

Abstract
The Rho family GTPase effector IRSp53 has essential roles in filopodia formation and neuronal development, but its regulatory mechanism is poorly understood. IRSp53 contains a membrane-binding BAR domain followed by an unconventional CRIB motif that overlaps with a proline-rich region (CRIB–PR) and an SH3 domain that recruits actin cytoskeleton effectors. Using a fluorescence reporter assay, we show that human IRSp53 adopts a closed inactive conformation that opens synergistically with the binding of human Cdc42 to the CRIB–PR and effector proteins, such as the tumor-promoting factor Eps8, to the SH3 domain. The crystal structure of Cdc42 bound to the CRIB–PR reveals a new mode of effector binding to Rho family GTPases. Structure-inspired mutations disrupt autoinhibition and Cdc42 binding in vitro and decouple Cdc42- and IRSp53-dependent filopodia formation in cells. The data support a combinatorial mechanism of IRSp53 activation.
The ability to reshape the plasma membrane underlies vital cellular processes such as cell migration, phagocytosis, axonal guidance and tumor metastasis1. Bin-Amphiphysin-Rvs (BAR)-domain proteins that sit at the interface between the plasma membrane and the actin cytoskeleton, have prominent roles in the regulation of these processes2,3. The inverted BAR (I-BAR) protein IRSp53 is a prototypical example of such proteins, linking Rho family GTPase signaling to remodeling of the plasma membrane and the actin cytoskeleton4,5. The N-terminal 230 amino acids (aa) of IRSp53 form the I-BAR domain (referred to here as the BAR domain), which is an α-helical, antiparallel dimerization and membrane-binding fold6,7. Following the BAR domain is an unconventional Cdc42 and Rac interactive binding (CRIB) motif, which is unique in that it contains only three N-terminal consensus residues, whereas its C-terminal half consists of a proline-rich (PR) sequence featuring a canonical SH3 domain–binding site (Fig. 1a,b). Here, we refer to the combined partial CRIB and PR sites as the CRIB–PR domain. An SH3 domain lies 83 aa C terminal to the CRIB–PR domain and is followed by a variable, isoform-specific C-terminal tail8,9. The region between the CRIB–PR and SH3 domains contains several phosphorylation sites that have a role in binding Tiam1 (ref. 10) and 14-3-3 (refs. 11–13), offering alternative pathways for IRSp53 regulation.
Figure 1.

Autoinhibition and activation of IRSp53 by Cdc42. (a) Domain organization of IRSp53 and constructs used in this study (WWB, WW domain–binding site; PDZB, PDZ domain–binding site). (b) Comparison of IRSp53’s CRIB–PR to canonical CRIB motifs (top) and targets of the SH3 domain of IRSp53 (bottom). Highlighted in orange are residues conserved in the CRIB motifs of human PAK1 (UniProt Q13153), PAK4 (UniProt O96013), PAK6 (UniProt Q9NQU5), N-WASP (UniProt O00401), ACK1 (UniProt Q07912) and PAR6B (UniProt Q9BYG5). Highlighted in cyan are proline residues conserved in SH3-binding targets: human Eps8 (UniProt Q12929), E. coli EspFU species (UniProt Q8X482 and C6UYI3) and human atrophin-1 (UniProt P54259). (c,d) ITC experiments in which 200 (c) or 400 (d) μM GTPases (color coded) were titrated into a cell containing 8 μM FL or 15 μM CRIB–PR, respectively. The stoichiometry, N, and dissociation constant, Kd, are shown for each titration. (e) ITC experiments in which 1.3 mM SH3 domain was titrated into a cell containing 50 μM FL or CRIB–PR. (f) Steady-state fluorescence of 0.5 μM FL FRET reporter (Supplementary Fig. 1b) as a function of GMP-PNP–Cdc42G12V concentration (0 to 50 μM). a.u., arbitrary units. (g) Normalized decrease in donor fluorescence for 0.5 μM FL FRET reporter (solid squares) or 0.5 μM BAR–SH3 FRET reporter (open squares) as a function of increasing concentrations of GMP-PNP–Cdc42G12V (red), GDP–Cdc42G12V (blue), GMP-PNP–Rac1 (purple) and SH3 domain (green). (h) Comparison of the total change in FRET for the titrations shown in g. The differences in FRET calculated at zero and saturating ligand concentrations are shown normalized and scaled relative to the total change in FRET for GMP-PNP–Cdc42G12V. Data are presented as mean ± s.d. (n = 3 titrations).
Underscoring the intense interest in IRSp53 is an ever-growing list of cytoskeletal effectors that bind to its SH3 domain, including Eps8 (refs. 14,15), Mena16, VASP17, N-WASP18, WAVE2 (refs. 19,20), mDia1 (refs. 20,21), espin22, PSD-95 (ref. 23), Shank3 (ref. 24), IQ-ArfGEF (BRAG1)25 and atrophin-1 (ref. 26). Like many important cytoskeletal proteins27,28, IRSp53 is also a target for bacterial pathogens, including enterohemorrhagic Escherichia coli, whose secreted scaffold EspFu is localized at the plasma membrane through interaction with IRSp53 (refs. 29,30). As a result of its interactions with downstream cytoskeletal effectors, IRSp53 has a critical role in the formation of filopodia and lamellipodia in processes such as neurite extension31, dendritic spine morphogenesis23,24,32,33, cell motility14, tumor invasiveness13,34,35, establishment of cell polarity12, eye lens formation36, myogenic differentiation37 and pathogen invasion20,29.
The activities of IRSp53 depend on interaction with Rho family GTPases16,18,19,31. Autoinhibition and GTPase-dependent activation and localization is a fundamental regulatory mechanism among cytoskeletal proteins38–40. However, there are few examples of proteins for which the molecular mechanism for autoinhibition and activation has been fully demonstrated, including WASP41,42, formins43,44 and the WAVE complex45,46. Here, to elucidate the regulatory mechanism of IRSp53, we studied its autoinhibited conformation and synergistic activation by Cdc42 and downstream effectors, in vitro and in cells.
RESULTS
Autoinhibition and activation by Cdc42
Cdc42 is required for filopodia formation in cells expressing full-length IRSp53 but not in cells expressing the isolated BAR domain, suggesting that IRSp53 is autoinhibited16. Compared to canonical CRIB motifs, the partial CRIB of IRSp53 is interrupted by a PR sequence, 276-PLPVPP-281 (Fig. 1a,b), containing a canonical SH3 domain–binding site31. This PR sequence could bind the SH3 domain of IRSp53 itself, suggesting a mechanism for autoinhibition47. Activation would then result from competitive binding of Rho family GTPases to the CRIB–PR16,18,31.
As a first step to directly test this model, we used isothermal titration calorimetry (ITC) to quantify the interactions of Rho family GTPases and the SH3 domain of IRSp53 with both the isolated CRIB–PR and full-length IRSp53 (FL, Fig. 1a and Supplementary Fig. 1a). In these experiments, we used a constitutively active mutant (G12V) of the two GTPases suggested to activate IRSp53: Cdc42 (refs. 16,31) and Rac1 (ref. 19). We exchanged the nucleotide on the GTPases with either GDP or GMP-PNP and confirmed the nucleotide state before each ITC experiment by HPLC (as described in ref. 48). GMP-PNP–Cdc42G12V bound FL with a dissociation constant (Kd) of 16.1 μM. The affinity of GMP-PNP–Cdc42G12V for the isolated CRIB–PR was three-fold higher (Kd = 5.0 μM) (Fig. 1c,d), a result suggesting that the CRIB–PR is partially occluded in the full-length protein, presumably by intramolecular inhibitory interactions. Of note, these affinities are lower than those of Cdc42 and Rac for canonical CRIB motifs, including those of ACK, PAK, PAR6 and WASP, typically ranging from 1 to 50 nM (refs. 42,49,50). Neither GDP–Cdc42G12V nor GMP-PNP–Rac1G12V bound to FL or the isolated CRIB–PR, indicating that the interaction is both GTPase and nucleotide specific.
The SH3 domain also bound to the isolated CRIB–PR domain (Fig. 1e), albeit with low affinity (Kd = 114 μM), which is common for autoinhibitory interactions that are enhanced by the proximity achieved within a single polypeptide42,51. This reaction did not reach saturation, and therefore the stoichiometry of the interaction could not be determined. The affinity of the SH3 domain for FL was too low to be accurately quantified (Fig. 1e), probably owing to competition with the endogenous SH3 domain. That both the SH3 domain and Cdc42G12V bind to the CRIB–PR is consistent with a model of autoinhibition and competitive activation.
Autoinhibition and competitive activation would also be consistent with a conformational change upon Cdc42 binding. Indeed, the three-fold-reduced enthalpy of Cdc42G12V binding to FL compared to CRIB–PR already suggested a reaction in which endothermic binding of Cdc42G12V is offset by an exothermic conformational change. To explore this possibility, we developed a fluorescence resonance energy transfer (FRET) assay, using two different reporters based on IRSp53 constructs FL and BAR–SH3 (Supplementary Fig. 1b). In both cases, we labeled pairs of cysteines with the donor probe fluorescein maleimide and the acceptor probe rhodamine maleimide. IRSp53 contains four endogenous cysteines, all within the BAR domain. By peptide mass fingerprinting, we found that only one of these cysteines (Cys230) reacted with the fluorescence probe (Supplementary Fig. 1c). We added a second reactive cysteine by mutagenesis at the ends of constructs FL (S519C) and BAR–SH3 (S452C), such that changes in FRET were observed only for the full-length sensors. (Both proteins undergo partial C-terminal degradation.) To ensure that most of the donors were coupled to acceptors, we first under-labeled with the donor probe and then performed saturating labeling with the acceptor probe (Online Methods).
In the absence of Rho family GTPases, energy transfer (quenching) was present with both FRET reporters (Fig. 1f–h). Titration of GMP-PNP–Cdc42G12V, but not GDP–Cdc42G12V or GMP-PNP–Rac1G12V, reduced the energy transfer, consistent with a Cdc42-dependent conformational change in IRSp53 that increases the distance between fluorescence probes. By fitting the fluorescence decrease as a function of GMP-PNP–Cdc42G12V concentration, we found that Cdc42G12V bound with an affinity approximately seven-fold weaker to FL (Kd = 23.0 μM) than to BAR–SH3 (Kd = 3.2 μM). This suggests that the region C terminal to the SH3 domain also participates in autoinhibitory interactions. We also note that the affinity of GMP-PNP–Cdc42G12V for FL measured by the FRET reporter is remarkably similar to that measured by ITC (Fig. 1c), mutually validating both affinity measurements.
The energy transfer between probes was unchanged during titration of the isolated SH3 domain (Fig. 1g,h). This result is consistent with its lack of binding in ITC (Fig. 1e) and suggests that the SH3 domain interacts too weakly in trans to offset autoinhibitory interactions occurring intramolecularly.
Heterohexameric complex of IRSp53, Cdc42 and Eps8
The results described above suggested that activation of IRSp53 requires the detachment of the SH3 domain from the CRIB–PR domain. In principle, this detachment could also be achieved through the binding of an effector protein to the SH3 domain24. To test this hypothesis, we selected the IRSp53 effector Eps8, which is overexpressed in numerous cancers14,34. We titrated Eps8 into the BAR–SH3 FRET reporter (Fig. 2a). At saturation, Eps8 produced a total increase in donor fluorescence similar in magnitude to that produced by GMP-PNP–Cdc42G12V (Fig. 2a). The titration could not be fit to a single-site binding model, owing to its biphasic character. Indeed, the donor fluorescence first decreased and then increased, with the transition between phases taking place at an Eps8/BAR–SH3 molar ratio of ~1:1. (The concentration of BAR–SH3 in this experiment was 0.5 μM.) The second phase was best fit to a cooperative binding function, with Hill coefficient of 2.5 and Kd of 1.0 μM. The biphasic and cooperative character of the interaction suggested that one molecule of Eps8 binds to one molecule of IRSp53 or, more likely, two Eps8 molecules bind to the IRSp53 dimer.
Figure 2.
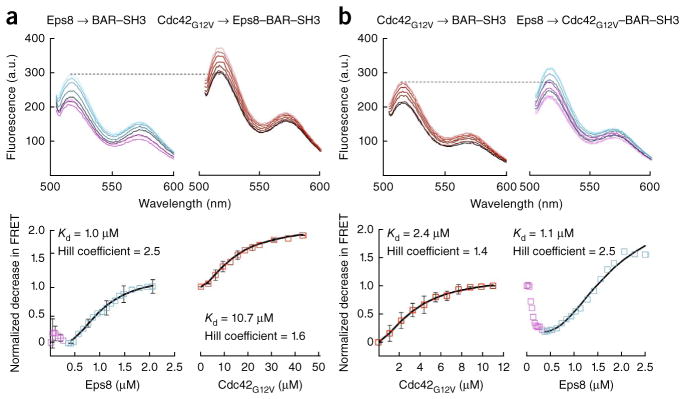
Formation of a heterohexameric complex between Cdc42, IRSp53 and Eps8. (a) Titration of 11 μM Eps8 into 0.5 μM BAR–SH3 FRET reporter (left), followed by titration of 200 μM GMP-PNP–Cdc42G12V (right). The total change in FRET for the first titration was normalized to one and used as a reference for scaling of the second titration. The normalized decrease in FRET for each titration was fitted to a cooperative binding equation (black lines). (b) Titration of 200 μM GMP-PNP–Cdc42G12V into 0.5 μM BAR–SH3 FRET reporter (left), followed by titration with 11 μM Eps8 (right) (normalized and fitted as in a). Data from repeated titrations are presented as mean ± s.d. (n = 3 titrations).
To more conclusively determine the stoichiometry of the Eps8–IRSp53 complex, we used size-exclusion chromatography combined with glycerol gradient sedimentation (Supplementary Fig. 2). This analysis showed that Eps8 is an elongated monomer in solution with a measured mass of 106,000 Da (theoretical mass, 114.8 Da), IRSp53 is a dimer as expected for a BAR-domain protein, and the two interact to form a 2:2 complex with a measured mass of 288,000 Da (theoretical mass, 298,600 Da).
To test whether IRSp53 could simultaneously bind Cdc42 and a downstream effector, we titrated the saturated Eps8–IRSp53 complex with Cdc42G12V. This second titration produced a conformational change in addition to that generated by Eps8 (Fig. 2a). To further investigate this point, we reversed the order of the experiment, by first titrating Cdc42G12V into IRSp53 and then titrating Eps8. The results confirmed the additive character of the conformational changes induced by these two proteins on IRSp53 (Fig. 2b). Cdc42G12V bound to IRSp53 and to the Eps8–IRSp53 complex in a similar manner, i.e., noncooperatively (Hill coefficient < 2.0), albeit with a somewhat lower affinity for the complex (Kd of 2.4 versus 10.7 μM). Eps8 also bound similarly to IRSp53 and the Cdc42G12V–IRSp53 complex; in both cases, the interaction was cooperative (Hill coefficient > 2.0), and the affinities were nearly identical (Kd of 1.0 versus 1.1 μM). Both Eps8 titrations displayed the characteristic biphasic binding described above.
These experiments showed that Cdc42 binding to the CRIB–PR and Eps8 binding to the SH3 domain both produce conformational changes in IRSp53, but these changes are of a different nature. Thus, independent of the order in which Cdc42G12V and Eps8 were titrated into IRSp53, the total change in energy transfer at equilibrium was similar for both titrations, and was greater than for either Eps8 or Cdc42G12V alone. These results suggest that Cdc42G12V and Eps8 can bind simultaneously to IRSp53 and additively alter its structure to produce full activation. Because one Cdc42G12V molecule binds to each of the CRIB–PR domains of IRSp53 (Fig. 1c,d), and Eps8 forms a 2:2 complex with IRSp53 (Supplementary Fig. 2), these results suggest that IRSp53, Cdc42 and Eps8 form a hexameric 2:2:2 complex.
Structure of GMP-PNP–Cdc42G12V bound to the CRIB–PR
We determined the crystal structure of GMP-PNP–Cdc42G12V bound to the CRIB–PR domain at 1.9-Å resolution (Table 1, Fig. 3a and Supplementary Video 1). Amino acids Ser263–Ser291 of the CRIB–PR were well defined in the electron density map (Fig. 3b,c). Three residues at the N terminus of the peptide were disordered in the structure and presumably do not interact with Cdc42. The structure reveals a new mode of GTPase binding to a CRIB-related sequence. Indeed, only the N-terminal portion of the CRIB–PR, up to residue Pro270, overlaps with classical CRIB motifs (Fig. 3d). Coincidentally, Pro270 is also the last residue of the CRIB–PR shared with canonical CRIB motifs (Fig. 1b). From Pro270 to Pro280, the CRIB–PR chain meanders along a hydrophobic surface on Cdc42 (Fig. 3c). Classical CRIB motifs take a different path, forming a β-strand that runs antiparallel to β-strand 2 of the GTPase and making hydrophobic and electrostatic contacts with switch I of the GTPase through their canonical FxHxxH C-terminal sequence that is missing in IRSp53 (Fig. 3d). In contrast, in the CRIB–PR, the all-critical interaction with the switch I–switch II region is established through a short helix formed by residues Pro281–Phe286 and the last six amino acids of the CRIB–PR, which run parallel to switch I of Cdc42. Of these, the last five amino acids of the CRIB–PR appear somewhat more flexible; they display higher temperature factors and are less well defined in the electron density map.
Table 1.
Data collection and refinement statistics
| GMP-PNP–Cdc42–CRIB–PR | |
|---|---|
| Data collection | |
| Space group | P42212 |
| Cell dimensions | |
| a, b, c (Å) | 67.23, 67.23, 79.75 |
| Resolution (Å) | 1.90–50.00 (1.90–1.97)a |
| Rmerge | 3.3 (52.6) |
| I / σI | 20.9 (3.3) |
| Completeness (%) | 100.0 (100.0) |
| Redundancy | 18.4 (17.8) |
| Refinement | |
| Resolution (Å) | 1.90–34.30 (1.90–2.04) |
| No. reflections | 14,959 (2,747) |
| Rwork / Rfree | 14.81 (16.13) / 19.87 (22.10) |
| No. atoms | |
| Protein | 1,693 |
| Ligand | 69 |
| Water | 151 |
| B factors | |
| Protein | 26.67 |
| Ligand | 33.16 |
| Water | 35.27 |
| r.m.s. deviations | |
| Bond lengths (Å) | 0.015 |
| Bond angles (°) | 1.689 |
Values in parentheses are for highest-resolution shell. The structure is based on a single crystal.
Figure 3.

Structure of the CRIB–PR bound to Cdc42. (a) Ribbon representation of the crystal structure of the CRIB–PR (orange and cyan) bound to GMP-PNP–Cdc42G12V (gray). Switch I (residues 30–38), switch II (residues 57–72) and the P loop (residues 9–16) are colored yellow, purple and green, respectively. (b) Close-up view of the 2Fo−Fc electron density map contoured at 1σ around the CRIB–PR. (c) Interactions of the side chains of the CRIB–PR on the electrostatic surface of Cdc42G12V (blue, red and white indicate positive, negative and neutral electrostatic potential, respectively). Pairs of CRIB–PR residues mutated in this study are labeled, corresponding to mutants I267A S268A (orange), L277E F286E (cyan) and P278D P281D (yellow). (d) Comparison of the structures of the CRIB–PR and the canonical CRIB motifs of PAK1, PAK4, PAK6 and PAR6 (PDB 2QME, 2OV2, 2ODB and 1NF3 (ref. 50), respectively).
Cellular and biochemical analysis of IRSp53 activation
A remarkable feature of the structure of the CRIB–PR bound to Cdc42G12V is that it shows that a PR sequence, featuring a canonical SH3 domain-binding site, can specifically interact with a Rho family GTPase, providing a mechanism for competitive activation. To further test this unexpected observation, we designed IRSp53 truncations and mutations with different predicted effects on Cdc42 binding and autoinhibition and characterized their cellular and in vitro activities. In cells, we quantified the number and length of filopodia in B16F1 mouse melanoma cells expressing mCherry-Cdc42G12V and IRSp53-GFP constructs. We defined filopodia as actin-filled, nontapered protrusions that emerged more than 1 μm from the cell periphery and had a width of 0.2–0.5 μm.
Individually, IRSp53 or Cdc42 did not increase the number or length of filopodia relative to those in control cells coexpressing GFP and RFP empty vectors (Supplementary Fig. 3a). Thus, cells expressing mCherry-Cdc42G12V and GFP showed Cdc42G12V uniformly enriched along the plasma membrane and diffusely distributed in the cytoplasm (Fig. 4a and Supplementary Fig. 3b), but the number and length of filopodia were mostly unchanged (Fig. 4i,j). Cells expressing FL-GFP and RFP showed IRSp53 localized in patches along the plasma membrane and in the cytoplasm (Fig. 4b and Supplementary Fig. 3c), but the number and length of filopodia did not change (Fig. 4i,j).
Figure 4.

Effect of IRSp53 mutations on filopodia formation in B16F1 melanoma cells as a function of Cdc42 coexpression. (a,b) Control cells coexpressing GFP and mCherry-Cdc42G12V (a) or FL-GFP and RFP (b) (scale bars, 10 μm). Zoomed-in regions (white box) are shown separately for Cdc42G12V, IRSp53 and actin. (c–h) Coexpression of mCherry-Cdc42G12V with the indicated IRSp53 constructs: FL (c), BAR (d), BAR–CRIB–PR (e), BAR–SH3 (f), I267A S268A (g) and L277E F286E (h). The main phenotype characterizing each construct is shown on the left. (i) Quantification of the average number of filopodia and filopodia-like protrusions per cell for each IRSp53 mutant coexpressed with either RFP (gray bars) or mCherry-Cdc42G12V (red bars). The data are presented as mean ± s.e.m. The total number of cells quantified is indicated inside each bar. (j) Filopodia length distribution presented as box-and-whisker plots, in which boxes represent the middle half of the data and are divided by the median value, whiskers encompass the data from the 5th to the 95th percentile, and a dot represents the mean value of all the data. (k) Percentage of filopodia that were filled (blue), partially filled (green) or not filled (orange) with actin.
In contrast, there was a dramatic increase in the number and length of filopodia in cells coexpressing FL-GFP and mCherry-Cdc42G12V (Fig. 4c,i,j). Moreover, in these cells IRSp53 became uniformly distributed along the plasma membrane (Supplementary Fig. 3d). Therefore, we classified construct FL as fully ‘regulated’, because its filopodia-inducing activity and proper plasma-membrane localization depended on coexpression with Cdc42G12V. Interestingly, as filopodia became longer, they frequently showed unusual characteristics: they became segmented, as indicated by periodic nodes of enhanced IRSp53 and Cdc42G12V colocalization, and occasionally they also formed branches at the sites of these nodes (Fig. 4c). We refer to these structures as filopodia-like protrusions.
By comparison, we classified constructs BAR and BAR–CRIB–PR as ‘unregulated’, because they produced a large increase in the number of filopodia or filopodia-like protrusions independently of coexpression with Cdc42G12V (Fig. 4d,e,i,j). Of these protrusions, 20–35% were not filled with actin (Fig. 4k) or were only partially filled. This result is consistent with the inability of constructs BAR and BAR–CRIB–PR to recruit downstream actin assembly effectors that bind to the SH3 domain. Curiously, however, filopodia length with these two constructs increased as a function of Cdc42G12V coexpression (Fig. 4j). Because only one of these constructs can bind Cdc42G12V (BAR–CRIB–PR), this result suggests that once filopodia are initiated Cdc42G12V controls their length through mechanisms independent of its interaction with IRSp53.
Construct BAR–SH3 displayed an intermediate phenotype between FL and BAR, and therefore we classified it as ‘partially regulated’. This construct increased the number and length of filopodia or filopodia-like protrusions (Fig. 4f,i,j), and the majority of these protrusions were actin filled (Fig. 4k), consistent with the ability of BAR–SH3 to recruit actin assembly factors through the SH3 domain. However, filopodia induction was only partially dependent on Cdc42G12V coexpression (Fig. 4i), suggesting that construct BAR–SH3 is not fully inhibited. This is consistent with our finding that sequences C terminal to the SH3 domain participate in autoinhibition (Fig. 1g).
Next, we tested the effect of point mutations within the CRIB–PR domain, which were suggested by the structure to disrupt Cdc42G12V binding (Fig. 3c) and thus to interfere with Cdc42-dependent activation in cells. A BAR–SH3 double mutant targeting two highly conserved residues in the N-terminal portion of the CRIB–PR, I267A S268A, failed to bind Cdc42G12V by ITC (Supplementary Fig. 4), confirming the importance of canonical CRIB residues for this interaction. Another double mutant, L277E F286E, targeting hydrophobic interactions of two residues in the PR region, also failed to bind Cdc42G12V (Supplementary Fig. 4). Moreover, the mutation F286E alone was sufficient to abrogate Cdc42G12V binding to IRSp53 (Supplementary Fig. 4), demonstrating the importance of the PR region for interaction with Cdc42G12V. Phe286 binds at the interface between switches I and II, suggesting that the PR region also has a key role in sensing the nucleotide state of the GTPase. Coexpression of Cdc42G12V with FL mutants I267A S268A (Fig. 4g) or L277E F286E (Fig. 4h) resulted in a phenotype similar to that of FL expressed alone (Fig. 4b); i.e., filopodia number and length were unchanged as compared to control cells, with or without Cdc42G12V (Fig. 4i,j). These results suggest that these two mutants are trapped in a constitutively inactive state. These results also confirm that filopodia initiation by full-length IRSp53 requires interaction with Cdc42 as a prerequirement for filopodia elongation that depends on Cdc42 but not on its interaction with IRSp53.
These results are consistent with previous cellular work16,18,31,52 that analyzed IRSp53 mutants, some of which were similar to those studied here (Fig. 4c–e,g). Some of these mutants are reproduced here as a control for additional structure-inspired mutagenesis addressing autoinhibition.
Cellular and biochemical analysis of IRSp53 autoinhibition
The data presented to this point address the mechanism of IRSp53 coactivation by Cdc42 and effectors such as Eps8. Next, we set out to test the mechanism of autoinhibition, which appears to involve an interaction between the CRIB–PR and SH3 domains (Fig. 1e and Supplementary Fig. 5a). To test this interaction, we designed CRIB–PR and SH3-domain mutants and characterized their in vitro and cellular activities.
The only canonical SH3 domain–binding PxxP site in IRSp53 is found within the CRIB–PR (278-PVPP-281). Interestingly, Pro278 and Pro281 are exposed in the structure and do not interact with Cdc42 directly, potentially allowing for the competitive binding of the SH3 domain of IRSp53 itself or that of a downstream effector (Supplementary Fig. 5a). We mutated these residues to aspartic acid to disrupt potential interactions with SH3 domains. As suggested by the structure, the affinity of Cdc42G12V for the BAR–SH3 double mutant P278D P281D was reduced only two-fold compared to that of wild type (Kd of 10.7 versus 4.7 μM) (Fig. 5a). However, as predicted by the model of autoinhibition, the binding of Cdc42G12V produced only a minor conformational change in the FRET reporter (Fig. 5b), suggesting that its conformation was already mostly open.
Figure 5.

Autoinhibitory interactions involve the CRIB–PR domain. (a) ITC titration of 400 μM GMP-PNP–Cdc42G12V into 15 μM BAR–SH3 (red) or mutant P278D P281D (black). This double mutant targets the PxxP site in the PR region of the CRIB–PR (Supplementary Fig. 5a). (b) Titration of 200 μM GMP-PNP–Cdc42G12V into 0.5 μM BAR–SH3 FRET reporter (red) or BAR–SH3 FRET reporter mutant P278D P281D (black). Data are presented as mean ± s.d. (n = 3 titrations). (c) Titration of 11 μM Eps8 into 0.5 μM BAR–SH3 FRET reporter mutant P278D P281D (left), followed by titration of 200 μM GMP-PNP–Cdc42G12V (right). Titrations were normalized and fitted as described in Figure 2. (d) Images of cells coexpressing FL mutant P278D P281D with RFP or with mCherry-Cdc42G12V (left) and quantification (right) of the number of filopodia per cell, filopodia length and the percentage of filopodia filled with actin (as described in Fig. 4). The data for filopodia per cell are presented as mean ± s.e.m. The total number of cells quantified is indicated inside each bar. *P < 0.005 by two-tailed unpaired Student’s t test.
To further explore the conformation of this mutant, we titrated Eps8 into the BAR–SH3 FRET reporter mutant. This resulted in a biphasic conformational transition that fitted to a cooperative binding curve with similar affinity to that of wild type (Figs. 2a and 5c). This result shows that the SH3 domain of Eps8 does not interact with the CRIB–PR of IRSp53, and the interaction probably involves the SH3 domain of IRSp53 and one of the PR sequences of Eps8 (refs. 14,15,34). In contrast, binding of Cdc42G12V to the Eps8–IRSp53 complex produced only a minor change in FRET (Fig. 5c). This finding reaffirms the notion that the conformational changes produced by the binding of Cdc42G12V to the CRIB–PR and of Eps8 to the SH3 domain are different in nature and additive in magnitude (Fig. 2).
In cells, the FL mutant P278D P281D displayed a significant increase in the basal number of filopodia (Fig. 5d). However, coexpression with Cdc42G12V did not increase the number of filopodia, suggesting that the two-fold-lower affinity of this mutant for Cdc42G12V observed in vitro (Fig. 5a) is sufficient to abrogate Cdc42G12V-dependent regulation in cells. In contrast, filopodia length increased with Cdc42G12V coexpression (Fig. 5d), and long filopodia-like protrusions were often segmented and buckled as described above.
To address the role of the SH3 domain in autoinhibition, we introduced the mutation P428L in the SH3 domain (Supplementary Fig. 5a). This amino acid is strictly conserved in the SH3 fold and has been mutated in cellular studies to abolish effector binding to IRSp53 (refs. 16,18). The P428L mutant behaved similarly to the P278D P281D double mutant, producing a high basal number of filopodia-like protrusions independently of Cdc42G12V coexpression. Filopodia length was only marginally increased with Cdc42G12V coexpression, and ~20% of these filopodia were not actin filled (Supplementary Fig. 5b). These results suggest that the P428L mutation disrupts autoinhibitory interactions and recruitment of downstream effectors in cells. In vitro, however, this mutant could not be analyzed, owing to instability and protein degradation.
To more directly test the role of the SH3 domain in autoinhibition, we swapped the SH3 domain of IRSp53 for that of Eps8 that was established here not to bind to the CRIB–PR (compare Figs. 2 and 5c). Although the two SH3 domains share low sequence identity (15%), their crystal structures have been determined, allowing for a precise definition of domain boundaries for swapping (Fig. 6a). By ITC, Cdc42G12V bound to the domain-swapped mutant (SH3-SWAP) with higher affinity than to FL (Kd of 3.8 versus 16.1 μM) but with similar affinity as to the isolated CRIB–PR (Fig. 1d), suggesting that this mutation breaks autoinhibitory interactions. Consistent with this result, the binding of Cdc42G12V to a FL FRET reporter carrying this mutation did not produce a conformational change, suggesting that its conformation was already open. In cells, this mutant displayed a phenotype similar to mutants P278D P281D and P428L, in that it produced a high basal number of filopodia (Fig. 6d). Cdc42G12V coexpression led to an increase in the length and number of filopodia, ~10% of which were not actin filled. The increase in the number of filopodia was unexpected and may result from the specific activity of the Eps8 SH3 domain.
Figure 6.

Autoinhibitory interactions involve the SH3 domain. (a) Top, superimposition of crystal structures resulting from swapping the SH3 domain of IRSp53 with that of Eps8. Bottom, structure-based sequence alignment showing the sequences that were swapped, which share only 15% identity. (b) ITC titration of 200 μM GMP-PNP–Cdc42G12V into 8 μM FL (red) or FL mutant SH3-SWAP (black). (c) Titration of 400 μM GMP-PNP–Cdc42G12V into 0.5 μM FL FRET reporter (red) or FL FRET reporter mutant SH3-SWAP (black). Data are presented as mean ± s.d. (n = 3 titrations). (d) Left, cells coexpressing FL mutant SH3-SWAP with RFP or with mCherry-Cdc42G12V. Right, quantification of the number of filopodia per cell, filopodia length and the percentage of filopodia filled with actin (as described in Fig. 4). The data for filopodia per cell are presented as mean ± s.e.m. The total number of cells quantified is indicated inside each bar. *P < 0.005 by two-tailed unpaired Student’s t test.
DISCUSSION
By linking signaling pathways to membrane and actin-cytoskeleton dynamics, I-BAR domain–containing proteins have critical roles in numerous cellular functions involving membrane remodeling3–5. Previous studies have addressed the molecular mechanism by which the BAR domain can sense and/or induce membrane curvature2,53. Much less is known about the mechanisms by which BAR domain–containing proteins are regulated. These proteins are characterized by their modular architecture comprising domains specialized in the recruitment of cytoskeletal effectors that enhance or modulate their membrane-deforming activities. Thus, several BAR-domain proteins interact with small GTPases and contain SH3 domains through which they recruit proline-rich effectors in a regulated manner2. In this work, we have presented evidence, both in vitro and in cells, that the unique GTPase-binding domain of IRSp53, which we have named CRIB–PR, serves a dual role: autoinhibition and Cdc42-dependent membrane recruitment and activation. Similarly, we have shown that the SH3 domain also serves a dual role: autoinhibition and effector recruitment.
Nucleotide and GTPase specificity of the CRIB–PR domain
The interaction of the CRIB–PR with Cdc42 differs substantially from that of canonical CRIB motifs; the CRIB–PR binds with weaker affinity and interacts with a different surface on the GTPase than do canonical CRIB motifs41,49,50,54. Only residues 267-ISDP-270 of the CRIB–PR, shared with canonical CRIB motifs, overlap with existing structures. Despite these differences, two basic principles of GTPase–effector binding are preserved: nucleotide-state sensing and exquisite GTPase specificity. Indeed, the C-terminal PR portion of the CRIB–PR interacts with the switch I–switch II region of Cdc42 that undergoes a nucleotide-dependent conformational change (Supplementary Fig. 6a). This conformational change explains the inability of GDP-Cdc42 to bind to the CRIB-PR. It thus emerges that the PR portion of the CRIB–PR, involved in autoinhibitory interactions with the SH3 domain, is also responsible for GTPase nucleotide-state sensing.
Rho family GTPases are highly conserved, with Cdc42 and Rac sharing 72% sequence identity. It was therefore surprising that Rac1 did not bind to the CRIB–PR, particularly given that IRSp53 was initially identified as a downstream effector of Rac19. Some CRIB motifs, including those of PAK protein kinases, appear to bind both Cdc42 and Rac54, whereas the CRIB motifs of ACK and WASP bind only Cdc42 (refs. 41,49). Therefore, the question arises as to how is specificity achieved. To address this question, we calculated the conservation scores of each residue among the three Rac (Rac1, Rac2 and Rac3) and two Cdc42 (Cdc42-1 and Cdc42-2) variants expressed in humans, by using the program Consurf55. A surface representation of the Cdc42 structure, colored according to residue conservation scores, highlights an area of high sequence variation that coincides with the site where the structure of the CRIB–PR diverges from those of canonical CRIB motifs (Fig. 3d and Supplementary Fig. 6b). It thus appears that the sequence and structural divergence between the CRIB–PR and canonical CRIB motifs coincides with a site of sequence variation between Cdc42 and Rac. This suggests that the CRIB–PR is specifically adapted for binding to Cdc42.
Model of IRSp53 inhibition and combinatorial activation
Coincident activation is a common theme among cytoskeletal proteins, whereby various inputs work synergistically for efficient activation and localization. For instance, N-WASP adopts a compact autoinhibited conformation in the resting state, with full activation requiring the concurrent binding of Cdc42, negatively charged membranes, and SH3-containing binding partners41,42. Similarly, some formins are maintained in an inactive state through intramolecular interaction of the C-terminal DAD and N-terminal DID domains. Activation and membrane localization is attained through multiple inputs, including binding of GTPases to a site near the DID43,44,56 and concurrent interaction with the plasma membrane through the N-terminal basic tail57,58. Another example is the WAVE complex, which is activated at the membrane by phosphorylation and simultaneous interactions with Rac and acidic phospholipids45,46.
We propose that, analogous to these examples, the activation and localization of IRSp53 is a combinatorial process involving dimerization, GTPase binding, effector binding, membrane binding, and cooperative clustering at the membrane (Fig. 7). IRSp53 is an antiparallel dimer, such that most of its interactions are enhanced by duplication. This includes autoinhibitory interactions that, as shown here, involve the CRIB–PR and SH3 domain (Figs. 1e, 5 and 6) as well as sequences C terminal to the SH3 domain (Figs. 1g and 4c,f). We also established that autoinhibitory interactions are weak (Fig. 1e), but are likely to be enhanced by the proximity of the interacting domains within a single polypeptide51. Moreover, autoinhibitory interactions must be reversible, so their weak affinity probably has a functional role. In the inhibited state, IRSp53 is not uniformly localized to the plasma membrane (Fig. 4b), suggesting that autoinhibitory interactions weaken membrane binding and/or IRSp53 clustering at the membrane59. Autoinhibition also occludes the SH3 domain, which is necessary for effector binding.
Figure 7.

Combinatorial activation model of IRSp53.
Membrane localization is enhanced by coexpression with Cdc42G12V (Fig. 4c), which is itself tethered to the membrane. Activation at the plasma membrane appears to occur as a two-step process culminating with the formation of a hexameric 2:2:2 complex between Cdc42, IRSp53 and downstream effectors such as Eps8. We have shown here that the binding site of Cdc42, comprising the entire CRIB–PR, overlaps with the binding site of the SH3 domain on the PR region, revealing the structural bases for competitive activation by Cdc42. However, as in other autoinhibitory interactions43,44, the overlap is not complete, because the PxxP motif is partially exposed in the structure (Supplementary Fig. 5a).
Cdc42 binding to the CRIB–PR and effector binding to the SH3 domain both produce conformational changes on IRSp53 that are similar in magnitude but different in nature, which when combined produce a more open and presumably fully activated state (Figs. 2 and 7). This may explain how coexpression of IRSp53 with either Cdc42 or some downstream effectors enhances IRSp53 localization at the plasma membrane13,16,18,19. Finally, BAR-BAR interactions may allow IRSp53 to form a coat at the membrane, necessary for membrane deformation6,59. In summary, we have shown that through a highly specialized modular structure, IRSp53 constitutes a node for integration of signaling and functional inputs at the interface between the plasma membrane and the actin cytoskeleton.
ONLINE METHODS
Protein expression and purification
Human IRSp53 constructs and mutants (Fig. 1a and Supplementary Table 1) were expressed in E. coli BL21(DE3) cells with the intein IMPACT expression system (New England BioLabs). Cells were grown in TB medium supplemented with 100 mg/ml ampicillin at 37 °C to an OD of 4. Protein expression was induced with 1 mM IPTG at 18 °C for 6 h. Cells were homogenized in 20 mM HEPES, pH 7.5, 500 mM NaCl, 5 mM EDTA, 4 mM benzamidine hydrochloride and 1 mM PMSF, lysed with a microfluidizer (Microfluidics), and centrifuged at 20,000g for 30 min. Proteins were purified first through a chitin affinity column. The affinity tag was removed on the column by self-cleavage of the intein, induced by incubation with 50 mM DTT overnight at room temperature. Proteins were eluted in 20 mM MES, pH 6.0, 300 mM NaCl, 4 mM benzamidine hydrochloride, 5% glycerol and 50 mM DTT. Proteins were 50% diluted in 20 mM MES, pH 6.0, and additionally purified through a HiTrap SP FF cation exchange column (GE Healthcare). A final purification step was performed on a Superdex 200 gel-filtration column (GE Healthcare) in 40 mM HEPES, pH 7.0, 300 mM NaCl, 1 mM MgCl2, 4 mM benzamidine hydrochloride, 5% glycerol and 1 mM DTT. Proteins were concentrated to ~4 mg/mL with an Amicon stirred cell (Millipore).
Human Cdc42 residues 1–178 and Rac1A residues 1–178 (Supplementary Table 1) bearing the constitutively active mutation G12V were expressed and purified as described above, in 50 mM Tris-HCl, pH 7.5, 500 mM NaCl, 5 mM MgCl2, 4 mM benzamidine hydrochloride and 1 mM PMSF. Self-cleavage of the intein was induced by incubation with 50 mM DTT for 48 h at room temperature. Proteins were additionally purified on a Superdex 200 gel-filtration column in 50 mM Tris-HCl, pH 7.5, 50 mM NaCl, 10 mM MgCl2, 4 mM benzamidine hydrochloride, 0.1 mM GDP and 5 mM DTT. Proteins were concentrated to ~50 mg/mL, and aliquots were flash frozen in liquid nitrogen.
The CRIB–PR domain (IRSp53 residues 260–291) was synthesized with N-terminal acetylation and C-terminal amide (P. Leavis, Tufts University). The lyophilized peptide was dissolved in 20 mM HEPES, pH 7.5, 50 mM NaCl, 1 mM MgCl2 and 1 mM DTT, to a concentration of ~5 mM.
Human Eps8 (Supplementary Table 1) was expressed and purified as described15.
Nucleotide exchange
Purified GTPases contained GDP bound, as we confirmed by HPLC analysis as described48. To obtain the GTP-bound state, GTPases at ~10 mg/mL were incubated with 4 U of calf intestinal alkaline phosphatase (New England BioLabs) in 10 μM ZnCl2, 200 mM NH4SO4 and a ten-fold molar excess of 5′-guanylyl imidodiphosphate (GMP-PNP, Sigma) for 5 h at 25 °C. To ensure that the exchange of the nucleotide was complete, we monitored the reaction by HPLC48. GMP-PNP GTPases were dialyzed against 20 mM HEPES, pH 7.0, 150 mM NaCl, 1 mM MgCl2, 4 mM benzamidine hydrochloride, 5% glycerol and 1 mM DTT.
Fluorescence resonance energy transfer
Two fluorescence reporters were made on the background of constructs BAR–SH3 and FL (Supplementary Fig. 1b). In both reporters, pairs of cysteine resides were labeled with the donor probe fluorescein maleimide and the acceptor probe rhodamine maleimide (Invitrogen). IRSp53 contains four endogenous cysteine residues, which are all located within the BAR domain. However, by peptide fingerprinting, we found that under saturating conditions (20:1 molar excess of probe to protein) only one of these cysteines (Cys230) reacted with fluorescein maleimide (Supplementary Fig. 1c). A second reactive cysteine was introduced by mutagenesis at the end of constructs FL (S519C) and BAR–SH3 (S452C). To ensure that most of the donor probes were coupled to acceptors, we first under-labeled the two IRSp53 constructs at 5 μM with the donor probe, by incubation with a 0.9 molar ratio of fluorescein maleimide for 30 min at 4 °C. Then, a five-fold molar excess of the acceptor probe was added for 1 h. Unreacted excess probes were removed by gel filtration with a PD10 desalting column (GE Healthcare). The extent of the labeling reaction was determined by measurement of the protein concentration by the Bradford assay (Bio-Rad) and comparison of the result to the dye concentration determined from the peak absorbance of the bound fluorophores, with extinction coefficients ε(492 nm) = 83,000 cm−1 M−1 for fluorescein-5-maleimide and ε(542 nm) = 91,000 cm−1 M−1 for tetramethyl-rhodamine-5-maleimide. The dye/protein ratio was determined to be 2:1 for the two reporters.
Steady-state fluorescence emission spectra were obtained at 20 °C by titration of IRSp53-binding ligands (GTPases, SH3 domain or Eps8) into both FRET reporters at 0.5 μM. Experiments were performed in 20 mM HEPES, pH 7.5, 150 mM NaCl, 4 mM benzamidine hydrochloride 1 mM MgCl2 and 0.1 mM TCEP, with a Varian Cary Eclipse fluorescence spectrophotometer (Agilent). Excitation was set at 496 nm (5-nm slit width), and the emission spectra were recorded at 520 nm (5-nm slit width). Titration curves were fit with IGOR (WaveMetrics) to either (as indicated) a noncooperative binding function F([ligand]) = Fmax × [ligand] × (Kd + [ligand])−1 or a cooperative binding function F([ligand]) = Fmax × [ligand]H × (KdH + [ligand]H)−1, where H is the Hill coefficient. Best fits were selected on the basis of residual analysis.
Isothermal titration calorimetry
Binding reactions were performed at 20 °C with a VP-ITC calorimeter (MicroCal). Proteins were dialyzed side by side against 20 mM HEPES, pH 7.5, 150 mM NaCl, 1 mM MgCl2, 4 mM benzamidine hydrochloride and 0.1 mM TCEP. Each titration consisted of 30 10-μL injections of 400 μM GTPases or 1.3 mM SH3 domain into a 1.475-mL reaction cell containing 8–15 μM IRSp53 constructs or 50 μM CRIB–PR. The duration of each injection was 20 s, with an interval of 4–5 min between injections. The heat of binding was corrected for the small exothermic heat of injection resulting from the titration of GTPases or the SH3 domain into buffer. ITC data from three independent experiments were analyzed with MicroCal’s custom scripts in Origin 7.0 (OriginLab). Best fits were selected on the basis of residual analysis. Integrated heats from all the binding reactions fit well to a single-site binding model.
Crystallography
GMP-PNP–Cdc42G12V was concentrated to 11 mg/mL in 20 mM HEPES, pH 7.5, 50 mM NaCl, 1 mM MgCl2, and 1 mM DTT, and incubated with a 1.5-fold molar excess of the CRIB–PR domain. Crystallization was performed at 18 °C, with the hanging-drop vapor-diffusion method. A typical 2-μL drop consisted of a 1:1 (v/v) mixture of protein solution and a well solution containing 18–20% PEG 4000, 200 mM MgCl2, and 4 mM betaine hydrochloride. Crystals appeared after 1 d and were flash frozen in a solution containing 20% PEG 400 added to the well solution. Data sets were collected at beamline 17-ID at the Advanced Photon Source (APS, Argonne). Data indexation and scaling were done with HKL-2000 (refs. 60,61). The structure was determined by molecular replacement, with a search model of the structure of the complex of Cdc42 with Par6 (PDB 1NF3 (ref. 50)). Par6 was removed before molecular replacement. Refinement and model building were carried out with PHENIX62 and Coot63, respectively. Data collection and refinement statistic are listed in Table 1. Representations of the structure and Supplementary Video 1 were prepared with PyMOL (http://www.pymol.org/).
Cell biology
B16F1 mouse melanoma cells were cultured as described52. IRSp53-GFP constructs were cotransfected with N-terminally fused RFP or mCherry human full length Cdc42G12V with Lipofectamine LTX and Plus reagents (Invitrogen). On day 2 after transfection, the cells were fixed with 4% paraformaldehyde in PBS, permeabilized with 1% Triton X-100 in PBS for 5 min, and stained with Alexa Fluor-680 phalloidin for 30 min. Cells were mounted on a slide with ProLong Gold Antifade Reagent (Invitrogen). Light microscopy was performed with an Eclipse TE2000-U inverted microscope (Nikon) equipped with a Planapo 100 × 1.3 NA objective and a Cascade 512B CCD camera (Photometrics) controlled by the Metamorph imaging software (Molecular Devices). All the cells were imaged with the same exposure time. Filopodia were quantified manually with ImageJ64.
Statistics
Quantifications were based on at least three independent experiments. FRET data are presented as mean ± s.d. Filopodia quantification is shown as mean ± s.e.m. Two-tailed unpaired P values were calculated with Student’s t test.
Supplementary Material
Acknowledgments
This work was supported by the US National Institutes of Health (NIH) grant R01 MH087950 to R.D. D.J.K. was supported by NIH grant T32 AR053461 and American Cancer Society grant PF-13-033-01-DMC. T.S. and C.Y. were supported by NIH grant GM095977. G.S. and A.D. were supported by the Associazione Italiana per la Ricerca sul Cancro grant IG-2013-14104. Use of IMCA-CAT beamline 17-ID was supported by the Industrial Macromolecular Crystallography Association through a contract with the Hauptman-Woodward Medical Research Institute. The Advanced Photon Source was supported by the US Department of Energy Contract DE-AC02-06CH11357. We thank P. Leavis (Tufts University) for the synthesis of the CRIB–PR peptide.
Footnotes
Accession codes. Atomic coordinates and structure factors for CRIB–PR bound to GMP-PNP–Cdc42G12V have been deposited in the Protein Data Bank under accession number 4JS0.
Note: Any Supplementary Information and Source Data files are available in the online version of the paper.
AUTHOR CONTRIBUTIONS
D.J.K. designed and purified proteins, determined crystal structure, designed and performed FRET and ITC experiments, analyzed filopodia from cell images and participated in the writing of the manuscript. C.Y. transfected and imaged cells. A.D. prepared Eps8 and performed cosedimentation assays. M.B. and Y.M. participated in construct design and protein preparation. G.S. and T.S. participated in project design and data analysis. R.D. was responsible for the overall design of the project and participated in structure determination, data analysis and preparation of the manuscript.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
Reprints and permissions information is available online at http://www.nature.com/reprints/index.html.
References
- 1.Insall RH, Machesky LM. Actin dynamics at the leading edge: from simple machinery to complex networks. Dev Cell. 2009;17:310–322. doi: 10.1016/j.devcel.2009.08.012. [DOI] [PubMed] [Google Scholar]
- 2.Frost A, Unger VM, De Camilli P. The BAR domain superfamily: membrane-molding macromolecules. Cell. 2009;137:191–196. doi: 10.1016/j.cell.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Suetsugu S, Gautreau A. Synergistic BAR-NPF interactions in actin-driven membrane remodeling. Trends Cell Biol. 2012;22:141–150. doi: 10.1016/j.tcb.2012.01.001. [DOI] [PubMed] [Google Scholar]
- 4.Scita G, Confalonieri S, Lappalainen P, Suetsugu S. IRSp53: crossing the road of membrane and actin dynamics in the formation of membrane protrusions. Trends Cell Biol. 2008;18:52–60. doi: 10.1016/j.tcb.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 5.Ahmed S, Goh WI, Bu W. I-BAR domains, IRSp53 and filopodium formation. Semin Cell Dev Biol. 2010;21:350–356. doi: 10.1016/j.semcdb.2009.11.008. [DOI] [PubMed] [Google Scholar]
- 6.Millard TH, et al. Structural basis of filopodia formation induced by the IRSp53/ MIM homology domain of human IRSp53. EMBO J. 2005;24:240–250. doi: 10.1038/sj.emboj.7600535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee SH, et al. Structural basis for the actin-binding function of missing-in-metastasis. Structure. 2007;15:145–155. doi: 10.1016/j.str.2006.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mattila PK, Salminen M, Yamashiro T, Lappalainen P. Mouse MIM, a tissue-specific regulator of cytoskeletal dynamics, interacts with ATP-actin monomers through its C-terminal WH2 domain. J Biol Chem. 2003;278:8452–8459. doi: 10.1074/jbc.M212113200. [DOI] [PubMed] [Google Scholar]
- 9.Hori K, Konno D, Maruoka H, Sobue K. MALS is a binding partner of IRSp53 at cell-cell contacts. FEBS Lett. 2003;554:30–34. doi: 10.1016/s0014-5793(03)01074-3. [DOI] [PubMed] [Google Scholar]
- 10.Connolly BA, Rice J, Feig LA, Buchsbaum RJ. Tiam1-IRSp53 complex formation directs specificity of rac-mediated actin cytoskeleton regulation. Mol Cell Biol. 2005;25:4602–4614. doi: 10.1128/MCB.25.11.4602-4614.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mackie S, Aitken A. Novel brain 14–3-3 interacting proteins involved in neurodegenerative disease. FEBS J. 2005;272:4202–4210. doi: 10.1111/j.1742-4658.2005.04832.x. [DOI] [PubMed] [Google Scholar]
- 12.Cohen D, Fernandez D, Lazaro-Dieguez F, Musch A. The serine/threonine kinase Par1b regulates epithelial lumen polarity via IRSp53-mediated cell-ECM signaling. J Cell Biol. 2011;192:525–540. doi: 10.1083/jcb.201007002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Robens JM, Yeow-Fong L, Ng E, Hall C, Manser E. Regulation of IRSp53-dependent filopodial dynamics by antagonism between 14–3-3 binding and SH3-mediated localization. Mol Cell Biol. 2010;30:829–844. doi: 10.1128/MCB.01574-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Funato Y, et al. IRSp53/Eps8 complex is important for positive regulation of Rac and cancer cell motility/invasiveness. Cancer Res. 2004;64:5237–5244. doi: 10.1158/0008-5472.CAN-04-0327. [DOI] [PubMed] [Google Scholar]
- 15.Disanza A, et al. Regulation of cell shape by Cdc42 is mediated by the synergic actin-bundling activity of the Eps8–IRSp53 complex. Nat Cell Biol. 2006;8:1337–1347. doi: 10.1038/ncb1502. [DOI] [PubMed] [Google Scholar]
- 16.Krugmann S, et al. Cdc42 induces filopodia by promoting the formation of an IRSp53:Mena complex. Curr Biol. 2001;11:1645–1655. doi: 10.1016/s0960-9822(01)00506-1. [DOI] [PubMed] [Google Scholar]
- 17.Boczkowska M, Rebowski G, Dominguez R. Glia maturation factor (GMF) interacts with Arp2/3 complex in a nucleotide state-dependent manner. J Biol Chem. 2013;288:25683–25688. doi: 10.1074/jbc.C113.493338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lim KB, et al. The Cdc42 effector IRSp53 generates filopodia by coupling membrane protrusion with actin dynamics. J Biol Chem. 2008;283:20454–20472. doi: 10.1074/jbc.M710185200. [DOI] [PubMed] [Google Scholar]
- 19.Miki H, Yamaguchi H, Suetsugu S, Takenawa T. IRSp53 is an essential intermediate between Rac and WAVE in the regulation of membrane ruffling. Nature. 2000;408:732–735. doi: 10.1038/35047107. [DOI] [PubMed] [Google Scholar]
- 20.Goh WI, et al. mDia1 and WAVE2 proteins interact directly with IRSp53 in filopodia and are involved in filopodium formation. J Biol Chem. 2012;287:4702–4714. doi: 10.1074/jbc.M111.305102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fujiwara T, Mammoto A, Kim Y, Takai Y. Rho small G-protein-dependent binding of mDia to an Src homology 3 domain-containing IRSp53/BAIAP2. Biochem Biophys Res Commun. 2000;271:626–629. doi: 10.1006/bbrc.2000.2671. [DOI] [PubMed] [Google Scholar]
- 22.Sekerková G, et al. Novel espin actin-bundling proteins are localized to Purkinje cell dendritic spines and bind the Src homology 3 adapter protein insulin receptor substrate p53. J Neurosci. 2003;23:1310–1319. doi: 10.1523/JNEUROSCI.23-04-01310.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Soltau M, et al. Insulin receptor substrate of 53 kDa links postsynaptic shank to PSD-95. J Neurochem. 2004;90:659–665. doi: 10.1111/j.1471-4159.2004.02523.x. [DOI] [PubMed] [Google Scholar]
- 24.Soltau M, Richter D, Kreienkamp HJ. The insulin receptor substrate IRSp53 links postsynaptic shank1 to the small G-protein cdc42. Mol Cell Neurosci. 2002;21:575–583. doi: 10.1006/mcne.2002.1201. [DOI] [PubMed] [Google Scholar]
- 25.Sanda M, et al. The postsynaptic density protein, IQ-ArfGEF/BRAG1, can interact with IRSp53 through its proline-rich sequence. Brain Res. 2009;1251:7–15. doi: 10.1016/j.brainres.2008.11.061. [DOI] [PubMed] [Google Scholar]
- 26.Okamura-Oho Y, Miyashita T, Ohmi K, Yamada M. Dentatorubral-pallidoluysian atrophy protein interacts through a proline-rich region near polyglutamine with the SH3 domain of an insulin receptor tyrosine kinase substrate. Hum Mol Genet. 1999;8:947–957. doi: 10.1093/hmg/8.6.947. [DOI] [PubMed] [Google Scholar]
- 27.Gruenheid S, Finlay BB. Microbial pathogenesis and cytoskeletal function. Nature. 2003;422:775–781. doi: 10.1038/nature01603. [DOI] [PubMed] [Google Scholar]
- 28.Day B, Henty JL, Porter KJ, Staiger CJ. The pathogen-actin connection: a platform for defense signaling in plants. Annu Rev Phytopathol. 2011;49:483–506. doi: 10.1146/annurev-phyto-072910-095426. [DOI] [PubMed] [Google Scholar]
- 29.de Groot JC, et al. Structural basis for complex formation between human IRSp53 and the translocated intimin receptor Tir of enterohemorrhagic E. coli. Structure. 2011;19:1294–1306. doi: 10.1016/j.str.2011.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ruetz TJ, Lin AE, Guttman JA. Enterohaemorrhagic Escherichia coli requires the spectrin cytoskeleton for efficient attachment and pedestal formation on host cells. Microb Pathog. 2012;52:149–156. doi: 10.1016/j.micpath.2011.12.001. [DOI] [PubMed] [Google Scholar]
- 31.Govind S, Kozma R, Monfries C, Lim L, Ahmed S. Cdc42Hs facilitates cytoskeletal reorganization and neurite outgrowth by localizing the 58-kD insulin receptor substrate to filamentous actin. J Cell Biol. 2001;152:579–594. doi: 10.1083/jcb.152.3.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abbott MA, Wells DG, Fallon JR. The insulin receptor tyrosine kinase substrate p58/53 and the insulin receptor are components of CNS synapses. J Neurosci. 1999;19:7300–7308. doi: 10.1523/JNEUROSCI.19-17-07300.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Choi J, et al. Regulation of dendritic spine morphogenesis by insulin receptor substrate 53, a downstream effector of Rac1 and Cdc42 small GTPases. J Neurosci. 2005;25:869–879. doi: 10.1523/JNEUROSCI.3212-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu PS, Jong TH, Maa MC, Leu TH. The interplay between Eps8 and IRSp53 contributes to Src-mediated transformation. Oncogene. 2010;29:3977–3989. doi: 10.1038/onc.2010.144. [DOI] [PubMed] [Google Scholar]
- 35.Suetsugu S, et al. The RAC binding domain/IRSp53-MIM homology domain of IRSp53 induces RAC-dependent membrane deformation. J Biol Chem. 2006;281:35347–35358. doi: 10.1074/jbc.M606814200. [DOI] [PubMed] [Google Scholar]
- 36.Chauhan BK, et al. Cdc42- and IRSp53-dependent contractile filopodia tether presumptive lens and retina to coordinate epithelial invagination. Development. 2009;136:3657–3667. doi: 10.1242/dev.042242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Misra A, et al. Insulin receptor substrate protein 53kDa (IRSp53) is a negative regulator of myogenic differentiation. Int J Biochem Cell Biol. 2012;44:928–941. doi: 10.1016/j.biocel.2012.02.020. [DOI] [PubMed] [Google Scholar]
- 38.Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420:629–635. doi: 10.1038/nature01148. [DOI] [PubMed] [Google Scholar]
- 39.Heasman SJ, Ridley AJ. Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol. 2008;9:690–701. doi: 10.1038/nrm2476. [DOI] [PubMed] [Google Scholar]
- 40.Symons M, Settleman J. Rho family GTPases: more than simple switches. Trends Cell Biol. 2000;10:415–419. doi: 10.1016/s0962-8924(00)01832-8. [DOI] [PubMed] [Google Scholar]
- 41.Abdul-Manan N, et al. Structure of Cdc42 in complex with the GTPase-binding domain of the ‘Wiskott–Aldrich syndrome’ protein. Nature. 1999;399:379–383. doi: 10.1038/20726. [DOI] [PubMed] [Google Scholar]
- 42.Kim AS, Kakalis LT, Abdul-Manan N, Liu GA, Rosen MK. Autoinhibition and activation mechanisms of the Wiskott–Aldrich syndrome protein. Nature. 2000;404:151–158. doi: 10.1038/35004513. [DOI] [PubMed] [Google Scholar]
- 43.Lammers M, Rose R, Scrima A, Wittinghofer A. The regulation of mDia1 by autoinhibition and its release by Rho•GTP. EMBO J. 2005;24:4176–4187. doi: 10.1038/sj.emboj.7600879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rose R, et al. Structural and mechanistic insights into the interaction between Rho and mammalian Dia. Nature. 2005;435:513–518. doi: 10.1038/nature03604. [DOI] [PubMed] [Google Scholar]
- 45.Lebensohn AM, Kirschner MW. Activation of the WAVE complex by coincident signals controls actin assembly. Mol Cell. 2009;36:512–524. doi: 10.1016/j.molcel.2009.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen Z, et al. Structure and control of the actin regulatory WAVE complex. Nature. 2010;468:533–538. doi: 10.1038/nature09623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Alvarez CE, Sutcliffe JG, Thomas EA. Novel isoform of insulin receptor substrate p53/p58 is generated by alternative splicing in the CRIB/SH3-binding region. J Biol Chem. 2002;277:24728–24734. doi: 10.1074/jbc.M202512200. [DOI] [PubMed] [Google Scholar]
- 48.Smith SJ, Rittinger K. Preparation of GTPases for structural and biophysical analysis. Methods Mol Biol. 2002;189:13–24. doi: 10.1385/1-59259-281-3:013. [DOI] [PubMed] [Google Scholar]
- 49.Mott HR, et al. Structure of the small G protein Cdc42 bound to the GTPase-binding domain of ACK. Nature. 1999;399:384–388. doi: 10.1038/20732. [DOI] [PubMed] [Google Scholar]
- 50.Garrard SM, et al. Structure of Cdc42 in a complex with the GTPase-binding domain of the cell polarity protein, Par6. EMBO J. 2003;22:1125–1133. doi: 10.1093/emboj/cdg110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hertzog M, et al. Molecular basis for the dual function of Eps8 on actin dynamics: bundling and capping. PLoS Biol. 2010;8:e1000387. doi: 10.1371/journal.pbio.1000387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yang C, Hoelzle M, Disanza A, Scita G, Svitkina T. Coordination of membrane and actin cytoskeleton dynamics during filopodia protrusion. PLoS ONE. 2009;4:e5678. doi: 10.1371/journal.pone.0005678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Peter BJ, et al. BAR domains as sensors of membrane curvature: the amphiphysin BAR structure. Science. 2004;303:495–499. doi: 10.1126/science.1092586. [DOI] [PubMed] [Google Scholar]
- 54.Morreale A, et al. Structure of Cdc42 bound to the GTPase binding domain of PAK. Nat Struct Biol. 2000;7:384–388. doi: 10.1038/75158. [DOI] [PubMed] [Google Scholar]
- 55.Ashkenazy H, Erez E, Martz E, Pupko T, Ben-Tal N. ConSurf 2010: calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 2010;38 (suppl):W529–W533. doi: 10.1093/nar/gkq399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Watanabe N, Kato T, Fujita A, Ishizaki T, Narumiya S. Cooperation between mDia1 and ROCK in Rho-induced actin reorganization. Nat Cell Biol. 1999;1:136–143. doi: 10.1038/11056. [DOI] [PubMed] [Google Scholar]
- 57.Ramalingam N, et al. Phospholipids regulate localization and activity of mDia1 formin. Eur J Cell Biol. 2010;89:723–732. doi: 10.1016/j.ejcb.2010.06.001. [DOI] [PubMed] [Google Scholar]
- 58.Gorelik R, Yang C, Kameswaran V, Dominguez R, Svitkina T. Mechanisms of plasma membrane targeting of formin mDia2 through its amino terminal domains. Mol Biol Cell. 2011;22:189–201. doi: 10.1091/mbc.E10-03-0256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Saarikangas J, et al. Molecular mechanisms of membrane deformation by I-BAR domain proteins. Curr Biol. 2009;19:95–107. doi: 10.1016/j.cub.2008.12.029. [DOI] [PubMed] [Google Scholar]
- 60.Minor W, Cymborowski M, Otwinowski Z, Chruszcz M. HKL-3000: the integration of data reduction and structure solution–from diffraction images to an initial model in minutes. Acta Crystallogr D Biol Crystallogr. 2006;62:859–866. doi: 10.1107/S0907444906019949. [DOI] [PubMed] [Google Scholar]
- 61.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 62.Adams PD, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Luan Q, Nolen BJ. Structural basis for regulation of Arp2/3 complex by GMF. Nat Struct Mol Biol. 2013;20:1062–1068. doi: 10.1038/nsmb.2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.