

Abstract
Somatostatin receptor subtype 2 (SSTR2) is the most frequently expressed SSTR subtype in normal human tissues. SSTR2 expression is differentially regulated in various tumor types and therapeutic somatostatin analogs binding to SSTR2 are in clinical use. In prostate cancers highly contradictory results in terms of SSTR2 expression and its consequences have been published over the past years. The aim of this study was to clarify prevalence and clinical significance of SSTR2 expression in prostate cancer. Therefore, quantitative immunohistochemistry (IHC) using a tissue microarray containing samples from 3,261 prostate cancer patients with extensive clinical and molecular cancer characteristics and oncological follow-up data was performed. IHC data was compared to publicly available Gene Expression Omnibus datasets of human prostate cancer gene expression arrays. While membranous SSTR2 staining was always seen in normal prostate epithelium, SSTR2 staining was absent in more than half (56.1%) of 2,195 interpretable prostate cancer samples. About 13% of all analyzed prostate cancers showed moderate to strong cytoplasmic and membranous SSTR2 staining. Staining intensities were inversely correlated with high Gleason grade, advanced pT category, high tumor cell proliferation (p<0.0001 each), high pre-operative PSA levels, (p = 0.0011) and positive surgical margins (p = 0.006). In silico analysis confirmed lower SSTR2 gene expression in prostate cancers vs. normal adjacent tissue (p = 0.0424), prostate cancer metastases vs. primary cancers (p = 0.0011) and recurrent vs. non-recurrent prostate cancers (p = 0.0438). PSA-free survival gradually declined with SSTR2 staining intensity (p<0.0001). SSTR2-negative cancers were more likely to develop metastases over time (p<0.05). In conclusion, most prostate cancers are indeed SSTR2-negative and loss of SSTR2 strongly predicts an unfavorable tumor phenotype and poor prognosis. Therefore, SSTR2 expression seems an important factor in the pathogenesis of prostate cancer and re-introduction of the receptor in SSTR2-negative prostate cancers may feature a promising target for novel gene therapy approaches.
Introduction
In Western males, prostate cancer is the most frequently diagnosed malignant tumor with the second highest rate of cancer-attributed death [1]. Prostate cancers feature considerably variable courses of disease from slow local growth to aggressive and invasive metastatic proliferation. Therefore, a thorough molecular characterization of different prostate cancer subtypes is essential in order to discriminate between aggressive and non-aggressive cancer phenotypes.
Somatostatin (SST) is a cyclic neuroendocrine hormone which was originally isolated from sheep hypothalamus as in vitro inhibitor of growth hormone action (GH, [2]). SST is produced by neuroendocrine cells throughout the whole body and is therefore widely expressed. This comprises the central and peripheral nervous system and a variety of cells within the digestive, genitourinary and reproductive tracts [3].
SST exists in two biologically active isoforms (SST-14 and SST-28) which bind to 5 distinct membrane surface receptors (SSTR1-5) with variable binding affinities [4]. Upon activation by SST-14/-28 all SSTRs inhibit the generation of cyclo-AMP thereby diminishing mitogen-activated (MAP) kinase mediated cell proliferation in a broad variety of cell types [3]. The most widely expressed SSTR subtype in normal tissues [3] is SSTR2.
Various synthetic SSTR agonists are available, of which the anti-proliferative octapeptide octreotide is the most popular representative that is now routinely used for detection and therapy of neuroendocrine tumors [5]. In addition, by agonist profiling octreotide was identified as most potent binding partner of SSTR subtype 2 (SSTR2, [4]).
Using labeled octreotide binding assays, in situ hybridization, immunohistochemistry and RNA-binding assays SSTR2 expression has been shown to be also expressed in a variety of malignant and non-malignant tumors such as pituitary adenoma, meningeoma, neuroblastoma, Non-Hodgkin’s lymphoma, neuroendocrine carcinoid tumors, breast tumors, renal, pancreatic and small cell lung cancers [3]. In these cancers SSTR2 activation leads to an inhibition of tumor cell proliferation, mostly mediated via growth arrests [3].
In prostate cancer there are multiple contradictory reports regarding the role and expression of SSTR2 (for a summary see [6]): Hansson et al., for instance, suggest an up-regulation of SSTR2 expression in prostate cancers [7], Morichetti and coworkers found increased SSTR2 immunostaining in 80% of incidental prostate cancers [8]. Other groups found diminished or absent SSTR2 expression in prostate cancers [9], [10].
In the light of these conflicting reports, we analyzed SSTR2 expression by immunohistochemistry on a large (3,261 tumors) prostate cancer tissue microarray (TMA) and by publicly available Gene Expression Omnibus (GEO) datasets of human prostate cancer gene expression arrays in order to comprehensively clarify prevalence and clinical significance of SSTR2 expression in prostate cancers.
Materials and Methods
Tissue microarray construction
The prostate cancer prognosis tissue microarray (TMA) consisted of cancer samples from 3,261 patients distributed over 7 paraffin blocks. The sampling and constructions have been previously described in detail [11]. In brief, specimens from radical prostatectomies performed between 1992 and 2005 at the Department of Urology, University Medical Center Hamburg-Eppendorf were paraffin-embedded and afterwards matched with clinico-pathological data. TNM classification (American Joint Committee on Cancer, 2002, 6th edition) was used for tumor staging in order to define primary tumor size and local invasiveness (pT), regional lymph node affection (pN) and distant spreading/metastatic disease (pM). Grading of cancers was performed using the Modified Gleason Score [12] and by evaluation of cancer-free surgical margins [13]. In all patients undergoing radical prostatectomy, prostate specific antigen (PSA, [14]) concentrations were measured at the time of diagnosis and for post-operative surveillance quarterly in the first year followed by biannual measurements in the second and annual measurements after the third year following surgery. Recurrence was defined as a postoperative PSA of 0.2 ng/ml. Time of recurrence was defined by the first PSA value above or equal to 0.2 ng/ml. Patients without evidence of tumor recurrence were censored at last follow-up. No patient of the cohort received neo-adjuvant or adjuvant therapy. For TMA construction, representative tissue cylinders with a diameter of 600 µm were punched from tumor areas of a paraffin-embedded donor tissue block and transferred to the corresponding coordinates on the recipient paraffin block in a half-automated process using precision instruments. Four-micrometer thick sections of each microarray block were transferred to adhesive slides for immunohistochemistry analyses.
Use of tissues and clinical data was approved by the ethics committee of the Hamburg Chamber of Physicians and in accordance with local law (Hamburgisches Krankenhausgesetz, HmbKHG) and the Declaration of Helsinki. In agreement with HmbKG, §12, 1–3 and §12a, 1–5 specific informed consent was neither required nor obtained for the present study. All patient records/information were anonymized and de-identified prior to analyses.
Immunohistochemistry (IHC)
Freshly cut TMA sections were stained in one experiment on a single day. TMA sections were de-paraffinized followed by heat-induced antigen retrieval in an autoclave in acetate buffer pH 6.0 for 5 min. Primary polyclonal rabbit anti-SSTR2 antibody (HPA007264, Atlas Antibodies, Stockholm, Sweden) was used in a final dilution of 1∶150. SSTR2 expression was visualized utilizing the Envision System (DAKO, Glostrup, Denmark).
Membranous and cytoplasmatic staining was evaluated separately for each spot. The staining intensity (negative = 0, weak = 1+, intermediate = 2+, strong = 3+) and the fraction of positive tumor cells (in %) were recorded for each tissue spot. A final score was built from these two parameters as previously described [15], [16]. In brief, negative scores had staining intensity of 0, weak scores had staining intensity of 1+ in ≤70% of tumor cells or staining intensity of 2+ in ≤30% of tumor cells; moderate scores had staining intensity of 1+ in >70% of tumor cells, staining intensity of 2+ in >30% and ≤70% of tumor cells or staining intensity of 3+ in ≤30% of tumor cells and strong scores had staining intensity of 2+ in >70% of tumor cells or staining intensity of 3+ in >30% of tumor cells. Ki67 IHC data generated on the same TMA were available from a previous study [17].
In silico cDNA microarray analysis
A Gene Expression Omnibus (GEO) search (www.ncbi.nlm.nih.gov/gds) was conducted for human gene array datasets with information on SSTR2 expression using the string “prostate cancer somatostatin receptor 2 homo sapiens”. Additional requirements were: expression data within the same dataset for comparison of (1) healthy prostate and prostate cancer, (2) primary prostate cancers and metastatic prostate cancers, or (3) non-recurrent and recurrent prostate cancers. Furthermore, suitable datasets needed to include a minimum of n = 22 per group in order to accomplish a Type I error probability of 0.05 to reach statistical power of 0.9 and a difference in groups of one standard deviation in a non-paired sample setting.
Out of a total of 158 datasets identified with expression data on SSTR2 in human prostate cancers only 2 datasets fulfilled all of the criteria above, namely GDS2545 [18], [19] and GDS4109 [20].
GDS2545 contains expression data from 65 primary prostate cancers, 63 normal tissues adjacent to prostate cancer and 25 prostate cancer metastases hybridized to the Affymetrix Human Genome U95 Version 2 Array platform (GPL8300). GDS4109 contains expression data from 39 recurrent and 49 non-recurrent primary prostate cancers hybridized to the Affymetrix Human Genome U133A Array platform (GPL96).
Normalized gene expression values for SSTR2 (GDS2545+ GDS4109) as well as SSTR3, 4 and 5 (GDS2545 only) were extracted and analyzed using the GEO Dataset Browser data analysis online tools. No information was available for SSTR1 from GDS2545.
Statistical analysis
Statistical analyses were accomplished using JMP 5.0.1 software (SAS Institute Inc., Cary, NC, USA) performing Pearson’s chi-square test for contingency tables. Analysis of Variances (ANOVA) was used to test the association of Ki67 labeling index (LI) and SSTR2 staining, Kolmogorov-Smirnov test for determination of normal distribution and subsequent unpaired t-test or Mann-Whitney-U test calculations were performed for in silico cDNA expression analysis using PRISM 6 (Graphpad Software, Inc, La Jolla, CA, USA). Survival curves were calculated by Kaplan–Meier analysis and compared with log rank test. Multivariate analysis utilized the Cox regression model to identify independence of clinical parameters and SSTR2 immunohistochemistry to predict PSA-recurrence, cancer-specific survival and time to onset of metastatic disease after radical prostatectomy.
Results
Baseline characteristics of prostate cancer patients
A TMA was constructed from cancer tissues after radical prostatectomies from 3,261 patients treated at the Department of Urology, University Medical Center Hamburg-Eppendorf between 1992 and 2005. Follow-up data for biochemical cancer relapse were available for 2,385 patients (73.1%) with a mean observation period of 34.7 months (range: 1 to 143 months, see Table 1 for baseline characteristics). TMA cores of 2,363 patients contained prostate cancer cells (72.5%). These cores were included in this study. The group of interpretable cases for statistical analysis was formed by 2,195 spots (67.3%) with immunohistochemical information regarding SSTR2 expression.
Table 1. Baseline Characteristics of 2,363 prostate cancer patients included in the IHC study*.
| Parameter | Variable | n = | % |
| Age at diagnosis [y] | <50 | 57 | 2.6 |
| 50–60 | 701 | 31.8 | |
| 60–70 | 1,317 | 59.7 | |
| >70 | 131 | 5.9 | |
| Pre-operative PSA [ng/ml] | <4 | 339 | 15.4 |
| 4–10 | 1,178 | 53.5 | |
| 10–20 | 493 | 22.4 | |
| >20 | 191 | 8.7 | |
| Gleason score | ≤3+3 | 932 | 41.6 |
| 3+4 | 1,002 | 44.7 | |
| 4+3 | 257 | 11.5 | |
| ≥4+4 | 49 | 2.2 | |
| pT category (AJCC 2002) | pT2 | 1,405 | 62.8 |
| pT3a | 495 | 22.1 | |
| pT3b | 305 | 13.6 | |
| pT4 | 31 | 1.5 | |
| pN category | pN0 | 1,150 | 51.5 |
| pN+ | 79 | 3.5 | |
| pNx | 1,005 | 45.0 | |
| Surgical margins | Negative | 1,749 | 78.2 |
| Positive | 488 | 21.8 | |
| Total: | 2,363* |
*Deviations from total are due to missing data in the subcategories.
(AJCC = American Joint Committee on Cancer, PSA = Prostate-specific antigen).
Loss of SSTR2 expression in prostate cancer is strongly linked with biologically aggressive cancers
SSTR2 expression as determined by IHC was absent in 1,231 (56.1%) of interpretable prostate cancer samples. Weak SSTR2 staining was detected in 680 of these tumors (31.0%). Moderate and strong staining occurred in 187 (8.5%) and 97 prostate cancers (4.4%), respectively. Staining pattern was mostly both membranous and cytoplasmic throughout positive TMA spots. For comparison, normal prostate tissue always showed a positive receptor staining (Figure 1A–D).
Figure 1. SSTR2 immunohistochemistry in normal prostate tissue and prostate cancer.

Microphotographs of tissue microarray cores showing normal prostate and prostate cancer tissues: SSTR2-positive normal epithelium (A), SSTR2-negative (B) and SSTR2-positive prostate cancer tissue (C) as well as SSTR2-negative cancer cells next to strongly SSTR2-positive normal epithelial cells (D).
Contingency analysis of tumor phenotype and clinical features revealed significant, inverse associations of SSTR2 staining intensity with de-differentiation of tumors (as indicated by high Gleason grade, p<0.0001), advanced tumor stage of prostate cancers (as indicated by advanced pT category, p<0.0001, and cancer-positive surgical margins, p = 0.006) as well as high pre-operative PSA levels (p = 0.0011). Statistical details on the correlation of clinico-pathological factors with SSTR2 IHC intensities and frequencies are given in Table 2.
Table 2. SSTR2 IHC intensities and frequencies in patients with radical prostatectomy.
| Parameter | Variable | n = | negative (%) | weak (%) | moderate (%) | strong (%) | p-value |
| Pre-operative PSA[ng/ml] | <4 | 304 | 53.0 | 33.2 | 9.2 | 4.6 | 0.0011 |
| 4–10 | 1,091 | 53.5 | 31.5 | 9.8 | 5.1 | ||
| 10–20 | 462 | 58.4 | 31.0 | 6.9 | 3.7 | ||
| >20 | 183 | 71.6 | 21.3 | 4.9 | 2.2 | ||
| Gleason score | ≤3+3 | 858 | 51.5 | 31.9 | 10.4 | 6.2 | <0.0001 |
| 3+4 | 930 | 55.4 | 32.4 | 8.3 | 4.0 | ||
| 4+3 | 243 | 71.6 | 23.5 | 4.1 | 0.8 | ||
| ≥4+4 | 46 | 78.3 | 17.4 | 4.4 | 0.0 | ||
| pT stage | pT2 | 1,277 | 51.5 | 32.0 | 11.0 | 5.6 | <0.0001 |
| pT3a | 470 | 63.0 | 28.3 | 5.5 | 3.2 | ||
| pT3b | 297 | 63.3 | 31.0 | 4.0 | 1.7 | ||
| pT4 | 32 | 78.1 | 21.9 | 0.0 | 0.0 | ||
| pN stage | pN0 | 1,093 | 57.9 | 32.3 | 6.2 | 3.6 | 0.0963 |
| pN+ | 76 | 65.8 | 29.0 | 5.2 | 0.0 | ||
| Surgical MarginStatus | Negative | 1,613 | 54.3 | 32.0 | 9.2 | 4.5 | 0.0060 |
| Positive | 461 | 62.9 | 26.9 | 6.1 | 4.1 |
Deviations from total are due to missing data in the subcategories and rounding.
The same strong inverse correlation with SSTR2 staining intensity was detected with tumor cell proliferation as determined by Ki67 LI. Ki67 LI increased with decreasing SSTR2 IHC intensity in prostate cancer cells from 2.8±0.4 (strong SSTR2 IHC) over 3.8±0.4 (intermediate SSTR2 IHC) and 5.0±0.2 (weak SSTR2 IHC) to 5.9±0.2 in SSTR2-negative prostate cancers (arithmetical mean ± standard error of the mean, p<0.0001, Figure 2).
Figure 2. Ki67 labeling index and SSTR2 immunohistochemistry (IHC) in prostate cancer cells.
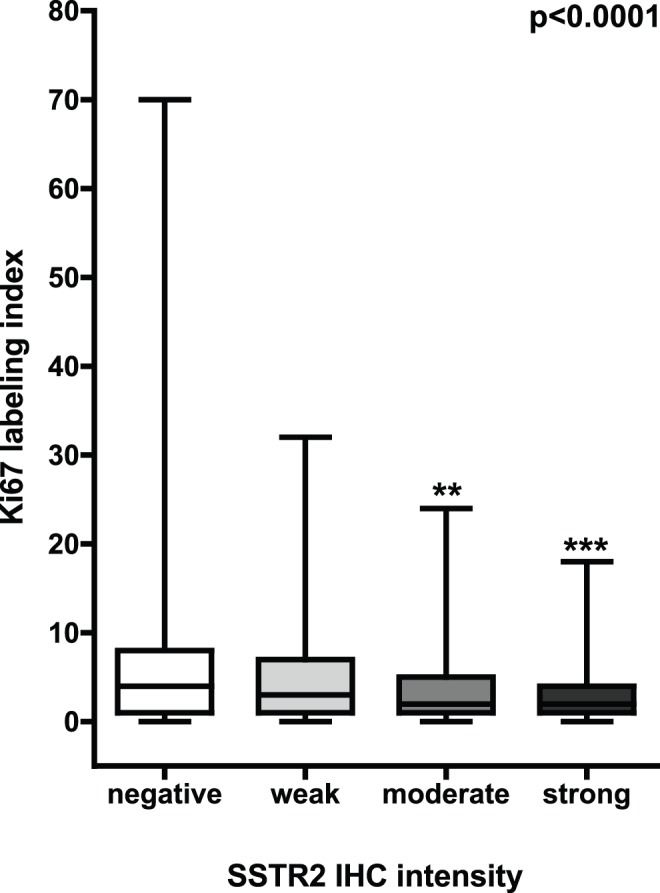
Ki67 labeling index shows a strong inverse correlation with SSTR2 staining intensities (p<0.0001, ANOVA, Dunn’s multiple comparison post-hoc test, box-and-whiskers graph plotting median, 25th and 75th percentile, ** = p<0.01 vs. negative SSTR2 IHC, *** = p<0.001 vs. negative SSTR2 IHC).
To test if the changes in SSTR2 expression detected by IHC could be confirmed on the transcriptional level, we performed in silico expression analysis utilizing two publicly available GEO datasets of human prostate cancer gene expression arrays (GDS2545 and GDS4109).
Using the GDS2545 dataset, we compared expression of SSTR subtypes 2, 3, 4 and 5 in normal prostate tissue with physically adjacent primary prostate cancer in an unpaired fashion. While expression of SSTR3 (mean normalized expression value ± SD: 59.7±30.1 [normal] vs. 76.2±89.1 [cancer], p>0.05) and SSTR5 (226.3±47.1 [normal] vs. 236.1±50.6 [cancer], p>0.05) did not differ significantly, SSTR4 expression was increased (189.7±65.8 [normal] vs. 217.9±73.1 [cancer], p = 0.0236) and SSTR2 expression was significantly lower in prostate cancers (109.5±67.4 [normal] vs. 87.2±65.3 [cancer], p = 0.0424, Figure 3A).
Figure 3. In silico analysis of SSTR gene expression in prostate cancers.

Gene expression analysis of SSTR subtypes in primary prostate cancer tissues versus normal adjacent prostate tissues shows increased SSTR4 (p = 0.0236) and confirms lower SSTR2 expression (p = 0.0424) in primary prostate cancers when comparing all available samples with each other (A, n = 65 vs. 63 patients) or corresponding samples from the same patients only (B, n = 58). Levels of SSTR3 and SSTR5 expression are unchanged (p>0.05). A heatmap of relative SSTR2 gene expression in prostate cancer tissue (tumor) and the surrounding adjacent normal prostate (norm. adj.) per individual patient (column) is shown in (C). SSTR2 expression is also lower in prostate cancer metastases vs. primary tumor (D, p = 0.0011, n = 25) and in recurrent compared with non-recurrent prostate cancers (E, p = 0.0438, n = 39 vs. 40 patients). Standardized expression values were extracted from the identified GEO datasets GDS2545 [18], [19] and GDS4109 [20] and compared as described in the Methods section (Data given as Mean ± SEM; * = p<0.05 vs. corresponding control, ** = p<0.01 vs. corresponding control).
This was also the case when comparing SSTR2 expression in prostate cancer with the corresponding normal adjacent tissue from the same patient using paired analysis (112.5±67.9 [normal] vs. 89.8±66.9 [cancer], p = 0.0486, n = 58 patients, Figure 3B). In total, prostate cancers from 36 patients (62.1%) showed lower SSTR2 expression than their corresponding normal adjacent tissue (Figure 3C). Prostate cancer metastases had even lower SSTR2 expression values than primary prostate cancers (109.5±67.4 [primaries] vs. 55.4±23.9 [metastases], p = 0.0011, Figure 3D). In addition, using the GDS4109 dataset, recurrent prostate cancer showed lower SSTR2 expression compared to non-recurrent cancers (386.5±140.5 [non-recurrent] vs. 340.2±137.0 [recurrent], p = 0.0438, Figure 3E).
Loss of SSTR2 in prostate cancers predicts metastatic and biochemical cancer relapse
In Kaplan-Meier analysis, all tested clinical and pathological features were strongly linked to PSA recurrence, prostate cancer specific survival and time to onset of metastatic disease. This applied for Gleason score, pT stage, pN Stage, pre-operative PSA levels and surgical margin status (all p<0.0001, Table 3 and Figure 4A–C).
Table 3. Association of clinico-pathological features of prostate cancer samples included in this study with PSA recurrence, cancer-specific survival and time to onset of metastatic disease.
| Log-Rank test (Kaplan-Meier analysis) | ||||||
| Parameters | PSA recurrence | Cancer-specific survival | Metastasis-free survival | |||
| χ2 | P value | χ2 | P value | χ2 | P value | |
| Pre-operative PSA [ng/ml] | 111.0 | <0.0001 | 10.4 | 0.0157 | 20.7 | 0.0001 |
| Gleason score | 425.3 | <0.0001 | 96.1 | <0.0001 | 166.1 | <0.0001 |
| pT category (AJCC 2002) | 418.0 | <0.0001 | 63.2 | <0.0001 | 103.8 | <0.0001 |
| pN category | 182.6 | <0.0001 | 9.2 | 0.0024 | 25.5 | <0.0001 |
| Surgical margin status | 111.7 | <0.0001 | 28.1 | <0.0001 | 31.4 | <0.0001 |
Figure 4. pT stage and event-free survival in prostate cancer patients.

Kaplan-Meier curves showing pT stage dependence of PSA-recurrence free survival (A), metastasis-free survival (B) and cancer-specific survival (C, all p<0.0001, Log-Rank test) in radically prostatectomized prostate cancer patients.
Except for prostate cancer-specific survival (p = 0.5942, not shown) SSTR2 staining was inversely correlated with biochemical recurrence and metastasis-free survival: Respectively, PSA free survival (p = 0.0009, Figure 5A) and metastasis-free survival (p = 0.0452, Figure 5B) gradually declined from strong SSTR2 staining over medium and weak to negative.
Figure 5. Clinical impact of SSTR2 staining on event-free survival.
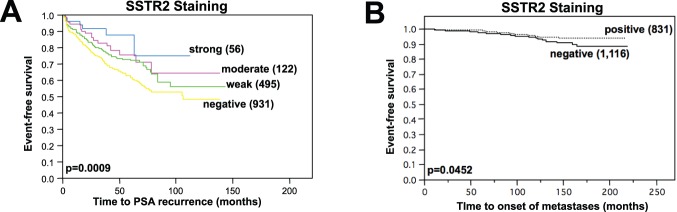
PSA recurrence-free survival gradually declines from strong staining of cancer spots over moderate and weak to SSTR2-negative prostate cancers (A, p = 0.0009, Kaplan-Meier analysis with Log-Rank test). Prostatectomized patients with SSTR2-negative prostate cancers also have impaired metastasis-free survival (B, p = 0.0452, Kaplan-Meier analysis with Log-Rank test).
In multivariate analyses, all tested parameters (Gleason score, pT stage, pN Stage, surgical margin status and pre-operative PSA levels, all p<0.001) but not SSTR2 staining intensity (p = 0.6938) were identified as independent risk factors for biochemical relapse. Comparable results were found for metastasis-free survival (SSTR2 staining, p = 0.8443).
Discussion
Immunohistochemically detectable cytoplasmic and membranous SSTR2 protein was seen in 44% of our 2,195 interpretable prostate cancer samples. Previous studies had analyzed smaller patient cohorts (14–45 cases) and found highly variable results including lower [9], [10] and higher [7], [8] numbers of “SSTR2-positive” prostate cancers as compared to our data. By RT-PCR, Halmos et al. only found three SSTR2-positive prostate cancers out of 22 samples [9]. Cariaga-Martinez et al. report diminished or absent SSTR2 expression in 40 out of 45 prostate cancers using immunohistochemistry [10]. Hansson et al. suggested an up-regulation of SSTR2 expression in 12 out of 14 prostate cancer samples based on RNA in-situ hybridization [7]. Morichetti et al. recently reported a weak to intermediate cytoplasmic SSTR2 IHC staining in approx. 80% of prostate cancers from radical prostatectomies [8].
The comparison of normal and cancerous prostate epithelium in the present study by IHC demonstrated that SSTR2 is not overexpressed, but instead generally downregulated in prostate cancers. Our IHC data were confirmed by two independent cDNA microrarray datasets evaluated for SSTR2 gene expression. Furthermore, our findings are also supported by early data from studies using in vitro receptor autoradiography to compare SSTR2 expression between normal and neoplastic prostate epithelium [21]. Our data show, that SSTR2 downregulation is strongly linked to unfavorable tumor phenotype, early PSA relapse and onset of metastatic disease. This observation is also consistent with a previous study showing IHC positivity in 100% of 12 cases with Gleason grade 1 or 2 but in only 20% of 20 Gleason 4 or 5 cancers [10].
The strong association between high Ki67 LI and downregulated SSTR2 protein found in our study implies that SSTR2 downregulation exerts unfavorable biologic effects upon prostate epithelial cells through diminished cell proliferation control. Several studies have indeed suggested a role of SSTR2 in the regulation of tumor cell proliferation in various tumors, since tumors with reduced SSTR2 protein levels revealed increased cell proliferation [22]–[29]. For example, Qui et al. found significantly higher Ki67 LI in colorectal cancer cells with absent SSTR2 [25]. Over-expression of SSTR2 in MCF-7 breast cancer cells, which naturally express low levels of SSTR2, led to increased apoptosis and cell cycle arrest [29]. In C6 glioma cells, proliferation was inhibited by activation of SSTR2 as measured by [3H]thymidine incorporation assays [28], [30]. Additionally, infection of pancreatic and non-small cell lung cancer cells with SSTR2 expressing adenoviral vectors significantly decreased tumor growth and proliferation rate [27].
The mechanism, by which SSTR2 is able to confer its anti-proliferative effects, has recently been investigated. Zou et al. found a strong inhibitory effect of SSTR2 on the cell cycle in the aforementioned pancreatic cancer cells [27]. In their animal model for pancreatic adenocarcinoma, SSTR2 overexpression led to strong up-regulation of cyclin-dependent kinase inhibitor p16, which then inhibited tumor cell cycle progression from G1 to S phase. Recent data suggest that methylation may be a relevant mechanism for controlling SSTR2 expression in cancer. Torrisani et al. reported the regulation of human SSTR2 expression in various (cancer) cell lines by epigenetic modifications [31]. Their data showed that DNA methylation and histone acetylation can regulate the activity of the SSTR2 promoter and demonstrated that promoter activity directly correlates with SSTR2 expression in breast cancer, pancreatic cancer, hepatic cancer, melanoma and retinoblastoma cell lines in an inverse fashion. Moreover, treatment of these cells with demethylating agents and acetylase inhibitors rescued SSTR2 mRNA expression [31].
As a surface membrane receptor, SSTR2 is suitable as a therapeutic target. Since somatostatin’s short half-life limits its therapeutic use, synthetic analogs have been developed since the 1980s [32]. Octreotide is the best-characterized analog, and it binds to SSTR2 with higher affinity and much higher therapeutic potency than somatostatin [32]. Several other therapeutic ligands of SSTR2 are currently available or under clinical testing, including long-acting formulations (lancreotide, vapreotide, seglitide and pasireotide/SOM230) [33], and chimeric molecules coupled with cytotoxic agents (e.g. AN-238, a doxorubicin/somatostatin conjugate, [34]).
Octreotide and its derivatives have been used routinely for the detection and therapy of neuroendocrine tumors for years [35]. In C6 glioma, activation of SSTR2 by various somatostatin analogs led to a strong inhibition of in vivo cancer cell proliferation, intratumoral neoangiogenesis and Ki-67 expression [30].
Moreover, experimental treatment with the cytotoxic somatostatin analog AN-238 strongly inhibited tumor proliferation in a broad variety of SSTR2 positive cancer models such as Non-Hodgkin’s lymphoma [34], malignant melanoma [36], pheochromocytoma [37], endometrial [38], ovarian [39], colon [40] and gastric carcinomas [41] as well as small and non-small cell lung carcinoma [42], [43].
In concordance with our data, human pancreatic adenocarcinomas lose SSTR2 expression [44]. Re-introduction of SSTR2 in pancreas cancer by gene transfer robustly inhibited tumor cell proliferation and tumorigenicity [24], [45]. In the SSTR2-negative pancreatic and non-small lung cell cancer models of Zou and colleagues, re-expression of SSTR2 in the cancer cells led to significantly impaired tumor growth which was aggravated in a dose-dependent manner by application of octreotide and its derivate vapreotide (RC-160, [27]).
Lastly, we show that loss of SSTR2 was linked to metastatic progression of prostate cancers. Patients with SSTR2-negative prostate cancers had impaired metastasis-free survival, and metastases of prostate cancers expressed even lower levels of SSTR2 mRNA than primary prostate cancers. Similar findings have been described for colorectal cancers [46] and seminonas [47]. In addition, gene transfer of SSTR2 inhibited metastatic progression in two different pancreatic carcinoma models [48], [49].
How SSTR2 might regulate tumor spreading is less clear, but published data suggests a mechanism promoting cell-cell adhesions. In pancreatic cancer the Src homology region 2 domain-containing tyrosine phosphatase 1 (SHP-1) is able to de-phosphorylate the epithelial cell adhesion molecule E-cadherin, thereby stabilizing inter-epithelial cell junctions in a SSTR2-dependent fashion [48], [50]. SHP-1 has also been shown to be downregulated in biologically aggressive prostate cancers [10]. This mechanism is further supported by work of Lahlou et al. showing that SSTR2 facilitated restoration of functional gap junctions in pancreatic adenocarcinoma cells through up-regulation of connexins Cx26 and Cx43 [51].
Most interestingly, in prostate cancer cells SSTR2 is also able to inhibit another feature of cancer metastasis, cell migration [52]. Mechanistically, this seems to be mediated by ligand-dependent activation of the Y-27632-sensitive Rho-GTPase pathway [52]. Data from normal, healthy, primary keratinocytes corroborate this mechanism and suggest underlying SSTR2- (amongst others) mediated inhibition of Rac1 activity [53].
Conclusions
We have been able to clarify that loss of SSTR2 is strongly linked to an aggressive tumor phenotype and predicts poor prognosis of prostate cancers. Indeed, most prostate cancers are SSTR2 negative and SSTR2 expression seems to be an important factor in the pathogenesis of prostate cancer. Although, the suitability of SSTR2 as a target for gene therapy needs to be evaluated, loss of SSTR2 is strongly linked to invasiveness, early PSA relapse, and metastatic spread in prostate cancer. Our data suggest that mechanistically these effects are mediated by increased cancer cell proliferation in cells with down-regulated SSTR2.
Acknowledgments
The authors would like to thank Christina Koop for technical assistance.
Funding Statement
This work was funded by the University Medical Center Hamburg-Eppendorf. JKH received a research fellowship from the “Hubertus Wald Tumorzentrum / University Cancer Center Hamburg” during this work. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Siegel R, Ma J, Zou Z, Jemal A (2014) Cancer statistics, 2014. CA Cancer J Clin 64: 9–29 10.3322/caac.21208 [DOI] [PubMed] [Google Scholar]
- 2. Brazeau P, Vale W, Burgus R, Ling N, Butcher M, et al. (1973) Hypothalamic polypeptide that inhibits the secretion of immunoreactive pituitary growth hormone. Science 179: 77–79. [DOI] [PubMed] [Google Scholar]
- 3. Patel YC (1999) Somatostatin and its receptor family. Front Neuroendocrinol 20: 157–198 10.1006/frne.1999.0183 [DOI] [PubMed] [Google Scholar]
- 4. Patel YC, Srikant CB (1994) Subtype selectivity of peptide analogs for all five cloned human somatostatin receptors (hsstr 1–5). Endocrinology 135: 2814–2817. [DOI] [PubMed] [Google Scholar]
- 5. Kwekkeboom D, Krenning EP, de Jong M (2000) Peptide receptor imaging and therapy. J Nucl Med 41: 1704–1713. [PubMed] [Google Scholar]
- 6. Mazzucchelli R, Morichetti D, Santinelli A, Scarpelli M, Bono AV, et al. (2011) Immunohistochemical expression and localization of somatostatin receptor subtypes in androgen ablated prostate cancer. Cell Oncol (Dordr) 34: 235–243 10.1007/s13402-011-0031-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hansson J, Bjartell A, Gadaleanu V, Dizeyi N, Abrahamsson P-A (2002) Expression of somatostatin receptor subtypes 2 and 4 in human benign prostatic hyperplasia and prostatic cancer. The Prostate 53: 50–59 10.1002/pros.10121 [DOI] [PubMed] [Google Scholar]
- 8. Morichetti D, Mazzucchelli R, Stramazzotti D, Santinelli A, Lopez-Beltran A, et al. (2010) Immunohistochemical expression of somatostatin receptor subtypes in prostate tissue from cystoprostatectomies with incidental prostate cancer. BJU Int 106: 1072–1080 10.1111/j.1464-410X.2010.09238.x [DOI] [PubMed] [Google Scholar]
- 9. Halmos G, Schally AV, Sun B, Davis R, Bostwick DG, et al. (2000) High expression of somatostatin receptors and messenger ribonucleic acid for its receptor subtypes in organ-confined and locally advanced human prostate cancers. J Clin Endocrinol Metab 85: 2564–2571. [DOI] [PubMed] [Google Scholar]
- 10.Cariaga-Martinez AE, Lorenzati MA, Riera MA, Cubilla MA, La Rossa De A, et al. (2009) Tumoral prostate shows different expression pattern of somatostatin receptor 2 (SSTR2) and phosphotyrosine phosphatase SHP-1 (PTPN6) according to tumor progression. Adv Urol: 723831. doi:10.1155/2009/723831. [DOI] [PMC free article] [PubMed]
- 11. Schlomm T, Iwers L, Kirstein P, Jessen B, Köllermann J, et al. (2008) Clinical significance of p53 alterations in surgically treated prostate cancers. Mod Pathol 21: 1371–1378 10.1038/modpathol.2008.104 [DOI] [PubMed] [Google Scholar]
- 12. Epstein JI, Allsbrook WC, Amin MB, Egevad LL (2005) ISUP Grading Committee (2005) The 2005 International Society of Urological Pathology (ISUP) Consensus Conference on Gleason Grading of Prostatic Carcinoma. Vol. 29: 1228–1242. [DOI] [PubMed] [Google Scholar]
- 13. Tan PH, Cheng L, Srigley JR, Griffiths D, Humphrey PA, et al. (2011) International Society of Urological Pathology (ISUP) Consensus Conference on Handling and Staging of Radical Prostatectomy Specimens. Working group 5: surgical margins. Vol. 24: 48–57 10.1038/modpathol.2010.155 [DOI] [PubMed] [Google Scholar]
- 14. Trock BJ (2014) Circulating biomarkers for discriminating indolent from aggressive disease in prostate cancer active surveillance. Curr Opin Urol 24: 293–302 10.1097/MOU.0000000000000050 [DOI] [PubMed] [Google Scholar]
- 15.Tennstedt P, Fresow R, Simon R, Marx A, Terracciano L, et al. (2012) RAD51 overexpression is a negative prognostic marker for colorectal adenocarcinoma. Int J Cancer. doi:10.1002/ijc.27907. [DOI] [PubMed]
- 16. Minner S, Enodien M, Sirma H, Luebke AM, Krohn A, et al. (2011) ERG status is unrelated to PSA recurrence in radically operated prostate cancer in the absence of antihormonal therapy. Clin Cancer Res 17: 5878–5888 10.1158/1078-0432.CCR-11-1251 [DOI] [PubMed] [Google Scholar]
- 17. Minner S, Jessen B, Stiedenroth L, Burandt E, Köllermann J, et al. (2010) Low level HER2 overexpression is associated with rapid tumor cell proliferation and poor prognosis in prostate cancer. Clin Cancer Res 16: 1553–1560 10.1158/1078-0432.CCR-09-2546 [DOI] [PubMed] [Google Scholar]
- 18. Yu YP, Landsittel D, Jing L, Nelson J, Ren B, et al. (2004) Gene expression alterations in prostate cancer predicting tumor aggression and preceding development of malignancy. Journal of Clinical Oncology 22: 2790–2799 10.1200/JCO.2004.05.158 [DOI] [PubMed] [Google Scholar]
- 19. Chandran UR, Ma C, Dhir R, Bisceglia M, Lyons-Weiler M, et al. (2007) Gene expression profiles of prostate cancer reveal involvement of multiple molecular pathways in the metastatic process. BMC Cancer 7: 64 10.1186/1471-2407-7-64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sun Y, Goodison S (2009) Optimizing molecular signatures for predicting prostate cancer recurrence. The Prostate 69: 1119–1127 10.1002/pros.20961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Reubi JC, Waser B, Schaer JC, Markwalder R (1995) Somatostatin receptors in human prostate and prostate cancer. J Clin Endocrinol Metab 80: 2806–2814. [DOI] [PubMed] [Google Scholar]
- 22. Taylor JE, Theveniau MA, Bashirzadeh R, Reisine T, Eden PA (1994) Detection of somatostatin receptor subtype 2 (SSTR2) in established tumors and tumor cell lines: evidence for SSTR2 heterogeneity. Peptides 15: 1229–1236. [DOI] [PubMed] [Google Scholar]
- 23. Plonowski A, Schally AV, Nagy A, Sun B, Szepeshazi K (1999) Inhibition of PC-3 human androgen-independent prostate cancer and its metastases by cytotoxic somatostatin analogue AN-238. Cancer Res 59: 1947–1953. [PubMed] [Google Scholar]
- 24. Rochaix P, Delesque N, Estève JP, Saint-Laurent N, Voight JJ, et al. (1999) Gene therapy for pancreatic carcinoma: local and distant antitumor effects after somatostatin receptor sst2 gene transfer. Hum Gene Ther 10: 995–1008 10.1089/10430349950018391 [DOI] [PubMed] [Google Scholar]
- 25. Qiu C-Z, Wang C, Huang Z-X, Zhu S-Z, Wu Y-Y, et al. (2006) Relationship between somatostatin receptor subtype expression and clinicopathology, Ki-67, Bcl-2 and p53 in colorectal cancer. World J Gastroenterol 12: 2011–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhou T, Xiao X, Xu B, Li H, Zou Y (2009) Overexpression of SSTR2 inhibited the growth of SSTR2-positive tumors via multiple signaling pathways. Acta Oncol 48: 401–410 10.1080/02841860802314746 [DOI] [PubMed] [Google Scholar]
- 27. Zou Y, Xiao X, Li Y, Zhou T (2009) Somatostatin analogues inhibit cancer cell proliferation in an SSTR2-dependent manner via both cytostatic and cytotoxic pathways. Oncol Rep 21: 379–386. [PubMed] [Google Scholar]
- 28. Barbieri F, Pattarozzi A, Gatti M, Aiello C, Quintero A, et al. (2009) Differential efficacy of SSTR1, −2, and −5 agonists in the inhibition of C6 glioma growth in nude mice. Am J Physiol Endocrinol Metab 297: E1078–E1088 10.1152/ajpendo.00292.2009 [DOI] [PubMed] [Google Scholar]
- 29. He Y, Yuan X-M, Lei P, Wu S, Xing W, et al. (2009) The antiproliferative effects of somatostatin receptor subtype 2 in breast cancer cells. Acta Pharmacol Sin 30: 1053–1059 10.1038/aps.2009.59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Barbieri F, Pattarozzi A, Gatti M, Porcile C, Bajetto A, et al. (2008) Somatostatin receptors 1, 2, and 5 cooperate in the somatostatin inhibition of C6 glioma cell proliferation in vitro via a phosphotyrosine phosphatase-eta-dependent inhibition of extracellularly regulated kinase-1/2. Endocrinology 149: 4736–4746 10.1210/en.2007-1762 [DOI] [PubMed] [Google Scholar]
- 31. Torrisani J, Hanoun N, Laurell H, Lopez F, Maoret J-J, et al. (2008) Identification of an upstream promoter of the human somatostatin receptor, hSSTR2, which is controlled by epigenetic modifications. Endocrinology 149: 3137–3147 10.1210/en.2007-1525 [DOI] [PubMed] [Google Scholar]
- 32. Appetecchia M, Baldelli R (2010) Somatostatin analogues in the treatment of gastroenteropancreatic neuroendocrine tumours, current aspects and new perspectives. J Exp Clin Cancer Res 29: 19 10.1186/1756-9966-29-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Grozinsky-Glasberg S, Shimon I, Korbonits M, Grossman AB (2008) Somatostatin analogues in the control of neuroendocrine tumours: efficacy and mechanisms. Endocr Relat Cancer 15: 701–720 10.1677/ERC-07-0288 [DOI] [PubMed] [Google Scholar]
- 34. Keller G, Engel JB, Schally AV, Nagy A, Hammann B, et al. (2005) Growth inhibition of experimental non-Hodgkin’s lymphomas with the targeted cytotoxic somatostatin analogue AN-238. Int J Cancer 114: 831–835 10.1002/ijc.20806 [DOI] [PubMed] [Google Scholar]
- 35. Kwekkeboom DJ, Kam BL, van Essen M, Teunissen JJM, van Eijck CHJ, et al. (2010) Somatostatin-receptor-based imaging and therapy of gastroenteropancreatic neuroendocrine tumors. Endocr Relat Cancer 17: R53–R73 10.1677/ERC-09-0078 [DOI] [PubMed] [Google Scholar]
- 36. Keller G, Schally AV, Nagy A, Baker B, Halmos G, et al. (2006) Effective therapy of experimental human malignant melanomas with a targeted cytotoxic somatostatin analogue without induction of multi-drug resistance proteins. Int J Oncol 28: 1507–1513. [PubMed] [Google Scholar]
- 37. Ziegler CG, Brown JW, Schally AV, Erler A, Gebauer L, et al. (2009) Expression of neuropeptide hormone receptors in human adrenal tumors and cell lines: antiproliferative effects of peptide analogues. P Natl Acad Sci Usa 106: 15879–15884 10.1073/pnas.0907843106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Engel JB, Schally AV, Halmos G, Baker B, Nagy A, et al. (2005) Targeted therapy with a cytotoxic somatostatin analog, AN-238, inhibits growth of human experimental endometrial carcinomas expressing multidrug resistance protein MDR-1. Cancer 104: 1312–1321 10.1002/cncr.21327 [DOI] [PubMed] [Google Scholar]
- 39. Buchholz S, Keller G, Schally AV, Halmos G, Hohla F, et al. (2006) Therapy of ovarian cancers with targeted cytotoxic analogs of bombesin, somatostatin, and luteinizing hormone-releasing hormone and their combinations. P Natl Acad Sci Usa 103: 10403–10407 10.1073/pnas.0602971103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Szepeshazi K, Schally AV, Halmos G, Armatis P, Hebert F, et al. (2002) Targeted cytotoxic somatostatin analogue AN-238 inhibits somatostatin receptor-positive experimental colon cancers independently of their p53 status. Cancer Res 62: 781–788. [PubMed] [Google Scholar]
- 41. Szepeshazi K, Schally AV, Nagy A, Wagner BW, Bajo AM, et al. (2003) Preclinical evaluation of therapeutic effects of targeted cytotoxic analogs of somatostatin and bombesin on human gastric carcinomas. Cancer 98: 1401–1410 10.1002/cncr.11649 [DOI] [PubMed] [Google Scholar]
- 42. Kiaris H, Schally AV, Nagy A, Szepeshazi K, Hebert F, et al. (2001) A targeted cytotoxic somatostatin (SST) analogue, AN-238, inhibits the growth of H-69 small-cell lung carcinoma (SCLC) and H-157 non-SCLC in nude mice. Eur J Cancer 37: 620–628. [DOI] [PubMed] [Google Scholar]
- 43. Szereday Z, Schally AV, Szepeshazi K, Bajo AM, Hebert F, et al. (2003) Effective treatment of H838 human non-small cell lung carcinoma with a targeted cytotoxic somatostatin analog, AN-238. Int J Oncol 22: 1141–1146. [PubMed] [Google Scholar]
- 44. Buscail L, Saint-Laurent N, Chastre E, Vaillant JC, Gespach C, et al. (1996) Loss of sst2 somatostatin receptor gene expression in human pancreatic and colorectal cancer. Cancer Res 56: 1823–1827. [PubMed] [Google Scholar]
- 45. Vernejoul F, Faure P, Benali N, Calise D, Tiraby G, et al. (2002) Antitumor effect of in vivo somatostatin receptor subtype 2 gene transfer in primary and metastatic pancreatic cancer models. Cancer Res 62: 6124–6131. [PubMed] [Google Scholar]
- 46. Evangelou I, Petraki C, Msaouel P, Scorilas A, Sdrolia E, et al. (2012) Immunohistochemical expression of somatostatin receptor subtypes 2 and 5 in colorectal cancer. Eur J Clin Invest 42: 777–783 10.1111/j.1365-2362.2012.02648.x [DOI] [PubMed] [Google Scholar]
- 47. Port M, Wang Y, Schmelz HU, Pottek T, Meineke V, et al. (2011) A gene signature of primary tumor identifies metastasized seminoma. Urol Oncol 29: 764–773 10.1016/j.urolonc.2009.08.008 [DOI] [PubMed] [Google Scholar]
- 48. Benali N, Cordelier P, Calise D, Pages P, Rochaix P, et al. (2000) Inhibition of growth and metastatic progression of pancreatic carcinoma in hamster after somatostatin receptor subtype 2 (sst2) gene expression and administration of cytotoxic somatostatin analog AN-238. P Natl Acad Sci Usa 97: 9180–9185 10.1073/pnas.130196697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Feng Y, Huang T, Gao J, Chang Q, Qin R (2006) Inhibition of metastatic progression of SSTR2 gene transfection mediated by adenovirus in human pancreatic carcinoma cells. J Huazhong Univ Sci Technol Med Sci 26: 68–71. [DOI] [PubMed] [Google Scholar]
- 50. Lopez F, Estève JP, Buscail L, Delesque N, Saint-Laurent N, et al. (1997) The tyrosine phosphatase SHP-1 associates with the sst2 somatostatin receptor and is an essential component of sst2-mediated inhibitory growth signaling. J Biol Chem 272: 24448–24454. [DOI] [PubMed] [Google Scholar]
- 51. Lahlou H, Fanjul M, Pradayrol L, Susini C, Pyronnet S (2005) Restoration of functional gap junctions through internal ribosome entry site-dependent synthesis of endogenous connexins in density-inhibited cancer cells. Mol Cell Biol 25: 4034–4045 10.1128/MCB.25.10.4034-4045.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tang X, Takekoshi S, Itoh J, Umemura S, Shoji S, et al. (2010) Somatostatin analogue inhibits the mobility of prostate carcinoma cells: a new therapeutic method for advanced prostate carcinoma. Int J Oncol 37: 1077–1083 10.3892/ijo00000759 [DOI] [PubMed] [Google Scholar]
- 53. Vockel M, Pollok S, Breitenbach U, Ridderbusch I, Kreienkamp H-J, et al. (2011) Somatostatin inhibits cell migration and reduces cell counts of human keratinocytes and delays epidermal wound healing in an ex vivo wound model. PLoS ONE 6: e19740 10.1371/journal.pone.0019740 [DOI] [PMC free article] [PubMed] [Google Scholar]