain expression values based on the assembled Boechera reference transcriptome, short read data was processed as described in [13]. Gene-wise expression values were then defined as the sum of the expression values of individual transcript variants. Expression values based on the A. thaliana reference genome (TAIR10) were likewise calculated as described in [13].
Defining closest Arabidopsis homologues for Boechera gene models
To identify potential homologues of known genes from A. thaliana in the assembled reference transcriptome of B. gunnisoniana we used BLAT (version 34, [54]). Sequences from Boechera were aligned to Arabidopsis cDNAs (TAIR10), allowing for a maximal intron size (-maxIntron) of 2 kb. Individual alignment scores (bitScore) and lengths between a given pair of Boechera and Arabidopsis sequences were then summed up. For each gene of interest from Arabidopsis, the Boechera homologue was then defined as the gene with the highest bitScore sum (or none if no alignments were reported or the total alignment length was below 100 bp).
Analysis of sequence divergence
To estimate the extent of sequence divergence between a certain set of genes from A. thaliana and B. gunnisoniana we used ClustalX (version 2.1, [91]) with default settings (complete alignment, draw tree). Tree files were then used to cluster the genes in the heatmap plots (R packages ape, version 3.0-8 [92] and gplots, version 2.11.0, cran.r-project.org/web/packages/gplots/index.html).
BgPANP
Microarray data were processed as described in [12], except using an updated annotation of the ATH1 microarray (brainarray.mbni.med.umich.edu, TAIRG, version 14), and an alternative list of probesets for the background estimation (“negative probes”). Probe sequences were aligned to the assembled Boechera reference transcriptome using bowtie (version 0.12.7, [93]), allowing three mismatches. Probes without any alignments were considered as “negative probe” for the PANP algorithm [11].
Gene set enrichment studies using NOISeq and EdgeR
We used the NOISeq-sim algorithm (downloaded in April 2012, http://bioinfo.cipf.es/noiseq/doku.php, [57]) to analyse differential expression of genes between RNA-Seq samples of the Boechera germline (apo_initial3, egg_cell2, central_cell2, synergid_cell2). Reads were aligned to the Boechera reference transcriptome. The normalization method was set to tmm (Trimmed mean of M, [94]), no correction for feature length was applied, and default settings were used for all other parameters, including q = 0.9 as threshold to determine differentially expressed genes. Genes identified as significantly upregulated in all three pairwise comparisons of one cell type with the other three Boechera germline samples were described as enriched in the cell type. For higher stringency analysis EdgeR was used with the biological coefficient of variation (bcv) set to 0.8 and Benjamini-Hochberg multiple testing corrections. Genes with an adjusted p value (FDR) below 0.05 were considered to be significantly differentially expressed. To identify genes differentially expressed between egg cell and central cell we applied an unadjusted p value<0.001.
Identification of genes with evidence of expression only in Boechera or Arabidopsis
See Supporting Methods S1.
Mappings
Gene Ontology (GO) terms associated with A. thaliana genes were extracted from the functional descriptions and GOSLIM mappings available on TAIR (ftp.arabidopsis.org/home/tair/Proteins/TAIR10_functional_descriptions, and (ftp.arabidopsis.org/home/tair/Ontologies/Gene_Ontology/ATH_GO_GOSLIM.txt). GO terms associated with the genes of Boechera were obtained with b2g4pipe (version 2.5, [54]). Protein family (PFAM) and gene family (FAM) annotation was used as described [95].
GO, PFAM and FAM analyses
We used the Bioconductor package topGO [96] for gene ontology analysis. To test for overrepresentation of GO terms we used a Fisher's exact test in combination with the function “weight”. As gene universe in the test for Arabidopsis MMC the whole ATH1 array genome was used, otherwise all genes annotated in the respective GO annotation were used. We used a two-sided Fisher's exact test and comparison against the gene universe as defined above to test for misrepresentation of protein family domains (PFAM) and gene families (FAM).
Heatmap clustering
Heatmaps were generated using the Bioconductor package gplots [97]. Hierachical agglomerative clustering (complete linkage) and euclidean distance was used. Normalization of RNA-Seq reads was done with the Bioconductor package DESeq [98]. Heatmaps were based on normalized log2-transformed total read counts for RNA-Seq data or log2-scale expression values generated by RMA for microarray data as previously described [12].
Venn diagrams
Venn diagrams were made with the online tool BioVenn (http://www.cmbi.ru.nl/cdd/biovenn/).
Supporting Information
Cytological characterization of reproductive development in B. gunnisoniana. (A, B) Development of the B. gunnisoniana AIC. A low percentage of AICs did not seem to divide (C) and likely arrested their development (D). (E) Dyad, (F) dyad with enlarged parietal cell or triad, (G) tetrad (artificially coloured in blue; based on the development of the integuments this tetrad is likely arrested or developmentally delayed), and (H) functional megaspore (FMS). (I, J) Mature gametophytes with unfused and fused polar nuclei, respectively. (K) Rarely more than one female gametophyte (artificially coloured in blue and pink) developed. (L–P) Seed development in young siliques after fertilization with embryo and endosperm development (L, M), young embryo developing in the absence of endosperm development (N), endosperm development without embryo development (O), and seed coat development in the absence of embryo or endosperm development (P). Black arrows point to AICs, stars mark (putative) parietal cells, white arrows point to dyads (or potential triads). Abbreviations: cen, central cell; egg, egg cell; syn, synergid cells; PN, polar nuclei; emb, embryo; end, endosperm. Scale bars are 40 µm. (Q) Summary of megaspore formation in B. gunnisoniana. In total 224 ovules were analysed. (R) Summary of mature gametophyte development in B. gunnisoniana. The percentages of mature gametophytes, gametophytes arrested at early developmental stages, gametophytes with an unexpected number of nuclei, and double gametophytes are given as analysed in 353 ovules.
(TIF)
Transcriptome analysis of the Boechera female gametes isolated by laser-assisted microdissection. (A) 6 µm thin section of a Boechera ovule harbouring the mature female gametophyte composed of egg cell, central cell, and synergid cells. Scale bar is 20 µm. (B, C) Venn diagrams showing the overlap of predicted expression in the Boechera and Arabidopsis female gametes. (B) Comparison of gene expression in the egg cell. Genes expressed in the Arabidopsis egg cell have been described before [11], [ reanalysed in 12] and were identified by RNA-Seq. Genes with evidence of expression in the Boechera egg cell were identified either by a P call with BgPANP for the egg_cell1 sample or by at least 5 reads for homologues genes when mapped to the Boechera reference transcriptome. (C) Comparison of gene expression in the central cell. Genes expressed in the Arabidopsis central cell were previously identified using RNA-Seq [13]. Genes expression in the Boechera central cell was analysed by heterologous hybridization to the ATH1 microarray (central_cell1) or by RNA-Seq (central_cell2) by mapping the reads to the Boechera reference transcriptome and identification of Arabidopsis homologues.
(TIF)
Schematic alignment of B. gunnisoniana and A. thaliana genes selected for in situ hybridization. Schematic representation of gene exon and intron structures in B. gunnisoniana and A. thaliana for 5 genes selected for in situ hybridization. Arabidopsis gene and Boechera gene identifiers are given. The region selected for in situ probe design is indicated in red. Scaling is given in kb.
(TIF)
Heatmap of read counts for genes involved in silencing and small RNA pathways. Hierarchical clustering of log2 transformed read counts for 69 Arabidopsis genes homologues in Boechera involved in small RNA and gene silencing pathways (as used in [12]). RNA-Seq data from different Arabidopsis and B. gunnisoniana cell- and tissue types were used [13], [62]–[66]. The hierarchical clustering of samples and genes was based on euclidean distance and hierarchical agglomerative clustering. Colours are scaled per row. Red denotes high and black denotes low expression.
(TIF)
Heatmap of expression of AGO genes. (A) Hierarchical clustering of log2 transformed read counts of AtAGO genes and Boechera [13], [62]–[66]. (B) Hierarchical clustering of log2 scale expression values of AtAGO genes in Arabidopsis as analysed by the RMA algorithm [12]. (A, B) The hierarchical clustering of samples and genes was based on euclidean distance and hierarchical agglomerative clustering. Colours were scaled by row. Red denotes high and black low expression.
(TIF)
Analysis of sequence divergence and heatmap of expression. Analysis of sequence divergence of members of the ARI (A) and AGO (B) gene family and read counts assigned.
(TIF)
Heatmap of expression of ARI genes. (A) Hierarchical clustering of log2 transformed read counts of AtARI genes and Boechera homologues including datasets from different transcriptional studies [13], [62]–[66]. (B) Hierarchical clustering of log2 scale expression values of AtARI genes in Arabidopsis as analysed by the RMA algorithm [12]. The hierarchical clustering of samples and genes was based on euclidean distance and hierarchical agglomerative clustering. Colours were scaled by row. Red denotes high and black low expression.
(TIF)
Annotation of B. gunnisoniana genes. Boechera genes were annotated using Blast2GO.
(ZIP)
Assignment of Arabidopsis homologues to Boechera genes.
(TXT)
BgPANP expression calls. Datasheet with BgPANP present (P) and absent (A) calls and p values in the AIC (apo_initial1, apo_initial2), the surrounding nucellus (sporo_nucellus1, and _2), egg cell (egg_cell1), central cell (central_cell), and synergid cells (synergid_cell) of Boechera.
(XLS)
Expression values of individual variants of Boechera genes aligned to the reference transcriptome.
(TXT)
Expression of genes selected for independent data confirmation by in situ analysis. P/A calls as analysed with BgPANP for microarray samples and read counts for B. gunnisoniana homologues generated by mapping to the B. gunnisoniana reference transcriptome.
(PDF)
Gene ontology (GO) analysis. (A) Molecular functions identified to be up-regulated based on 3'509 genes identified to be up-regulated in the egg cell as compared to the AIC, central cell and synergid cells. (B) Biological Processes identified to be up-regulated based on 1'806 genes identified to be up-regulated in the egg cell as compared to the AIC, central cell and synergid cells.
(PDF)
Gene ontology (GO) analysis. Biological processes significantly upregulated in 142 genes enriched identified by EdgeR analysis in the B. gunnisoniana AIC as compared to the cell types of the mature female gametophyte.
(PDF)
Analysis of protein family (PFAM) enrichment. Analysis of PFAM domains enriched in 852 genes with evidence of expression in the Arabidopsis MMC but not in the B. gunnisoniana AIC as tested by a two sided Fisher test. P values≤0.01 were considered significant.
(PDF)
Analysis of protein family (PFAM) enrichment. Analysis of PFAM domains enriched in 2'146 genes with evidence of expression in the Arabidopsis but not in the B. gunnisoniana central cell as tested by a two sided Fisher test. P values≤0.01 were considered significant.
(PDF)
Analysis of PFAM domains enriched in the apomictic Boechera germline. Significant enrichment of PFAM domains based on 901, 5'273 and 4'902 genes with evidence of expression in the AIC, egg cell and central cell of Boechera but not in the corresponding cell types of sexual Arabidopsis as analysed by two sided Fisher's exact test (p value<0.01).
(PDF)
Enrichment of gene families in the Boechera female gametes. Significant enrichment of gene families based on 5'273 and 4'902 genes with evidence of expression in the egg cell and central cell of Boechera but not in the corresponding cell types of sexual Arabidopsis as analysed by two sided Fisher's exact test (p value<0.01).
(PDF)
Analysis of expression of core cell cycle genes. Lists of core cell cycle genes only found to be expressed in the AIC or the MMC, but not other way round.
(XLS)
Primers and templates used for cloning of in situ probes.
(PDF)
Methods description on the generation of the B. gunnisoniana reference transcriptome. Methods description on the identification of genes with evidence of expression only in Boechera or in Arabidopsis.
(PDF)
Alignment of in situ probe sequences from Arabidopsis to the homologues B. gunnisoniana genes generated by BLAT [54].
(ZIP)
Description of genes involved in the small RNA pathway or in the DNA methylation pathway only detected in Arabidopsis or in Boechera and description on the influence of sequence similarities between Arabidopsis and Boechera homologues on the distribution of count data. As examples the ARI and the AGO gene families are discussed.
(PDF)
Acknowledgments
We are grateful to Samuel E. Wüst (University of Zürich) for helpful discussions, providing tools for data analysis, and for critical reading of the manuscript. We thank Catharine Aquino (Functional Genomics Center Zürich) for help with sequencing the reference transcriptome, Bitty Roy (ETH Zürich; presently University of Oregon) for providing B. gunnisoniana seeds and plants, Charles Spillane and Amal J. Johnston (University of Zürich; presently NUI Galway and University of Heidelberg, respectively) for maintaining and propagating B. gunnisoniana, Valeria Gagliardini, Arturo Bolanos, Christof Eichenberger and Peter Kopf (University of Zürich) for general lab support, and Christian Frey and Karl Huwiler (University of Zürich) for plant care and greenhouse maintenance.
Funding Statement
This project was supported by the University of Zürich, the Marie Curie project IDEAGENA (to AS), and grants from the “Staatssekretariat für Bildung und Forschung” in the framework of COST action FA0903 (to AS and UG) and the Swiss National Science Foundation (to UG). PR was supported by the DFG Cluster of excellence Inflammation at Interfaces (CL Nucleotide lab). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Nogler GA (1984) Gametophytic apomixis. In: Embryology of Angiosperms. Edited by Johri BM. Berlin: Springer; pp. 475–518. [Google Scholar]
- 2. Savidan Y (2000) Apomixis: Genetics and breeding. Plant Breeding Reviews 18: 13–86. [Google Scholar]
- 3. Spillane C, Steimer A, Grossniklaus U (2001) Apomixis in agriculture: The quest for clonal seeds. Sex Plant Reprod 14: 179–87. [DOI] [PubMed] [Google Scholar]
- 4. Bicknell RA, Koltunow AM (2004) Understanding apomixis: Recent advances and remaining conundrums. Plant Cell 16: 228–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Spillane C, Curtis MD, Grossniklaus U (2004) Apomixis technology development - virgin births in farmers' fields? Nat Biotechnol 22: 687–691. [DOI] [PubMed] [Google Scholar]
- 6. Koltunow A, Grossniklaus U (2003) Apomixis: A developmental perspective. Annu Rev Plant Biol 54: 547–574. [DOI] [PubMed] [Google Scholar]
- 7.Grossniklaus U (2001) From sexuality to apomixis: molecular and genetic approaches. In: The Flowering of Apomixis: From Mechanisms to Genetic Engineering. Edited by Savidan Y, Carman J, Dresselhaus T. Mexico DF: CIMMYT; pp.168–211. [Google Scholar]
- 8. Sprunck S, Gross-Hardt R (2011) Nuclear behavior, cell polarity, and cell specification in the female gametophyte. Sex Plant Reprod 24: 123–136. [DOI] [PubMed] [Google Scholar]
- 9. Koltunow AM (1993) Apomixis: Embryo sacs and embryos formed without meiosis or fertilization in ovules. Plant Cell 5: 1425–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rodkienwicz B (1970) Callose in cell walls during megasporogenesis in angiosperms. Planta 93: 39–47. [DOI] [PubMed] [Google Scholar]
- 11. Wüst SE, Vijverberg K, Schmidt A, Weiss M, Gheyselinck J, et al. (2010) Arabidopsis female gametophyte gene expression map reveals similarities between plant and animal gametes. Curr Biol 20: 506–512. [DOI] [PubMed] [Google Scholar]
- 12. Schmidt A, Wüst SE, Vijverberg K, Baroux C, Kleen D, et al. (2011) Transcriptome analysis of the Arabidopsis megaspore mother cell uncovers the importance of RNA helicases for plant germline development. PLoS Biol 9: e1001155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schmid MW, Schmidt A, Klostermeier UC, Barann M, Rosenstiel P, et al. (2012) A powerful method for transcriptional profiling of specific cell types in eukaryotes: laser-assisted microdissection and RNA sequencing. PLoS ONE 7: e29685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Grossniklaus U, Nogler GA, van Dijk PJ (2001) How to avoid sex: the genetic control of gametophytic apomixis. Plant Cell 2001 13: 1491–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Savidan Y (1982) Nature et hérédité de l' apomixie chez Panicum maximum Jacq. Trav. & Doc. Orstom 153: 1–159. [Google Scholar]
- 16. Nogler GA (1984) Genetics of apospory in apomictic Ranunculus auricomus . V Conclusion Bot Helv 94: 411–422. [Google Scholar]
- 17. Sherwood RT, Berg CC, Young BA (1994) Inheritance of apospory in buffelgrass. Crop Sci 34: 1490–1494. [Google Scholar]
- 18. Grimanelli D, Leblanc O, Espinosa E, Perotti E, González de León D, et al. (1998) Mapping diplosporous apomixis in tetraploid Tripsacum: one gene or several genes? Heredity 80: 33–39. [DOI] [PubMed] [Google Scholar]
- 19. Ozias-Akins P, Roche D, Hanna WW (1998) Tight clustering and hemizygosity of apomixis-linked molecular markers in Pennisetum squamulatum implies genetic control of apospory by a divergent locus that may have no allelic form in sexual genotypes. Proc Natl Acad Sci U S A 95: 5127–5132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Noyes RD, Rieseberg LH (2000) Two independent loci control agamospermy (apomixis) in the triploid flowering plant Erigeron annuus . Genetics 155: 379–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Valle CB, Miles JW (2001) Breeding of apomictic species. In: The Flowering of Apomixis: From Mechanisms to Genetic Engineering. Edited by Savidan Y, Carman J, Dresselhaus T. Mexico DF: CIMMYT; pp. 137–152. [Google Scholar]
- 22. Cáceres ME, Matzk F, Busti A, Pupilli F, Arcioni S (2001) Apomixis and sexuality in Paspalum simplex: characterization of the mode of reproduction in segregating progenies by different methods. Sex Plant Reprod 14: 201–206. [DOI] [PubMed] [Google Scholar]
- 23. van Dijk PJ, Bakx-Schotman JM (2004) Formation of unreduced megaspores (diplospory) in apomictic dandelions (Taraxacum officinale, s.l.) is controlled by a sex-specific dominant locus. Genetics 166: 483–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Koltunow AM, Johnson SD, Rodrigues JC, Okada T, Hu Y, Tsuchiya T, et al. (2011) Sexual reproduction is the default mode in apomictic Hieracium subgenus Pilosella, in which two dominant loci function to enable apomixis. Plant J 66: 890–902. [DOI] [PubMed] [Google Scholar]
- 25. Schranz ME, Kantama L, de Jong H, Mitchell-Olds T (2006) Asexual reproduction in a close relative of Arabidopsis: a genetic investigation of apomixis in Boechera (Brassicaceae). New Phytol 171: 425–438. [DOI] [PubMed] [Google Scholar]
- 26. Grimanelli D, Leblanc O, Perotti E, Grossniklaus U (2001) Developmental genetics of gametophytic apomixis. Trends Genet 17: 597–604. [DOI] [PubMed] [Google Scholar]
- 27. Sharbel TF, Voigt ML, Corral JM, Thiel T, Varshney A, et al. (2009) Molecular signatures of apomictic and sexual ovules in the Boechera holboellii complex. Plant J 104: 14026–14031. [DOI] [PubMed] [Google Scholar]
- 28. Sharbel TF, Voigt ML, Corral JM, Galla G, Kumlehn J, et al. (2010) Apomictic and sexual ovules of Boechera display heterochronic global gene expression patterns. Plant Cell 22: 655–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Leblanc O, Armstead I, Pessino S, Ortiz JP, Evans C, et al. (1997) Non-radioactive mRNA fingerprinting to visualise gene expression in mature ovaries of Brachiaria hybrids derived from B. brizantha, an apomictic tropical forage. Plant Sci 126: 49–58. [Google Scholar]
- 30. Rodrigues JC, Carbral GB, Dusi DM, de Mello LV, Rigden DJ, et al. (2003) Identification of differentially expressed cDNA sequences in ovaries of sexual and apomictic plants of Brachiaria brizantha . Plant Mol Biol 53: 745–757. [DOI] [PubMed] [Google Scholar]
- 31. Okada T, Hu Y, Tucker MR, Taylor JM, Johnson SD, et al. (2013) Enlarging cells initiating apomixis in Hieracium praealtum transition to an embryo sac program prior to entering mitosis. Plant Physiol 163: 216–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vielle-Calzada JP, Nuccio ML, Budiman MA, Burson BL, Hussey MA, et al. (1996) Comparative gene expression in sexual and apomictic ovaries of Pennisetum ciliare (L.) Link. Plant Mol Biol 32: 1085–1092. [DOI] [PubMed] [Google Scholar]
- 33. Sahu PP, Gupta S, Malaviya DR, Roy AK, Kaushal P, et al. (2012) Transcriptome analysis of differentially expressed genes during embryo sac development in apomeiotic non-parthenogenetic interspecific hybrid of Pennisetum glaucum . Mol Biotechnol 51: 262–271. [DOI] [PubMed] [Google Scholar]
- 34. Pessino SC, Espinoza F, Martinez EJ, Ortiz JP, Valle EM, et al. (2001) Isolation of cDNA clones differentially expressed in flowers of apomictic and sexual Paspalum notatum . Hereditas 134: 35–42. [DOI] [PubMed] [Google Scholar]
- 35. Polegri L, Calderini O, Arcioni S, Pupilli F (2010) Specific expression of apomixis-linked alleles revealed by comparative transcriptomic analysis of sexual and apomictic Paspalum simplex Morong flowers. J Exp Bot 61: 1869–83. [DOI] [PubMed] [Google Scholar]
- 36. Ochogavía AC, Seijo JG, González AM, Podio M, Duarte Silveira E, et al. (2011) Characterization of retrotransposon sequences expressed in inflorescences of apomictic and sexual Paspalum notatum plants. Sex Plant Reprod 24: 231–246. [DOI] [PubMed] [Google Scholar]
- 37. Barcaccia G, Varotto S, Meneghetti S, Albertini E, Porceddu A, et al. (2001) Analysis of gene expression during flowering in apomeiotic mutants of Medicago spp.: cloning ESTs and candidate genes for apomeiosis. Sex Plant Reprod 14: 233–238. [DOI] [PubMed] [Google Scholar]
- 38. Chen L, Miyazaki C, Kojima A, Saito A, Adachi T (1999) Isolation and characterization of a gene expressed during early embryo sac development in apomictic Guinea grass (Panicum maximum). J Plant Physiol 154: 55–62. [Google Scholar]
- 39. Albertini E, Marconi G, Barcaccia G, Raggi L, Falcinelli M (2004) Isolation of candidate genes for apomixis in Poa pratensis L. Plant Mol Biol 56: 879–894. [DOI] [PubMed] [Google Scholar]
- 40. Albertini E, Marconi G, Reale L, Barcaccia G, Porceddu A, et al. (2005) SERK and APOSTART. Candidate genes for apomixis in Poa pratensis . Plant Physiol 138: 2185–2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tucker MR, Araujo AC, Paech NA, Hecht V, Schmidt ED, et al. (2003) Sexual and apomictic reproduction in Hieracium subgenus pilosella are closely interrelated developmental pathways. Plant Cell 15: 1524–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Olmedo-Monfil V, Durán-Figueroa N, Arteaga-Vázquez M, Demesa-Arévalo E, Autran D, et al. (2010) Control of female gamete formation by a small RNA pathway in Arabidopsis . Nature 464: 628–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Garcia-Aguilar M, Michaud C, Leblanc O, Grimanelli D (2010) Inactivation of a DNA methylation pathway in maize reproductive organs results in apomixis-like phenotypes. Plant Cell 22: 3249–3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Singh M, Goel S, Meeley RB, Dantec C, Parrinello H, et al. (2011) Production of viable gametes without meiosis in maize deficient for an ARGONAUTE protein. Plant Cell 23: 443–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ravi M, Marimuthu MP, Siddiqi I (2008) Gamete formation without meiosis in Arabidopsis . Nature 451: 1121–1124. [DOI] [PubMed] [Google Scholar]
- 46. d'Erfurth I, Jolivet S, Froger N, Catrice O, Novatchkova M, et al. (2009) Turning meiosis into mitosis. PLoS Biol 7: e1000124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Matzk F, Meister A, Schubert I (2010) An efficient screen for reproductive pathways using mature seeds of monocots and dicots. Plant J 21: 97–108. [DOI] [PubMed] [Google Scholar]
- 48. Roy BA (1995) The breeding system of six species of Arabis (Brassicaceae). Am J Bot 82: 869–877. [Google Scholar]
- 49. Taskin KM, Turgut K, Scott RJ (2004) Apomictic development in Arabis gunnisoniana . Isr J Plant Sci 52: 155–160. [Google Scholar]
- 50. Aliyu OM, Schranz ME, Sharbel TF (2010) Quantitative variation for apomictic reproduction in the genus Boechera (Brassicaceae). Am J Bot 97: 1719–1731. [DOI] [PubMed] [Google Scholar]
- 51.Johnston AJ (2007) Functional genomics of sexual and asexual reproduction in Arabidopsis and relatives. PhD Thesis, University of Zürich, Switzerland. [Google Scholar]
- 52. Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, et al. (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nature Biotechnol 29: 644–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, et al. (2005) Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21: 3674–3676. [DOI] [PubMed] [Google Scholar]
- 54. Kent WJ (2002) BLAT-the BLAST-like alignment tool. Genome Res 12: 656–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bar-Or C, Czosnek H, Koltai H (2007) Cross-species microarray hybridizations: a developing tool for studying species diversity. Trends Genet 23: 200–207. [DOI] [PubMed] [Google Scholar]
- 56. Tang F, Barbacioru C, Wang Y, Nordman E, Lee C, et al. (2009) mRNA-Seq whole-transcriptome analysis of a single cell. Nat Methods 6: 377–382. [DOI] [PubMed] [Google Scholar]
- 57. Tarazona S, García-Alcalde F, Dopazo J, Ferrer A, Conesa A (2011) Differential expression in RNA-seq: a matter of depth. Genome Res 21: 2213–2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Robinson MD, McCarthy DJ, Smyth GK (2010) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26: 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wijnker E, Schnittger A (2013) Control of the meiotic cell division program in plants. Plant Reprod 26: 143–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Vandepoele K, Raes J, De Veylder L, Rouzé P, Rombauts S, et al. (2002) Genome-wide analysis of core cell cycle genes in Arabidopsis . Plant Cell 14: 903–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Gutierrez C (2009) The Arabidopsis cell division cycle. Arabidopsis Book 7: e0120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Nodine MD, Bartel DP (2010) MicroRNAs prevent precocious gene expression and enable pattern formation during plant embryogenesis. Genes Dev 24: 2678–2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Chen C, Farmer AD, Langley RJ, Mudge J, Crow JA, et al. (2010) Meiosis-specific gene discovery in plants: RNA-Seq applied to isolated Arabidopsis male meiocytes. BMC Plant Biol 10: 280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Torti S, Fornara F, Vincent C, Andrés F, Nordström K, et al. (2012) Analysis of the Arabidopsis shoot meristem transcriptome during floral transition identifies distinct regulatory patterns and a leucine-rich repeat protein that promotes flowering. Plant Cell 24: 444–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Filichkin SA, Priest HD, Givan SA, Shen R, Bryant DW (2010) Genome-wide mapping of alternative splicing in Arabidopsis thaliana . Genome Res 20: 45–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Lister R, O'Malley RC, Tonti-Filippini J, Gregory BG, Berry CC, et al. (2008) Highly integrated single-base resolution maps of the epigenome in Arabidopsis . Cell 133: 523–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Koszegi D, Johnston AJ, Rutten T, Czihal A, Altschmied L, et al. (2011) Members of the RKD transcription factor family induce an egg cell-like gene expression program. Plant J 67: 280–91. [DOI] [PubMed] [Google Scholar]
- 68. Imai A, Matsuyama T, Hanzawa Y, Akiyama T, Tamaoki M, et al. (2004) Spermidine synthase genes are essential for survival of Arabidopsis . Plant Physiol 135: 1565–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wu XB, Wang J, Liu JH, Deng XX (2009) Involvement of polyamine biosynthesis in somatic embryogenesis of Valencia sweet orange (Citrus sinensis) induced by glycerol. J Plant Physiol 166: 52–62. [DOI] [PubMed] [Google Scholar]
- 70. De-la-Peña C, Galaz-Avalos RM, Loyola-Vargas VM (2008) Possible role of light and polyamines in the onset of somatic embryogenesis of Coffea canephora . Mol Biotechnol 39: 215–24. [DOI] [PubMed] [Google Scholar]
- 71. Dutra NT, Silveira V, de Azevedo IG, Gomes-Neto LR, Façanha AR, et al. (2013) Polyamines affect the cellular growth and structure of pro-embryogenic masses in Araucaria angustifolia embryogenic cultures through the modulation of proton pump activities and endogenous levels of polyamines. Physiol Plant 148: 121–32. [DOI] [PubMed] [Google Scholar]
- 72. Ha HC, Sirisoma NS, Kuppusamy P, Zweier JL, Woster PM, et al. (1998) The natural polyamine spermidine functions directly as free radical scavenger. Proc Natl Acad Sci USA 95: 11140–11145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Hörandl E, Hadacek F (2013) The oxidative damage initiation hypothesis for meiosis. Plant Reprod DOI 10.1007s0049701302347/s00497-013-0234-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Martin MV, Fiol DF, Sundaresan V, Zabaleta EJ, Pagnussat GC (2013) oiwa, a female gametophytic mutant impaired in a mitochondrial manganese-superoxide dismutase, reveals crucial roles for reactive oxygen species during embryo sac development and fertilization in Arabidopsis . Plant Cell 25: 1573–1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Conner JA, Goel S, Gunawan G, Cordonnier-Pratt MM, Johnson VE, et al. (2008) Sequence analysis of bacterial artificial chromosome clones from the apospory-specific genomic region of Pennisetum and Cenchrus . Plant Physiol 147: 1396–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Guilfoyle TJ, Ulmasov T, Hagen G (1998) The ARF family of transcription factors and their role in plant hormone-responsive transcription. Cell Mol Life Sci 54: 619–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Rademacher EH, Möller B, Lokerse AS, Llavata-Peris CI, van den Berg W, et al. (2011) A cellular expression map of the Arabidopsis AUXIN RESPONSE FACTOR gene family. Plant J 68: 597–606. [DOI] [PubMed] [Google Scholar]
- 78. Craig KL, Tyers M (1999) The F-box: a new motif for ubiquitin dependent proteolysis in cell cycle regulation and signal transduction. Prog Biophys Mol Biol 72: 299–328. [DOI] [PubMed] [Google Scholar]
- 79. Lechner E, Achard P, Vansiri A, Potuschak T, Genschik P (2006) F-box proteins everywhere. Curr Opin Plant Biol 9: 631–638. [DOI] [PubMed] [Google Scholar]
- 80. Amiteye S, Corral JM, Vogel H, Sharbel TF (2011) Analysis of conserved microRNAs in floral tissues of sexual and apomictic Boechera species. BMC Genomics 12: 500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Schallau A, Arzenton F, Johnston AJ, Hähnel U, Koszegi D, et al. (2010) Identification and genetic analysis of the APOSPORY locus in Hypericum perforatum L. Plant J 62: 773–784. [DOI] [PubMed] [Google Scholar]
- 82. Mladek C, Guger K, Hauser MT (2003) Identification and characterization of the ARIADNE gene family in Arabidopsis. A group of putative E3 ligases. Plant Physiol 131: 27–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Marimuthu MP, Jolivet S, Ravi M, Pereira L, Davda JN, et al. (2011) Synthetic clonal reproduction through seeds. Science 331: 876. [DOI] [PubMed] [Google Scholar]
- 84. Crismani W, Girard C, Mercier R (2013) Tinkering with meiosis. J Exp Bot 64: 55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Cook GS, Grønlund AL, Siciliano I, Spadafora N, Amini M, et al. (2013) Plant WEE1 kinase is cell cycle regulated and removed at mitosis via the 26S proteasome machinery. J Exp Bot 64: 2093–2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Heckmann S, Lermontova I, Berckmans B, De Veylder L, Bäumlein H, et al. (2011) The E2F transcription factor family regulates CENH3 expression in Arabidopsis thaliana . Plant J 68: 646–656. [DOI] [PubMed] [Google Scholar]
- 87. Zeng Y, Conner J, Ozias-Akins P (2011) Identification of ovule transcripts from the Apospory-Specific Genomic Region (ASGR)-carrier chromosome. BMC Genomics 12: 206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Grossniklaus U, Schneitz K (1998) The molecular and genetic basis of ovule and megagametophyte development. Semin Cell Dev Biol 9: 227–238. [DOI] [PubMed] [Google Scholar]
- 89. Tucker MR, Okada T, Johnson SD, Takaiwa F, Koltunow AM (2012) Sporophytic ovule tissues modulate the initiation and progression of apomixis in Hieracium . J Exp Bot 63: 3229–3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Vielle-Calzada J-P, Thomas J, Spillane C, Coluccio A, Hoeppner MA, Grossniklaus U (1999) Maintenance of genomic imprinting at the Arabidopsis MEDEA locus requires zygotic DDM1 activity. Genes Dev 13: 2971–2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, et al. (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23: 2947–2948. [DOI] [PubMed] [Google Scholar]
- 92. Paradis E, Claude J, Strimmer K (2004) APE: Analysis of phylogenetics and evolution in the R language. Bioinformatics 20: 289–290. [DOI] [PubMed] [Google Scholar]
- 93. Langmead B, Trapnell C, Pop M, Salzberg SL (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Robinson MD, Oshlack A (2010) A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol 11: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Wüst SE, O'Maoileidigh DS, Rae L, Kwasniewska K, Raganelli A, Hanczaryk K, et al. (2012) Molecular basis for the specification of floral organs by APETALA3 and PISTILLATA. Proc Natl Acad Sci U S A 109: 13452–13457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Alexa A, Rahnenführer J (2009) Gene set enrichment analysis with topGO. (www.bioconductor.org).
- 97.Warnes G, Bolker B, Lumley T (2010) gplots: Various R programming tools for plotting data. (www.bioconductor.org).
- 98. Anders S, Huber W (2010) Differential expression analysis for sequence count data. Genome Biol 11: R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Cytological characterization of reproductive development in B. gunnisoniana. (A, B) Development of the B. gunnisoniana AIC. A low percentage of AICs did not seem to divide (C) and likely arrested their development (D). (E) Dyad, (F) dyad with enlarged parietal cell or triad, (G) tetrad (artificially coloured in blue; based on the development of the integuments this tetrad is likely arrested or developmentally delayed), and (H) functional megaspore (FMS). (I, J) Mature gametophytes with unfused and fused polar nuclei, respectively. (K) Rarely more than one female gametophyte (artificially coloured in blue and pink) developed. (L–P) Seed development in young siliques after fertilization with embryo and endosperm development (L, M), young embryo developing in the absence of endosperm development (N), endosperm development without embryo development (O), and seed coat development in the absence of embryo or endosperm development (P). Black arrows point to AICs, stars mark (putative) parietal cells, white arrows point to dyads (or potential triads). Abbreviations: cen, central cell; egg, egg cell; syn, synergid cells; PN, polar nuclei; emb, embryo; end, endosperm. Scale bars are 40 µm. (Q) Summary of megaspore formation in B. gunnisoniana. In total 224 ovules were analysed. (R) Summary of mature gametophyte development in B. gunnisoniana. The percentages of mature gametophytes, gametophytes arrested at early developmental stages, gametophytes with an unexpected number of nuclei, and double gametophytes are given as analysed in 353 ovules.
(TIF)
Transcriptome analysis of the Boechera female gametes isolated by laser-assisted microdissection. (A) 6 µm thin section of a Boechera ovule harbouring the mature female gametophyte composed of egg cell, central cell, and synergid cells. Scale bar is 20 µm. (B, C) Venn diagrams showing the overlap of predicted expression in the Boechera and Arabidopsis female gametes. (B) Comparison of gene expression in the egg cell. Genes expressed in the Arabidopsis egg cell have been described before [11], [ reanalysed in 12] and were identified by RNA-Seq. Genes with evidence of expression in the Boechera egg cell were identified either by a P call with BgPANP for the egg_cell1 sample or by at least 5 reads for homologues genes when mapped to the Boechera reference transcriptome. (C) Comparison of gene expression in the central cell. Genes expressed in the Arabidopsis central cell were previously identified using RNA-Seq [13]. Genes expression in the Boechera central cell was analysed by heterologous hybridization to the ATH1 microarray (central_cell1) or by RNA-Seq (central_cell2) by mapping the reads to the Boechera reference transcriptome and identification of Arabidopsis homologues.
(TIF)
Schematic alignment of B. gunnisoniana and A. thaliana genes selected for in situ hybridization. Schematic representation of gene exon and intron structures in B. gunnisoniana and A. thaliana for 5 genes selected for in situ hybridization. Arabidopsis gene and Boechera gene identifiers are given. The region selected for in situ probe design is indicated in red. Scaling is given in kb.
(TIF)
Heatmap of read counts for genes involved in silencing and small RNA pathways. Hierarchical clustering of log2 transformed read counts for 69 Arabidopsis genes homologues in Boechera involved in small RNA and gene silencing pathways (as used in [12]). RNA-Seq data from different Arabidopsis and B. gunnisoniana cell- and tissue types were used [13], [62]–[66]. The hierarchical clustering of samples and genes was based on euclidean distance and hierarchical agglomerative clustering. Colours are scaled per row. Red denotes high and black denotes low expression.
(TIF)
Heatmap of expression of AGO genes. (A) Hierarchical clustering of log2 transformed read counts of AtAGO genes and Boechera [13], [62]–[66]. (B) Hierarchical clustering of log2 scale expression values of AtAGO genes in Arabidopsis as analysed by the RMA algorithm [12]. (A, B) The hierarchical clustering of samples and genes was based on euclidean distance and hierarchical agglomerative clustering. Colours were scaled by row. Red denotes high and black low expression.
(TIF)
Analysis of sequence divergence and heatmap of expression. Analysis of sequence divergence of members of the ARI (A) and AGO (B) gene family and read counts assigned.
(TIF)
Heatmap of expression of ARI genes. (A) Hierarchical clustering of log2 transformed read counts of AtARI genes and Boechera homologues including datasets from different transcriptional studies [13], [62]–[66]. (B) Hierarchical clustering of log2 scale expression values of AtARI genes in Arabidopsis as analysed by the RMA algorithm [12]. The hierarchical clustering of samples and genes was based on euclidean distance and hierarchical agglomerative clustering. Colours were scaled by row. Red denotes high and black low expression.
(TIF)
Annotation of B. gunnisoniana genes. Boechera genes were annotated using Blast2GO.
(ZIP)
Assignment of Arabidopsis homologues to Boechera genes.
(TXT)
BgPANP expression calls. Datasheet with BgPANP present (P) and absent (A) calls and p values in the AIC (apo_initial1, apo_initial2), the surrounding nucellus (sporo_nucellus1, and _2), egg cell (egg_cell1), central cell (central_cell), and synergid cells (synergid_cell) of Boechera.
(XLS)
Expression values of individual variants of Boechera genes aligned to the reference transcriptome.
(TXT)
Expression of genes selected for independent data confirmation by in situ analysis. P/A calls as analysed with BgPANP for microarray samples and read counts for B. gunnisoniana homologues generated by mapping to the B. gunnisoniana reference transcriptome.
(PDF)
Gene ontology (GO) analysis. (A) Molecular functions identified to be up-regulated based on 3'509 genes identified to be up-regulated in the egg cell as compared to the AIC, central cell and synergid cells. (B) Biological Processes identified to be up-regulated based on 1'806 genes identified to be up-regulated in the egg cell as compared to the AIC, central cell and synergid cells.
(PDF)
Gene ontology (GO) analysis. Biological processes significantly upregulated in 142 genes enriched identified by EdgeR analysis in the B. gunnisoniana AIC as compared to the cell types of the mature female gametophyte.
(PDF)
Analysis of protein family (PFAM) enrichment. Analysis of PFAM domains enriched in 852 genes with evidence of expression in the Arabidopsis MMC but not in the B. gunnisoniana AIC as tested by a two sided Fisher test. P values≤0.01 were considered significant.
(PDF)
Analysis of protein family (PFAM) enrichment. Analysis of PFAM domains enriched in 2'146 genes with evidence of expression in the Arabidopsis but not in the B. gunnisoniana central cell as tested by a two sided Fisher test. P values≤0.01 were considered significant.
(PDF)
Analysis of PFAM domains enriched in the apomictic Boechera germline. Significant enrichment of PFAM domains based on 901, 5'273 and 4'902 genes with evidence of expression in the AIC, egg cell and central cell of Boechera but not in the corresponding cell types of sexual Arabidopsis as analysed by two sided Fisher's exact test (p value<0.01).
(PDF)
Enrichment of gene families in the Boechera female gametes. Significant enrichment of gene families based on 5'273 and 4'902 genes with evidence of expression in the egg cell and central cell of Boechera but not in the corresponding cell types of sexual Arabidopsis as analysed by two sided Fisher's exact test (p value<0.01).
(PDF)
Analysis of expression of core cell cycle genes. Lists of core cell cycle genes only found to be expressed in the AIC or the MMC, but not other way round.
(XLS)
Primers and templates used for cloning of in situ probes.
(PDF)
Methods description on the generation of the B. gunnisoniana reference transcriptome. Methods description on the identification of genes with evidence of expression only in Boechera or in Arabidopsis.
(PDF)
Alignment of in situ probe sequences from Arabidopsis to the homologues B. gunnisoniana genes generated by BLAT [54].
(ZIP)
Description of genes involved in the small RNA pathway or in the DNA methylation pathway only detected in Arabidopsis or in Boechera and description on the influence of sequence similarities between Arabidopsis and Boechera homologues on the distribution of count data. As examples the ARI and the AGO gene families are discussed.
(PDF)