

Abstract
Duchenne muscular dystrophy (DMD) is a recessive disease caused by a dystrophin gene mutation. Myoblast transplantation permits the introduction of the dystrophin gene into dystrophic muscle fibers. However, this strategy has so far produced limited results. Modulation of transforming growth factor β (TGF-β) superfamily signaling promotes skeletal muscle differentiation and growth and myogenic regeneration. We investigated the possibility that the combination of TGF-β superfamily signaling inhibition with myoblast transplantation might be an effective therapeutic approach in dystrophin deficient patients. In vitro, blocking myostatin and other ligands with a soluble form of the extracellular domain of the activin IIB receptor (ActRIIB/Fc) up-regulated the expression of myogenic differentiation factors and increased human myoblast fusion. In vivo, systemic inhibition of activin IIB receptor signalling by delivery of ActRIIB/Fc increased the success of the myoblast transplantation. This effect was further increased by forcing the mice to swim weekly to induce cycles of muscle degeneration and regeneration. Treatment of dystrophic mice with ActRIIB/Fc led to increased body weight, increased skeletal muscle mass and improved myoblast transplantation. Thus ActRIIB/Fc represents an effective therapeutic strategy for muscular dystrophies, and its effects are enhanced when combined with muscle exercise.
Keywords: Myoblasts transplantation, Duchenne muscular dystrophy, TGF-β super-family, activin receptor
INTRODUCTION
Duchenne muscular dystrophy (DMD) is a severe degenerative disorder of skeletal and cardiac muscles that affect ~ 1 in 3500 male births (10). DMD patients characteristically display progressive muscle weakness, which begins in early childhood (9). Although the mutation in the gene encoding dystrophin responsible for DMD is present at birth, clinical symptoms are not evident until 3–5 years of age (18). New insights into the pathophysiology of dystrophic muscle, the identification of compensating proteins, and the discovery of new binding partners are paving the way for novel therapeutic strategies to treat this fatal muscle disease. Dystrophin is required for the assembly of the dystrophin-glycoprotein complex and provides a mechanically strong link between the cytoskeleton and the extracellular matrix.
So far there is no corrective therapy for the DMD, but several potential therapies have been considered to increase muscle mass, improve muscle physiology and reduce fibrosis (24). A particularly attractive strategy is the inhibition of transforming growth factor (TGF)-β signaling pathways. TGF-β superfamily members, including TGF-β1 and myostatin, are known to negatively regulate muscle growth (21). Myostatin, also known as growth/differentiation factor-8 (GDF-8), is predominantly expressed in skeletal muscle tissue, where it is secreted and circulated as a serum protein (21). Myostatin inhibition has been shown to ameliorate the phenotype of mouse models of muscular dystrophy (mdx mice), by increasing muscle growth and regeneration (6,27,37). The mdx phenotype was caused by a point mutation (A to G) in exon 23 of the dystrophin gene. The development of myostatin inhibitors, such as follistatin (2,22), follistatin-related gene (15) GDF-associated serum protein-1 (16), anti-myostatin antibodies (6,35) and the myostatin propeptide (22,36) has offered a multifaceted approach to the treatment of muscle degenerative diseases currently under pre-clinical or clinical investigation. They range from suppression of myostatin activity to inhibition of myostatin activation. Interestingly, Lee et al. demonstrated that a soluble version of the activin type IIB receptor (ActRIIB/Fc), which competes with the natural receptor for circulating agonists of this receptor, promotes skeletal muscle growth in wild type mice (23). The in vivo ligands for ActRIIB include activin, inhibin, members of the bone morphogenetic protein (BMP) family, myostatin and growth and differentiation factor 11 (GDF11) (3,34). Sako et al. demonstrated that both myostatin and GDF-11 bind the extracellular domain of ActRIIB with high affinities comparable with those of activin-A, whereas the affinities of BMP-2 and BMP-7 for ActRIIB are lower by orders of magnitude (29).
Myoblast transplantation represents a potential approach to restore the missing dystrophin (30,32). However, this technique requires multiple close injection trajectories due to limited migration of transplanted muscle precursor cells (myoblasts) and their fusion only with damaged host fibers (28,31,33). Several approaches have been developed to resolve these problems, among them, the blockade of myostatin and homologous proteins in mdx mice or in normal human myoblasts by follistatin up-regulation or by dnActRIIB expression. These approaches increased the extent of muscle repair and improved the success of myoblast transplantation (measured by the number of dystrophin positive fibers present in mdx mouse muscles) (4,5,11).
The aim of the present study was to investigate whether systemic administration of ActRIIB/Fc improves the transplantation of human myoblasts.
MATERIAL AND METHODS
Isolation of human myoblasts
Primary myoblast cultures were derived from a muscle biopsy of the Quadriceps femoris of a human cadaveric donor aged 13 months following informed consent of the family and approval by the Human Ethic Committee of the Research Center Hospital of Laval University (CRCHUL). The muscle samples were dissociated with collagenase and trypsin as previously described (20) and cultured in MB-1 growth medium (Hyclone, South Logan, UT, USA) with 15% fetal bovine serum (FBS), 1% of penicillin-streptomycin (Gibco, Burlington, Ontario, Canada) and 10 ng/ml of basic Fibroblast Growth Factor (Strathmann Biotec AG, Hamburg, Germany) in a humidified atmosphere with 5% CO2 at 37°. Desmin labeling and a cell viability test with Trypan blue were done before using the myoblasts for in vitro and in vivo experiments.
Expression and purification of ActRIIB/Fc
The ActRIIB/Fc is a fusion protein containing the extracellular domain of ActRIIB linked to a murine Fc domain. The methods for expression and purification of ActRIIB/Fc fusion protein have been previously described (19,23)
Cell differentiation assay
Myoblast differentiation was evaluated by counting the percentage of nuclei inside myotubes, i.e., syncytia with three or more nuclei. For this test, myoblasts were cultured in MB-1 for 24 h, and the proliferation medium was replaced for 72 h with a fusion medium (Dulbecco’s modified Eagle medium, St. Louis, MO, Canada) containing only 2% FBS in the presence or absence of ActRIIB/Fc (500 ng/ml) and of human recombinant myostatin (500 ng/ml). Myotubes were visualized by myosin heavy chain (MyHC) immunodetection. The cells were fixed with 95% ethanol, washed twice with phosphate-buffered saline (PBS) for 10 min and blocked with 10% FBS in PBS for 1 h. The samples were incubated 2 h at room temperature with a mouse anti-MyHC mAb (MF20, DSHB, Iowa City, IA) (1:100) and then for 1 h with an anti-mouse Ig Alexa 546-conjugated secondary antibody (Invitrogen inc., Burlington, ON, Canada) diluted 1:100. Finally, the cells were washed with PBS and mounted with PBS/glycerol. 4′, 6-Diamidino-2-phenylindole (DAPI) staining was done with PBS to label the nuclei. The number of nuclei in the myotubes was counted and normalized with the total number of nuclei in the culture well as a measure of terminal differentiation.
Quantitative RT-PCR
Quantitative RT-PCR (Q-RT-PCR) was used to examine the mRNA expression levels of myogenic regulator factors (MRFs) (myogenic differentiation [MyoD], myogenic factor 5 [Myf5], myogenic regulatory factor 4 [MRF4] and Myogenin) in normal myoblasts incubated or not with ActRIIB/Fc (500 ng/ml) and in presence or absence of recombinant myostatin (500 ng/ml). The mRNA was extracted using an RNeasy Plus kit (Qiagen, Mississauga, ON Canada). The cDNA templates for Q-RT-PCR were synthesized by an inverse transcriptase with 1 μg RNA in superscript II RNAse H-RT reaction (Invitrogen inc.). Q-RT-PCR was carried out in a Light Cycler 480 (Roche Diagnostics, Mississauga, ON, Canada) with SYBR Green light Cycler Fast Start DNA Master Plus (Roche Diagnostics) as a detector. All target gene expressions were normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) levels. The primer pairs were designed with GeneTools logiciels (Biotools Inc., Madrid, Spain) and are shown in Table 1.
Table 1.
Primer pair design
| Gene Symbol | Description | GenBank | Region | Size (pb) | T ann. (°C) | Primer sequences 5′→3′ F/R |
|---|---|---|---|---|---|---|
| MYOD1 | Homo sapiens myogenic differentiation 1 (MYOD1) | NM_002478.4 | 1152–1324 | 173 | 63 | CGAACCCCAACCCGATATACCA/ACTTCAGTTCTCCCGCCTCTC |
| MYF5 | Homo sapiens myogenic factor 5 (MYF5) | NM_005593 | 609–839 | 231 | 65 | CCCCACCTCCAACTGCTCTGA/CTGGCAACTGGAGAGAGAGAAGC |
| MYOG | Homo sapiens myogenic factor 4 (myogenin) | NM_002479.4 | 1220–1584 | 365 | 57 | AGCGCCCCCTCGTGTATG/TGTCCCCGGCAACTTCAGC |
| MRF4 | Homo sapiens myogenic factor 6 (herculin) (MYF6) | NM_002469 | 733–948 | 216 | 62 | TAGCCTTCGATGCCTTTCTTCC/CCCTTTCAGCAACTTTTCGGTCT |
| G6PD | Homo sapiens glucose-6-phosphate dehydrogenase (G6PD), nuclear gene encoding mitochondrial protein | NM_000402 | 2255–2375 | 121 | 65 | GATGTCCCCTGTCCCACCAACTCTG/GCAGGGCATTGAGGTTGGGAG |
Animals
All experiments were performed in Rag−/− mdx mice, which were 30 days old for the first experiment and 48-days-old for the second experiment. This stable mouse line was produced in our animal facilities by crossing mdx/mdx mice (a DMD model due to a mutation leading to dystrophin deficiency) with Rag−/− mice (an immunodeficient mouse that accepts human grafts). To establish a stable mouse line, we crossed female homozygous mdx with male homozygous rag mice. The F1 mice were genotype by PCR amplification and sequencing of the dystrophin exon 23 and of the rag gene. For Rag genotyping, we used the protocol described in this link: http://jaxmice.jax.org/protocolsdb/f?p=116:2:4118722926490498::NO:2:P2_MASTER_PROTOCOL_ID,P2_JRS_CODE:329,003145. We crossed the F1 females that were heterozygous mdx and heterozygous mdx with males that were mdx and heterozygous rag. In the F2 generation, one female, which was homozygous rag and homozygous mdx (1 female out of 8 had this genotype) was crossed with a male that was homozygous rag and mdx (1 male out of 4 had this genotype). All the mice resulting from that cross were homozygous for rag−/− and mdx genes. The mice were maintained under pathogen-free conditions. All the experiments with mice were conducted in accordance with the Laboratory Animal Care and use Ethics Committee of Laval University.
Cell transplantation and ActRIIB/Fc treatment
Half million human myoblasts resuspended in 10 μl of HBSS were transplanted at day 0 into both Tibialis anterior muscles (TAs) of 12 mice using a glass capillary along 12 transversal axis trajectories. All mice were sacrificed 30 days after the transplantation. Two experiments were done to evaluate the effect of ActRIIB/Fc on the myoblast transplantation success. In each experiment, two groups of mice (n=3) were treated or not with ActRIIB/Fc (i.p., 10 mg/kg). In the second experiment, to induce cycles of muscle degeneration and regeneration, mice were forced to swim for 10 min on days 0, 7, 14 and 21 post-transplantation. For this swimming, mice were placed inside a Plexiglass box filled with warm water (28°C). All contacts with box bottom and top were avoided. All mice were killed 30 days after the myoblast transplantation for muscle analysis. Thus, the four groups of mice are the following: Group 1A(noSwim/noActRIIB/Fc): mice were injected on days 0 and 7 post-transplantation with sterile PBS and not forced to swim; Group 1B(noSwim/ActRIIB/Fc 2wk): mice were injected on days 0 and 7 post-transplantation with ActRIIB/Fc and not forced to swim; Group 2A(Swim/noActRIIB/Fc): mice were injected on days −5, 0, 7, 14 and 21 post-transplantation with sterile PBS and were forced to swim; Group 2B(Swim/ActRIIB/Fc 5wk): mice were injected on days −5, 0, 7, 14 and 28 post-transplantation with ActRIIB/Fc and were forced to swim.
Muscle Morphometry
For measurement of muscle weights, individual TA from both sides of the animal was dissected and the average weight was used for each group. The muscles were mounted in embedding medium and snap-frozen in liquid nitrogen. Serial 12 μm cross-sections were obtained in a cryostat at −25°C. Sections were then processed for histological examination by haematoxylin and eosin-phloxine staining (H&E), and by immunohistochemistry with an anti-human dystrophin antibody (7F7, MRIC Biochemistry Group, Wrexham, UK) and with anti-lamin A/C (Vector laboratory, USA) to visualise human nuclei as previously described (11). Morphometric measurements (i.e. single fiber area, total number of nucleus lamin A/C, number of nuclei lamin A/C positive per fibers, total number of centro-nucleated fibers) were made using the Image J software (rsbweb.nih.gov/ij).
Statistical Analysis
The experimental values are presented as means + SD. Statistical analyses were performed by analysis of variance (one-way ANOVA) with Fisher’s PLSD test and Tukey’s test for multiple comparisons post-hoc in the StatView statistical package (StatView 5, SAS Institute Inc. Cary, NC). Differences were considered statistically significant at p<0.05.
RESULTS
ActRIIB/Fc promotes the differentiation of human myoblasts even in the presence of recombinant myostatin
The activation of satellite cells in skeletal muscle leads to their proliferation as myoblasts and to their differentiation and fusion with existing muscle fibers, which is essential for muscle hypertrophy. Several members of the TGF-β family inhibit myoblast differentiation. The effect of ActRIIB/Fc, which binds to several members of this family, was thus evaluated on the in vitro myogenic differentiation of human myoblasts in the presence or absence of recombinant myostatin. When myoblasts were transferred to a differentiation medium in the absence of myostatin, the presence of ActRIIB/Fc for 3 days did not significantly increase the number of myotubes (Figure 1a) or the fusion index (Figure 1b). The presence of myostatin alone in the differentiation medium significantly decreased the number of myotubes, with the fusion index being reduced by about 5-fold to 8%. However, this reduction in the number of myotubes and the fusion index by myostatin was completely blocked by the addition of ActRIIB/Fc to the cells.
Figure 1. ActRIIB/Fc promotes the differentiation of human myoblasts even in presence of recombinant myostatin.

Myoblasts treated or not with ActRIIB/Fc at 500 ng/ml, were differentiated in myotubes for 3 days in the presence or absence of recombinant myostatin (rMSTN) at 500 ng/ml in the differentiation medium (Dulbecco’s modified Eagle’s medium (DMEM) with 2% serum). (a) Myotubes were stained in red with anti-myosin heavy chain (MyHC) and nucleus were stained in bleu with 4′,6-diamidino-2-phenylindole. The fusion is significantly reduced by rMSTN but this effect is blocked by ActRIIB/Fc. Scale bar = 0.2 mm. (b) The same effects are observed when the fusion index was calculated. Means + SD from three independent experiments are illustrated. *p < 0.05.
Expression of MyoD family mRNA in ActRIIB/Fc treated myoblasts
To analyze further the effect of myostatin and ActRIIB on myoblast differentiation in these cultures, we carried out Q-RT-PCR analyses of Myf5, myogenin, MRF4 and MyoD. As shown in Figure 2a, myostatin and ActRIIB had opposite effects on MyoD expression, with myostatin causing a decrease and ActRIIB/Fc causing an increase in expression levels. In cells treated with both myostatin and ActRIIB/Fc, the two molecules appeared to neutralize each other in their effects on MyoD expression. Similar results were obtained with myogenin (Figure 2c); however, the ActRIIB/Fc and/or rMSTN treatment did not change the expression of Myf5 and MRF4 (Figures. 2b and d).
Figure 2. Expression of MyoD family mRNA in ActRIIB/Fc treated myoblasts.

The total RNA was extracted from control and ActRIIB/Fc treated myoblasts incubated during 2 days without or with recombinant myostatin (500 ng/ml) in the culture medium. The mRNA expression of MyoD (a), Myf5 (b), myogenin (c) and MRF4 (d) was quantified by Q-RT-PCR. ActRIIB/Fc significantly increased the expression of MyoD and myogenin. The data presented are means + SD (n = 3), *p <0.05.
ActRIIB/Fc long-term treatment increases the mouse body weight
Next, we examined the effect of ActRIIB/Fc following transplantation of myoblasts in vivo (Figures 3 and 4). All mice were weighed in the days 0 and 30 post-transplantation and the results are plotted in figures 3a and b. A two week treatment with ActRIIB/Fc did not affect the percentage increase in the mouse body weight of Group 1B(noSwim/ActRIIB/Fc 2wk) versus Group 1A(noSwim/noActRIIB/Fc) (Figure 3a). However, a five week treatment with ActRIIB/Fc combined with force swimming significantly increased the weight of Group 2B(Swim/ActRIIB/Fc 5wk) compared with Group 2A(Swim/noActRIIB/Fc) (Figure 4). Indeed, the mean percentage increase in body weight over that time period compared to controls mice was 40% (Figure 3b).
Figure 3. Effect of ActRIIB/Fc on body and muscle weights of Rag−/−/mdx mice.

(a) Means and SD of whole body weights (in grams) of control Rag−/−/mdx (Group 1A) mice (n=3) and ActRIIB/Fc treated Rag−/−/mdx (Group 1B) mice (n=3) in the no swimming group at the time of transplantation (D0) and 1 month post-transplantation (D30). (b) Means and SD of whole body weights (in grams) of control Rag−/−/mdx (Group 2A) mice (n=3) and ActRIIB/Fc treated Rag−/−/mdx (Group 2B) mice (n=3) in the weekly swimming groups at the time of transplantation and 1 month post-transplantation. (c) Means and SD (n=6) of TA muscle weights (in mg) of Rag−/−/mdx mice treated or not with ActRIIB/Fc, and forced or not to swim. (d) The ratio of muscle weight to body weight of each group of mice.* p < 0.05.
Figure 4. Increased muscle in mice treated with ActRIIB/Fc.

The mouse of group 2A was not treated with ActRIIB/Fc but was forced to swim during 4 weeks. The mouse of group 2B was injected with ActRIIB/Fc and forced to swim during 4 weeks. The muscles are clearly hypertrophied in the mouse that received the ActRIIB/Fc (arrows).
ActRIIB/Fc long-term treatment accentuate the increase of the muscle weight
The TA muscles of all experimental mice were dissected, trimmed of excess connective tissues and weighed to determine the effect of ActRIIB/Fc treatment on muscle mass (Figure 3c). The effect of ActRIIB/Fc long-term treatment was evident in the weight of the TA muscle, which was significantly increased in Group 2B(Swim/ActRIIB/Fc 5wk). This increase was 71% when compared with the TA weight of Group 2A(Swim/noActRIIB/Fc) and 85% when compared with the mice of Group 1A(noSwim/noActRIIB/Fc). However, the TA weight of Group 1B(noSwim/ActRIIB/Fc 2wk) was increased by only 22% compared to the TA of its control Group 1A(noSwim/noActRIIB/Fc), which did not reach statistical significance. There are no differences in muscle weights of mice in the control groups, namely, Group 2A(Swim/noActRIIB/Fc) versus Group 1A(noSwim/noActRIIB/Fc).
When TA muscle weights were expressed relative to body mass, significant increases with ActRIIB/Fc treatment were observed only for Group 2B(Swim/ActRIIB/Fc 5wk) compared to Group 2A(Swim/noActRIIB/Fc) and to Group 1A(noSwim/noActRIIB/Fc) (Figure 3d). The magnitude of the effects was blunted after normalization to body weights, which was expected given that ActRIIB/Fc has been shown previously to cause increases in muscle mass throughout the body (23) and given that skeletal muscle accounts about one-third of total body weight.
ActRIIB/Fc treatment improved success of human myoblast transplantation in mice
Following their transplantation, human myoblasts fused with the existing mouse muscle fibers leading to the expression of human dystrophin in the resulting hybrid fibers. To evaluate the success of the myoblast transplantation, human dystrophin was thus detected by immunohistochemistry in the 4 groups of mice. In Group 1B(noSwim/ActRIIB/Fc 2wk), the number of dystrophin-positive myofibers present in muscle cross-sections was more than double (135 ± 26) that observed in Group 1A(noSwim/noActRIIB/Fc) (59± 23) (Figure 5a). A very significant increase of the number of dystrophin positive fibers was also observed in the second experiment that involved swimming. Specifically, the TA muscle of Group 2B(Swim/ActRIIB/Fc 5wk) contained 185 dystrophin positive fibers per muscle cross-section, which represented an increase of 77% compared to Group 2A(Swim/noActRIIB/Fc). Swimming alone also seemed to increase the success of transplantation, as the number of dystrophin positive fiber in mice in Group 2A(Swim/noActRIIB/Fc) was more than double that seen in mice in Group 1A(noSwim/noActRIIB/Fc). ActRIIB/Fc and forced swimming appeared to have an additive effect, leading to a tripling of dystrophin positive fibers in Group 2B(Swim/ActRIIB/Fc 5wk) compared to Group 1A(noSwim/noActRIIB/Fc).
Figure 5. Increased success of human myoblast transplantation in mice treated with ActRIIB/Fc.

(a) Graphical representation of the mean number (+SD, N=6) of dystrophin-positive myofibers per cross-section of TA transplanted with human myoblasts in Rag−/− mdx mice treated or not with ActRIIB/Fc and forced or not to swim. (b) The mean numbers (+SD, N=6) of lamin A/C positive nuclei per cross-section of TA muscles transplanted with human myoblasts and treated or not with ActRIIB/Fc and forced or not to swim. * p<0.05.
ActRIIB/Fc treatment changed the total number of human nuclei per muscle cross-section
To confirm that the increased muscle mass and number of dystrophin positive fibers resulted in part from fusion of the transplanted myoblasts to myofibers, we counted the number of human nuclei in the myofibers using an antibody directed against human lamin A/C. As shown in Figure 5b, mice in Group 2A(Swim/ActRIIB/Fc 5wk) had about twice as many human nuclei per muscle cross-section compared to mice in the two control groups, Group 1A(noSwim/noActRIIB/Fc) and Group 2A(Swim/noActRIIB/Fc.
ActRIIB/Fc treatment increased the number of lamin A/C nuclei in individual dystrophin positive fiber
To verify the effect of ActRIIB/Fc on the fusion of human myoblasts with mouse myofibers, the number of lamin A/C nuclei was quantified in each individual muscle fiber expressing human dystrophin. All transplanted TAs of mice contained a mixed population of fibers incorporating one or more human nuclei. In the experimental groups of mice that received ActRIIB/Fc, i.e., Group 1B(noSwim/ActRIIB/Fc 2wk) and Group 2B(Swim/ActRIIB/Fc 5wk), the percentage of fibers with only one human nucleus was lower than in their respective control groups that did not receive ActRIIB/Fc, i.e. Group 1A(noSwim/noActRIIB/Fc) and Group 2A(Swim/noActRIIB/Fc) (Figures. 6a and b). Moreover, the percentage of fibers with more than 3 human nuclei was higher in muscles of ActRIIB/Fc treated mice compared to muscles of untreated mice.
Figure 6. ActRIIB/Fc treatments increased the number of lamin A/C nuclei in individual dystrophin positive fiber and induced hypertrophy of the dystrophin positive fibers.

(a) Repartition of dystrophin positive fibers based on the number of lamin A/C nuclei inside their cross-section in the no swimming groups treated or not with ActRIIB/Fc (2 injections/week). (b) Repartition of dystrophin positive fibers based on the number of lamin A/C nuclei inside their cross-section in the weekly swimming groups treated or not with ActRIIB/Fc (5 injections/week). (c) Cross-sectional area of human-dystrophin positive myofibers, measured in the TAs of Rag−/− mdx mice treated or not with ActRIIB/Fc and forced or not to swim. The means are represented (+ SD, N=6). * p<0.05
ActRIIB/Fc treatment induced dystrophin positive fibers hypertrophy
We then investigated whether the inhibition of signaling of myostatin and other TGF-β family members with ActRIIB/Fc was sufficient to trigger hypertrophy of the dystrophin positive fibers. As shown in Figure 6c, significant hypertrophy was observed in the hybrid muscle fibers of both ActRIIB/Fc treated groups. Specifically, Group 2A(noSwim/ActRIIB/Fc 2wk) had a 70% hypertrophy relative to Group 1A(noSwim/noActRIIB/Fc), and Group 2B(Swim/ActRIIB/Fc 5wk) had a 350% hypertrophy compared with Group 2A(Swim/noActRIIB/Fc). Swimming did not by itself induce a significant hypertrophy of the dystrophin positive muscle fibers since Group 2A(Swim/noActRIIB/Fc) was indeed not significantly different from Group 1A(noSwim/noActRIIB/Fc).
ActRIIB/Fc treatment does not affect the percentage of centrally nucleated fibers
Finally, we compared the percentage of centrally-nucleated fibers in the various groups of mice. ActRIIB/Fc treatment in the first experiment did not significantly increase the number of centro-nucleated fibers (Figure 7). However, swimming by itself increased the percentage of centrally nucleated fibers in Group 2A(Swim/noActRIIB/Fc) compared to Group 1A(noSwim/noActRIIB/Fc). Similarly, muscles from Group 2B(Swim/ActRIIB/Fc 5wk) contained more centro-nucleated fibers than those of either Group 1A(noSwim/noActRIIB/Fc) or Group 1B(noSwim/ActRIIB/Fc 2wk).
Figure 7. ActRIIB/Fc treatment does not affect the percentage of centrally nucleated fibers.
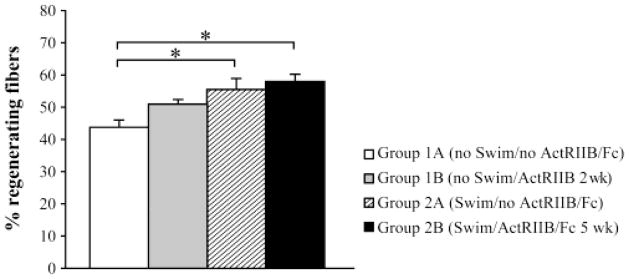
Percentage of regenerating fibers based on the presence of a central nucleus in group 1A (noSwim/noActRIIB/Fc), group1B (noSwim/ActRIIB/Fc 2 wk), group2A (Swim/noActRIIB/Fc) and group 2B (Swim/ActRIIB/Fc 5 wk). Swimming increased the regeneration. The means are represented (+ SD, N=6). * p<0.05.
DISCUSSION
Several studies have already demonstrated an increase of body and/or muscle growth in different mouse disease models after ActRIIB/Fc treatment (1,25,26). Our study confirms this effect of activin receptor blockade in a model of DMD. The increased skeletal muscle mass observed was due to hypertrophy and is comparable to prior studies of myostatin inhibition studies in mdx mice (7,37). Hypertrophy was even observed in our study in the dystrophin positive fibers resulting in part from the transplantation of normal human myoblasts into Rag−/−/mdx mice. Souza et al. demonstrated that depletion of circulating BMP-9 and BMP-10 by ActRIIB/Fc may indirectly contribute to the increase in muscle mass by maintaining a higher number of myogenic stem cells (satellite cells) in the skeletal muscle (34). In our studies, we found the proportion of centrally nucleated fibers, an indicator of regenerated fibers, to be unchanged in untreated and ActRIIB/Fc-treated TAs. Thus, our observations suggest that blocking this signalling pathway did not alter regeneration in TAs of mdx mice (25). However, when the ActRIIB/Fc treatment was combined to exercise, the number of centrally nucleated fibers increased significantly. Moreover, forced swimming alone appears to be inducing sufficient fiber damage to cause increased regeneration.
A major finding of our study is that ActRIIB/Fc improves the success of human myoblast transplantation by 3-fold as evaluated by the number of human dystrophin-positive myofibers. This improvement was the highest compared to the other studies (8,11). Our in vitro study indicated that ActRIIB/Fc increased the differentiation of human myoblasts through an up-regulation of the regulatory genes responsible for myogenesis, such as myogenin and MyoD, leading to myoblast fusion and myotube maturation (38). Despite its murine origin, the soluble form of the ActRIIB competed with the natural human receptors for ligand in the TGF-β family, which includes myostatin, activin-A and BMP9/10 (34). This is explained by the exceptional conservation of the ActRIIB extracellular domain sequence, with only one amino acid difference between mouse and human (13).
Following human myoblast transplantation, the mice treated with ActRIIB/Fc presented more dystrophin-positive myofibers and human nuclei than their respective control mice. This increase of human nuclei was, more significant in Group 2B(Swim/ActRIIB/Fc 5wk).
Different cellular therapy problems were responsible for the low number of dystrophin-positive fibers observed in clinical trial, among other, at least 75% of the transplanted myoblasts die in the first 3 days after transplantation (12,14,17). The ActRIIB Fc treatment increased early survival of the transplanted human myoblasts and improved myoblast transplantation success.
This enhanced success of myoblast transplantation may be due to an up-regulation myogenic gene expression and to increased proliferation, differentiation and/or fusion of the transplanted myoblasts with damaged fibers. The effect of ActRIIB/Fc on enhancing incorporation of transplanted myoblasts was observed in both experimental paradigms, i.e. with or without forced swimming. Thus, the ActRIIB/Fc treatment during the first two weeks after myoblast transplantation was sufficient to produce a beneficial effect on graft success, but exercise combined with a long-term ActRIIB/Fc treatment further enhanced myoblast transplantation success and fiber hypertrophy. Swimming is a physiological method to induce partial muscle fiber damage, and our results confirm that myoblasts are able to repair damaged myofibers, giving rise to more hybrid fibers (8).
In summary, inhibition of myostatin and other negative regulators of skeletal muscle growth by ActRIIB/Fc is a potential therapy for the treatment of diseases affecting skeletal muscle, such as muscular dystrophies. Our study suggests that combining myostatin inhibition and muscle exercise with myoblast transplantation may be an effective therapeutic strategy for delivering normal copies of the dystrophin gene and restoring muscle function to patients with DMD.
Acknowledgments
We thank the AFM “association française contre les myopathies” and Jessee Journey. Work in S-JL’s laboratory was supported by grants from the National Institutes of Health (NIH R01AR060636) and the Jain Foundation. Under a licensing agreement between MetaMorphix, Inc. (MMI), Pfizer, Inc., and the Johns Hopkins University, S.-J.L. is entitled to a share of royalty received by the University on sales of products related to myostatin. S.J.L. and the University own MMI stock, which is subject to certain restrictions under University policy. S-J.L., who is the scientific founder of MMI, is a consultant to MMI on research areas related to the study described in this paper.
Footnotes
The terms of these arrangements are being managed by the University in accordance with its conflict of interest policies.
References
- 1.Akpan I, Goncalves MD, Dhir R, Yin X, Pistilli EE, Bogdanovich S, Khurana TS, Ucran J, Lachey J, Ahima RS. The effects of a soluble activin type IIB receptor on obesity and insulin sensitivity. Int J Obes. 2009;33(11):1265–1273. doi: 10.1038/ijo.2009.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Amthor H, Nicholas G, McKinnell I, Kemp CF, Sharma M, Kambadur R, Patel K. Follistatin complexes Myostatin and antagonises Myostatin-mediated inhibition of myogenesis. Dev Biol. 2004;270(1):19–30. doi: 10.1016/j.ydbio.2004.01.046. [DOI] [PubMed] [Google Scholar]
- 3.Attisano L, Wrana JL, Cheifetz S, Massague J. Novel activin receptors: distinct genes and alternative mRNA splicing generate a repertoire of serine/threonine kinase receptors. Cell. 1992;68(1):97–108. doi: 10.1016/0092-8674(92)90209-u. [DOI] [PubMed] [Google Scholar]
- 4.Benabdallah BF, Bouchentouf M, Rousseau J, Bigey P, Michaud A, Chapdelaine P, Scherman D, Tremblay JP. Inhibiting myostatin with follistatin improves the success of myoblast transplantation in dystrophic mice. Cell Transplant. 2008;17(3):337–350. doi: 10.3727/096368908784153913. [DOI] [PubMed] [Google Scholar]
- 5.Benabdallah BF, Bouchentouf M, Tremblay JP. Improved success of myoblast transplantation in mdx mice by blocking the myostatin signal. Transplantation. 2005;79(12):1696–1702. doi: 10.1097/01.tp.0000167379.27872.2b. [DOI] [PubMed] [Google Scholar]
- 6.Bogdanovich S, Krag TO, Barton ER, Morris LD, Whittemore LA, Ahima RS, Khurana TS. Functional improvement of dystrophic muscle by myostatin blockade. Nature. 2002;420(6914):418–421. doi: 10.1038/nature01154. [DOI] [PubMed] [Google Scholar]
- 7.Bogdanovich S, Perkins KJ, Krag TO, Whittemore LA, Khurana TS. Myostatin propeptide-mediated amelioration of dystrophic pathophysiology. FASEB J. 2005;19(6):543–549. doi: 10.1096/fj.04-2796com. [DOI] [PubMed] [Google Scholar]
- 8.Bouchentouf M, Benabdallah BF, Mills P, Tremblay JP. Exercise improves the success of myoblast transplantation in mdx mice. Neuromuscul Disord. 2006;16(8):518–529. doi: 10.1016/j.nmd.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 9.Dubowitz V. Neuromuscular disorders in childhood. Old dogmas, new concepts. Arch Dis Child. 1975;50(5):335–346. doi: 10.1136/adc.50.5.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Emery AE. Population frequencies of inherited neuromuscular diseases--a world survey. Neuromuscul Disord. 1991;1(1):19–29. doi: 10.1016/0960-8966(91)90039-u. [DOI] [PubMed] [Google Scholar]
- 11.Fakhfakh R, Michaud A, Tremblay JP. Blocking the Myostatin Signal With a Dominant Negative Receptor Improves the Success of Human Myoblast Transplantation in Dystrophic Mice. Mol Ther. 2011;19(1):204–10. doi: 10.1038/mt.2010.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fan Y, Maley M, Beilharz M, Grounds M. Rapid death of injected myoblasts in myoblast transfer therapy. Muscle Nerve. 1996;19(7):853–860. doi: 10.1002/(SICI)1097-4598(199607)19:7<853::AID-MUS7>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 13.Garg RR, Bally-Cuif L, Lee SE, Gong Z, Ni X, Hew CL, Peng C. Cloning of zebrafish activin type IIB receptor (ActRIIB) cDNA and mRNA expression of ActRIIB in embryos and adult tissues. Mol Cell Endocrinol. 1999;153(1–2):169–181. doi: 10.1016/s0303-7207(99)00044-1. [DOI] [PubMed] [Google Scholar]
- 14.Guerette B, Skuk D, Celestin F, Huard C, Tardif F, Asselin I, Roy B, Goulet M, Roy R, Entman M, Tremblay JP. Prevention by anti-LFA-1 of acute myoblast death following transplantation. J Immunol. 1997;159(5):2522–2531. [PubMed] [Google Scholar]
- 15.Hill JJ, Davies MV, Pearson AA, Wang JH, Hewick RM, Wolfman NM, Qiu Y. The myostatin propeptide and the follistatin-related gene are inhibitory binding proteins of myostatin in normal serum. J Biol Chem. 2002;277(43):40735–40741. doi: 10.1074/jbc.M206379200. [DOI] [PubMed] [Google Scholar]
- 16.Hill JJ, Qiu Y, Hewick RM, Wolfman NM. Regulation of myostatin in vivo by growth and differentiation factor-associated serum protein-1: a novel protein with protease inhibitor and follistatin domains. Mol Endocrinol. 2003;17(6):1144–1154. doi: 10.1210/me.2002-0366. [DOI] [PubMed] [Google Scholar]
- 17.Huard J, Acsadi G, Jani A, Massie B, Karpati G. Gene transfer into skeletal muscles by isogenic myoblasts. Hum Gene Ther. 1994;5(8):949–958. doi: 10.1089/hum.1994.5.8-949. [DOI] [PubMed] [Google Scholar]
- 18.Jennekens FG, ten Kate LP, de Visser M, Wintzen AR. Diagnostic criteria for Duchenne and Becker muscular dystrophy and myotonic dystrophy. Neuromuscul Disord. 1991;1(6):389–391. doi: 10.1016/0960-8966(91)90001-9. [DOI] [PubMed] [Google Scholar]
- 19.Kaufman RJ. Vectors used for expression in mammalian cells. Methods Enzymol. 1990;185:487–511. doi: 10.1016/0076-6879(90)85041-l. [DOI] [PubMed] [Google Scholar]
- 20.Kinoshita I, Roy R, Dugre FJ, Gravel C, Roy B, Goulet M, Asselin I, Tremblay JP. Myoblast transplantation in monkeys: control of immune response by FK506. J Neuropathol Exp Neurol. 1996;55(6):687–697. doi: 10.1097/00005072-199606000-00002. [DOI] [PubMed] [Google Scholar]
- 21.Lee SJ, McPherron AC. Myostatin and the control of skeletal muscle mass. Curr Opin Genet Dev. 1999;9(5):604–607. doi: 10.1016/s0959-437x(99)00004-0. [DOI] [PubMed] [Google Scholar]
- 22.Lee SJ, McPherron AC. Regulation of myostatin activity and muscle growth. Proc Natl Acad Sci USA. 2001;98(16):9306–9311. doi: 10.1073/pnas.151270098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee SJ, Reed LA, Davies MV, Girgenrath S, Goad ME, Tomkinson KN, Wright JF, Barker C, Ehrmantraut G, Holmstrom J, Trowell B, Gertz B, Jiang MS, Sebald SM, Matzuk M, Li E, Liang LF, Quattlebaum E, Stotish RL, Wolfman NM. Regulation of muscle growth by multiple ligands signaling through activin type II receptors. Proc Natl Acad Sci USA. 2005;102(50):18117–18122. doi: 10.1073/pnas.0505996102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li ZB, Kollias HD, Wagner KR. Myostatin directly regulates skeletal muscle fibrosis. J Biol Chem. 2008;283(28):19371–19378. doi: 10.1074/jbc.M802585200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morine KJ, Bish LT, Selsby JT, Gazzara JA, Pendrak K, Sleeper MM, Barton ER, Lee SJ, Sweeney HL. Activin IIB receptor blockade attenuates dystrophic pathology in a mouse model of Duchenne muscular dystrophy. Muscle Nerve. 2010;42(5):722–730. doi: 10.1002/mus.21743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morrison BM, Lachey JL, Warsing LC, Ting BL, Pullen AE, Underwood KW, Kumar R, Sako D, Grinberg A, Wong V, Colantuoni E, Seehra JS, Wagner KR. A soluble activin type IIB receptor improves function in a mouse model of amyotrophic lateral sclerosis. Exp Neurol. 2009;217(2):258–268. doi: 10.1016/j.expneurol.2009.02.017. [DOI] [PubMed] [Google Scholar]
- 27.Qiao C, Li J, Jiang J, Zhu X, Wang B, Xiao X. Myostatin propeptide gene delivery by adeno-associated virus serotype 8 vectors enhances muscle growth and ameliorates dystrophic phenotypes in mdx mice. Hum Gene Ther. 2008;19(3):241–254. doi: 10.1089/hum.2007.159. [DOI] [PubMed] [Google Scholar]
- 28.Richard PL, Gosselin C, Laliberte T, Paradis M, Goulet M, Tremblay JP, Skuk D. A first semimanual device for clinical intramuscular repetitive cell injections. Cell Transplant. 2010;19(1):67–78. doi: 10.3727/096368909X478812. [DOI] [PubMed] [Google Scholar]
- 29.Sako D, Grinberg AV, Liu J, Davies MV, Castonguay R, Maniatis S, Andreucci AJ, Pobre EG, Tomkinson KN, Monnell TE, Ucran JA, Martinez-Hackert E, Pearsall RS, Underwood KW, Seehra J, Kumar R. Characterization of the ligand binding functionality of the extracellular domain of activin receptor type IIb. J Biol Chem. 2010;285(27):21037–21048. doi: 10.1074/jbc.M110.114959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Skuk D, Goulet M, Roy B, Chapdelaine P, Bouchard JP, Roy R, Dugre FJ, Sylvain M, Lachance JG, Deschenes L, Senay H, Tremblay JP. Dystrophin expression in muscles of duchenne muscular dystrophy patients after high-density injections of normal myogenic cells. J Neuropathol Exp Neurol. 2006;65(4):371–386. doi: 10.1097/01.jnen.0000218443.45782.81. [DOI] [PubMed] [Google Scholar]
- 31.Skuk D, Goulet M, Tremblay JP. Use of repeating dispensers to increase the efficiency of the intramuscular myogenic cell injection procedure. Cell Transplant. 2006;15(7):659–663. doi: 10.3727/000000006783981648. [DOI] [PubMed] [Google Scholar]
- 32.Skuk D, Roy B, Goulet M, Chapdelaine P, Bouchard JP, Roy R, Dugre FJ, Lachance JG, Deschenes L, Helene S, Sylvain M, Tremblay JP. Dystrophin expression in myofibers of Duchenne muscular dystrophy patients following intramuscular injections of normal myogenic cells. Mol Ther. 2004;9(3):475–482. doi: 10.1016/j.ymthe.2003.11.023. [DOI] [PubMed] [Google Scholar]
- 33.Skuk D, Tremblay JP. Progress in myoblast transplantation: a potential treatment of dystrophies. Microsc Res Tech. 2000;48(3–4):213–222. doi: 10.1002/(SICI)1097-0029(20000201/15)48:3/4<213::AID-JEMT9>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 34.Souza TA, Chen X, Guo Y, Sava P, Zhang J, Hill JJ, Yaworsky PJ, Qiu Y. Proteomic identification and functional validation of activins and bone morphogenetic protein 11 as candidate novel muscle mass regulators. Mol Endocrinol. 2008;22(12):2689–2702. doi: 10.1210/me.2008-0290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sunada Y. Therapeutic strategies for muscular dystrophy by myostatin inhibition. Rinsho Shinkeigaku. 2006;46(11):942–944. [PubMed] [Google Scholar]
- 36.Thies RS, Chen T, Davies MV, Tomkinson KN, Pearson AA, Shakey QA, Wolfman NM. GDF-8 propeptide binds to GDF-8 and antagonizes biological activity by inhibiting GDF-8 receptor binding. Growth Factors. 2001;18(4):251–259. doi: 10.3109/08977190109029114. [DOI] [PubMed] [Google Scholar]
- 37.Wagner KR, McPherron AC, Winik N, Lee SJ. Loss of myostatin attenuates severity of muscular dystrophy in mdx mice. Ann Neurol. 2002;52(6):832–836. doi: 10.1002/ana.10385. [DOI] [PubMed] [Google Scholar]
- 38.Weintraub H, Dwarki VJ, Verma I, Davis R, Hollenberg S, Snider L, Lassar A, Tapscott SJ. Muscle-specific transcriptional activation by MyoD. Genes Dev. 1991;5(8):1377–1386. doi: 10.1101/gad.5.8.1377. [DOI] [PubMed] [Google Scholar]