

Abstract
Aim
Following toll-like receptor (TLR) engagement, lipopolysaccharide (LPS) can stimulate the expression of pro-inflammatory cytokines thus activating the innate immune response. The production of inflammatory cytokines results, in part, from the activation of kinase-induced signaling cascades and transcriptional factors. Of the four distinct classes of mitogen-activated protein (MAP) kinases described in mammals, p38, c-Jun N-terminal activated kinases (JNK1-3) and extracellular activated kinases (ERK1,2) are the best studied. Previous data has established that p38 MAPK signaling is required for inflammation and bone loss in periodontal disease preclinical animal models.
Materials and Methods
In the present study, we obtained healthy and diseased periodontal tissues along with clinical parameters and microbiological parameters. Excised fixed tissues were immunostained with total and phospho-specific antibodies against p38, JNK, and ERK kinases.
Results
Intensity scoring from immunostained tissues was correlated with clinical periodontal parameters. Rank correlations with clinical indices were statistically significantly positive (p-value < 0.05) for total p38 (correlations ranging 0.49–0.68), phospho-p38 (range 0.44–0.56), and total ERK (range 0.52–0.59) levels, and correlations with JNK levels also supported association (range 0.42–0.59). Phospho-JNK and phospho-ERK showed no significant positive correlation with clinical parameters of disease.
Conclusion
These data strongly implicate p38 MAPK as a major MAPK involved in human periodontal inflammation and severity.
Keywords: mitogen activating protein kinases, periodontitis, inflammation, cell signaling
Introduction
In the periodontal microenvironment, bacterial constituents including gram-negative derived lipopolysaccharide (LPS) can initiate inflammatory bone loss as appreciated in periodontal diseases. LPS can stimulate the expression of IL-1β, TNF-α, IL-6 and receptor activator of NFκB Ligand (RANKL) by activating the innate immune response as well as non-immune cells such as gingival fibroblasts (Nakashima et al., 2000, Jiang et al., 2002, Kikuchi et al., 2003). The production of inflammatory cytokines results from the activation of kinase-induced signaling cascades and transcriptional factors. Within periodontal diseased tissues monocytes, macrophages, and fibroblasts all produce cytokines such as TNF-α, IL-1β, PGE2, and IL-6 (Reddi et al., 1996, Lee et al., 1995) and have all been found to be significantly elevated in diseased periodontal sites compared to healthy or inactive sites (Ejeil et al., 2003, Gamonal et al., 2000, Geivelis et al., 1993, Gorska et al., 2003, Stashenko et al., 1991). These cytokines orchestrate the cascade of destructive events that occur in the periodontal tissues, and trigger the production of an array of inflammatory enzymes and mediators including MMPs and prostaglandins. Moreover, pro-inflammatory cytokines directly or indirectly recruit and activate osteoclasts through RANKL-dependent and independent pathways, resulting in irreversible periodontal bone destruction (Graves, 1999, Assuma et al., 1998). Therefore, knowledge regarding the molecular pathways that govern expression of many inflammatory mediators may have therapeutic significance in the management of inflammatory diseases including chronic periodontitis.
Bacterial LPS initiates the innate immune signaling cascade through interaction with cell surface protein expressed CD14 as well as toll-like receptors (TLRs), mainly TLR-2 and TLR-4 (Yang et al., 2000, Kirschning and Bauer, 2001, Kirschning et al., 1998). Expression of both TLR-2 and TLR-4 is increased in periodontal disease tissues, suggesting that these receptors have an increased capacity to signal and influence downstream cytokine expression (Mori et al., 2003, Beklen et al., 2008). TLR-4 signaling activates MyD88-dependent pathways to subsequent activation of interleukin-1R-associated kinase (IRAK), tumor necrosis factor receptor-associated factor-6 (TRAF6) and ultimately nuclear factor kappa B (NF-κB) required for cytokine induction. Also, TRAF6-dependent pathways are required for recruitment of different adaptor proteins and activation of various mitogen activating protein kinases (MAPK) and activation of nuclear factor kappa B (NF-κB). The transcription factors NF-κB and activator protein-1 (AP-1) control expression of some of the most common inflammatory mediators present in periodontal inflammation, including IL-1, IL-6, TNF-α and MMPs (reviewed in (Kirkwood and Rossa, 2009)).
Although both MAPK pathways and NF-κB signaling pathways are well established in regulation of inflammatory cytokines and MMP genes in periodontal diseases, considerable less information is known about MAPK than NF-κB signaling in human disease. MAPKs phosphorylate a number of intracellular targets, including transcription factors (e.g. AP-1 and CHOP) and RNA binding proteins (e.g. tristetraprolin and HuR), which tightly regulate expression of inflammatory genes including at transcriptional and posttranscriptional levels. Target genes downstream of MAPK signaling include many common inflammatory mediators such as IL-1, IL-6, TNF-α, COX-2, and MMPs (recently reviewed in (Palanisamy et al., 2012)).
Understanding the cell signaling processes involved in periodontal disease progression and destruction is key in the development of new therapeutic approaches to prevent or attenuate periodontal destruction. Preclinical periodontal disease models indicate that there is a strong positive correlation between activation of MAPKs, inflammation and bone loss (Kirkwood et al., 2007a, Kirkwood and Rossa, 2009, Li et al., 2012, Li et al., 2011, Rogers et al., 2007). However, no data regarding the significance of MAPK activation in periodontal disease in humans has been evaluated. In the study, clinical periodontal parameters were correlated with the level of activation of MAPKs in subjects with chronic periodontal disease compared to healthy controls. Data obtained strongly indicate that total p38 and ERK have a significant positive correlation with periodontal disease severity while only phosphorylated p38 only correlates with clinical parameters of periodontal disease progression.
MATERIALS AND METHODS
This study was approved by the Institutional Review Board for the Health Sciences at the University of Michigan, Ann Arbor, MI, USA. A total of twenty-one tissue samples from human subjects were included in the study (13 females and 8 males). The subjects were divided into two groups: healthy (10 subjects) and diseased (11 subjects). Informed consent was obtained with all patients prior to initiating study.
Prior to surgery, clinical parameters were measured at the same sites where tissues were harvested including: Plaque Index (PI) on a scale of 0-3 (0-no plaque, 1-w/probe, 2-visible, 3-abundant) (Loe, 1967), Gingival Index (GI) on a scale of 0-3 (0-no inflammation, 1-mild, 2-moderate w/BOP, 3-severe, spontaneous bleeding on probing (BOP), pocket depth (PD), BOP, gingival recession (REC) and clinical attachment level (CAL). Based on these parameters the inclusion criteria for the diseased group consisted of: at least 1 site with PD>4mm, GI 1-3 and PI 1-3. For the healthy controls acceptable parameters were: PD≤4mm, GI≤1, and PI≤2. The exclusion criteria for both groups included: smokers, unstable systemic diseases or chronic disorders (diabetes, rheumatoid arthritis), patients using steroids, antibiotics, NSAIDS and/or other host modulators. The procured samples were from tissues that would have been otherwise discarded after periodontal surgery and or extraction sites. When clinically indicated, procured tissues included connective tissue near the sulcular epithelium.
Microbial assessments: BANA Test
The BANA test was used to determine the presence of “red-complex” periodontal pathogens (Porphyromonas gingivalis, Treponema denticola, and Tannerella forsythia) (Loesche et al., 1990) due to the unique ability of these pathogens to hydrolyze a synthetic trypsin substrate, benzoyl-DL-arginine-naphthylamide (BANA). Plaque was collected for BANA analysis from the surgical site prior to surgery. A curette was used for plaque collection from the site. After incubation for 5 minutes at 35°C with Evan’s black dye solution, naphthylamine was released as a result of the presence of any one of the “red-complex” pathogens and formed a permanent blue-black color. The test was read as positive or negative based on the relative intensity of the blue color.
Tissue processing and Immunohistochemistry
Immediately following procurement the tissue samples were stored in vials containing 4% vol formaldehyde and phosphatase inhibitor cocktail (PhosSTOP, Roche Diagnostic, Germany). After 24–36 hours they were transferred into 70% ethanol, embedded in paraffin and then sections were prepared. An attempt was made to cut the sections in such a manner so that every sample would have both the epithelium and underlying connective tissue whenever possible. Immunohistochemical staining for p38, phospho-p38, ERK, phospho-ERK, JNK and phospho-JNK was performed. Deparaffinized ethanol-dehydrated tissue sections were placed in a pressure chamber (Biocare Medical, Concord, CA) for 15 min in an antigen retrieval buffer (DAKO, Glostrup, Denmark) and allowed to cool to room temperature. Primary antibodies for p38, phospho-p38, ERK, phospho-ERK, JNK and phospho-JNK (Cell Signaling, Beverly, MA; R&D Systems, Minneapolis, MN and Abcam, Cambridge, MA) were used to evaluate the extent of expression, detected using Vectastain Elite ABC reagent and Nova Red (Vector Laboratories, Burlingame, CA) per manufacturer’s instructions. Control sections were incubated as well with pre-immune serum or control IgG. An independent oral pathologist (NJD), blinded to the periodontal status of the subjects, completed the immunohistochemical staining scoring analysis of the inflammatory infiltrate in the connective tissue as previously described (Kirkwood et al., 2007b, Mitra et al., 2008). Due to high background staining, the epithelium was not scored. The examiner first went through all the slides for a particular stain and selected slides that represented each of the four levels of intensity. All slides were scored using reference slides as the standard. All slides were assigned a score of 0, 1, 2, or 3, where “0 = none”, “1 = low”, “2 = medium/moderate”, and “3 = high/severe” positive stain.
Inflammation Intensity Scoring
Serial sections from periodontal tissue samples were sectioned for standard hematoxylin and eosin (H&E) staining to correlate with IHC staining. Specimens were assessed and read for the degree of inflammatory infiltrate by a blinded oral pathologist (NJD). Infiltrate intensity was scored as: none (less than mild), mild, moderate or severe.
Statistical Analysis
All statistical analyses were performed using R version 2.12.1(Team., 2010). The raw immunohistochemical staining scores and periodontal outcomes of gingival index (GI) and periodontal index (PI) are presented in table format. PD measurements associated with the tooth from which the tissue sample was taken were summarized using the periodontal inflammatory burden index, PIBI, defined as the number of PD measurements of 4 or 5mm plus twice the number of PD measurements ≥ 6mm (Lindy et al., 2008, Leppilahti et al., 2011). We defined a summary clinical attachment level index, CALI, in an analogous fashion as the number of CAL measurements of 4 or 5mm plus twice the number of CAL measurements ≥ 6mm. The clinical summaries PIBI and CALI of the disease status of the area where tissue was taken range from 0 to 12, with higher values indicating more severe disease. Associations between the ordinal immunohistochemical staining outcomes for p38, phospho-p38, ERK, phospho-ERK, JNK and phospho-JNK and clinical periodontal outcomes were quantified using the Spearman rank correlation (computed using the function rcorr from the R package Hmisc (Harrell Jr, 2010). Confidence intervals for the correlations were obtained by back transforming normality-based confidence intervals for the Fisher transformation (Fisher, 1915) of the estimated correlation. Because Fisher’s transformation is nonlinear, this back transformation of the symmetric normality-based confidence interval results in an asymmetric confidence interval for the raw (untransformed) correlation.
RESULTS
Subsets of MAPK Expression Correlates with Inflammation and Microbiological Assessments
Twenty-one samples of periodontal tissues were collected from healthy subjects and chronic periodontitis patients. In the healthy group, nine samples out of ten (90%) were graded as “none - no inflammatory infiltrate observed” with scattered focally present lymphocytes and occasionally plasma cells. One sample was graded as mild and in that case focally present lymphocytes, plasma cells and macrophages were observed. The diseased group consisted of no samples in the “none” category, four (36%) in the “mild”, six (55%) in the “moderate”, and one (9%) in the “severe”. The diseased group exhibited significantly more inflammatory infiltrate, and lymphocytes, plasma cells and macrophages were present in almost all the samples. Occasionally bacterial colonies were observed and interpreted as dental calculus as well as extravasated erythrocytes. Immunohistochemical (IHC) staining was performed on tissues following extensive optimization for each of the following antibodies: total p38, JNK, and ERK along with phosphorylated (active) p38, JNK, and ERK. Representative examples of total and phospho-p38 (Figure 1), total and phospho-JNK (Figure 2) and total and phospho-ERK (Figure 3) are presented.
Figure 1.
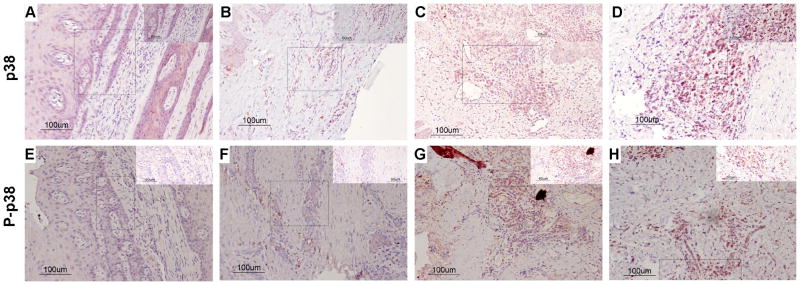
Total (A–D) and Phospho (E–H) p38 MAPK immunohistochemistry scoring. The left column (panels A, B, E, and F) illustrates a “0” score (no staining) of immunostained tissue samples. The right column (panels C, D, G, and H) illustrates a high/severe level of “3” immunostained tissue samples. “E” refers to epithelium and “CT” refers to connective tissue. Arrows indicate representative areas of the inflammatory infiltrate scored.
Figure 2.
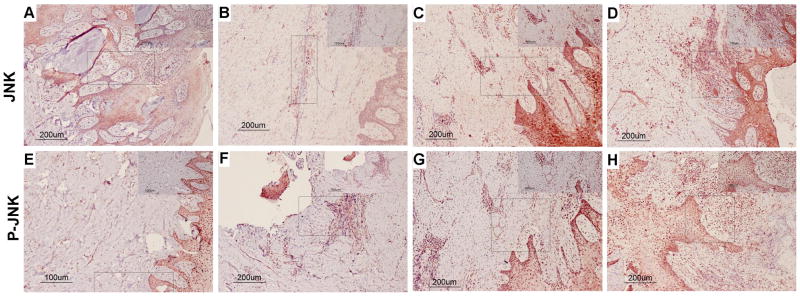
Total (A–D) and Phospho (E–H) JNK MAPK immunohistochemistry scoring. The left column (panels A, B, E, and F) illustrates a “0” score (no staining) of immunostained tissue samples. The right column (panels C, D, G, and H) illustrates a high/severe level of “3” immunostained tissue samples. “E” refers to epithelium and “CT” refers to connective tissue. Arrows indicate representative areas of the inflammatory infiltrate scored.
Figure 3.
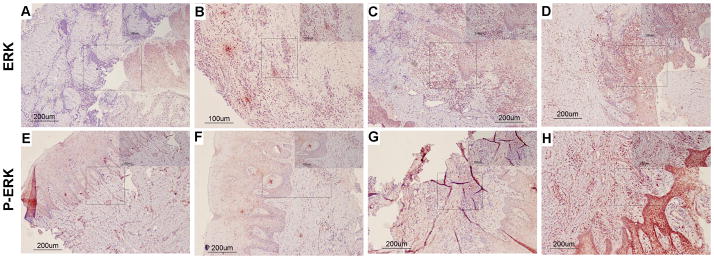
Total (A–D) and Phospho (E–H) ERK MAPK immunohistochemistry scoring. The left column (panels A, B, E, and F) illustrates a “0” score (no staining) of immunostained tissue samples. The right column (panels C, D, G, and H) illustrates a high/severe level of “3” immunostained tissue samples. “E” refers to epithelium and “CT” refers to connective tissue. Arrows indicate representative areas of the inflammatory infiltrate scored.
The intensity of the IHC scoring and the intensity of the inflammatory infiltrate are both positively correlated with the clinical parameters (Tables 1 and 2). BANA scores showed a striking correlation with clinical disease parameters and IHC scoring (p-values ≤ 0.02 for p38, phospho-p38, ERK and JNK; BANA scores were not significantly correlated with phospho-ERK and phospho-JNK).
TABLE 1.
Rank correlations for the inflammatory infiltrate scores and BANA test with MAPK intensities and clinical indices.
| MAPK | Infiltrate | BANA | ||
|---|---|---|---|---|
| Rank Corr | p-value | Rank Corr | p-value | |
| p38 | 0.60 | 0.004 | 0.65 | 0.001 |
| p.p38 | 0.53 | 0.012 | 0.50 | 0.020 |
| ERK | 0.62 | 0.003 | 0.62 | 0.003 |
| p.ERK | 0.43 | 0.051 | 0.39 | 0.084 |
| JNK | 0.58 | 0.006 | 0.56 | 0.008 |
| p.JNK | 0.29 | 0.196 | 0.19 | 0.411 |
| Clinical Indices | Infiltrate | BANA | ||
| PI | 0.96 | 1e-11 | 0.88 | 2e-07 |
| GI | 0.91 | 8e-09 | 0.96 | 7e-12 |
| PIBI | 0.87 | 4e-07 | 0.90 | 4e-08 |
| CALI | 0.84 | 2e-06 | 0.89 | 5e-08 |
TABLE 2.
Rank correlations between MAPK scoring and clinical indices.
| PI | GI | PIBI | CALI | |||||
|---|---|---|---|---|---|---|---|---|
| Rank Corr | p-value | Rank Corr | p-value | Rank Corr | p-value | Rank Corr | p-value | |
| p38 | 0.57 | 0.007 | 0.68 | 0.001 | 0.49 | 0.023 | 0.62 | 0.003 |
| p.p38 | 0.44 | 0.047 | 0.47 | 0.034 | 0.45 | 0.039 | 0.56 | 0.009 |
| ERK | 0.52 | 0.015 | 0.58 | 0.005 | 0.56 | 0.008 | 0.58 | 0.006 |
| p.ERK | 0.35 | 0.116 | 0.40 | 0.071 | 0.25 | 0.269 | 0.34 | 0.135 |
| JNK | 0.45 | 0.042 | 0.55 | 0.009 | 0.42 | 0.060 | 0.59 | 0.005 |
| p.JNK | 0.31 | 0.165 | 0.15 | 0.530 | 0.17 | 0.450 | 0.20 | 0.386 |
Figure 4 depicts the estimated correlations between the clinical parameters and IHC scoring together with 95% confidence intervals computed as described in the Methods section. The confidence intervals reinforce the pattern of association between the clinical parameters and MAPK expression. Specifically, the clinical parameters are consistently, significantly, and positively associated with the IHC scoring for p38, phospho-p38, ERK and JNK (with the sole exception that the confidence interval for the association between JNK and the summary index PIBI just covers zero; p-value = 0.06 from Table 2), and lack statistically significant correlation with phospho-ERK and phospho-JNK. Thus MAPK expression, inflammatory infiltrate and BANA exhibit similar patterns of association with the clinical parameters.
Figure 4.
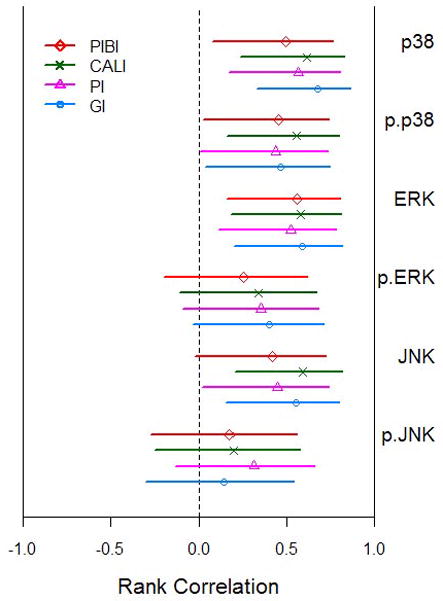
Association between the clinical periodontal indices (PI = plaque index, GI = gingival index, PIBI = periodontal inflammatory burden index, and CALI = clinical attachment level index) and expression of MAPKs. The Spearman rank correlation and 95% confidence interval are shown.
DISCUSSION
Pro-inflammatory cytokines have long been identified as major pathogenic mediators involved in the pathobiology of both rheumatoid arthritis periodontitis, inducing and propagating a chronic inflammatory process leading towards tissue and bone destruction (Kirkwood et al., 2007b). In humans, MAPK activation has been evaluated in tissues from rheumatoid arthritis patients which has been well characterized as a chronic inflammatory disease where inflammatory, immunologic, and physical stimuli ultimately leading to tissue destruction (Zvaifler, 1995b) (Zvaifler, 1995a, Firestein, 1999). MAPK signaling is vital for synthesis and amplification of pro-inflammatory mediators and MMPs by synovial cells, chemoattraction of mononuclear cells, and angiogenesis of endothelial cells, as well as in synovial cell apoptosis (Schett et al., 2000), and p38α and p38β seemed to be predominantly activated in the inflamed tissue (Korb et al., 2006). Given the similarities in cytokine networks between RA and periodontitis, we sought to understand if some of the major MAPK signaling pathways are operative during periodontal disease progression.
The present study evaluated and compared the expression and activation of p38, ERK and JNK MAPKs in gingival tissues from normal healthy subjects and subjects with chronic periodontitis. Activation of total p38, ERK and JNK in diseased tissues was evident but an even more significant finding was the observation of more intense immunoreactivity for phosphorylated p38 in diseased tissues compared to controls (Tables 1, 2 and Figure 4) suggesting increased phosphorylation of p38 in human chronic periodontitis. These findings are in agreement with results from studies that demonstrated activation of p38, ERK and JNK in samples from patients with rheumatoid arthritis, and that p38, specifically isoforms α and β dominate in chronic inflammation (Korb et al., 2006, Schett et al., 2000). Although initial etiology is different between periodontitis and rheumatoid arthritis, the signaling pathways used in response to bacterial challenge or proinflammatory cytokines are comparable. In fact, many of the same cytokines (including TNF-α, IL-1β, and IL-6) are associated with progression in both diseases. Data from periodontal disease preclinical models using a p38α specific inhibitor suggest that at least p38α may be a major p38 isoform involved in inflammatory signaling in chronic periodontitis (Kirkwood et al., 2007b).
Although this study is the first to address MAPK activation in human periodontitis, previous studies addressed NF-κB (p50/p65) and inhibitor of kappa B (IκB) expression in chronic periodontitis patients where activation of NF-κB (p50/p65) was more significantly expressed in diseased periodontal tissues (Ambili et al., 2005). NF-κB and MAPK activation have also been shown to be instrumental in progression of inflammatory diseases including rheumatoid arthritis, cardiovascular disease (Cook et al., 1999, Schett et al., 2000), Alzheimer’s disease (Culbert et al., 2006) and cancer progression (Hanahan and Weinberg, 2000).
Clinical data and histological grading of periodontal lesions presented agrees with the historical literature in clinical periodontics (Page and Schroeder, 1976) providing assurance that correlative data presented here relative to MAPK expression and activation is compelling. Intensity scoring from immunostained tissues presented in Tables 1 and 2 along with graphical representation of these data in Figure 4 was correlated with clinical periodontal parameters. Rank correlations with clinical indices were significantly positive for total p38, phospho-p38, and total ERK expression levels, along with total JNK levels. Phospho-JNK and phospho-ERK showed no significant positive correlation with clinical parameters of disease or BANA positivity. Since there was an increase in the inflammatory infiltrate in the diseased versus healthy sites, there was an increase in the number of nuclei within these sites. Thus, the total amount of p38, JNK and ERK expression maybe positively correlated with clinical indices since there were more cells within the periodontal lesions. The lack of correlative data with p-JNK and p-ERK suggests that although there is more total JNK and ERK, these MAPKs are not significantly active during periodontal disease progression in humans. One of the potential shortcoming of these data reside in the inability to express the phosphorylated to non-phosphorylated MAPKs ratios due to lack of sufficient amount of quality serial samples. However, it should be noted again, that the connective tissue was only scored and reported here since there was high background immunostaining with the epithelium.
It is important to note that diseased samples evaluated for inflammation, BANA and MAPK expression in the present study all had initial periodontal therapy including scaling and root planning prior to tissue procurement. Thus, the levels of inflammation observed are most likely lower then what would be expected in an untreated periodontal lesion, suggesting that the significance of the data presented here is highly significant. However, although BANA data correlated with periodontal disease status, these studies cannot conclude that all periodontal pathogens affect MAPK signaling pathways to the same extent or manner.
With emerging treatment modalities used to manage periodontitis that have been designed to modulate the host response as it has been recognized that the host response, not the bacterial infection, is primarily responsible for the connective tissue destruction in chronic periodontitis. The present study confirms the findings from studies in small animal models demonstrating increased activation of MAPKs in periodontal disease progression (Kirkwood et al., 2007b), indicating that both increased MAPK expression and activation occurs with increased severity of periodontal disease in human tissues. Moreover, this is the first time that the levels of activation of MAPKs have been correlated with clinical parameters as well as BANA microbiological test results. These findings suggest that MAPK signaling plays a significant part of the ontogeny of inflammation in periodontal disease ultimately resulting in alveolar bone loss.
Clinical Relevance.
Scientific Rationale for Study: This study provides basic biochemical data regarding the expression of mitogen activating protein kinase (MAPK) in diseased and healthy human periodontal tissues. MAPKs are activated via phosphorylation in response to bacterial exposure and are critical mediators of chronic inflammation through the production of pro-inflammatory cytokines. Previous data from the project laboratory have shown that blocking specific MAPK reduced experimental periodontitis. Principal Findings: This study shows that certain MAPK are more highly activated in clinically diseased versus healthy tissue counterparts. Practical Implications: These data may have clinically important information since MAPK can be targeted for therapeutic benefit.
Acknowledgments
Source of Funding: NIH DE018290, DE021423, DE019513, and 2P20RR017696 supported funding for this work.
The authors acknowledge the support of the Histology Core Facility at the University of Michigan School of Dentistry, the MUSC Center for Oral Health Research, and Ms. Bethany Herbert for her assistance with digital imaging.
Footnotes
Conflict of Interest
The authors have no conflict of interest to declare.
References
- Ambili R, Santhi WS, Janam P, Nandakumar K, Pillai MR. Expression of activated transcription factor nuclear factor-kappaB in periodontally diseased tissues. Journal of periodontology. 2005;76:1148–1153. doi: 10.1902/jop.2005.76.7.1148. [DOI] [PubMed] [Google Scholar]
- Assuma R, Oates T, Cochran D, Amar S, Graves DT. IL-1 and TNF antagonists inhibit the inflammatory response and bone loss in experimental periodontitis. J Immunol. 1998;160:403–409. [PubMed] [Google Scholar]
- Beklen A, Hukkanen M, Richardson R, Konttinen YT. Immunohistochemical localization of Toll-like receptors 1–10 in periodontitis. Oral Microbiology and Immunology. 2008;23:425–431. doi: 10.1111/j.1399-302X.2008.00448.x. [DOI] [PubMed] [Google Scholar]
- Cook SA, Sugden PH, Clerk A. Activation of c-Jun N-terminal kinases and p38-mitogen-activated protein kinases in human heart failure secondary to ischaemic heart disease. Journal of molecular and cellular cardiology. 1999;31:1429–1434. doi: 10.1006/jmcc.1999.0979. [DOI] [PubMed] [Google Scholar]
- Culbert AA, Skaper SD, Howlett DR, Evans NA, Facci L, Soden PE, Seymour ZM, Guillot F, Gaestel M, Richardson JC. MAPK-activated protein kinase 2 deficiency in microglia inhibits pro-inflammatory mediator release and resultant neurotoxicity. Relevance to neuroinflammation in a transgenic mouse model of Alzheimer disease. The Journal of biological chemistry. 2006;281:23658–23667. doi: 10.1074/jbc.M513646200. [DOI] [PubMed] [Google Scholar]
- Ejeil AL, Gaultier F, Igondjo-Tchen S, Senni K, Pellat B, Godeau G, Gogly B. Are cytokines linked to collagen breakdown during periodontal disease progression? J Periodontol. 2003;74:196–201. doi: 10.1902/jop.2003.74.2.196. [DOI] [PubMed] [Google Scholar]
- Firestein GS. Starving the synovium: angiogenesis and inflammation in rheumatoid arthritis. The Journal of clinical investigation. 1999;103:3–4. doi: 10.1172/JCI5929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher RA. Frequency distribution of the values of the correlation coefficient in samples of an indefinitely large population. Biometrika (Biometrika Trust) 1915;10:507–521. [Google Scholar]
- Gamonal J, Acevedo A, Bascones A, Jorge O, Silva A. Levels of interleukin-1 beta, -8, and -10 and RANTES in gingival crevicular fluid and cell populations in adult periodontitis patients and the effect of periodontal treatment. J Periodontol. 2000;71:1535–1545. doi: 10.1902/jop.2000.71.10.1535. [DOI] [PubMed] [Google Scholar]
- Geivelis M, Turner DW, Pederson ED, Lamberts BL. Measurements of interleukin-6 in gingival crevicular fluid from adults with destructive periodontal disease. J Periodontol. 1993;64:980–983. doi: 10.1902/jop.1993.64.10.980. [DOI] [PubMed] [Google Scholar]
- Gorska R, Gregorek H, Kowalski J, Laskus-Perendyk A, Syczewska M, Madalinski K. Relationship between clinical parameters and cytokine profiles in inflamed gingival tissue and serum samples from patients with chronic periodontitis. J Clin Periodontol. 2003;30:1046–1052. doi: 10.1046/j.0303-6979.2003.00425.x. [DOI] [PubMed] [Google Scholar]
- Graves DT. The potential role of chemokines and inflammatory cytokines in periodontal disease progression. Clin Infect Dis. 1999;28:482–490. doi: 10.1086/515178. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Harrell F., Jr Hmisc: Harrell Miscellaneous. R package version 3.8–3 2010 [Google Scholar]
- Jiang Y, Mehta CK, Hsu TY, Alsulaimani FF. Bacteria induce osteoclastogenesis via an osteoblast-independent pathway. Infect Immun. 2002;70:3143–3148. doi: 10.1128/IAI.70.6.3143-3148.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi T, Yoshikai Y, Miyoshi J, Katsuki M, Musikacharoen T, Mitani A, Tanaka S, Noguchi T, Matsuguchi T. Cot/Tpl2 is essential for RANKL induction by lipid A in osteoblasts. J Dent Res. 2003;82:546–550. doi: 10.1177/154405910308200712. [DOI] [PubMed] [Google Scholar]
- Kirkwood KL, Cirelli JA, Rogers JE, Giannobile WV. Novel host response therapeutic approaches to treat periodontal diseases. Periodontology 2000. 2007a;43:294–315. doi: 10.1111/j.1600-0757.2006.00166.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkwood KL, Li F, Rogers JE, Otremba J, Coatney DD, Kreider JM, D’Silva NJ, Chakravarty S, Dugar S, Higgins LS, Protter AA, Medicherla S. A p38alpha selective mitogen-activated protein kinase inhibitor prevents periodontal bone loss. The Journal of pharmacology and experimental therapeutics. 2007b;320:56–63. doi: 10.1124/jpet.106.112466. [DOI] [PubMed] [Google Scholar]
- Kirkwood KL, Rossa C., Jr The potential of p38 MAPK inhibitors to modulate periodontal infections. Current drug metabolism. 2009;10:55–67. doi: 10.2174/138920009787048347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirschning CJ, Bauer S. Toll-like receptors: cellular signal transducers for exogenous molecular patterns causing immune responses. Int J Med Microbiol. 2001;291:251–260. doi: 10.1078/1438-4221-00128. [DOI] [PubMed] [Google Scholar]
- Kirschning CJ, Wesche H, Merrill Ayres T, Rothe M. Human toll-like receptor 2 confers responsiveness to bacterial lipopolysaccharide. J Exp Med. 1998;188:2091–2097. doi: 10.1084/jem.188.11.2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korb A, Tohidast-Akrad M, Cetin E, Axmann R, Smolen J, Schett G. Differential tissue expression and activation of p38 MAPK alpha, beta, gamma, and delta isoforms in rheumatoid arthritis. Arthritis and rheumatism. 2006;54:2745–2756. doi: 10.1002/art.22080. [DOI] [PubMed] [Google Scholar]
- Lee HJ, Kang IK, Chung CP, Choi SM. The subgingival microflora and gingival crevicular fluid cytokines in refractory periodontitis. J Clin Periodontol. 1995;22:885–890. doi: 10.1111/j.1600-051x.1995.tb01788.x. [DOI] [PubMed] [Google Scholar]
- Leppilahti JM, Ahonen MM, Hernandez M, Munjal S, Netuschil L, Uitto VJ, Sorsa T, Mantyla P. Oral rinse MMP-8 point-of-care immuno test identifies patients with strong periodontal inflammatory burden. Oral diseases. 2011;17:115–122. doi: 10.1111/j.1601-0825.2010.01716.x. [DOI] [PubMed] [Google Scholar]
- Li Q, Valerio MS, Kirkwood KL. MAPK usage in periodontal disease progression. Journal of signal transduction. 2012;2012:308943. doi: 10.1155/2012/308943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Yu H, Zinna R, Martin K, Herbert B, Liu A, Rossa C, Jr, Kirkwood KL. Silencing mitogen-activated protein kinase-activated protein kinase-2 arrests inflammatory bone loss. The Journal of pharmacology and experimental therapeutics. 2011;336:633–642. doi: 10.1124/jpet.110.172395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindy O, Suomalainen K, Makela M, Lindy S. Statin use is associated with fewer periodontal lesions: A retrospective study. BMC oral health. 2008;8:16. doi: 10.1186/1472-6831-8-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loe H. The Gingival Index, the Plaque Index and the Retention Index Systems. Journal of periodontology. 1967;38(Suppl):610–616. doi: 10.1902/jop.1967.38.6.610. [DOI] [PubMed] [Google Scholar]
- Loesche WJ, Bretz WA, Kerschensteiner D, Stoll J, Socransky SS, Hujoel P, Lopatin DE. Development of a diagnostic test for anaerobic periodontal infections based on plaque hydrolysis of benzoyl-DL-arginine-naphthylamide. Journal of clinical microbiology. 1990;28:1551–1559. doi: 10.1128/jcm.28.7.1551-1559.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra RS, Goto M, Lee JS, Maldonado D, Taylor JM, Pan Q, Carey TE, Bradford CR, Prince ME, Cordell KG, Kirkwood KL, D’Silva NJ. Rap1GAP promotes invasion via induction of matrix metalloproteinase 9 secretion, which is associated with poor survival in low N-stage squamous cell carcinoma. Cancer research. 2008;68:3959–3969. doi: 10.1158/0008-5472.CAN-07-2755. [DOI] [PubMed] [Google Scholar]
- Mori Y, Yoshimura A, Ukai T, Lien E, Espevik T, Hara Y. Immunohistochemical localization of Toll-like receptors 2 and 4 in gingival tissue from patients with periodontitis. Oral Microbiol Immunol. 2003;18:54–58. doi: 10.1034/j.1399-302x.2003.180109.x. [DOI] [PubMed] [Google Scholar]
- Nakashima T, Kobayashi Y, Yamasaki S, Kawakami A, Eguchi K, Sasaki H, Sakai H. Protein expression and functional difference of membrane-bound and soluble receptor activator of NF-kappaB ligand: modulation of the expression by osteotropic factors and cytokines. Biochem Biophys Res Commun. 2000;275:768–775. doi: 10.1006/bbrc.2000.3379. [DOI] [PubMed] [Google Scholar]
- Page RC, Schroeder HE. Pathogenesis of inflammatory periodontal disease. A summary of current work. Laboratory investigation; a journal of technical methods and pathology. 1976;34:235–249. [PubMed] [Google Scholar]
- Palanisamy V, Jakymiw A, Van Tubergen EA, D’Silva NJ, Kirkwood KL. Control of Cytokine mRNA Expression by RNA-binding Proteins and microRNAs. Journal of dental research. 2012;91:651–658. doi: 10.1177/0022034512437372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddi K, Wilson M, Nair S, Poole S, Henderson B. Comparison of the pro-inflammatory cytokine-stimulating activity of the surface-associated proteins of periodontopathic bacteria. J Periodontal Res. 1996;31:120–130. doi: 10.1111/j.1600-0765.1996.tb00473.x. [DOI] [PubMed] [Google Scholar]
- Rogers JE, Li F, Coatney DD, Otremba J, Kriegl JM, Protter TA, Higgins LS, Medicherla S, Kirkwood KL. A p38 mitogen-activated protein kinase inhibitor arrests active alveolar bone loss in a rat periodontitis model. Journal of periodontology. 2007;78:1992–1998. doi: 10.1902/jop.2007.070101. [DOI] [PubMed] [Google Scholar]
- Schett G, Tohidast-Akrad M, Smolen JS, Schmid BJ, Steiner CW, Bitzan P, Zenz P, Redlich K, Xu Q, Steiner G. Activation, differential localization, and regulation of the stress-activated protein kinases, extracellular signal-regulated kinase, c-JUN N-terminal kinase, and p38 mitogen-activated protein kinase, in synovial tissue and cells in rheumatoid arthritis. Arthritis and rheumatism. 2000;43:2501–2512. doi: 10.1002/1529-0131(200011)43:11<2501::AID-ANR18>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Stashenko P, Jandinski JJ, Fujiyoshi P, Rynar J, Socransky SS. Tissue levels of bone resorptive cytokines in periodontal disease. J Periodontol. 1991;62:504–509. doi: 10.1902/jop.1991.62.8.504. [DOI] [PubMed] [Google Scholar]
- Team RDC. R: A language and environment for statistical computing. R Foundation for Statistical Computing; Vienna, Austria: 2010. [Google Scholar]
- Yang H, Young DW, Gusovsky F, Chow JC. Cellular events mediated by lipopolysaccharide-stimulated toll-like receptor 4. MD-2 is required for activation of mitogen-activated protein kinases and Elk-1. J Biol Chem. 2000;275:20861–20866. doi: 10.1074/jbc.M002896200. [DOI] [PubMed] [Google Scholar]
- Zvaifler NJ. Macrophages and the synovial lining. Scandinavian journal of rheumatology Supplement. 1995a;101:67–75. doi: 10.3109/03009749509100904. [DOI] [PubMed] [Google Scholar]
- Zvaifler NJ. Rheumatoid arthritis. The multiple pathways to chronic synovitis. Laboratory investigation; a journal of technical methods and pathology. 1995b;73:307–310. [PubMed] [Google Scholar]