

Abstract
Dendritic cells (DCs) are specialized sentinels responsible for coordinating adaptive immunity. This function is dependent upon coupled sensitivity to environmental signs of inflammation and infection to cellular maturation—the programmed alteration of DC phenotype and function to enhance immune cell activation. Although DCs are thus well equipped to respond to pathogens, maturation triggers are not unique to infection. Given that immune cells are exquisitely sensitive to the biological functions of DCs, we now appreciate that multiple layers of suppression are required to restrict the environmental sensitivity, cellular maturation, and even life span of DCs to prevent aberrant immune activation during the steady state. At the same time, steady-state DCs are not quiescent but rather perform key functions that support homeostasis of numerous cell types. Here we review these functions and molecular mechanisms of suppression that control steady-state DC maturation. Corruption of these steady-state operatives has diverse immunological consequences and pinpoints DCs as potent drivers of autoimmune and inflammatory disease.
Keywords: Toll-like receptors, MyD88, autoimmunity, colitis, costimulatory molecules, A20
INTRODUCTION
In the public announcement awarding the 2011 Nobel Prize in Physiology or Medicine to Dr. Ralph Steinman for his 1973 identification of dendritic cells (DCs), the Nobel Prize Assembly described DCs as “gatekeepers of the immune system” (1, 2). The work by Steinman and others has championed this title for DCs, such that today we appreciate the novelties of DC biology, the heterogeneity and specialization of DC subsets, and DC functional responses as well as how these components translate into the control and coordination of immunity (3). Central to the gatekeeping functions of DCs is their ability to couple a survey of the microenvironment, in the form of antigen uptake and responsiveness to environmental cues, to a cellular differentiation program termed “maturation” that enhances their abilities to activate immune cells (4). Cellular differentiation upon DC maturation involves upregulated cell surface expression of immunostimulatory molecules such as peptide–MHC class I and II molecules as well as T cell costimulatory molecules such as CD80 and CD86 (5–8). These molecules are collectively referred to as maturation markers, and DCs expressing high levels of these markers are often referred to as having undergone phenotypic maturation. Additional changes include altered endocytosis, migration into T cell zones of lymphoid organs, and cytokine expression; DCs that have undergone the complete or minimal set of cellular differentiation to enable immunostimulatory functions are referred to as functionally mature (9–11). In adaptive immunity, a distinction can be made between phenotypic and functional DC maturation, particularly in the discriminatory expression of cytokines (12–17). Although many environmental stimuli may induce phenotypic maturation, one critical component required to drive differentiation into functional maturity is the direct sensing of microbial compounds by DCs (reviewed in 11). To this end, DCs employ a diversity of microbial sensors and other probes that upon ligand binding initiate intracellular signaling cascades that drive both phenotypic and functional maturation. In essence, DCs and the intracellular signaling cascades transduced within them comprise the key that opens the gate toward adaptive immunity.
Per the functional analogy of gatekeeping, if opening the gates brings about productive adaptive immunity, what is constrained by keeping the gate closed? How do DCs accomplish gate sealing, and how does it facilitate steady-state conditions and immune homeostasis? Which mechanisms and intracellular signals govern the steady-state gatekeeping functions of DCs? Are these signals distinct or related to maturation signals, and how are they overcome within DCs to induce maturation when necessary? With the advancement of our knowledge of the molecules expressed by DCs, the immunologic functions of DCs during steady state, and the genetic tool of DC-specific ablation of gene expression, we are beginning to piece together how the mirrored functions of DCs as gatekeepers of immune homeostasis are achieved. In this review, we present a description of key molecules, intracellular signals, and mechanisms that regulate DC maturation and DC control of immune homeostasis in vivo during steady-state conditions. We limit our discussion in general to those mechanisms whose perturbation specifically in DCs leads to clear changes in DC maturation and/or function, and we further describe how these changes are associated with disrupted immune homeostasis (Table 1). DCs as a family share expression of integrin CD11c, and the tendency has been to analyze the phenotype of all CD11c+ cells in any given organ. However, given the unique attributes of each DC subset and the likely unique signals sensed by DCs depending on their microenvironment, whenever corresponding information is available we report how steady-state signals affect different DC subsets and tissue-specific DCs.
Table 1.
Disruptions to DC populations, steady-state signals sensed by DCs, and their consequence for immune homeostasis
| Disruption | DC populations | DC phenotype | Initiating and intracellular signals | Functional consequence | Steady-state immune homeostasis | Reference(s) |
|---|---|---|---|---|---|---|
| Commensal | ||||||
| Germ free | Spleen, pLN: NC mLN: ↓ CD8α+ |
NC | Commensals | NC | No disruption | 53, 54 |
| TLR signals | ||||||
| Myd88−/− Ticam1−/− | NC | NC | TLR ligands | NC | Increase in α-commensal serum Ig | 54, 64 |
| TLR7 transgenic overexpression | ↑ cDC, pDC | ↑ MHC class II, CD80 | TLR7 ligands MyD88 |
↑ IRF5, SOCS1, TNF-α, CCL2, IL-6 | Splenomegaly Myeloid expansion Autoantibody T cell activation Anemia leading to death |
56 |
| TLR9 mutant mislocalized to cell surface | Bone marrow: ↑ CD11c+ | NC | Irradiation-induced DNA MyD88 |
↑ TNF-α, IL-1β NC: Type I IFN |
Myeloid expansion Bone marrow failure Anemia leading to death |
27 |
| C-type lectins | ||||||
| DC-specific SHP-1 deficient | Spleen: ↑ cDC, pDC LN: NC |
↑ CD86, CCR7 NC: CD40, CD80, MHC class II |
ITIM-dependent cross-regulation of activating signals? | ↑ Phospho-Tyr ↑ TNF-α, IL-6, IL-10, IL-1β, IFN-β |
T and B cell activation and expansion ↑ Th1 Autoantibody Glomerulonephritis Interstitial pneumonitis |
89 |
| Clec9a−/− | NC | NC | F-actin exposed on necrotic cells Syk |
Cross-presentation of necrotic cargo | NC during steady state ↓ Immunity to necrotic cell antigens |
104–106 |
| DCIR1 deficient | LN: ↑ CD11c+ | NC | Unknown ligands SHP-1, 2? | ND | Autoantibody IgM rheumatoid factor Arthritis ↑ Collagen-induced arthritis |
108 |
| NLRs | ||||||
| Nod2−/− | NC | GvHD: ↑ CD80, CD86, CD40 | Bacterial peptidoglycan | GvHD: ↑ T cell activation | Increased intestinal bacteria ↑ DSS colitis |
130, 131, 134 |
| NOD2-human Crohn’s disease variants | ND | ND | Bacterial peptidoglycan | ↓ Autophagy ↓ MHC class II ↓ Intracellular killing of bacteria |
Crohn’s disease-associated variants ↑ DSS colitis |
128, 133 |
| DC life span | ||||||
| DC-specific p35 transgenic | Spleen: ↑ cDC, pDC | NC | — | p35-dependent caspase inhibition ↑ Survival ↓ Fas-induced death |
Autoantibody T and B cell activation (not expansion) ↑ Disease in MRL. Faslpr mice |
137 |
| Bim deficient | Spleen: ↑ cDC, pDC | NC | ND | ↑ Survival | Autoantibody | 37 |
| DC-specific Fas−/− | Spleen: ↑ CD11c+ | ↑CD86 | Fas-L | ↑ Survival | Splenomegaly Liver inflammation Autoantibody Mislocalization of splenic DC |
138 |
| DC ablation | ||||||
| DC ablation | ↓ cDC NC: pDC, LC |
— | — | ↑ FLT3-L (DC extrinsic) | Splenomegaly Lymphadenopathy Myeloid expansion Nonmalignant, nonfatal myeloid proliferative disorder |
140 |
| DC ablation | ↓ cDC, pDC, LC | — | — | Compromised negative selection ↑ CD4 single-positive thymocytes |
Splenomegaly Lymphadenopathy Myeloid expansion T cell activation, Th1, Th17 ↑ Ig, autoantibody Inflammation of kidney, liver, intestine |
141 |
| DC-ablated MRL. Faslpr mice | ↓ cDC, pDC, LC | — | MyD88? | ND | ↓ T cell expansion, IFN-γ production ↓ Plasmablasts ↓ Autoantibody ↓ Nephritis, dermatitis |
60, 62 |
| Transcription factors | ||||||
| Foxo3−/− (C57BL/6 mice) | Spleen: ↑ cDC, pDC | cDC, pDC: ↑ CD80, CD86 NC: MHC class II, MHC class I, CD40 |
CTLA-4 Oxidative stress? GM-CSF? |
↑ IL-6, TNF-α, CCL2 | Myeloid expansion ↑ T cell survival (IL-6-dependent) during immune response |
171, 178 |
| Nf-κb1−/− | Spleen: ↓ cDC, pDC | cDC: NC pDC: ↓ MHC class II |
ND | GM-CSF BMDCs: ↓ MAPK, ↑ TNF-α, IL-6 pDC: ↓ IL-6 |
NC during steady state ↓ Th2 immunity TNF-α-dependent disruption of CD8+ T cell tolerance to pancreatic antigens |
180, 187, 188 |
| RAG2−/− DC-specific T-bet−/− (BALB/c mice) | ND | ND | Commensal dependent MyD88 independent |
↑ TNF-α Fostered outgrowth of colitogenic commensals |
TNF-α-dependent ulcerative colitis and colorectal cancer Transmissible colitis via commensals |
198, 199 |
| MHC class II–related DC responses | ||||||
| Ii deficient | NC | cDC, pDC: ↓ MHC class II | — | ↓ Ii-dependent activation of myosin light chain | ↑ DC motile velocity Migration patterns unresponsive to maturation signals |
207 |
| MARCH1 deficient | NC | cDC: ↑ MHC class II, CD86 NC: CD80 |
ND | ↓ Ubiquitination of MHC class II and CD86 ↓ MHC class II antigen-presentation |
NC during steady state MHC II-dependent impotency of CD40 and TLR-induced cytokine production |
211, 212, 217, 218 |
| MHC class II deficient | NC | Absent MHC class II | — | ↓ MHC class II-dependent Btk activation ↓ TLR-induced NF-κB and cytokine production |
↓ Susceptibility to endotoxin shock | 226 |
| DC-expressed receptors, T cell costimulatory molecules, and MHC class II | ||||||
| DC-specific IL-15 receptor alpha deficient | NC | NC | ND | Inability to trans-present IL-15 | ↓ NK cell survival ↓ Memory phenotype CD8+ T cells |
142 |
| DC-specific CD70 transgenic overexpression | NC | cDC: ↑ CD70 | — | ↑ CD27-dependent T cell costimulation | Splenomegaly T cell activation (not expansion) Progressive lymphopenia Antigen-dependent T cell expansion and effector function despite tolerant setting |
154 |
| DC-specific CD80, CD86, MHC class II | — | Absent CD80, CD86 or MHC class II | ND | Lack of CD80 and CD86 costimulation or MHC II interaction | ↓ Peripheral Treg ↓ Treg proliferation in lymphoid organs and skin ↓ MHC class II–dependent interaction and suppression by Treg |
220, 232, 233 |
| TGF-β and DC signals regulating steady-state Treg populations | ||||||
| DC-specific ↑vβ8 deficient | NC | NC | ND | ↓ DC-dependent TGF-β activation |
Splenomegaly Lymphadenopathy ↓ Colonic Treg ↑ T cell activation, Th1, Th2 ↑ IgG1, IgE, IgA Autoantibody Colitis Liver inflammation |
238 |
| IDO1 function in pDCs | NC | ND | TGF-β IFN-γ CTLA-4 |
↑ IDO1 expression and catabolism pDC: TGF-β-dependent IDO1-mediated SHP activation, ↑ noncanonical NF-κB, ↑ IFN-α |
TGF-β-dependent, pDC-dependent ↑ Treg and long-term tolerance to peptide sensitization |
247 |
| DC-specific β-catenin deficient | Spleen: NC Intestine: ↓ CD103+ | ND | Commensal- independent source of Wnt-ligands | ↑ IL-6, IL-23 ↓ IL-10, TGF-β ↓ Vitamin A metabolism |
Intestine: ↓ Treg, ↑ Th1, Th17 ↑ DSS colitis |
249 |
| Regulators of steady-state maturation signals | ||||||
| Il6−/− | Spleen, LN: NC Liver: ↑ pDC |
LN: ↑ MHC class II, CD86 Liver: ↑ MHC class II, CD86, CD80, CCR7 Spleen: NC |
Liver: commensals Tissue-to-LN migration- inducing signals? STAT3 |
↑ Stability of intracellular MHC class II dimers Liver DCs: ↓ IRAK-M, ↓ IL-10, ↑ TNF-α |
Suppress DC maturation to commensals and TLR ligands | 251, 252, 254 |
| DC-specific Stat3−/− | cDC: NC ↓ pDC |
NC | Numerous cytokines, the most potent likely IL-10 | Lack of IL-10- induced anti-inflammatory state, potentially to MyD88 signals | Intestinal inflammation, predominantly colitis | 257 |
| DC-specific Socs1−/− | ↑ Thymic DC Spleen: ↑ cDC, pDC ↑ Atypical CD8+CD11clow |
↑ CD80, CD86, CD40 | IFN-γ TLRs | ↑ Thymic single positives ↑ BAFF |
Splenomegaly Lymphadenopathy ↑ T and B cell expansion (not activation) ↑ Ig, autoantibody Glomerulonephritis Dermatitis Arthritis |
259, 265, 267 |
| TAM receptor deficient | Spleen: ↑ CD11c+ | ↑ MHC class I, MHC class II, CD86 | TLRs Apoptotic cells Gas6 ProS IFN-α STAT1 STAT3 |
TLR signals: ↑ TRAF3, TRAF6 ubiquitination, ↑ IL-6, TNF-α ↓ SOCS1, SOCS3 ↑ BAFF ↓ Apoptotic cell–induced suppression ↓ Competition with IFN-α-STAT1 inflammatory responses |
Splenomegaly Lymphadenopathy B and T cell activation and expansion Autoantibody Glomerulonephritis Arthritis |
272–277 |
| DC-specific deletion of A20, exon 4–5 | Spleen: ↑ CD11clow CD11b+ SIRP1 α+, ↓ pDC |
CD11clow CD11b+: ↑ CD86, CD40 pDC: NC |
TLR signals MyD88 Apoptotic cells? |
↑ NF-κB ↑ IL-6, TNF-α, BAFF, FLT3-L ↑ Survival |
Splenomegaly Lymphadenopathy Myeloid expansion T and B cell activation, expansion ↑ Germinal center B cells, plasma cells ↑ Phagocytosis of apoptotic cells ↓ DC suppression by apoptotic cells Autoantibody Glomerular Ig deposits |
284 |
| DC-specific deletion of A20, exon2 | Spleen: cDC, pDC NC | cDC: ↑ CD80, CD40 pDC: ↑ MHC class II, CD80, CD86, CD40 |
TLR signals MyD88 MyD88-independent signals |
↑ NF-κB ↑ IL-6, TNF-α ↓ Survival |
Splenomegaly Lymphadenopathy Myeloid expansion Antigen-independent T cell activation and expansion, ↑ Th1 T cell–dependent colitis Seronegative spondyloarthritis |
63 |
| DC-specific deletion of Myd88 and A20, exon2 | Spleen: cDC, pDC NC | cDC: ↑ CD80, CD40 pDC: ↑ MHC class II, CD80, CD86, CD40 |
TRIF-dependent TLR signals? Non-TLR signals? |
↑ NF-κB to MyD88- independent signals ↓ IL-6, TNF-α as compared with MyD88+A20, exon2-deleted DC Survival? |
Rescue of lymphadenopathy T cell activation, not expansion Colitis? Spondyloarthritis? |
63 |
Abbreviations: BAFF, B cell–activating factor; cDC, conventional dendritic cell; CTLA-4, cytotoxic T lymphocyte antigen-4; DC, dendritic cell; DCIR1, dendritic cell immunoreceptor 1; DSS, dextran sodium sulfate; FLT3-L, fms-like tyrosine kinase receptor-3 ligand; Gas6, growth arrest gene 6; GM-CSF, granulocyte-macrophage colony-stimulating factor; GvHD, graft-versus-host disease; IDO1, indoleamine 2,3-dioxygenase 1; Ig, immunoglobulin; Ii, invariant chain; IRAK-M, IL-1 receptor-associated kinase M; IRF, interferon regulatory factor; ITIM, immunoreceptor tyrosine inhibitory motif; LC, Langerhans cell; LN, lymph node; MAPK, mitogen-activated protein kinase; MARCH1, membrane-associated RING-CH protein 1; mLN, mesenteric lymph node; MyD88, myeloid differentiation primary response gene 88; NC, no change; ND, not determined; NK cell; natural killer cell; NLR, nucleotide oligomerization domain (NOD)-like receptor; pDC, plasmacytoid dendritic cell; pLN, peripheral lymph node; phospho-Tyr, tyrosine phosphorylation; ProS, protein S; SHP, Src homology phosphatase; SOCS1, suppressor of cytokine signaling 1; STAT, signal transducer and activator of transcription; Syk, spleen tyrosine kinase; TGF-β, transforming growth factor β; Th1, T helper cell type 1; Th2, T helper cell type 2; Th17, T helper cell type 17; TLR, Toll-like receptor; TRAF, TNF receptor-associated factor; Treg, T regulatory cell; TRIF, TIR-domain-containing adaptor-inducing IFN-β.
PATTERN-RECOGNITION RECEPTORS
Pattern-recognition receptors (PRRs) consist of Toll-like receptors (TLRs), C-type lectin receptors (CLRs), nucleotide oligomerization domain (NOD)-like receptors (NLRs), and retinoic acid–inducible gene (RIG)-I-like receptors (RLRs) (18, 19). Although best known as probes employed by DCs to sense pathogens and other environmental stimuli, many PRRs also transduce potent maturation signals. As such, researchers must consider that PRR ligands are not unique to pathogenic infection. Ligands are expressed by host commensals and can even be of host origin. How DCs may sense these ligands during steady state and yet maintain immune homeostasis is an area of intense investigation, and our knowledge of these mechanisms is limited. Because the roles of PRRs in DC functions can be diverse and the consequences of misregulated PRR signaling in DCs do not yield overlapping phenotypes, we first present a general outline of the intracellular signals transduced by PRRs and describe conceptually how they may affect steady-state DC gatekeeping functions. The known molecular regulators of steady-state PRR signals in DCs are then discussed individually rather than grouped by PRR. Importantly, regulatory molecules and the stimulus upstream of their functions can be independent of PRRs, and for some, the upstream signals that govern their activity are not known. Thus, we have categorized the regulatory pathways on the basis of their mode of molecular action within DCs.
Toll-Like Receptors
TLRs are ligand-dependent detection modules that facilitate key aspects of innate and adaptive immunity and are also considered “gatekeepers of the immune system” (2, 18, 20), leading the Nobel Assembly to recognize in 2011 not only Dr. Steinman’s work on DCs but also the work of Jules Hoffman and Bruce Beutler on the innate immune functions of TLRs in flies and mammals (21, 22). Each TLR recognizes a unique set of ligands, which vary in origin from bacterial cell wall components to nucleic acids, the specificity of which is dictated by the TLR ligand binding domain and the preferential localization of the receptor within the cell (23–28). TLRs share myeloid differentiation primary response gene 88 (MyD88) and/or TIR domain–containing adaptor-inducing IFN-β (TRIF) (also known as TICAM-1) as receptor-proximal signaling molecules that activate IL-1R-associated kinase family members (IRAK), the E3 ubiquitin ligases TNFR-associated factor 6 (TRAF6) and TRAF3, to drive mitogen-activated protein kinase (MAPK), IFN regulatory factor (IRF), NF-κB, and other intracellular signals. We direct the reader to several current reviews for detailed information on TLR ligand specificities and signaling properties (19, 25, 28–30).
Each DC subset expresses a defined set of TLRs, and TLR signals within DCs are critical for productive immune responses, with some exceptions given the nature of the immunogen (13, 20, 31–35). TLR ligands induce phenotypic and functional maturation and can also modulate cellular metabolism and life span (36–40), cytoskeletal and organelle dynamics (7, 41), autophagy (42–46), and intracellular sorting and processing of endocytosed/phagocytosed antigens (47–49). Although TLR ligands are present and trigger intracellular signals during steady state (50, 51), the outcomes of these steady-state TLR signals in DCs have not been easy to decipher. Indeed, DC subsets, steady-state patterns of migration, and maturation marker expression are normal in mice void of TLR signals (Myd88−/− Ticam1−/−) and in germ-free mice (52–54). Thus, TLR signals and other signals originating from commensals do not drive steady-state DC maturation, and a role for PRR signals in DC maturation is detectable only upon overt stimulation with TLR ligands or infection. Steady-state expression of maturation markers may be imprinted during DC development, perhaps by transcription factors that direct DC subset lineage commitments (discussed below), as the main DC subsets in lymphoid organs—conventional DCs (cDCs), and plasmacytoid DCs (pDCs)—can be easily distinguished by their levels of expression of MHC class II and costimulatory molecules (55). Additionally, as we discuss in this review, cytokines, intracellular ubiquitin ligases, and other molecules may afford the proper regulation for steady-state expression of maturation markers by DCs.
Even though TLRs may not drive steady-state DC maturation, multiple lines of evidence indicate that TLR signals are active in DCs, and TLR ligands may be constitutively and systemically sensed by DCs during steady-state conditions. For example, duplication of the TLR7 gene leads to DC expansion, maturation, and systemic autoimmunity (56). In irradiated mouse recipients, overexpression and mislocalization of TLR9 to the cell surface of hematopoietic stem cells (HSCs) lead to bone marrow expansion of CD11c+ cells and fatal inflammation (27). Additionally, psoriasis in human patients is associated with the upregulation of host molecules that enhance nucleic acid sensing by TLRs expressed by pDCs (57–59). Significant insight into cellular sensing of steady-state TLR signals has come from analysis of MyD88-deficient mice crossed to disease-prone genetic backgrounds, such as MRL. Faslpr mice and lyn−/− mice, which develop autoimmune diseases comparable to systemic lupus erythematosus (SLE) in humans. MyD88 deficiency protects these mice from autoimmune disease and is associated with reduced expansion, activation, and cytokine production by DCs (60, 61). Elimination of DCs from MRL. Faslpr mice is also protective against SLE because of the lack of DC-dependent expansion of pathogenic T and B cells (62), an aberrant DC function that is driven by MyD88 signals in DCs (63). Steady-state TLR signals can also have a protective role for the host, as has been demonstrated in the maintenance of intestinal homeostasis and host-commensal mutualism (50, 64). Direct probing of the intestinal lumen by DCs can be enhanced by MyD88 signals, and commensal sampling may protect the host from colitis and intestinal pathogens (50, 65–71); analogous function has been noted in the prevention of diabetes in NOD mice (72). The mechanisms by which commensal sampling by DCs confers disease protection and intestinal homeostasis and the intracellular signaling cascades that drive these DC functions require further investigation.
As our knowledge has grown about how TLR signals are transduced and negatively regulated, it has become clear that steady-state TLR signals in DCs are actively suppressed to maintain immune homeostasis. We restrict our discussion to those molecules that negatively regulate steady-state TLR signals (as opposed to those that are involved in overt stimulation of TLR ligands), their molecular mechanism of action, the consequences for phenotypic and functional DC maturation, and immune homeostasis.
C-Type Lectin Receptors
CLRs are a diverse family of transmembrane molecules containing the C-type lectin protein domain that enables binding of Ca2+ and/or carbohydrate ligands of self, viral, bacterial, and fungal origin. We refer the reader to recent reviews for a comprehensive description of the functions, ligand specificities, and signaling capacities of this large family of receptors (73–75). Like TLRs, expression of most CLRs is not restricted to DCs; however, the repertoire of CLR expression varies among distinct DC subsets, and often, CLR expression is the unique identifier of any given DC. Such is the case for Langerin: In humans, it is exclusive to Langerhans cells of the skin (with wider distribution on mouse DCs) and gives rise to unique endosomal compartments known as Birbeck granules, a defining characteristic of Langerhans cells (76, 77).
As a family, CLRs are involved in endocytosis, phagocytosis, antigen sorting into MHC class II or cross-presented MHC class I peptide-processing pathways, immunoreceptor tyrosine activation motif (ITAM)-mediated spleen tyrosine kinase (Syk) activation, or immunoreceptor tyrosine-based inhibitory motif (ITIM)-mediated Src-homology phosphatase (SHP) activation (reviewed in 75). Cross-presentation, a specialized biological process that delivers extracellular antigens into the MHC class I antigen processing pathway, is a feature shared by many endocytic CLRs including DEC205, mannose receptor, dendritic cell immunoreceptor (DCIR) 1, and DCIR2 (78–82). Notably, however, ligand engagement of CLRs on DCs does not necessarily lead to DC maturation, even though it may activate Syk and/or productively direct the antigen loading of MHC molecules. In most cases, CLRs downregulate DC functions. For example, BDCA-2-Syk signals in human pDCs restrict type I IFN production (83, 84). Signals from macrophage galactose-type lectin, whose ligands are highly expressed in dermis and on lymph node high endothelial venules, restrict DC migration (85, 86). For endocytic CLRs, ligand engagement induces antigen uptake by DCs, but in the absence of maturation stimuli, these signals lead to antigen-specific T cell tolerance rather than to immunity; DEC205 is the best-studied CLR in this category (78, 80, 87). These properties suggest that CLRs may mediate key functions in tolerance, though mice deficient in these antigen-uptake molecules do not have perturbed immune homeostasis, suggesting that, in a tolerogenic capacity, steady-state CLRs may have overlapping functions. There is no evidence that steady-state antigen uptake, cross-presentation, or signals transduced by CLRs, including those that lead to DC maturation, necessarily require negative regulation in DCs. However, ITIM-containing CLRs that activate SHP phosphatases can negatively cross-regulate maturation signaling cascades when both types of pathways are triggered simultaneously. SHP-1 deficiency specifically in DCs leads to autoimmunity, indicating a nonredundant function of SHP-1 phosphatase in negative regulation of steady-state DC maturation and maintenance of immune homeostasis (88, 89). The speculated CLRs and/or other ITIM-containing receptors upstream of SHP-1 that are critical to suppress steady-state DC maturation are not defined but may be required to negatively cross-regulate MyD88 signals induced by commensals (90).
The unique functions of individual CLRs and disparate expression of CLR family members on DC subsets suggest that the ligand-CLR network may provide tailored regulation of DC functions in a manner consistent with the specialized role of DC subsets in immune homeostasis and/or immune responses. Such specialization extends beyond cross-presentation, as numerous reports indicate that DC subsets have differential abilities to activate T cells and may functionally participate only during distinct phases of an immune response (80, 91–95). Heath and colleagues (96) illustrated this principle in a comparative analysis of lung-derived DCs and lymphoid-resident DCs. Following influenza infection of the lung, lung-derived DCs were found to have a potent ability to activate naive T cells, yet their ability to reactivate these T cells after proceeding into memory T cells was dramatically reduced. By contrast, lymphoid-resident DCs did not show such discrimination, suggesting that lung-derived DCs are specialized to initiate naive T cell responses and actively discriminate against reactivation of memory T cells. Such discrimination may be afforded in part by differential expression of CLRs that may positively or negatively regulate T cell activation mediated by migratory, rather than nonmigratory, lymphoid-resident DCs. Accordingly, T cells, in naive or memory form, may display distinct carbohydrate moieties that engage CLRs and drive Syk, SHP, or other immunomodulatory signals. Evidence for a CLR/T cell ligand regulatory network that governs DC functions comes from research on Dectin-1, which activates DCs in response to β-glucans from microbial cell walls and also recognizes an unknown ligand expressed by host T cells (97). Additionally, macrophage galactose C-type lectin recognizes ligands of viral origin as well as CD45, with preference for CD45RB, the isoform highly expressed by antigen-experienced effector and memory T cells (98). For most CLRs, the corresponding endogenous, nonpathogen-associated ligand is unknown, and further research is required to determine whether a network comprising DCs, CLRs, and T cells controls steady-state DC responses and mediates immune homeostasis.
Consideration of the microbial-recognition function of CLRs and downstream augmentation of DC functions has spurred interest about whether and how CLRs expressed by DCs in the intestine regulate host-commensal mutualism. Limited information exists regarding the repertoire and subset specificities of CLR expression in intestinal DCs, yet some studies illustrate the potential of a commensal-CLR regulatory network. In human DCs, species of lactobacilli, an abundant intestinal commensal commonly used in probiotics, is recognized by DC-SIGN and induces DC polarization of regulatory T cells (Tregs) (99, 100). Further support for a lactobacilli-CLR regulatory network is found in mice whose DCs cocultured with lactobacilli prevented T cell–dependent colitis (68). Carbohydrate ligands recognized by SIGN-R1 induce CD11c+CD11b+ lamina propria (LP) DCs to produce IL-10 and induce IL-10-producing Tregs to confer oral tolerance and protection from food-related anaphylaxis (101). Although both SIGN-R1 and Dectin-1 are expressed by CD11c+CD11b+ LPDCs (101), only SIGN-R1 confers protection against colitis induced by dextran sodium sulfate (DSS) (102, 103), a chemical agent that drives intestinal inflammation by damaging intestinal epithelial cells and flooding the host with gut commensals. Discrepant roles for individual CLRs in DSS colitis suggest that each CLR may play a distinct role in DC regulation of intestinal homeostasis. Such a fine-tailored commensal-CLR network could maintain communities of host-beneficial microorganisms, prevent inflammatory bowel disease, and afford pathogen detection and elimination.
Among endogenous ligands, many CLRs recognize ligands expressed by apoptotic or necrotic cells and facilitate uptake of these dead cells by DCs, although the bulk of the responsibility may be assumed by macrophages and neutrophils (75). CLR-mediated clearance of apoptotic cells, which is a noninflammatory mode of cell death, can lead to immune tolerance and reduction of cellular responses. Although the clearance of dead cells is proposed as key to preventing the development of antinuclear antigens and autoimmune disease, mice deficient in CLRs that recognize dead cells do not have obvious perturbations to immune homeostasis. This suggests that these receptors may have redundant functions or that other mechanisms that facilitate DC-mediated immune tolerance may have precedence.
In contrast, CLRs that recognize necrotic cell death are associated with cellular activation and immunogenic responses. CLEC9a (also known as DNGR-1) has recently been described as a nonredundant receptor expressed by CD8α+ DCs to recognize necrotic cells (104). The CLEC9a ligand, F-actin, is exposed on the cell surface upon necrotic but not apoptotic cell death (105). CLEC9a-Syk signals do not induce DC activation but instead shunt antigens expressed within necrotic cells into the cross-presentation pathway to generate immunogenic peptide–MHC class I complexes (106). The immunostimulatory, rather than tolerogenic, capacity of CD8α+ DCs that have received antigen in a CLEC9a-dependent manner is due to the “adjuvant” effect of necrotic cells, which do not exist during steady state. Mincle, another receptor for necrotic cells, has an obviously proinflammatory function (107), further demonstrating the atypical and immune-excitable influence of the presence of necrotic cells. It is unknown how DCs with CLEC9a-necrotic cargo antigens are made immunogenic in the absence of clear signs of maturation, what molecular differences distinguish CLEC9a–necrotic cell–matured DCs from conventionally matured DCs, and whether misregulation of this unique type of DC-acquired immunogenicity may disrupt immune homeostasis.
C-Type Lectin Receptor Dendritic Cell Immunoreceptor-1
DCIR1 is the only CLR whose genetic deletion in mice induces spontaneous disease and disruption of immune homeostasis (108). DCIR1 is one of four mouse homologs of human DCIR. DCIR1 contains an ITIM and is proposed to deliver negative regulatory signals via SHP activation, though DCIR1 ligands and DCIR1 signaling properties have not been characterized. By 4–6 months of age, DCIR1-deficient mice develop IgM rheumatoid factor and antinuclear antibodies. Arthritis develops in ~30% of mice. Curiously, disease incidence does not correlate with detectable disruptions to immune cell populations. Only at 12 months of age, long after the onset of disease, do DCIR1-deficient mice contain a modestly increased number of CD11c+ cells. The DC subsets that may expand in DCIR1-deficient mice have not been characterized. The number of activated CD4+ T cells also increases, consistent with a role for human DCIR in T cell functions during inflammatory and arthritic diseases (109, 110). DCIR1-deficient DCs do not intrinsically possess an enhanced ability to activate T cells, but DCIR1-deficient mice are more susceptible to collagen-induced arthritis, a lymphocyte-dependent model of rheumatoid arthritis induced by immunization with type II collagen/Freund’s complete adjuvant.
Which activating signals are suppressed by DCIR1, and what are the mechanisms of disease in DCIR1-deficient mice? Granulocyte-macrophage colony-stimulating factor (GM-CSF)-STAT5 signals are enhanced in DCIR1-deficient cells, presumably as a result of a lack of DCIR1-SHP negative cross-regulation. Thus, enhanced GM-CSF-STAT5 signals likely confer susceptibility for DCIR1-deficient mice to rheumatoid and collagen-induced arthritis, in which GM-CSF is pathogenic (111–115). Whether exaggerated GM-CSF-STAT5 signals can drive DC expansion in DCIR1-deficient mice is questionable because GM-CSF and its receptor can be genetically deleted with little consequence to lymphoid organ DCs during steady-state and inflammatory conditions (116, 117). Because these studies were reported in F1 generations of 129 and C57BL/6 mice, the role of DCIR1 in steady-state DC functions and the molecular mechanisms by which DCIR restricts GM-CSF-STAT5 signals may be understood via analysis on a pure genetic background, cell-specific DCIR1 deletion, or deletion of GM-CSF. In human DCs, DCIR inhibits TLR-induced cytokine production (118, 119). TLR signals may also play a role in driving disease in DCIR1-deficient mice, yet cross-regulation of TLR signals by mouse DCIR1 has not been described. SHP-1 deficiency compromises phagosome/lysosome biogenesis, and the absence of DCIR1-SHP endocytic/cross-presentation function may abrogate DC-mediated T cell tolerance (120). Immune tolerance strictly dependent on CLR-mediated antigen presentation is an intriguing notion, but it has yet to be reported. DCIR1 and DCIR2 have seemingly identical mechanisms of endocytosis/cross-presentation, but deletion of DCIR2, which also contains an ITIM, is not associated with immune pathology (80, 81). These potential mechanisms of disease add intrigue to our understanding of the identity of DCIR1 ligands, as the steady-state functions of these ligands may confer protection from autoimmune and arthritic disease (121)—an exciting prospect that may yield disease-modulatory treatments with therapeutic value.
Nod-Like and Rig-I-Like Receptors
NLRs and RLRs are cytoplasmic receptors that use their caspase recruitment domains to activate NF-κB or inflammasome processing of IL-1β and IL-18 (18, 25, 122, 123). These signaling pathways are independent of MyD88, but as with TLR signals, ubiquitin conjugation plays a key regulatory role in signal transduction. No role for steady-state RLRs or immune pathology due to hyperresponsiveness of DCs to RLR signals has been described, and the most dominant influence of these receptors may be found in antiviral immunity rather than steady-state homeostasis. In contrast, NLR mutations and hyperactive signals are associated with multiple autoimmune and inflammatory diseases in humans (reviewed in 124). The exact role of DCs in NLR-associated diseases has not been described, but indiscriminant inflammasome activation in DCs could severely impact immune homeostasis (122, 125). Nod2, which recognizes bacterial peptidoglycans, regulates some aspects of DC biology, although the most prominent role for Nod2 seems to be the regulation of Paneth cell functions (126). The ligand binding domain of Nod2 is frequently mutated in patients with Crohn’s disease, a chronic inflammatory bowel disease that may be driven by improper immune responses to intestinal commensals (127). Knockin mice expressing a Nod2 variant associated with Crohn’s disease demonstrate gain-of-function and increased inflammatory cytokine production that confer increased susceptibility to DSS colitis (128). Although this result suggests that the hyperresponsiveness of Nod2 signals drives disease, Nod2-deficient mice, which lack Nod2 signals, are highly susceptible to colitis, aberrantly respond to commensals, and have increased intestinal bacteria (68, 129–132). These findings instead suggest that Nod2 ligands have a protective function in intestinal homeostasis. The nature of Nod2 variants associated with Crohn’s disease and how these variants drive disease remain unclear. Nonetheless, DC-intrinsic disruptions are associated with Nod2 function. Monocyte-derived DCs from Crohn’s disease patients expressing Nod2 variants are inefficient at autophagy-dependent bacterial killing and antigen processing, a feature that may contribute to intestinal inflammation (133). In graft-versus-host disease (GvHD), Nod2-deficient DCs demonstrate enhanced phenotypic and functional maturity (134), suggesting that, in some settings, Nod2 may negatively regulate DC maturation, an attribute that may contribute to the increased susceptibility of patients with Nod2 genetic polymorphisms to GvHD following HSC transplant (135, 136). Although the mechanism remains unclear, these findings suggest a potential role for misregulated Nod2 signals in DCs in the pathogenesis of Crohn’s disease and perturbation of immune homeostasis.
DENDRITIC CELL LIFE SPAN AND IMMUNE CELL POPULATIONS
One early finding linking steady-state DC biology with immune homeostasis was that the life span of DCs, i.e., their programmed cell death at the appropriate time, is necessary to prevent autoimmunity. DCs whose life spans are extended via transgenic overexpression of antiapoptotic genes (137), deficiency in proapoptotic Bim (37), or a lack of responsiveness to Fas-induced death signals (138) are linked to the production of autoantibodies and spontaneous T cell activation. These disruptions all increase DC life span and DC numbers, but only Fas deficiency also leads to an increase in DC phenotypic maturation. Conversely, deficiency in prosurvival factors Bcl2 or Akt limits the life span and immunogenicity of DCs (36, 139).
DCs are not required to maintain steady-state populations of conventional T or B cells (140, 141), but they are essential to support steady-state populations of natural killer cells (142), CD8+ memory T cell subsets (142), and Foxp3+ Tregs (the latter are discussed below). DCs also indirectly control myeloid cell populations, which undergo cellular expansion in the absence of DCs, owing to an increased abundance of fms-like tyrosine kinase receptor-3 ligand (FLT3-L) growth factor, which is normally consumed by DCs (140, 141). Inflammatory cytokines produced by DCs can also regulate myeloid populations (63, 143).
DCs contribute to thymic selection (144–146), but the two independent reports of mice in which DC populations have been ablated produced disparate conclusions regarding whether DCs are absolutely required for efficient negative selection and removal of autoreactive T cells. Whereas Birnberg et al. (140) described normal negative selection, Ohnmacht et al. (141) reported increased frequencies of single-positive CD4 T cells and impaired negative selection. Mice in both studies were void of cDCs, but differed in ablation of pDCs, which persisted in the report by Birnberg et al. (140). Recent documentation of steady-state thymic immigration of pDCs and pDC-mediated negative selection against peripheral antigens may explain the discrepant findings in reports of DC-ablated mice (147). The two studies utilized similar CRE-dependent strategies of DC-specific ablation, and the cause for disparate effects on pDC ablation may be the copy number of CRE transgenes in mice from either report. Ablation of both DC populations is likely required to compromise negative selection fully. By 8 weeks of age, mice lacking both cDCs and pDCs have massive cellular infiltrates into multiple organs and develop fatal autoimmunity (141). Open questions include the makeup of the antigenic repertoire expressed by cDCs and pDCs and whether a division of labor exists between DC subsets in thymic negative selection (148).
DENDRITIC CELL EXPRESSION OF COSTIMULATORY MOLECULES
Most DCs maintain a low level of costimulatory molecules, and DCs are generally considered to be phenotypically and functionally immature during steady state (9, 149, 150). Even though DCs with increased expression of maturation markers can be coincident with human and mouse autoimmune diseases, upregulation of T cell costimulatory molecules on DCs may be insufficient to disrupt immune homeostasis. Moreover, unlike PRR- or CD40-activation signals that induce expression of maturation markers in a concerted yet simultaneous fashion, DCs that appear spontaneously mature at steady state are usually found to have upregulated some, but not all, costimulatory molecules (151–153). Additionally, immune disruptions and diseases can occur without obvious changes in DC maturation, such as in mice bearing DCs with an exaggerated life span. Nonetheless, because T cells are exquisitely sensitive to costimulation, high expression of these molecules on DCs may drive aberrant T cell activation and, in the right context, also disrupt T cell tolerance. This has been best described for CD70: Transgenic overexpression of CD70 on DCs results in CD27-dependent T cell activation (154). However, T cell expansion by CD70-overexpressing DCs remains antigen dependent, and instead of spontaneous T cell activation culminating in autoimmune disease, afflicted mice progressively develop lymphopenia and immunodeficiency, likely because T cells undergo cell death in the absence of proper antigenic or inflammatory stimuli. Thus, because DC phenotypic maturation is not always consistent with immunostimulation (16, 155), upregulated expression of costimulatory molecules by DCs during steady state most likely must be accompanied by other gains or losses of function within DCs to drive autoimmune or inflammatory disease.
TRANSCRIPTION FACTORS IN DENDRITIC CELL–MEDIATED CONTROL OF IMMUNE HOMEOSTASIS
Cytokines, growth factors, and transcription factors that support DC development have been the topics of several excellent recent reviews (156–159). Some pathways such as FLT3-STAT3 and a more modest contribution from GM-CSF-STAT5 are universally required for the development of cDCs and pDCs. Although GM-CSF has been proposed to drive inflammation-dependent monocyte conversion into DCs (inflammatory DCs) (160), recent reports (117) indicate that inflammatory DCs instead require macrophage-colony stimulating factor, and as with steady-state DCs, GM-CSF is dispensable for their development and function. These reports call into question the true nature of GM-CSF-cultured, bone marrow–derived dendritic cells (BMDCs) and whether the biology of these cells genuinely reflects that of DC populations in vivo. FLT3-L bone marrow culture yields cDCs and pDCs, and because cellular responses differ between FLT3-L and GM-CSF cultures, use of FLT3-L may be a more appropriate approach for in vitro analysis of DC functions.
In contrast to lymphoid organ DCs, the development of many tissue-specific DC subsets is FLT-3 independent. These include Langerhans cells of the skin, which depend on TGF-β and GM-CSF, as well as intestinal LPDCs, which have diverse hematopoietic origins (157, 161, 161). Other transcription factors and their signaling modules align with specific DC subsets. Included in this latter category are E2-2, which drives pDC development (162); RelB, which drives CD8α− cDC development (163); and Batf3, which drives CD8α+ cDCs (164). Among all transcription factors currently identified, only Batf3 seems to have exclusive expression and function in DCs. This specificity may reflect the unique abilities of CD8α+ cDCs to cross-present extracellular antigens. Separate from their role in driving DC development, and as predicted given the response-ready nature of DCs to activating stimuli, transcription factors such as those of the NF-κB family also drive the expression of maturation markers and functional responses of DCs (165–168). Below, we discuss recent findings regarding DC-induced immunological disruptions triggered by deficiencies in transcription factors (Figure 1).
Figure 1.

Transcription factors Foxo3, T-bet, and NF-κB1 in dendritic cell (DC)–mediated control of immune homeostasis: upstream signals that control transcription factor activation and the resulting downstream transcription factor–mediated suppression and/or induction of DC responses. Foxo3 activation in DCs is induced by CTLA-4-B7 signals and possibly also by GM-CSF, metabolic or oxidative stress, and commensals (excluding TLR signals). Foxo3 activation includes post-translational modifications such as phosphorylation and acetylation, which induces its nuclear translocation and transcriptional functions. T-bet expression in DCs may be induced or regulated by commensals. Depicted are the functions of T-bet in DCs from T-bet−/−/RAG2−/− mice. The Cdcs1 locus regulates colitis susceptibility of T-bet−/−/RAG2−/− mice on a BALB/c or C57BL/6 genetic background. Whether T-bet directly regulates this locus is unknown. In the cytoplasm, full-length NF-κB1 (p105) enhances MAPK activation and sequesters other NF-κB subunits to prevent their activation. Proteolysis of p105 into p50 form is induced by IL-10, TLR signals, and potentially commensals. p50 homodimers are anti-inflammatory: They suppress TLR-induced inflammatory cytokines and also induce transcription of the anti-inflammatory cytokine IL-10. TLR signals also induce the activation of other NF-κB subunits (c-Rel, RelA, and RelB), which, when heterodimerized with p50, drive inflammatory cytokine gene transcription. pDCs are more dependent on p50 heterodimers for maturation and cytokine production than are cDCs or BMDCs. The relative contribution of p105, p50 homo- and heterodimers to the reduced cell number, aberrant functions, and increased potential of NF-κB1-deficient DCs to induce CD8+ T cell autoimmunity is unclear. (Abbreviations: Cdcs1, cytokine deficiency-induced colitis susceptibility-1; CTLA-4, cytotoxic T lymphocyte antigen 4; GM-CSF, granulocyte-macrophage colony-stimulating factor; IκBα, nuclear factor of kappa-light polypeptide gene enhancer in B cells inhibitor, alpha; TLR, Toll-like receptor; MAPK, mitogen-activated protein kinase.)
Transcription Factor Foxo3
Family members of the class O of forkhead box transcription factors (Foxo) regulate multiple cellular processes such as cell cycle arrest, gluconeogenesis, and oxidative stress responses (169, 170). During steady state, global Foxo3 deficiency (Foxo3−/−) results in twofold cellular expansion of splenic cDCs and pDCs and modest overexpression of CD80/86 (171). Although these DC perturbations have no consequence on T cell homeostasis, interrogation of the immunostimulatory capacity of DCs from Foxo3−/− mice revealed that T cell survival following antigen-dependent activation was enhanced owing to exaggerated IL-6 production by Foxo3−/− DCs. Foxo3 restriction of IL-6 (and to a lesser extent TNF-α and CCL2) in DCs may explain the increase in myeloid cells in Foxo3−/− mice, which, unlike DCs, do not seem to possess enhanced cytokine production in the absence of Foxo3. Foxo3 negative regulation of DC cytokine production may be part of an immunoregulatory network between B7 molecules (CD80/86) expressed on mature DCs and cytotoxic T lymphocyte antigen-4 (CTLA-4) expressed by T cells (172) (Figure 1). CTLA-4-immunoglobulin (CTLA-4-Ig) cross links B7 molecules on DCs, induces Foxo3 nuclear translocation, and inhibits splenic DC responses to TLR ligands (173). Negative regulation of CTLA-4-B7 signals on TLR responses is null in Foxo3-deficient DCs; this deficiency may severely compromise immune homeostasis in Foxo3−/− mice on a 129 genetic background (171, 174, 175). Foxo3−/− 129 mice suffer spontaneous T cell activation and lymphoproliferative disease not observed in Foxo3−/− C57BL/6 mice (176). However, in 129 mice, Foxo3-deficient T cells have altered cellular responses not found in Foxo3-deficient T cells from the C57BL/6 background. Whether the absence of CTLA-4-B7 suppression drives DC expansion in Foxo3−/− mice or whether T cells are required for DC expansion is unknown.
Insulin and oxidative stress signals activate Foxo proteins in other hematopoietic cells, but the nature of the signals that drive Foxo3 activation or deactivation in DCs remains undefined (169, 177). Interestingly, Foxo3 is not activated by TLR signals (171), suggesting that Foxo3-inducing stimuli serve to antagonize steady-state DC maturation, the suppression of which is overridden upon encounter with TLR or other activating signals. Unlike splenic DCs, Foxo3 is constitutively activated in GM-CSF-cultured BMDCs, possibly induced by GM-CSF (171, 178), implicating GM-CSF as a candidate for Foxo3 activation in vivo. In this context, Foxo3 activation in DCs may correlate with disease states driven by pathological GM-CSF. How cellular stress, GM-CSF, or T cell–derived signals regulate steady-state DC functions in a Foxo3-dependent or -independent manner presents intriguing questions.
Transcription Factor NF-κB1
Analogous to Foxo3 function, the NF-κB family member NF-κB1 (also known as p105) also restricts DC production of TNF-α (179, 180). In a tolerance model of rat insulin promoter–based antigen (RIP-antigen), adoptively transferred, antigenic peptide–pulsed NF-κB1-deficient, GM-CSF-cultured BMDCs activated autoreactive CD8+ T cells and induced diabetes. Nf-κb1−/− mice are healthy and do not develop diabetes when crossed to the RIP-antigen model of tolerance (Nf-κb1−/−/RIP-antigen). However, increasing the number of autoreactive CD8+ T cells in RIP-antigen-tolerant Nf-κb1−/− mice resulted in diabetic symptoms in ~50% of mice. Here and in the case of adoptively transferred NF-κB1-deficient BMDCs, disease was dependent on TNF-α. Immune tolerance may be particularly susceptible to the influence of adoptively transferred GM-CSF-cultured BMDCs, as these cells can induce antigen-dependent and -independent autoimmune and inflammatory diseases even in wild-type mice (181–184). Thus, the absence of spontaneous disease in Nf-κb1−/−/RIP-antigen mice likely reflects differences in steady-state DC and GM-CSF-cultured BMDC functions, cellular responses, and receptivity to suppressive signals. Additionally, during in vitro culture, BMDCs process bovine proteins from fetal calf serum into peptide-MHC class II complexes. Upon adoptive transfer of BMDCs into mouse recipients, these foreign peptide-MHC class II complexes activate CD4+ T cells, thus adding key “helper” CD4+ T cell products that aid the initiation and disease pathogenesis of CD8+ T cell autoimmunity (185, 186). What, then, is the constellation of changes in NF-κB1-deficient DCs that disrupts CD8+ T cell tolerance? Steady-state phenotypic maturation is not enhanced in NF-κB1-deficient DCs (187, 188), but the maturation of endogenous DCs in Nf-κb1−/−/RIP-antigen mice with and without diabetes has not been assessed. In particular, analysis of DCs of the pancreas and pancreatic lymph nodes, sites where RIP-antigen is presented and autoreactive immune responses are amplified (189), is required to determine whether overproduction of TNF-α is the only aspect of DC maturation restricted by NF-κB1 and whether TNF-α overproduction alone can disrupt peripheral CD8+ T cell tolerance to RIP-antigen or other autoantigens.
How NF-κB1 suppresses TNF-α production or functional maturation of DCs is unclear. Full-length NF-κB1/p105 can retain other NF-κB family members in the cytoplasm and can also regulate MAPK activation (190, 191) (Figure 1). Thus, overproduction of TNF-α in NF-κB1-deficient DCs may result from loss of suppressive cytoplasmic NF-κB1 functions. Proteolysis of NF-κB1/p105 into its active, DNA-binding p50 form can either activate or suppress gene transcription, depending on whether p50 heterodimerizes with other NF-κB members or forms p50 homodimers, the latter of which can suppress gene transcription of inflammatory cytokines (192–194). Conversely, p50/NF-κB heterodimers activate inflammatory gene transcription. Consistent with this function, NF-κB1 is critical for immune responses and the maturation and steady-state homeostasis of pDCs (187, 195). Whether defective pDC functions contribute to disease in Nf-κb1−/−/RIP-antigen mice remains unclear.
By contrast, cDCs are seemingly less dependent on NF-κB1. Maturation of NF-κB1-deficient cDCs is neither compromised nor globally enhanced, but it is associated with specific defects in priming of Th2 T cell responses in addition to overproduction of TNF-α (180, 188). The steady-state signals sensed by DCs that induce p50/NF-κB heterodimers or are negatively regulated by p105 or p50 homodimers are currently undefined. However, they may include TLR signals because TLR ligand–matured DCs can disrupt tolerance and drive diabetic disease in similar models of RIP-antigen tolerance (186). Furthermore, upon overt stimulation of macrophages with TLR ligands, p50 homodimers suppress transcription of inflammatory cytokines, and induce transcription of anti-inflammatory IL-10 and NF-κB1 is upregulated in endotoxin-tolerant macrophages, suggesting a similar suppressive function of NF-κB1 in steady-state TLR signals in DCs (179, 194, 196). Additionally, p50 homodimers support IL-10-mediated suppression of human monocyte responses to TLR ligands (197). In DCs, however, it remains to be determined whether NF-κB1, in p105 or p50 form, acts as a negative regulator of steady-state maturation signals or whether it acts as a downstream effector of steady-state suppressive signals such as IL-10. Of further interest in this context is determining whether NF-κB1 is also required to induce nonresponsiveness of intestinal DCs to TLR4 agonists.
Transcription Factor T-Bet
One of the most striking findings pertaining to transcription factor–mediated control of DC functions relates to T-bet-dependent control of DC maintenance of intestinal tolerance. Examining T-bet−/−/RAG2−/− mice in this context, a series of reports by Garrett et al. (198–200) describe a novel function of T-bet (a transcription factor most noted for its role in T cell effector differentiation). Intestinal commensals trigger T-bet-deficient DCs to overexpress TNF-α and drive spontaneous ulcerative colitis with progressive development to colorectal cancer. Restoration of T-bet expression in DCs, antibody-mediated blockade of TNF-α, antibiotic depletion of commensals, or adoptive transfer of Tregs all effectively abrogate colitis and colorectal cancer in T-bet−/−/RAG2−/− mice. Whether disease or T-bet deficiency is also associated with DC expansion or phenotypic maturation has not been reported. Although commensal dependent, the intracellular signaling cascades triggered upon commensal recognition and the corresponding transcriptional program normally suppressed/modulated by T-bet (other than TNF-α) have not been defined. MyD88-dependent signals are reportedly not upstream of T-bet−/−/RAG2−/− DC-driven colitis; however, the poor survival and immunological paucity of mice lacking both MyD88 and lymphocytes obscure this conclusion (64, 199). Nevertheless, commensal sampling and signals in DCs that lead to colitis can be either MyD88-dependent or -independent, so T-bet may preferentially regulate transcriptional programs initiated by signals from the latter (69, 201–203). T-bet−/−/RAG2−/− DC-driven colitis is most severe in the BALB/c genetic background compared with that of C57BL/6, as a result of the congenic differences in the Cdcs1 locus (cytokine deficiency–induced colitis susceptibility-1), a largely uncharacterized disease-susceptibility region that also plays a role in colitis associated with IL-10 deficiency (204, 205). Further analysis of Cdcs1 gene products, the signaling pathways that induce transcription of the Cdcs1 locus in intestinal DCs, and whether T-bet regulates this locus should provide insight into the colitogenic responses of T-bet-deficient DCs.
Surprisingly, the regulatory network set into motion by T-bet control of DC function shapes the intestinal microbiota and maintains commensal balance such that, in the absence of T-bet, the composition of intestinal microbiota becomes colitogenic and induces disease when transferred into wild-type mice (198, 200). How T-bet-deficient DCs foster the outgrowth of colitogenic commensals is unclear. Whether T-bet functions as a positive regulator of DC responses to beneficial commensals or whether it is required to suppress inflammatory and growth-fostering responses to colitogenic commensals requires further investigation. T-bet function in DCs is a particularly intriguing aspect of DC immunobiology because of the relationship between T-bet-mediated suppression of DC-produced TNF-α and TNF-α-dependent progression of colitis and because human patients suffering inflammatory bowel disease are often successfully treated with anti-TNF-α therapy.
Perspectives in Transcription Factor–Dependent Control of Dendritic Cells
A useful resource for future studies would be a detailed analysis of transcriptomic changes in DCs that result from a deficiency in the transcription factors described above. Consideration of the illustrations described above, all of which note upregulation of DC production of TNF-α, makes clear that overproduction of cytokines by DCs does not yield overlapping immunological disruptions. This aberration of functional maturation most likely must be accompanied by other phenotypic or functional changes. Especially because the steady-state signals upstream of these transcription factors have not been characterized, transcriptomic analysis of DCs isolated from disease-relevant tissues are needed to provide significant mechanistic insight into how the misregulated responses of DCs can induce autoimmune and inflammatory diseases. Such analysis may also reveal novel mechanisms of DC-mediated control of immune homeostasis.
MHC CLASS II MOLECULES AND DENDRITIC CELL FUNCTIONS
MHC class II expression by DCs has many unique biological properties that confer specialized antigen-presenting functions. Notably, immature DCs maintain a large store of intracellular MHC class II and peptide-loading chaperones [e.g., H2-M, invariant chain (Ii)] as well as antigen-processing enzymes in atypical vesicles with features characteristic of late endosomes/early lysosomes (5–7, 206). Upon DC activation, these vesicles are acidified, internalized antigens are proteolyzed, and peptide-loaded MHC class II molecules are exported to the cell surface. The outstanding features of DC–MHC class II biology has inspired several questions: How and for what purpose are MHC class II molecules maintained in intracellular vesicles? Which molecules and mechanisms couple DC activation and cell surface MHC II expression? Below, we highlight current findings that shed light on these questions of DC–MHC class II biology, with a focus on those that result in disrupted DC functions in vivo. Collectively, several lines of research indicate that the link between the unique features of DC–MHC class II expression and DC functions in innate and adaptive immunity is tighter than previously expected.
Invariant Chain and Dendritic Cell Migration
The abilities of DCs to migrate within tissues and to traffic into lymphoid organs enable their sentinel functions and initiate adaptive immune responses. Faure-André and colleagues (207) recently noted that DC migration is regulated by Ii, a key intracellular chaperone of MHC class II molecules that directs MHC class II endosomal localization and enables peptide loading (208). Adoptively transferred Ii-deficient DCs migrate to draining lymph nodes more quickly than do wild-type DCs. Likewise, in microchannels, which assess the movement of cells in constricting environments such as those found in tissues, the velocity of Ii-deficient DCs was enhanced and more constant, a marked difference from the slow-down phases and velocity variations of wild-type DCs. Maturation slows motile velocity and increases the frequency of directional changes. This functional component of DC differentiation is Ii dependent and is regulated by Ii-mediated association and activation of myosin light chain, which mediates cell migration and may enable DCs to effectively probe the tissue microenvironment (209). Positive regulation of this antigen-processing chaperone and of cellular motility—a seemingly distant aspect of DC biology—suggests that the specialized function of intracellular retention and peptide loading of MHC class II molecules in DCs is intimately and perhaps evolutionarily linked to DC sentinel function.
MHC Class II Molecules, MARCH1 Ubiquitination, and Dendritic Cell Responses
The intracellular retention of MHC class II molecules prior to activation is a defining feature of DC biology, and researchers have long speculated on how immunity and immune tolerance would be impacted should this feature be lost. The conjugation of ubiquitin chains to a single lysine residue present in MHC class II β-chain is a newly appreciated mechanism by which MHC class II molecules remain intracellular in immature DCs (210–213). Upon DC activation, ubiquitinated MHC class II species are no longer detected and MHC class II molecules accumulate at the cell surface, demonstrating that deubiquitination or the cessation of ubiquitin conjugation to MHC class II molecules is coupled to DC maturation (210, 213). The ubiquitin-conjugating molecule that confers this coupling function is MARCH1, a member of the membrane-associated RING-CH (MARCH) family of E3-ubiquitin ligases (214). MARCH1 is highly expressed in immature DCs, associates with intracellular MHC class II molecules, and is downregulated upon DC activation, characteristics that well match the profile of MHC class II ubiquitination (212, 215, 216). MARCH1-mediated ubiquitination also regulates CD86 expression. Thus, MARCH1-deficient cDCs and BMDCs express tenfold higher levels of MHC class II molecules and CD86 (but not CD80) (217, 218). Spontaneous overexpression of these molecules in MARCH1-deficient mice does not appear to affect immune homeostasis or immune tolerance (211, 217). However, functional analysis of DCs in MARCH1-deficient mice revealed an unexpected regulatory network involving MARCH1, steady-state MHC class II expression, and DC functions. Surprisingly, MARCH1-deficient DCs have significantly attenuated cytokine responses to LPS and CD40 and diminished capacity to present MHC class II antigens (217). These defects are contingent on MHC class II overexpression; that is, deletion of MHC class II molecules from MARCH1-deficient DCs rescues their functional impairment, suggesting that DC functional maturation is made impotent by the lack of ubiquitinated MHC class II molecules and their spontaneous overexpression at the cell surface. MHC class II–impotency feedback does not exist in MARCH1-deficient B cells (although MARCH1-deficient B cells also overexpress MHC class II molecules) (211), pDCs (95), or FLT3-L-cultured BMDCs (217), indicating that unique in vivo requirements dictate MHC class II–impotency feedback on cDC functions. The mechanism of this feedback could involve cell-intrinsic or -extrinsic suppression of inappropriate MHC class IIhigh DCs, perhaps transduced by MHC class II molecules (219, 220). cDCs of MARCH1-deficient mice may be analogous to hypothetical “exhausted” DCs (221–223) or to endotoxin tolerant macrophages, which are unresponsive to endotoxin stimulation partly as a result of altered NF-κB activation, chromatin modifications, and upregulated expression of negative regulators of signal transduction (179, 224, 225). Alternatively, the intracellular localization of ubiquitinated MHC class II species may positively regulate TLR and CD40 signals. Because MHC class II–impotency feedback is specific to MARCH1 deficiency and does not universally occur in DCs that spontaneously overexpress MHC class II molecules, the mechanism may involve other molecular targets for MARCH1 ubiquitination. Signaling cascades, corresponding ubiquitin modifications of signaling molecules, and expression of negative regulators in MARCH1-deficient versus MARCH1-MHC class II double-deficient DCs have not been described. Future studies are required to determine how MARCH1-mediated maintenance of intracellular MHC class II molecules and DC immaturity drives differentiation into functional maturity.
MHC Class II, Btk, and Toll-Like Receptor Signals
The most unexpected relationship yet reported between intracellular MHC class II molecules and DC functions may be the positive regulation of MHC class II dimers on the transduction of TLR signals. Liu et al. (226) noted that MHC class II–deficient DCs and macrophages have reduced NF-κB, MAPK, and IRF activation as well as reduced cytokine production downstream of MyD88 and TRIF signals. The mechanism by which MHC class II molecules support TLR signals requires indirect association of intracellular MHC class II molecules (but not MHC class II molecules at the cell surface) with the tyrosine kinase Btk. Btk becomes phosphorylated upon TLR stimulation, binds MyD88 and TRIF, and enhances but is not essential for the transduction of TLR signals (226–228). Unexpectedly, the direct binding partner of Btk is CD40. Upon TLR stimulation, Btk–CD40–MHC class II complexes colocalize inside the cell and can be coimmunoprecipitated. Unlike MHC class II molecules, CD40 is not presynthesized and retained intracellularly, but its expression is induced by maturation signals. Thus, MHC class II molecules, CD40, and Btk may engage MyD88 and TRIF signaling complexes while en route to the cell surface. The absence of intracellular MHC class II–Btk signaling events may also contribute to the impotency observed in MARCH1-deficient cDCs. However, defects in Btk-driven TLR signals are apparent in BMDCs, whereas those of MARCH1-deficient DCs are not. Description of MHC class II–Btk signaling events in maturing DCs undergoing vesicular remodeling and export of peptide–MHC class II molecules is required to determine whether MHC class II–mediated amplification of TLR signals affects DC function in vivo.
DENDRITIC CELLS AND STEADY-STATE T REGULATORY CELLS
Foxp3+ Tregs suppress proliferation and cellular responses of immune cells and are essential for immune tolerance and homeostasis (229). In the absence of Tregs or when their functions are compromised, a number of autoimmune and inflammatory disorders can arise (230). Akin to how DC functions control the polarization of T cell effectors, DCs can also generate, expand, or maintain Tregs. During steady state, the number of DCs in spleen and lymph node is linked to the number of Tregs (231). In vivo expansion of DCs with exogenous FLT3-L causes increased Treg homeostatic proliferation and cellular expansion. Conversely, depletion of DCs reduces both the number of Tregs and their homeostatic proliferation. Although DC-ablated mice were initially reported to have normal Foxp3+ populations, it was later determined that CD25 expression on Tregs was dramatically reduced, indicative of reduced Treg function (232). Interestingly, MHC class II and CD80/86 expression by DCs is required to maintain peripheral Treg populations and their relative expression of Foxp3 (231, 232). Langerhans cells perform analogous functions in human skin (233). DC metabolism of vitamin A may play a key role in intestinal Treg and effector T cell populations, but these metabolic functions may not be required to maintain intestinal Treg populations (234, 235). The connections between the number of DCs, MHC class II and CD80/86 expression, and Tregs appear to be of an all-or-none character, meaning that only when these molecules are entirely absent on DCs do Treg populations suffer. Accordingly, in mice harboring DCs with exaggerated levels of costimulatory molecules, a proportional or preferential increase in Treg populations has not been observed. However, during inflammatory conditions such as in some mice bearing spontaneously mature DCs, Tregs may become less dependent on DCs for their homeostasis owing to an increased abundance of survival factors such as IL-2. Below, we discuss DC-specific disruptions to immune homeostasis resulting from an impaired ability to generate/maintain Treg populations.
Dendritic Cells, α vβ8, and TGF-β Activation
TGF-β, a key molecule required for Treg homeostasis, is expressed in a latent form that requires activation to achieve functionality (236, 237). Travis et al. (238) demonstrated that αvβ8 integrin expressed by DCs activates latent TGF-β and is critical for immune homeostasis. The pathologies of mice with DCs lacking αvβ8 and those of mice lacking αvβ8 on all immune cells overlap: They suffer splenomegaly, lymph-adenopathy, spontaneous T cell activation, autoantibodies, and colitis. DC-specific loss of αvβ8 did not result in global loss of Treg populations, but it was key to generate/maintain intestinal Treg populations. Among intestinal DC subsets, αvβ8 is most highly expressed by CD103+ DCs, which have an enhanced functional ability to generate Tregs, partially owing to their increased ability to activate latent TGF-β (239–241). TGF-β regulates the development, survival, and expansion of CD4 and CD8 T cell effectors. Thus, autoimmunity in mice with αvβ8-deficient DCs is likely also due to the dependence of non-Treg lymphocytes on DC-αvβ8-TGF-β activation as a physiological source of TGF-β (236, 242, 243).
What is the source of latent TGF-β in vivo? DCs (as well as many other cell types, including T cells) can secrete the inactive TGF-β precursor. Therefore, TGF-β may not be a limiting factor in DC-mediated regulation of T cells. Additional studies further suggest that DC–MHC class II–T cell–TCR interactions are required if αvβ8-mediated TGF-β activation is to have the most potent effect on T cell polarization (244). Such findings suggest that, rather than having a systemic effect, DC-mediated activation of TGF-β, much like costimulation, promotes a more intimate relationship between DCs and T cells, likely to afford an additional layer of control to prevent unwanted immune activation and immunopathology.
Plasmacytoid Dendritic Cells, TGF-β, and IDO
Another TGF-β-mediated means by which DCs control Treg populations occurs via the catabolic enzyme indoleamine 2,3-dioxygenase 1 (IDO1) (245). IDO family members catabolize tryptophan and regulate cellular responses by inducing tryptophan starvation and by generating biologically active tryptophan metabolites. Among DCs, IDO is most highly expressed by CD8α + cDCs and pDCs. Expression is upregulated by IFN-γ and TGF-β as well as by cross-linkage of B7 molecules (246). IDO expression correlates with downregulation of DC immunostimulatory functions in favor of immune tolerance. IDO is also implicated in the regulation of numerous immunological events, including antimicrobial immunity, autoimmunity, inflammation, and pregnancy. Although these regulatory functions have been ascribed to IDO catabolism, researchers (247) have reported a novel mechanism of TGF-β-mediated, IDO1-dependent regulation of pDCs that is independent of tryptophan metabolism. Pallotta et al. (247) reported that TGF-β, but not IFN-γ, induces phosphorylation of IDO1 ITIM domains, which then recruit and activate SHP-1 and SHP-2 phosphatases. Although these ITIM domains are conserved in IDO1 orthologs in other species, this was the first description of the functional role of IDO1 ITIMs. TGF-β-IDO1-SHP signals activate noncanonical NF-κB, which in turn upregulates pDC expression of IFN-α, TGF-β, and IDO1, thus establishing an IDO1 signaling amplification loop. IDO1 ITIM signaling, but not catabolism, was required for pDCs to induce T cell tolerance in an in vivo model of peptide sensitization (247). Tolerance required pDC upregulation of TGF-β and was associated with an increase in the number of Tregs and a corresponding decrease in IFN-γ-producing T cell effectors. These findings reinforce a relationship among TGF-β signals in pDCs, IDO1 activation, and immune tolerance via induction of Tregs.
Because pDCs are not universally immunosuppressive but in many cases stimulate immune responses, these findings suggest that TGF-β signals, both received and delivered by DCs, must be contextually regulated to coordinate tolerance or immune responses as appropriate. Reinforcing this notion is the observation that immune homeostasis is normal in IDO-deficient mice (248), suggesting that, unlike the dependence of Tregs on DCs as a source of active TGF-β, IDO-dependent DC functions are not required either for Treg generation/maintenance or for homeostasis of other immune cells. Thus, determining the immunological context in which the DC-IDO axis of tolerance is most pertinent requires further investigation.
β-Catenin Signals in Dendritic Cell Control of Intestinal Homeostasis
Wnt-signals, β-catenin activation/ transcription, DC functional responses, and Treg generation are connected via an intimate relationship (for this elegant description, see Reference 249). β-catenin and its activating receptors in the Wnt family form a conserved signaling module best recognized for regulating cellular differentiation, including that of HSCs. In vitro, β-catenin signals can induce DC activation and condition their functional ability to induce IL-10-producing CD4+ T cells (16). In vivo, β-catenin is constitutively active to varying degrees in most immune cell types, and among DC subsets, β-catenin activation is greatest in intestinal LPDCs, especially in the CD103+ subset (249). Levels of β-catenin activation in intestinal DCs correlate with an enhanced functional ability to generate Tregs. Conversely, in the absence of Wnt-β-catenin signals, LPDCs are significantly compromised in Treg generation. Thus, in mice lacking β-catenin specifically in DCs (β-catDC−/−), Treg populations in the intestine are decreased two- to fourfold. An intriguing consequence of this disability is the enhanced function of LPDCs to generate IFN-γ-producing and IL-17-producing CD4+ T cells; in the intestine, these inflammatory T cell effectors are increased nearly tenfold in β-catDC−/− mice. These disruptions were not observed in other immune organs, and no disease or spontaneous colitis was reported in β-catDC−/− mice. In comparison with other regulatory mechanisms of intestinal homeostasis such as IL-10 and TGF-β, the Wnt-β-catenin axis may be more selective in its governance of steady-state intestinal T cell populations, without systemic consequence to immune homeostasis.
Mechanistically, β-catenin signals in DCs upregulates TGF-β, IL-10, and genes pertaining to vitamin A metabolism. β-catenin concomitantly downregulates proinflammatory cytokines IL-6 and IL-23, the net effect of which enhances DC-mediated generation of Tregs and disfavors inflammatory T cell effectors. β-catenin may also support development or longevity of Treg-generating CD103+ DCs, akin to the relationship between Batf3 and the CD8α + DC subset in lymphoid organs. Wnt-β-catenin signals in the intestine may have a global anti-inflammatory effect on mononuclear phagocytes because intestinal macrophages of β-catDC−/− mice (a significant number of which were β-catenin-deficient due to off-target CRE recombination) displayed the same, and in some cases are even more dramatic, profile of skewed cytokine and metabolic enzyme expression as was found in intestinal DCs. Enhanced proinflammatory cytokine production by DCs and macrophages likely conveys susceptibility of β-catDC−/− mice to DSS colitis. However, alterations in these macrophages did not translate into significant changes in the ability of intestinal macrophages to generate Tregs or T cell effectors. Thus, β-catenin signals in DCs, but not macrophages, control their ability to regulate steady-state intestinal T cell homeostasis. Other molecules such as integrin αvβ8 or CD103 expression may reinforce the preferential influence of DCs on T cell homeostasis in the intestine (250). The steady-state intestinal architecture, including the ratio of Tregs to T cell effectors, may affect immune responses or increase the disease potential of incidental perturbations to intestinal homeostasis. It will be interesting to analyze the ability of β-catenin-deficient DCs to drive T cell–mediated colitis, such as is found in adoptive transfer of naive T cells into lymphocyte-deficient hosts or 2,4,6-trinitrobenzene sulfonic acid–mediated colitis.
What is the source of Wnt ligands? This question remains unanswered and has proven difficult to answer in most biological situations. Manicassamy et al. (249) reported that intestinal DCs as well as macrophages express various Wnt ligands and receptors. Although the expression levels of intestinal DCs exceeded those of splenic DCs, the authors did not draw conclusions regarding the source of Wnt ligands or the relevant receptors. Again, because macrophages and DCs differ in their dependence on β-catenin to control T cells, the levels of receptor or ligand expression do not seem to coincide with the unique regulatory functions of DCs. Additionally, antibiotic depletion of commensals does not alter β-catenin activation in DCs, indicating that commensals do not trigger Wnt-β-catenin signals in DCs. Identifying the source of Wnt ligands, as well as which ligands most affect DC regulation, may make novel therapies possible.
STEADY-STATE CYTOKINES, TOLL-LIKE RECEPTORS, AND THEIR SIGNALING CASCADES IN DENDRITIC CELL MATURATION
IL-6 and STAT3
The proinflammatory functions of IL-6 in innate and adaptive immunity have been well documented. At steady state, however, IL-6 has a suppressive function on DCs, and in Il6−/− mice, lymph node DCs spontaneously express increased levels of MHC class II and CD86 (251) (Figure 2). The gp130 component of the IL-6 receptor transduces signals via activation of SHP-2 and STAT3. Using mutant gp130 receptors deficient in their ability to activate one or the other pathway, Park et al. (251) demonstrated that STAT3-mediated transcription was required to suppress activation markers on DCs. Moreover, DCs can be categorized by the levels of cellular STAT3 phosphorylation/activation, the highest of which correlates with low expression of MHC class II. In the spleen, where DCs express modest levels of MHC class II, phospho-STAT3 levels are almost uniformly high. In contrast, in lymph nodes, where DC populations are more heterogeneous in their maturation state owing to the presence of migratory subsets, DCs can be segregated into high and low phospho-STAT3. The latter population coincides with epithelium-derived DC migrants. These differences in steady-state DC populations suggest that stimuli that induce DC activation/maturation or migration from tissues relieve DCs from IL-6-STAT3 suppression, which otherwise delivers tonic signals to suppress MHC class II and CD86 expression. Whether this is also true in pDCs, which in comparison to cDCs express significantly lower levels of MHC class II and T cell costimulatory molecules, is not known.
Figure 2.

Steady-state cytokines, TAM receptor ligands, TLRs, and their signaling cascades in dendritic cell (DC) maturation: suppression and/or induction of DC responses and negative feedback regulation by SOCS proteins. IL-6 and IL-10 signals both induce STAT3 activation/phosphorylation and upregulation of SOCS proteins. Steady-state IL-6-STAT3 suppresses maturation of lymph node and liver DCs, whereas steady-state IL-10-STAT3 induces a broad-acting, potent, anti-inflammatory state that is critical to prevent colitis. IL-6 signals are more susceptible to negative regulation by SOCS3 than are IL-10 signals. Commensals and TLR ligands are upstream of steady-state IL-6 and IL-10 signals. IFN-γ, type I IFN, and TAM receptors all induce STAT1 activation/phosphorylation and SOCS protein expression. TAM receptor–mediated STAT1 activation is dependent upon association with and “hijacking” of the IFNAR-STAT1 signaling module (linked circles). Traditional IFNAR-STAT1 signals induce a proinflammatory response in addition to SOCS1 and SOCS3, which provide negative feedback regulation of IFNAR signals. TAM-IFNAR-STAT1 selectively induces SOCS1 and SOCS3, which negatively regulate TLR signals. SOCS1-deficient DCs and TAM receptor–deficient DCs induce overlapping perturbations to immune homeostasis, suggesting that IFN-γ, which drives disease mediated by SOCS1-deficient DCs, may regulate TAM receptor or TAM ligand expression. TAM receptors also transduce inhibitory signals upon recognition of apoptotic cells; these signals suppress subsequent TLR-induced responses. TLR-induced responses upregulate TAM receptors and the TAM ligand Gas6, providing an intrinsic mechanism for negative feedback regulation of TLR signals in DCs. (Abbreviations: IFNAR, IFN-α receptor; IFNGR, IFN-γ receptor; IRAK-M, IL-1 receptor-associated kinase M; IRF, interferon regulatory factor; SOCS, suppressor of cytokine signaling; STAT, signal transducer and activator of transcription; TLR, Toll-like receptor.)
The IL-6-STAT3 axis may be particularly critical in liver DCs to suppress their activation to bacterial products that continuously arrive via the portal vein. Studies by Lunz et al. (252) indicate that steady-state STAT3 activation in liver cells exceeds that in other organs and that these levels are markedly reduced in the absence of IL-6. Similar to the findings of Park et al. (251), liver DCs were found to express higher levels of maturation markers in IL-6-deficient, versus wild-type, mice. Depletion of bacteria by oral antibiotics and consequent reduction in the amount of endotoxin in the portal vein significantly reduce IL-6 and phospho-STAT3 in the wild-type liver, indicating that commensal bacteria drive these signals in the liver. Interestingly, counter to what we understand about DC responses to bacterial products, commensal depletion enhances phenotypic maturation of liver DCs. Commensal depletion has no effect on phenotypic maturation of liver DCs in Il6−/− mice, thus highlighting a commensal-induced, IL-6-dependent regulatory network on DC phenotypic maturation. How commensals are sensed and whether IL-6 production in the liver is DC intrinsic or extrinsic are not known. Presumably, this network is required to minimize DC responses to the influx of bacterial products. In the intestine, where the heaviest burden of commensals reside, recent studies suggest that IL-6 signals in DCs may enable the discrimination of noncolitogenic and pathogenic bacteria; the latter generate more TNF-α than IL-6 and result in enhanced maturation and migration of LPDCs to mesenteric lymph nodes (253). However, DC expression of IL-6 can also drive pathogenic T cell expansion in the intestine (203). Thus, although steady-state IL-6 signals in DCs may suppress their maturation to maintain a balanced relationship with commensals, high IL-6 produced during inflammatory conditions or adaptive immune responses likely overrides this control and exerts pathological effects.
The molecular mechanism of IL-6-STAT3 suppression of DC maturation may be unique depending on the tissue of DC residence. For example, even though Park et al. (251) described increased phenotypic maturation of lymph node DCs in Il6−/− mice, splenic DCs from these mice manifested this difference only after LPS stimulation (251). In liver DCs, IL-6 signals increase DC expression of IRAK-M, a negative regulator in TLR signaling (252). One mechanism of action for IL-6 regulation of DCs in lymphoid tissues occurs in part via direct destabilization of intracellular H2-M, Ii, and MHC class II (254). Lymph node DCs in knockin mice bearing a mutated gp130 receptor that transduces exaggerated IL-6-STAT3 signals have reduced levels of intracellular MHC class II, confirming the mechanism by which steady-state IL-6-STAT3 signals suppress MHC class II expression in DCs.
IL-10 and STAT3
In deciphering the role of IL-6-STAT3 in regulating DC functions, we should consider the functions of IL-10 (Figure 2): Although it activates STAT3, the IL-10 receptor is independent of the IL-6 receptor gp130 that transduces negative regulatory signals (255). In vitro, pretreatment of BMDCs with either IL-6 or IL-10 downregulates their subsequent responses to LPS stimulation, but IL-10-STAT3 induces a far more potent and comprehensive anti-inflammatory state via transcriptional repression of inflammatory genes. Interestingly, IL-10-mediated suppression of MHC class II expression is largely dependent on IL-10-induced upregulation of MARCH1 (215, 218); whether IL-6 also increases MARCH1 levels is unknown. The critical roles of the anti-inflammatory functions of IL-10-STAT3 are readily apparent in Il10−/− mice and mice with cell type–specific deletion of STAT3, which all develop spontaneous inflammatory bowel disease and other inflammatory disruptions in mucosal tolerance that are not observed in Il6−/− mice (256).
Mice with DC-specific deletion of STAT3 develop colitis that resembles a less severe version of colitic disease in Il10−/− mice (257). With the exception of pDCs, CD11c expression and CRE-driven genetic deletion occurs after DC lineage commitment; thus, conditional STAT3 deletion does not interfere with the FLT3-STAT3 signals required for development. On a systemic level, whereas proinflammatory cytokines were increased, cDC numbers and their expression of maturation markers were normal. The composition and function of intestinal DC subsets, the integrity of which can compromise mucosal tolerance, have not been fully described in mice lacking STAT3 in DCs. However, T cell expansion and inflammation were restricted to areas involved in mucosal tolerance, such as the intestine, bronchials, and cervical lymph nodes. Nevertheless, how IL-6-STAT3 differs from IL-10-STAT3 is unclear. Because DCs lacking STAT3 are hyperinflammatory but not phenotypically mature, the predominant in vivo functions of the IL-10-STAT3 axis in DCs may be to suppress the production of cytokines and enable mucosal tolerance, whereas the IL-6-STAT3 axis suppresses MHC class II and CD86 expression.
IFN-γ, Toll-Like Receptors, and SOCS1
Suppressor of cytokine signaling-1 (SOCS1) is a key negative regulator of several cytokine signaling cascades (258). Suppression occurs by directly inhibiting JAK kinase function (mediated by the kinase inhibitory region) and also by directing proteins for proteasomal degradation (mediated by the SOCS box). SOCS1-deficient mice die within 3 weeks of birth, primarily as a result of rampant T cell activation and IFN-γ production. Using transgenes in Socs1−/− mice (SOCS1-Tg) to reintroduce SOCS1 expression in lymphocytes, Hanada et al. (259) described clear in vivo function for SOCS1-mediated negative regulation of DC maturation (Figure 2). Whereas SOCS1-deficient macrophages from these mice are phenotypically normal, DCs express a greater number of T cell costimulatory molecules and are numerically expanded in lymphoid organs. By 8 weeks of age, SOCS1-Tg mice develop a number of immunological disruptions, including T cell and B cell expansion and autoantibody production. In the absence of IFN-γ, these phenotypes are significantly delayed until 6 months of age, indicating that IFN-γ-STAT1 was a key trigger in these DC-driven immunological disruptions. DCs from SOCS1-Tg mice expressed exaggerated levels of BAFF and APRIL (key B cell survival and proliferation factors), and overproduction of these molecules by SOCS1-deficient DCs is the likely impetus behind the increase in germinal centers, glomerulonephritis, hypergammaglobulinemia, and autoantibody in SOCS1-Tg mice. Adoptive transfer of SOCS1-deficient BMDCs into wild-type mice also induces autoantibodies, thus highlighting the potent ability of SOCS1-deficient DCs to disrupt B cell homeostasis.
SOCS1-Tg mice also develop dermatitis, and a small percentage also develop arthritis, pathologies that suggest disruption of immune cells other than B cells. Although expanded, T cells in SOCS1-Tg mice were not hyperactivated in SOCS1-Tg mice. Nevertheless, multiple lines of evidence suggest that SOCS1-deficient DCs have enhanced ability to activate T cells. One notable study (260) assessed the ability of DCs to induce antitumor T cell responses that also translate into autoimmune pathology against normal cells with shared tumor-antigen expression. SOCS1-silenced BMDCs and human monocyte-derived DCs induce potent antitumor responses and break self-tolerance via a dual mechanism of exaggerated expression of IL-12 and hyperresponsiveness to autocrine IL-12-STAT4 signals (261, 262). Thus, steady-state IL-12 signals in DCs may drive T cell expansion and autoimmune and inflammatory disease in SOCS1-Tg mice.
TLRs have been a long-standing candidate for driving disease in Socs1−/− mice because Socs1−/− Ifnγ−/− mice are highly susceptible to endotoxin shock (263). SOCS1 negative regulation of TLR responses is mostly indirect, as autocrine cytokine receptor signals amplify the initial cellular response to TLR ligands (264). However, the adaptor Mal, a MyD88 homolog that transduces signals from TLR2 and TLR4, has been described as a target for SOCS1-mediated ubiquitination that leads to Mal degradation and restriction of TLR2 and TLR4 activation signals (265). SOCS1 may also negatively regulate TNF receptor signals, the mechanism of which is unknown (266). Although TNF-α and TLR signals may also trigger DC activation and contribute to disease in SOCS1-Tg mice, the roles of these signaling pathways in SOCS1-deficient DC-driven disease have not been described.
Whether SOCS1 expression differs in distinct DC subsets is not reported, yet SOCS1 deficiency is associated with the appearance of atypical CD8α+ DC subsets, the development of which may be a by-product of progressive inflammation in such mice (267). SOCS1 negatively regulates type I IFN signals: This cytokine plays a key role in the upregulation of maturation markers on cDCs and pDCs (32) as well as in pDC stimulation of B cell IgA production (268). Type I IFN also enhances the biological functions of CD8α+ DCs (269). An overabundance of IFN-α may also drive the development of atypical DC subsets (151), although no reports have indicated that increased responsiveness to IFN-α, such as in SOCS1-deficient cells, also influences DC lineage commitment. Thus, during steady state, SOCS1 likely restricts DC maturation in response to multiple cytokines as well as TLR ligands to maintain immune homeostasis and prevent inflammatory and autoimmune diseases.
TAM Receptors and STAT1
Tyro3, Axl, and Mer comprise the TAM family of receptor tyrosine kinases. These receptors share ligand specificity for growth arrest gene 6 (Gas6) and protein S (ProS) and are best known for their role in the recognition and phagocytosis of apoptotic cells (270, 271). In DCs, which express both Axl and Mer, TAM receptors transduce inhibitory signals upon recognition of apoptotic cells (273–275). They also negatively regulate TLR signals (272). Pretreatment of DCs with Gas6 or ProS inhibits proximal signaling events from TLRs, including the ubiquitination of key signaling molecules TRAF6 and TRAF3. The mechanism, described by Rothlin et al. (272), is surprisingly dependent on TAM receptor association with and “hijacking” of the IFNAR-STAT1 signaling module (Figure 2). Although IFNAR-STAT1 traditionally leads to the induction of type I IFN and IRFs, TAM-IFNAR-STAT1 does not induce these proinflammatory molecules, although it does upregulate the negative regulatory molecules SOCS1 and SOCS3. Thus, negative regulation of TLR signals by TAM ligands is dependent on STAT1-induced SOCS protein expression. The specialized version of TAM-IFNAR-induced STAT1 from conventional IFNAR-induced STAT1 is reminiscent of that described for IL-10-induced STAT3 (as opposed to that of IL-6-induced STAT3). However, the disparate anti-inflammatory potency of the IL-10 and IL-6 receptor signaling complexes may be explained in part according to their differential susceptibility to negative regulation by SOCS proteins (255). In the context of TAM-IFNAR-STAT1, tailored STAT1 competes with proinflammatory IFNAR-STAT1 and effectively decreases IRF production downstream of IFN-α stimulation. This suggests that STAT1 activated via TAM-IFNAR signals may have unique post-translational modifications that enable tailored gene transcription or recruitment of unique cofactors that direct its preferential anti-inflammatory functions.
Several disruptions to DCs and immune homeostasis resulting from TAM receptor deficiency are also found in SOCS1-Tg mice—for example, DC expansion, DC maturation, splenomegaly, and lymphadenopathy (259, 272, 276). Similarly, autoantibodies in Mer single-deficient, Tyro-Axl-Mer (i.e., TAM) triple-deficient, and SOCS1-Tg mice may all be attributed to BAFF overexpression by DCs (259, 277). This begs the question of whether IFN-γ, a key trigger of disease in Socs1−/− mice, upregulates TAM receptor expression and/or regulates the expression of TAM ligands Gas6 or ProS. What is the source of TAM ligands for DCs? DCs produce some Gas6, and DC-produced TAM ligands may provide autocrine downregulation of TLR responses. However, TAM ligands seem to be systemically abundant and may even be increased in SLE patients (278). Thus, identifying the relevant source of TAM ligands for DCs in vivo as well as the factors that regulate accessibility of DCs to TAM ligands is of considerable interest. It will also be interesting to determine the extent to which TLR signals, as opposed to other TAM functions such as recognition of apoptotic cells, drive autoimmune disease. Also of interest is determining whether TAM negative regulation of TLR signals disrupts intracellular processing of antigens derived from apoptotic bodies, a process that can be manipulated by TLR signals and may initiate autoimmunity (48).
DENDRITIC CELLS AND UBIQUITIN-MODIFYING ENZYME A20
A20, the product of the TNFAIP3 gene, restricts a number of innate immune signaling cascades that regulate DC functions, including TLR, NOD2, CD40, and TNF signals (279–283). All these signaling pathways can trigger distinct features of DC phenotypic and functional maturation; they can also alter DC survival. Hence, A20, a pleiotropically expressed protein, may play important cell-autonomous functions in DCs. In independent studies, investigators generated mice with deletion of A20 specifically in DCs (63, 284). Both mice strains revealed major spontaneous perturbations in myeloid as well as lymphoid immune homeostasis, demonstrating that regulation by A20 of intracellular signals in DCs is critical for steady-state immune homeostasis. Epistatic experiments with mice lacking MyD88 specifically in DCs indicate that A20 is required to regulate steady-state MyD88-dependent and -independent signaling pathways in DCs to preserve immune homeostasis (63; discussed below).
A20 exhibits dynamic interactions with proteins that mediate NF-κB activation (Figure 3). These interactions can be either direct or indirect; the latter property is conveyed via A20 binding of polyubiquitin chains generated and conjugated to signaling molecules during signal transduction. A20 contains two enzymatic domains, a deubiquitinase ovarian tumor (OTU) domain at its N terminus, and seven C-terminal zinc finger (Zf) domains, which bind polyubiquitin chains and other proteins and at least one of which exhibits E3 ligase function (Zf4) (280, 285–288). Because A20 mediates multiple protein-protein interactions and contains rivaling ubiquitin-enzymology functions, A20 may remove “activating” ubiquitin chains and add “degradative” ubiquitin chains on key signaling molecules to negatively regulate NF-κB activation (280). The functions of A20 are further complicated by additional protein-protein interactions, including ubiquitin binding; these functions may enable A20 to negatively regulate NF-κB independently of its enzymatic activities (288–290). A20 is also well recognized for its antiapoptotic functions, although how A20 domains collaborate to enhance cell survival is not well understood (279, 291).
Figure 3.
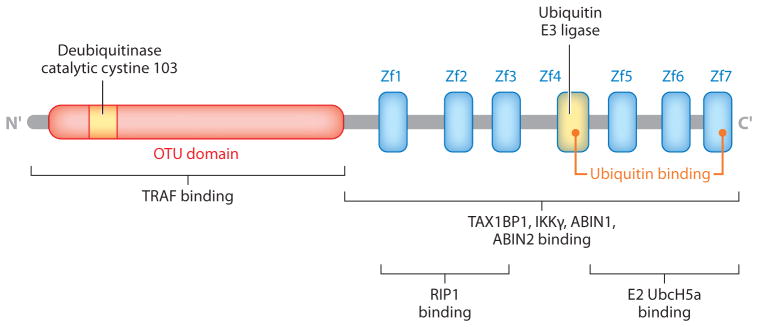
Structure and functional domains of A20. The ovarian tumor (OTU) domain (red) and seven zinc fingers (Zfs) are shown. The cystine 103 and Zf4 motifs implicated in ubiquitin-modifying activity are indicated in yellow. Orange circles point to ubiquitin-binding motifs. A20 interacts with multiple other proteins, including other ubiquitin-binding proteins, ubiquitin-modifying enzymes, and kinases.
CRE recombination in the cells analyzed by Kool et al. (284) led to the excision of exons4–5 (corresponding to the C terminus of the OTU domain and linker region before Zf1). In our studies, it led to excision of exon2 (within the OTU domain, including the catalytic cystine residue). We refer to these DCs as either A20-ex4,5-deleted or A20-ex2-deleted, respectively. Both genomic excisions were also designed to introduce frameshift mutations, and in both studies, full-length A20 protein was not detectable in CRE-expressing cells (assayed by A20 N terminus antiserum) (283, 292). The two studies were performed against a C57BL/6 genetic background (derived from C57BL/6 embryonic stem cells) and relied on genomic recombination mediated by the same CD11c-CRE transgene (293). They both highlight a key function for A20 in the regulation of DC biology, but they deviate as to the specific mechanisms by which A20 accomplishes proper DC-mediated immune homeostasis and the immune phenotype that results owing to a lack of A20 in DCs. Phenotypic differences between the two strains of mice thus suggest that important lessons may be learned about how A20 interacts with cellular proteins, regulates signaling cascades, and controls programmed cell death. From a genetic viewpoint, the diverse functional A20 domains and its complex modes of action raise the possibility that, should partial A20 proteins exist in A20-ex4,5-deleted or A20-ex2-deleted DCs, these truncated versions could contribute to diverse phenotypes pertaining to NF-κB signaling or cell survival. From an environmental perspective, differences between the two strains could be caused by differences in host microbiomes. Researchers are developing sophisticated tools to detect such differences between physically separated mice. Given the importance of A20 as a potential regulator of diverse immune diseases in humans, these environmental and genetic factors warrant further investigation.
A20 AND DENDRITIC CELL–INTRINSIC PHENOTYPE
During steady state, A20 is expressed nearly equivalently in cDCs and pDCs (63). In our study (63), we found that A20-ex2-deleted cDCs and pDCs were spontaneously phenotypically mature and expressed high levels of costimulatory molecules. Additionally, pDCs, which are MHC class IIlow during steady state, were predominantly MHC class IIhigh. Analysis of mice with targeted deletion of both A20-ex2 and MyD88 specifically in DCs revealed that steady-state MyD88-independent signals were sufficient to trigger phenotypic maturation of both pDCs and cDCs (63). By contrast, Kool et al. (284) did not observe differences in A20-ex4,5-deleted pDCs (they did not directly analyze cDC populations). pDCs express low levels of CD11c protein, and CD11c-CRE-mediated recombination is less efficient in these cells than it is in CD11chigh cDCs. However, the efficiency of A20 genomic deletion in pDCs appears equivalent in both studies and thus does not seem to account for the different results. In vivo analyses reported by Kool et al. (284) focused on CD11clowCD11b+ cells, which the authors likened to CD11clowCD11b+Mac3+ monocyte-derived DCs that can arise during inflammation (294). Upon A20-ex4,5-deletion, CD11clowCD11b+ cells were expanded and expressed high levels of CD40 and CD86. We did not detect CD11c+Mac3+ DCs in mice bearing A20-ex2-deleted DCs (G.E. Hammer, unpublished observations). The role of A20 in other DC subsets has not been reported. In summary, A20 likely restricts multiple signaling cascades triggered by steady-state stimuli to prevent DC maturation to physiological stimuli. Whether these stimuli and corresponding signaling pathways are distinct in pDCs, cDCs, or CD11clowCD11b+ cells, how these signals induce exaggerated expression of costimulatory molecules and how phenotypic maturation of distinct DC subsets may perturb immune homeostasis and drive the phenotypes of the two mice bearing A20-deleted DCs are among the issues that need to be addressed in the future.
As discussed above, loss or cellular expansion of DCs can have potent effects on immune homeostasis. Because A20 regulates the cell survival of fibroblasts, intestinal epithelial cells, and B cells—intriguingly in opposite ways—A20 may also regulate DC survival and their number in vivo (279, 283, 292). Kool et al. (284) reported an increase in total CD11c+ cells in the spleen but a decrease in the pDC subset. GM-CSF cultures of A20-ex4,5-deleted BMDCs have high mRNA expression of Bcl2 and Bcl-x, suggesting that these molecules may be involved in CD11c+ cellular expansion. These antiapoptotic molecules, alongside proapoptotic molecules such as Bim, are expressed during steady state and can change upon DC maturation in vivo. In addition, the relative expression of Bcl2 family proteins can regulate the size of the DC population as well as DC life span (36, 37). Thus far, reports indicate that Bcl2 family proteins have similar roles in both cDCs and pDCs, and any numerical changes in DC populations owing to a lack of these apoptotic regulators affects both DC subsets, so it is unclear why the A20-ex4,5-deleted pDCs reported by Kool et al. (284) would result in loss of this DC subset while other DC populations are numerically increased. Because these pDCs did not upregulate CD40, these cells may have received minimal CD40L survival signals that may have benefited other CD40highCD11c+ cells.
In contrast to A20-ex4,5-deleted pDCs, A20-ex2-deleted cDCs and pDCs showed increased CD40 levels. Neither DC subset was numerically increased, nor were pDC populations reduced, suggesting that the potential ability of increased CD40L signals to enhance DC survival and induce population expansion during steady state is low. Moreover, A20-ex2-deleted DCs had reduced life spans both in vivo and in vitro, suggesting that A20 is antiapoptotic in DCs (G.E. Hammer, unpublished observations). Although A20 is antiapoptotic in most cell types, inactivation of A20 may actually enhance B cell survival and the development of human B cell lymphomas, likely as a result of the prosurvival effects of exaggerated NF-κB signaling (283, 295, 296). Thus, how A20 may influence DC life span and expression of pro- and antiapoptotic Bcl2 family proteins during steady state or upon maturation is an intriguing issue requiring further investigation. Because NF-κB and programmed cell death signals are integrated differently in distinct cell types, and at different stages of differentiation, the roles of A20 in regulating DC survival may be complex.
AUTOIMMUNE AND AUTOINFLAMMATORY DISEASES IN MICE WITH A20-DELETED DENDRITIC CELLS
Perturbed functions of A20-ex2-deleted and A20-ex4,5-deleted DCs cause broad disruptions to immune homeostasis and drive immunological phenotypes resembling human autoimmune and autoinflammatory diseases. The spontaneous immunological phenotypes exhibited by both mice types highlight the overall significance of A20-dependent DC functions in controlling immune homeostasis. In addition, the differences between the two immunological phenotypes broaden our knowledge of the mechanistic spectrum by which DC functions control immune homeostasis and prevent disease.
Kool et al. (284) reported spontaneous B cell activation and expansion, autoantibodies, and glomerular deposition of IgG in mice >20 weeks of age. The CD11clowCD11b+ cells that were expanded in the spleen had heightened ability to phagocytose apoptotic cells. When combined with increased DC maturation, this property may disrupt self-tolerance. Additionally, incubation of BMDCs with apoptotic cells, which generally suppresses DC responses to TLR ligands, does not suppress cytokine responses of A20-ex4,5-deleted BMDCs and actually enhances IL-10 and IL-23 production. These disruptions to DC biology resulted in systemic autoimmune disease in the form of B cell expansion, T cell activation, and autoantibodies. Such outcomes are consistent with those of mice bearing DCs with increased life spans. These mice did not develop other disease pathologies such as diabetes or colitis.
Splenomegaly, lymphadenopathy, myeloid cell expansion, and T cell activation were noted in both reports of mice with A20-deleted DCs. Yet although A20-ex2-deleted DCs drove marked T cell and B cell expansion in peripheral lymph nodes, we did not detect any signs of antibody-related autoimmunity. In marked contrast to other mice bearing DCs with disrupted biology/functions, mice with A20-ex2-deleted DCs were free of all autoantibody-related pathology, including antinuclear antigens, anti-DNA, and immunoglobulin deposition on kidney glomeruli. Instead, these mice developed two autoimmune-inflammatory diseases, colitis and spondyloarthritis. These disease phenotypes may arise as a result of aberrant T cell activation by A20-ex2-deleted DCs. Independent of antigen, A20-ex2-deleted DCs drive potent activation and expansion of polyclonal T cells in the periphery. They also induce colitogenic T cell differentiation and can even override tolerizing signals that induce peripheral T cell deletion. The extent to which cDCs or pDCs contribute to these pathologies remains unknown. Different genetic backgrounds [as reported for Foxo3−/− and T-bet−/−/RAG2−/− mice on different backgrounds, discussed above (176, 205)] and differences in microbiota can govern disease severity or disease penetrance (72, 297, 298). These factors may also contribute to the different disease phenotypes in mice bearing either A20-ex4,5-deleted DCs or A20-ex2-deleted DCs. However, to our knowledge, neither genetic nor commensal disparities between mice have been shown to eliminate autoantibody-related autoimmune disease (as is induced by A20-ex4,5-deleted DCs) and instead cause the development of novel, autoinflammatory disease (as is induced by A20-ex2-deleted DCs). The differences in DC-intrinsic biology, the differential effect of A20 deletion on the maturation of DC subsets, and the nonoverlapping set of disease pathologies described in the two reports of mice bearing A20-deleted DCs all suggest that DCs with genomic deletion of exon2 or exons4–5 may express different truncated versions of A20 protein or may differ in whether they are A20-null. Another possibility is that intragenic miRNAs or other noncoding RNAs may be differentially mutated in the two strains. Future investigation of these potential differences and how they may regulate the biochemical functions of A20, NF-κB signaling, and cellular apoptosis could shed light on the distinct functions of A20 in DCs.
A20, TOLL-LIKE RECEPTORS, COLITIS, AND SPONDYLOARTHRITIS
As discussed in this review, hyperresponsiveness of DCs to TLRs correlates with spontaneous DC maturation and the development of autoimmunity. In our analyses, we determined that these phenotypes can be mutually exclusive. A20-ex2-deleted DCs were hyperresponsive to steady-state TLR signals, and MyD88-dependent signaling cascades in DCs induced exaggerated production of inflammatory cytokines IL-6 and TNF-α. Yet MyD88 signals were not required to induce the overexpression of costimulatory molecules on A20-ex2-deleted cDCs or pDCs, highlighting the regulatory function of A20 in both MyD88-dependent and MyD88-independent signaling cascades. The combined input of both types of signals drove promiscuous T cell activation and antigen-independent T cell proliferation. Thus, in the absence of MyD88 signals, phenotypically mature A20-ex2-deleted DCs induce aberrant T cell activation but fail to induce aberrant T cell expansion in peripheral lymph nodes. These findings provide a clear illustration of how DC functions are not solely dictated by their expression of maturation markers. Previously described in relation to antigen-specific adaptive immune responses, this notion holds true during steady state, in which MyD88-dependent and -independent signals confer distinctive functions upon mature DCs.
In the TLR signaling pathway, A20 expression downregulates TRAF6 ubiquitination to restrict both MyD88-dependent and -independent NF-κB signals (281, 299). The nature of the A20-regulated, MyD88-independent steady-state signals has not been completely defined, but it could include TRIF-dependent TLR signals, TNF, or CD40, as overt stimulation of these pathways can lead to DC maturation. Whether commensals provide the main stimulus that triggers these pathways is not known. Further investigation of whether MyD88 signals in A20-ex2-deleted DCs also drive spontaneous colitis may help clarify the role intestinal commensals play in driving this component of disease.
The absence of autoantibodies or antibody-related pathology in mice with A20-ex2-deleted DCs was surprising, given that both T and B cells are expanded by these DCs in lymph nodes. The mechanisms by which disrupted DC functions may compromise B cell homeostasis to facilitate the production of autoanti-bodies may involve overproduction of B cell survival cytokines, improper disposal of apoptotic bodies, or hyperactivation of T cells. In our analyses, the contribution of A20 to these components may not be of the necessary combination to facilitate the production of autoantibodies. Alternatively, a shortened life span of A20-ex2-deleted DCs may still enable their aberrant activation of T cells while not overstimulating B cells. We have detected a marked increase in serum and stool IgA (63; G.E. Hammer, unpublished observations), but not in other isotypes, indicating that the capacity of A20-ex2-deleted DCs to stimulate exaggerated immunoglobulin production is isotype selective and thus also likely to be context dependent. Increased IgA is indicative of perturbed intestinal homeostasis and is consistent with our findings that A20 function in DCs is particularly important to prevent DSS colitis and T cell–mediated colitis.
The coincidence of gut-joint disease is a common feature in the spondyloarthropathies (arthritis typically affecting the spinal column). This collection of heterogeneous seronegative arthritides is characterized by enthesitis (inflammation of the entheses—the site where tendon attaches to bone) and is strongly associated with the MHC class I allele HLA-B27 (300). Overexpression of HLA-B27 in rats and overexpression of TNF-α in mice can lead to coincident gut-joint disease characterized by enthesitis (301, 302). In human patients, spondyloarthropathies are distinguished from rheumatoid arthritis by the combined pathology of enthesitis and spinal and peripheral joint arthritis as well as by the absence of antibody-related pathology. The development of these pathologies in mice bearing A20-ex2-deleted DCs strongly indicates that compromised A20 function in DCs contributes to the development of coincident gut-joint pathology in humans. Mechanisms of disease that drive colitis may be distinct from those that drive arthritis, yet both conditions in human patients may be treated with anti-TNF-α therapy (302–305). In this regard, one key function of A20 in DCs may be to restrict the production of TNF-α and other inflammatory cytokines. Thus, the role of MyD88 in T cell–mediated colitis and arthritis in mice bearing A20-ex2-deleted DCs is of particular interest. The proinflammatory functions of DCs with compromised A20 functions may also enhance HLA-B27-associated immune pathology (306). Future studies investigating how A20 regulates steady-state DC functions and the nature of the intracellular signals that trigger DC-related pathology will provide significant insight regarding the mechanisms by which DCs may drive coincident gut-joint disease.
CONCLUSIONS
Analysis of steady-state signals and their control of DC functions and maturation demonstrates how changes in DC biology, whether altered expression of maturation markers, disrupted functions, or altered cytokine expression, do not evoke linear changes in immune homeostasis. For example, the level of expression of MHC class II or costimulatory molecules during steady state does not necessarily coincide with increased numbers of activated T cells, nor is an increase in these maturation markers required for spontaneous T cell activation to occur. Moreover, disruption of DC responses seems to require multiple hits to induce both aberrant T cell activation and expansion. Overexpression of any given cytokine by DCs, or the absence of DC functions, can cause distinct immunological disruptions depending on the tissue type and particular immunological setting in question. Considering the distinctions between DCs in vivo and DCs in culture, future research should focus on in vivo DC phenotypes, with consideration for the differential functions of distinct DC subsets and the types of stimuli driving DC functions and maturation. Such work will enable researchers to decipher which regulatory networks control DCs during steady state, how they are disrupted during disease, and how the integration of multiple intracellular signals translates into DC control of immune homeostasis in systemic and tissue-specific manners. Such studies may enable researchers to harness the therapeutic potential of DC “gate-sealing” functions to treat autoimmune and inflammatory diseases.
Acknowledgments
The authors thank Dr. Bao Duong, Dr. Barbara Malynn, and Julio Barrera for critical reading and contributions to the manuscript.
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
Contributor Information
Gianna Elena Hammer, Email: Gianna.Hammer@ucsf.edu.
Averil Ma, Email: Averil.Ma@ucsf.edu.
LITERATURE CITED
- 1.Steinman RM, Cohn ZA. Identification of a novel cell type in peripheral lymphoid organs of mice. I Morphology, quantitation, tissue distribution. J Exp Med. 1973;137(5):1142–62. doi: 10.1084/jem.137.5.1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nobel Assem. Karolinska Inst. The gatekeepers of the immune system. 2011 Nobel Prize in Physiology or Medicine: Popular Information. 2011:8. http://www.nobelprize.org/nobel_prizes/medicine/laureates/2011/popular-medicineprize2011.pdf.
- 3.Steinman RM. Decisions about dendritic cells: past, present, and future. Annu Rev Immunol. 2012;30:1–22. doi: 10.1146/annurev-immunol-100311-102839. [DOI] [PubMed] [Google Scholar]
- 4.Banchereau J, Brière F, Caux C, Davoust J, Lebecque S, et al. Immunobiology of dendritic cells. Annu Rev Immunol. 2000;18:767–811. doi: 10.1146/annurev.immunol.18.1.767. [DOI] [PubMed] [Google Scholar]
- 5.Cresswell P. Assembly, transport, and function of MHC class II molecules. Annu Rev Immunol. 1994;12:259–93. doi: 10.1146/annurev.iy.12.040194.001355. [DOI] [PubMed] [Google Scholar]
- 6.Turley SJ, Inaba K, Garrett WS, Ebersold M, Unternaehrer J, et al. Transport of peptide-MHC class II complexes in developing dendritic cells. Science. 2000;288(5465):522–27. doi: 10.1126/science.288.5465.522. [DOI] [PubMed] [Google Scholar]
- 7.Trombetta ES, Ebersold M, Garrett W, Pypaert M, Mellman I. Activation of lysosomal function during dendritic cell maturation. Science. 2003;299(5611):1400–3. doi: 10.1126/science.1080106. [DOI] [PubMed] [Google Scholar]
- 8.Greenwald RJ, Freeman GJ, Sharpe AH. The B7 family revisited. Annu Rev Immunol. 2005;23:515–48. doi: 10.1146/annurev.immunol.23.021704.115611. [DOI] [PubMed] [Google Scholar]
- 9.Kamath AT, Pooley J, O’Keeffe MA, Vremec D, Zhan Y, et al. The development, maturation, and turnover rate of mouse spleen dendritic cell populations. J Immunol. 2000;165(12):6762–70. doi: 10.4049/jimmunol.165.12.6762. [DOI] [PubMed] [Google Scholar]
- 10.Garrett WS, Chen LM, Kroschewski R, Ebersold M, Turley S, et al. Developmental control of endocytosis in dendritic cells by Cdc42. Cell. 2000;102(3):325–34. doi: 10.1016/s0092-8674(00)00038-6. [DOI] [PubMed] [Google Scholar]
- 11.Reis e Sousa C. Dendritic cells in a mature age. Nat Rev Immunol. 2006;6(6):476–83. doi: 10.1038/nri1845. [DOI] [PubMed] [Google Scholar]
- 12.Fujii S-I, Liu K, Smith C, Bonito AJ, Steinman RM. The linkage of innate to adaptive immunity via maturing dendritic cells in vivo requires CD40 ligation in addition to antigen presentation and CD80/86 costimulation. J Exp Med. 2004;199(12):1607–18. doi: 10.1084/jem.20040317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sato A, Iwasaki A. Induction of antiviral immunity requires Toll-like receptor signaling in both stromal and dendritic cell compartments. Proc Natl Acad Sci USA. 2004;101(46):16274–79. doi: 10.1073/pnas.0406268101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nolte MA, Leibundgut-Landmann S, Joffre O, Reis e Sousa C. Dendritic cell quiescence during systemic inflammation driven by LPS stimulation of radioresistant cells in vivo. J Exp Med. 2007;204(6):1487–501. doi: 10.1084/jem.20070325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Spörri R, Reis e Sousa C. Inflammatory mediators are insufficient for full dendritic cell activation and promote expansion of CD4+ T cell populations lacking helper function. Nat Immunol. 2005;6(2):163–70. doi: 10.1038/ni1162. [DOI] [PubMed] [Google Scholar]
- 16.Jiang A, Bloom O, Ono S, Cui W, Unternaehrer J, et al. Disruption of E-cadherin-mediated adhesion induces a functionally distinct pathway of dendritic cell maturation. Immunity. 2007;27(4):610–24. doi: 10.1016/j.immuni.2007.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rimoldi M, Chieppa M, Larghi P, Vulcano M, Allavena P, Rescigno M. Monocyte-derived dendritic cells activated by bacteria or by bacteria-stimulated epithelial cells are functionally different. Blood. 2005;106(8):2818–26. doi: 10.1182/blood-2004-11-4321. [DOI] [PubMed] [Google Scholar]
- 18.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140(6):805–20. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 19.Kawasaki T, Kawai T, Akira S. Recognition of nucleic acids by pattern-recognition receptors and its relevance in autoimmunity. Immunol Rev. 2011;243(1):61–73. doi: 10.1111/j.1600-065X.2011.01048.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5(10):987–95. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- 21.Lemaitre B, Nicolas E, Michaut L, Reichhart JM, Hoffmann JA. The dorsoventral regulatory gene cassette spätzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell. 1996;86(6):973–83. doi: 10.1016/s0092-8674(00)80172-5. [DOI] [PubMed] [Google Scholar]
- 22.Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282(5396):2085–88. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 23.Ewald SE, Lee BL, Lau L, Wickliffe KE, Shi G-P, et al. The ectodomain of Toll-like receptor 9 is cleaved to generate a functional receptor. Nature. 2008;456(7222):658–62. doi: 10.1038/nature07405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kagan JC, Su T, Horng T, Chow A, Akira S, Medzhitov R. TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-β. Nat Immunol. 2008;9(4):361–68. doi: 10.1038/ni1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barbalat R, Ewald SE, Mouchess ML, Barton GM. Nucleic acid recognition by the innate immune system. Annu Rev Immunol. 2011;29:185–214. doi: 10.1146/annurev-immunol-031210-101340. [DOI] [PubMed] [Google Scholar]
- 26.Fukui R, Saitoh S-I, Kanno A, Onji M, Shibata T, et al. Unc93B1 restricts systemic lethal inflammation by orchestrating Toll-like receptor 7 and 9 trafficking. Immunity. 2011;35(1):69–81. doi: 10.1016/j.immuni.2011.05.010. [DOI] [PubMed] [Google Scholar]
- 27.Mouchess ML, Arpaia N, Souza G, Barbalat R, Ewald SE, et al. Transmembrane mutations in Toll-like receptor 9 bypass the requirement for ectodomain proteolysis and induce fatal inflammation. Immunity. 2011;35(5):721–32. doi: 10.1016/j.immuni.2011.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moresco EMY, LaVine D, Beutler B. Toll-like receptors. Curr Biol. 2011;21(13):R488–93. doi: 10.1016/j.cub.2011.05.039. [DOI] [PubMed] [Google Scholar]
- 29.Zaru R, Ronkina N, Gaestel M, Arthur JSC, Watts C. The MAPK-activated kinase Rsk controls an acute Toll-like receptor signaling response in dendritic cells and is activated through two distinct pathways. Nat Immunol. 2007;8(11):1227–35. doi: 10.1038/ni1517. [DOI] [PubMed] [Google Scholar]
- 30.Kaisho T, Tanaka T. Turning NF-κB and IRFs on and off in DC. Trends Immunol. 2008;29(7):329–36. doi: 10.1016/j.it.2008.03.005. [DOI] [PubMed] [Google Scholar]
- 31.Asselin-Paturel C, Trinchieri G. Production of type I interferons: plasmacytoid dendritic cells and beyond. J Exp Med. 2005;202(4):461–65. doi: 10.1084/jem.20051395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Asselin-Paturel C, Brizard G, Chemin K, Boonstra A, O’Garra A, et al. Type I interferon dependence of plasmacytoid dendritic cell activation and migration. J Exp Med. 2005;201(7):1157–67. doi: 10.1084/jem.20041930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gay NJ, Gangloff M, Weber ANR. Toll-like receptors as molecular switches. Nat Rev Immunol. 2006;6(9):693–98. doi: 10.1038/nri1916. [DOI] [PubMed] [Google Scholar]
- 34.Hou B, Reizis B, DeFranco AL. Toll-like receptors activate innate and adaptive immunity by using dendritic cell-intrinsic and -extrinsic mechanisms. Immunity. 2008;29(2):272–82. doi: 10.1016/j.immuni.2008.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Haley K, Igyártó BZ, Ortner D, Bobr A, Kashem S, et al. Langerhans cells require MyD88-dependent signals for Candida albicans response but not for contact hypersensitivity or migration. J Immunol. 2012;188(9):4334–39. doi: 10.4049/jimmunol.1102759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hou W-S, Van Parijs L. A Bcl-2-dependent molecular timer regulates the life span and immuno-genicity of dendritic cells. Nat Immunol. 2004;5(6):583–89. doi: 10.1038/ni1071. [DOI] [PubMed] [Google Scholar]
- 37.Chen M, Huang L, Wang J. Deficiency of Bim in dendritic cells contributes to overactivation of lymphocytes and autoimmunity. Blood. 2007;109(10):4360–67. doi: 10.1182/blood-2006-11-056424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li X, Jiang S, Tapping RI. Toll-like receptor signaling in cell proliferation and survival. Cytokine. 2010;49(1):1–9. doi: 10.1016/j.cyto.2009.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fuertes Marraco SA, Scott CL, Bouillet P, Ives A, Masina S, et al. Type I interferon drives dendritic cell apoptosis via multiple BH3-only proteins following activation by PolyIC in vivo. PLoS ONE. 2011;6(6):e20189. doi: 10.1371/journal.pone.0020189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zanoni I, Ostuni R, Capuano G, Collini M, Caccia M, et al. CD14 regulates the dendritic cell life cycle after LPS exposure through NFAT activation. Nature. 2009;460(7252):264–68. doi: 10.1038/nature08118. [DOI] [PubMed] [Google Scholar]
- 41.West MA, Wallin RPA, Matthews SP, Svensson HG, Zaru R, et al. Enhanced dendritic cell antigen capture via Toll-like receptor-induced actin remodeling. Science. 2004;305(5687):1153–57. doi: 10.1126/science.1099153. [DOI] [PubMed] [Google Scholar]
- 42.Xu Y, Jagannath C, Liu X-D, Sharafkhaneh A, Kolodziejska KE, Eissa NT. Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity. 2007;27(1):135–44. doi: 10.1016/j.immuni.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shi C-S, Kehrl JH. MyD88 and TRIF target Beclin 1 to trigger autophagy in macrophages. J Biol Chem. 2008;283(48):33175–82. doi: 10.1074/jbc.M804478200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Delgado MA, Elmaoued RA, Davis AS, Kyei G, Deretic V. Toll-like receptors control autophagy. EMBO J. 2008;27(7):1110–21. doi: 10.1038/emboj.2008.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shi C-S, Kehrl JH. TRAF6 and A20 regulate lysine 63-linked ubiquitination of Beclin-1 to control TLR4-induced autophagy. Sci Signal. 2010;3(123):ra42. doi: 10.1126/scisignal.2000751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Into T, Inomata M, Takayama E, Takigawa T. Autophagy in regulation of Toll-like receptor signaling. Cell Signal. 2012;24(6):1150–62. doi: 10.1016/j.cellsig.2012.01.020. [DOI] [PubMed] [Google Scholar]
- 47.Blander JM, Medzhitov R. Regulation of phagosome maturation by signals from Toll-like receptors. Science. 2004;304(5673):1014–18. doi: 10.1126/science.1096158. [DOI] [PubMed] [Google Scholar]
- 48.Blander JM, Medzhitov R. Toll-dependent selection of microbial antigens for presentation by dendritic cells. Nature. 2006;440(7085):808–12. doi: 10.1038/nature04596. [DOI] [PubMed] [Google Scholar]
- 49.Blander JM, Medzhitov R. On regulation of phagosome maturation and antigen presentation. Nat Immunol. 2006;7(10):1029–35. doi: 10.1038/ni1006-1029. [DOI] [PubMed] [Google Scholar]
- 50.Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by Toll-like receptors is required for intestinal homeostasis. Cell. 2004;118(2):229–41. doi: 10.1016/j.cell.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 51.Marshak-Rothstein A, Rifkin IR. Immunologically active autoantigens: the role of Toll-like receptors in the development of chronic inflammatory disease. Annu Rev Immunol. 2007;25:419–41. doi: 10.1146/annurev.immunol.22.012703.104514. [DOI] [PubMed] [Google Scholar]
- 52.Kaisho T, Takeuchi O, Kawai T, Hoshino K, Akira S. Endotoxin-induced maturation of MyD88-deficient dendritic cells. J Immunol. 2001;166(9):5688–94. doi: 10.4049/jimmunol.166.9.5688. [DOI] [PubMed] [Google Scholar]
- 53.Walton KLW, He J, Kelsall BL, Sartor RB, Fisher NC. Dendritic cells in germ-free and specific pathogen-free mice have similar phenotypes and in vitro antigen presenting function. Immunol Lett. 2006;102(1):16–24. doi: 10.1016/j.imlet.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 54.Wilson NS, Young LJ, Kupresanin F, Naik SH, Vremec D, et al. Normal proportion and expression of maturation markers in migratory dendritic cells in the absence of germs or Toll-like receptor signaling. Immunol Cell Biol. 2008;86(2):200–5. doi: 10.1038/sj.icb.7100125. [DOI] [PubMed] [Google Scholar]
- 55.Shortman K, Liu Y-J. Mouse and human dendritic cell subtypes. Nat Rev Immunol. 2002;2(3):151–61. doi: 10.1038/nri746. [DOI] [PubMed] [Google Scholar]
- 56.Deane JA, Pisitkun P, Barrett RS, Feigenbaum L, Town T, et al. Control of Toll-like receptor 7 expression is essential to restrict autoimmunity and dendritic cell proliferation. Immunity. 2007;27(5):801–10. doi: 10.1016/j.immuni.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tian J, Avalos AM, Mao S-Y, Chen B, Senthil K, et al. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol. 2007;8(5):487–96. doi: 10.1038/ni1457. [DOI] [PubMed] [Google Scholar]
- 58.Lande R, Gregorio J, Facchinetti V, Chatterjee B, Wang Y-H, et al. Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature. 2007;449(7162):564–69. doi: 10.1038/nature06116. [DOI] [PubMed] [Google Scholar]
- 59.Ganguly D, Chamilos G, Lande R, Gregorio J, Meller S, et al. Self-RNA-antimicrobial peptide complexes activate human dendritic cells through TLR7 and TLR8. J Exp Med. 2009;206(9):1983–94. doi: 10.1084/jem.20090480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sadanaga A, Nakashima H, Akahoshi M, Masutani K, Miyake K, et al. Protection against autoimmune nephritis in MyD88-deficient MRL/lpr mice. Arthritis Rheum. 2007;56(5):1618–28. doi: 10.1002/art.22571. [DOI] [PubMed] [Google Scholar]
- 61.Silver KL, Crockford TL, Bouriez-Jones T, Milling S, Lambe T, Cornall RJ. MyD88-dependent autoimmune disease in Lyn-deficient mice. Eur J Immunol. 2007;37(10):2734–43. doi: 10.1002/eji.200737293. [DOI] [PubMed] [Google Scholar]
- 62.Teichmann LL, Ols ML, Kashgarian M, Reizis B, Kaplan DH, Shlomchik MJ. Dendritic cells in lupus are not required for activation of T and B cells but promote their expansion, resulting in tissue damage. Immunity. 2010;33(6):967–78. doi: 10.1016/j.immuni.2010.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hammer GE, Turer EE, Taylor KE, Fang CJ, Advincula R, et al. Expression of A20 by dendritic cells preserves immune homeostasis and prevents colitis and spondyloarthritis. Nat Immunol. 2011;12(12):1184–93. doi: 10.1038/ni.2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Slack E, Hapfelmeier S, Stecher B, Velykoredko Y, Stoel M, et al. Innate and adaptive immunity cooperate flexibly to maintain host-microbiota mutualism. Science. 2009;325(5940):617–20. doi: 10.1126/science.1172747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rescigno M, Urbano M, Valzasina B, Francolini M, Rotta G, et al. Dendritic cells express tight junction proteins and penetrate gut epithelial monolayers to sample bacteria. Nat Immunol. 2001;2(4):361–67. doi: 10.1038/86373. [DOI] [PubMed] [Google Scholar]
- 66.Niess JH, Brand S, Gu X, Landsman L, Jung S, et al. CX3CR1-mediated dendritic cell access to the intestinal lumen and bacterial clearance. Science. 2005;307(5707):254–58. doi: 10.1126/science.1102901. [DOI] [PubMed] [Google Scholar]
- 67.Chieppa M, Rescigno M, Huang AYC, Germain RN. Dynamic imaging of dendritic cell extension into the small bowel lumen in response to epithelial cell TLR engagement. J Exp Med. 2006;203(13):2841–52. doi: 10.1084/jem.20061884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Foligne B, Zoumpopoulou G, Dewulf J, Ben Younes A, Chareyre F, et al. A key role of dendritic cells in probiotic functionality. PLoS ONE. 2007;2(3):e313. doi: 10.1371/journal.pone.0000313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Arques JL, Hautefort I, Ivory K, Bertelli E, Regoli M, et al. Salmonella induces flagellin- and MyD88-dependent migration of bacteria-capturing dendritic cells into the gut lumen. Gastroenterology. 2009;137(2):579–87. e2. doi: 10.1053/j.gastro.2009.04.010. [DOI] [PubMed] [Google Scholar]
- 70.Schulz O, Jaensson E, Persson EK, Liu X, Worbs T, et al. Intestinal CD103+, but not CX3CR1+, antigen sampling cells migrate in lymph and serve classical dendritic cell functions. J Exp Med. 2009;206(13):3101–14. doi: 10.1084/jem.20091925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hall JA, Bouladoux N, Sun C-M, Wohlfert EA, Blank RB, et al. Commensal DNA limits regulatory T cell conversion and is a natural adjuvant of intestinal immune responses. Immunity. 2008;29(4):637–49. doi: 10.1016/j.immuni.2008.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wen L, Ley RE, Volchkov PY, Stranges PB, Avanesyan L, et al. Innate immunity and intestinal microbiota in the development of Type 1 diabetes. Nature. 2008;455(7216):1109–13. doi: 10.1038/nature07336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Buzás EI, György B, Pásztói M, Jelinek I, Falus A, Gabius H-J. Carbohydrate recognition systems in autoimmunity. Autoimmunity. 2006;39(8):691–704. doi: 10.1080/08916930601061470. [DOI] [PubMed] [Google Scholar]
- 74.García-Vallejo JJ, van Kooyk Y. Endogenous ligands for C-type lectin receptors: the true regulators of immune homeostasis. Immunol Rev. 2009;230(1):22–37. doi: 10.1111/j.1600-065X.2009.00786.x. [DOI] [PubMed] [Google Scholar]
- 75.Sancho D, Reis e Sousa C. Signaling by myeloid C-type lectin receptors in immunity and homeostasis. Annu Rev Immunol. 2012;30:491–529. doi: 10.1146/annurev-immunol-031210-101352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Valladeau J, Ravel O, Dezutter-Dambuyant C, Moore K, Kleijmeer M, et al. Langerin, a novel C-type lectin specific to Langerhans cells, is an endocytic receptor that induces the formation of Birbeck granules. Immunity. 2000;12(1):71–81. doi: 10.1016/s1074-7613(00)80160-0. [DOI] [PubMed] [Google Scholar]
- 77.Idoyaga J, Suda N, Suda K, Park CG, Steinman RM. Antibody to Langerin/CD207 localizes large numbers of CD8α+ dendritic cells to the marginal zone of mouse spleen. Proc Natl Acad Sci USA. 2009;106(5):1524–29. doi: 10.1073/pnas.0812247106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Steinman RM, Nussenzweig MC. Avoiding horror autotoxicus: the importance of dendritic cells in peripheral T cell tolerance. Proc Natl Acad Sci USA. 2002;99(1):351–58. doi: 10.1073/pnas.231606698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Burgdorf S, Kautz A, Böhnert V, Knolle PA, Kurts C. Distinct pathways of antigen uptake and intracellular routing in CD4 and CD8 T cell activation. Science. 2007;316(5824):612–16. doi: 10.1126/science.1137971. [DOI] [PubMed] [Google Scholar]
- 80.Dudziak D, Kamphorst AO, Heidkamp GF, Buchholz VR, Trumpfheller C, et al. Differential antigen processing by dendritic cell subsets in vivo. Science. 2007;315(5808):107–11. doi: 10.1126/science.1136080. [DOI] [PubMed] [Google Scholar]
- 81.Klechevsky E, Flamar A-L, Cao Y, Blanck J-P, Liu M, et al. Cross-priming CD8+ T cells by targeting antigens to human dendritic cells through DCIR. Blood. 2010;116(10):1685–97. doi: 10.1182/blood-2010-01-264960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Idoyaga J, Lubkin A, Fiorese C, Lahoud MH, Caminschi I, et al. Comparable T helper 1 (Th1) and CD8 T-cell immunity by targeting HIV gag p24 to CD8 dendritic cells within antibodies to Langerin, DEC205, and Clec9A. Proc Natl Acad Sci USA. 2011;108(6):2384–89. doi: 10.1073/pnas.1019547108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cao W, Zhang L, Rosen DB, Bover L, Watanabe G, et al. BDCA2/FcεRIγ complex signals through a novel BCR-like pathway in human plasmacytoid dendritic cells. PLoS Biol. 2007;5(10):e248. doi: 10.1371/journal.pbio.0050248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Röck J, Schneider E, Grün JR, Grützkau A, Küppers R, et al. CD303 (BDCA-2) signals in plasmacytoid dendritic cells via a BCR-like signalosome involving Syk, Slp65 and PLCγ2. Eur J Immunol. 2007;37(12):3564–75. doi: 10.1002/eji.200737711. [DOI] [PubMed] [Google Scholar]
- 85.van Vliet SJ, Paessens LC, Broks-van den Berg VCM, Geijtenbeek TBH, van Kooyk Y. The C-type lectin macrophage galactose-type lectin impedes migration of immature APCs. J Immunol. 2008;181(5):3148–55. doi: 10.4049/jimmunol.181.5.3148. [DOI] [PubMed] [Google Scholar]
- 86.Singh SK, Streng-Ouwehand I, Litjens M, Weelij DR, García-Vallejo JJ, et al. Characterization of murine MGL1 and MGL2 C-type lectins: distinct glycan specificities and tumor binding properties. Mol Immunol. 2009;46(6):1240–49. doi: 10.1016/j.molimm.2008.11.021. [DOI] [PubMed] [Google Scholar]
- 87.Hawiger D, Inaba K, Dorsett Y, Guo M, Mahnke K, et al. Dendritic cells induce peripheral T cell unresponsiveness under steady state conditions in vivo. J Exp Med. 2001;194(6):769–79. doi: 10.1084/jem.194.6.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhang J, Somani AK, Siminovitch KA. Roles of the SHP-1 tyrosine phosphatase in the negative regulation of cell signalling. Semin Immunol. 2000;12(4):361–78. doi: 10.1006/smim.2000.0223. [DOI] [PubMed] [Google Scholar]
- 89.Kaneko T, Saito Y, Kotani T, Okazawa H, Iwamura H, et al. Dendritic cell-specific ablation of the protein tyrosine phosphatase SHP1 promotes Th1 cell differentiation and induces autoimmunity. J Immunol. 2012;188(11):5397–407. doi: 10.4049/jimmunol.1103210. [DOI] [PubMed] [Google Scholar]
- 90.Croker BA, Lawson BR, Rutschmann S, Berger M, Eidenschenk C, et al. Inflammation and autoimmunity caused by a SHP1 mutation depend on IL-1, MyD88, and a microbial trigger. Proc Natl Acad Sci USA. 2008;105(39):15028–33. doi: 10.1073/pnas.0806619105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Allan RS, Smith CM, Belz GT, van Lint AL, Wakim LM, et al. Epidermal viral immunity induced by CD8α+ dendritic cells but not by Langerhans cells. Science. 2003;301(5641):1925–28. doi: 10.1126/science.1087576. [DOI] [PubMed] [Google Scholar]
- 92.Itano AA, McSorley SJ, Reinhardt RL, Ehst BD, Ingulli E, et al. Distinct dendritic cell populations sequentially present antigen to CD4 T cells and stimulate different aspects of cell-mediated immunity. Immunity. 2003;19(1):47–57. doi: 10.1016/s1074-7613(03)00175-4. [DOI] [PubMed] [Google Scholar]
- 93.Allan RS, Waithman J, Bedoui S, Jones CM, Villadangos JA, et al. Migratory dendritic cells transfer antigen to a lymph node-resident dendritic cell population for efficient CTL priming. Immunity. 2006;25(1):153–62. doi: 10.1016/j.immuni.2006.04.017. [DOI] [PubMed] [Google Scholar]
- 94.Mount AM, Smith CM, Kupresanin F, Stoermer K, Heath WR, Belz GT. Multiple dendritic cell populations activate CD4+ T cells after viral stimulation. PLoS ONE. 2008;3(2):e1691. doi: 10.1371/journal.pone.0001691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Young LJ, Wilson NS, Schnorrer P, Proietto A, ten Broeke T, et al. Differential MHC class II synthesis and ubiquitination confers distinct antigen-presenting properties on conventional and plasmacytoid dendritic cells. Nat Immunol. 2008;9(11):1244–52. doi: 10.1038/ni.1665. [DOI] [PubMed] [Google Scholar]
- 96.Belz GT, Bedoui S, Kupresanin F, Carbone FR, Heath WR. Minimal activation of memory CD8+ T cell by tissue-derived dendritic cells favors the stimulation of naive CD8+ T cells. Nat Immunol. 2007;8(10):1060–66. doi: 10.1038/ni1505. [DOI] [PubMed] [Google Scholar]
- 97.Ariizumi K, Shen GL, Shikano S, Xu S, Ritter R, et al. Identification of a novel, dendritic cell-associated molecule, dectin-1, by subtractive cDNA cloning. J Biol Chem. 2000;275(26):20157–67. doi: 10.1074/jbc.M909512199. [DOI] [PubMed] [Google Scholar]
- 98.van Vliet SJ, Gringhuis SI, Geijtenbeek TBH, van Kooyk Y. Regulation of effector T cells by antigen-presenting cells via interaction of the C-type lectin MGL with CD45. Nat Immunol. 2006;7(11):1200–8. doi: 10.1038/ni1390. [DOI] [PubMed] [Google Scholar]
- 99.Smits HH, Engering A, van der Kleij D, de Jong EC, Schipper K, et al. Selective probiotic bacteria induce IL-10-producing regulatory T cells in vitro by modulating dendritic cell function through dendritic cell-specific intercellular adhesion molecule 3-grabbing nonintegrin. J Allergy Clin Immunol. 2005;115(6):1260–67. doi: 10.1016/j.jaci.2005.03.036. [DOI] [PubMed] [Google Scholar]
- 100.Bron PA, van Baarlen P, Kleerebezem M. Emerging molecular insights into the interaction between probiotics and the host intestinal mucosa. Nat Rev Microbiol. 2012;10(1):66–78. doi: 10.1038/nrmicro2690. [DOI] [PubMed] [Google Scholar]
- 101.Zhou Y, Kawasaki H, Hsu S-C, Lee RT, Yao X, et al. Oral tolerance to food-induced systemic anaphylaxis mediated by the C-type lectin SIGNR1. Nat Med. 2010;16(10):1128–33. doi: 10.1038/nm.2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Saunders SP, Barlow JL, Walsh CM, Bellsoi A, Smith P, et al. C-type lectin SIGN-R1 has a role in experimental colitis and responsiveness to lipopolysaccharide. J Immunol. 2010;184(5):2627–37. doi: 10.4049/jimmunol.0901970. [DOI] [PubMed] [Google Scholar]
- 103.Heinsbroek SE, Oei A, Roelofs JJTH, Dhawan S, te Velde A, et al. Genetic deletion of dectin-1 does not affect the course of murine experimental colitis. BMC Gastroenterol. 2012;12:33. doi: 10.1186/1471-230X-12-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sancho D, Joffre OP, Keller AM, Rogers NC, Martínez D, et al. Identification of a dendritic cell receptor that couples sensing of necrosis to immunity. Nature. 2009;458(7240):899–903. doi: 10.1038/nature07750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ahrens S, Zelenay S, Sancho D, Hanč P, Kjær S, et al. F-actin is an evolutionarily conserved damage-associated molecular pattern recognized by DNGR-1, a receptor for dead cells. Immunity. 2012;36(4):635–45. doi: 10.1016/j.immuni.2012.03.008. [DOI] [PubMed] [Google Scholar]
- 106.Zelenay S, Keller AM, Whitney PG, Schraml BU, Deddouche S, et al. The dendritic cell receptor DNGR-1 controls endocytic handling of necrotic cell antigens to favor cross-priming of CTLs in virus-infected mice. J Clin Investig. 2012;122(5):1615–27. doi: 10.1172/JCI60644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Yamasaki S, Ishikawa E, Sakuma M, Hara H, Ogata K, Saito T. Mincle is an ITAM-coupled activating receptor that senses damaged cells. Nat Immunol. 2008;9(10):1179–88. doi: 10.1038/ni.1651. [DOI] [PubMed] [Google Scholar]
- 108.Fujikado N, Saijo S, Yonezawa T, Shimamori K, Ishii A, et al. Dcir deficiency causes development of autoimmune disease in mice due to excess expansion of dendritic cells. Nat Med. 2008;14(2):176–80. doi: 10.1038/nm1697. [DOI] [PubMed] [Google Scholar]
- 109.Eklöw C, Makrygiannakis D, Bäckdahl L, Padyukov L, Ulfgren A-K, et al. Cellular distribution of the C-type II lectin dendritic cell immunoreceptor (DCIR) and its expression in the rheumatic joint: identification of a subpopulation of DCIR+ T cells. Ann Rheum Dis. 2008;67(12):1742–49. doi: 10.1136/ard.2007.076976. [DOI] [PubMed] [Google Scholar]
- 110.Lambert AA, Imbeault M, Gilbert C, Tremblay MJ. HIV-1 induces DCIR expression in CD4+ T cells. PLoS Pathog. 2010;6(11):e1001188. doi: 10.1371/journal.ppat.1001188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hazenberg BP, Van Leeuwen MA, Van Rijswijk MH, Stern AC, Vellenga E. Correction of granulocytopenia in Felty’s syndrome by granulocyte-macrophage colony-stimulating factor. Simultaneous induction of interleukin-6 release and flare-up of the arthritis. Blood. 1989;74(8):2769–70. [PubMed] [Google Scholar]
- 112.Xu WD, Firestein GS, Taetle R, Kaushansky K, Zvaifler NJ. Cytokines in chronic inflammatory arthritis. II Granulocyte-macrophage colony-stimulating factor in rheumatoid synovial effusions. J Clin Investig. 1989;83(3):876–82. doi: 10.1172/JCI113971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Campbell IK, Rich MJ, Bischof RJ, Dunn AR, Grail D, Hamilton JA. Protection from collagen-induced arthritis in granulocyte-macrophage colony-stimulating factor-deficient mice. J Immunol. 1998;161(7):3639–44. [PubMed] [Google Scholar]
- 114.Cook AD, Braine EL, Campbell IK, Rich MJ, Hamilton JA. Blockade of collagen-induced arthritis post-onset by antibody to granulocyte-macrophage colony-stimulating factor (GM-CSF): requirement for GM-CSF in the effector phase of disease. Arthritis Res. 2001;3(5):293–98. doi: 10.1186/ar318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sonderegger I, Iezzi G, Maier R, Schmitz N, Kurrer M, Kopf M. GM-CSF mediates autoimmunity by enhancing IL-6-dependent Th17 cell development and survival. J Exp Med. 2008;205(10):2281–94. doi: 10.1084/jem.20071119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Vremec D, Lieschke GJ, Dunn AR, Robb L, Metcalf D, Shortman K. The influence of granulocyte/macrophage colony-stimulating factor on dendritic cell levels in mouse lymphoid organs. Eur J Immunol. 1997;27(1):40–44. doi: 10.1002/eji.1830270107. [DOI] [PubMed] [Google Scholar]
- 117.Greter M, Helft J, Chow A, Hashimoto D, Mortha A, et al. GM-CSF controls nonlymphoid tissue dendritic cell homeostasis but is dispensable for the differentiation of inflammatory dendritic cells. Immunity. 2012;36(6):1031–46. doi: 10.1016/j.immuni.2012.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Meyer-Wentrup F, Benitez-Ribas D, Tacken PJ, Punt CJA, Figdor CG, et al. Targeting DCIR on human plasmacytoid dendritic cells results in antigen presentation and inhibits IFN-α production. Blood. 2008;111(8):4245–53. doi: 10.1182/blood-2007-03-081398. [DOI] [PubMed] [Google Scholar]
- 119.Meyer-Wentrup F, Cambi A, Joosten B, Looman MW, de Vries IJM, et al. DCIR is endocytosed into human dendritic cells and inhibits TLR8-mediated cytokine production. J Leukoc Biol. 2009;85(3):518–25. doi: 10.1189/jlb.0608352. [DOI] [PubMed] [Google Scholar]
- 120.Gómez CP, Tiemi Shio M, Duplay P, Olivier M, Descoteaux A. The protein tyrosine phosphatase SHP-1 regulates phagolysosome biogenesis. J Immunol. 2012;189(5):2203–10. doi: 10.4049/jimmunol.1103021. [DOI] [PubMed] [Google Scholar]
- 121.Lorentzen JC, Flornes L, Eklöw C, Bäckdahl L, Ribbhammar U, et al. Association of arthritis with a gene complex encoding C-type lectin-like receptors. Arthritis Rheum. 2007;56(8):2620–32. doi: 10.1002/art.22813. [DOI] [PubMed] [Google Scholar]
- 122.Franchi L, Muñoz-Planillo R, Núñez G. Sensing and reacting to microbes through the inflammasomes. Nat Immunol. 2012;13(4):325–32. doi: 10.1038/ni.2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Rathinam VAK, Vanaja SK, Fitzgerald KA. Regulation of inflammasome signaling. Nat Immunol. 2012;13(4):333–32. doi: 10.1038/ni.2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Davis BK, Wen H, Ting JP-Y. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev Immunol. 2011;29:707–35. doi: 10.1146/annurev-immunol-031210-101405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Franchi L, Kamada N, Nakamura Y, Burberry A, Kuffa P, et al. NLRC4-driven production of IL-1β discriminates between pathogenic and commensal bacteria and promotes host intestinal defense. Nat Immunol. 2012;13(5):449–56. doi: 10.1038/ni.2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Biswas A, Petnicki-Ocwieja T, Kobayashi KS. NOD2: a key regulator linking microbiota to intestinal mucosal immunity. J Mol Med. 2012;90(1):15–24. doi: 10.1007/s00109-011-0802-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Strober W, Watanabe T. NOD2, an intracellular innate immune sensor involved in host defense and Crohn’s disease. Mucosal Immunol. 2011;4(5):484–95. doi: 10.1038/mi.2011.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Maeda S, Hsu L-C, Liu H, Bankston LA, Iimura M, et al. Nod2 mutation in Crohn’s disease potentiates NF-κB activity and IL-1β processing. Science. 2005;307(5710):734–38. doi: 10.1126/science.1103685. [DOI] [PubMed] [Google Scholar]
- 129.Watanabe T, Kitani A, Murray PJ, Wakatsuki Y, Fuss IJ, Strober W. Nucleotide binding oligomerization domain 2 deficiency leads to dysregulated TLR2 signaling and induction of antigen-specific colitis. Immunity. 2006;25(3):473–85. doi: 10.1016/j.immuni.2006.06.018. [DOI] [PubMed] [Google Scholar]
- 130.Watanabe T, Asano N, Murray PJ, Ozato K, Tailor P, et al. Muramyl dipeptide activation of nucleotide-binding oligomerization domain 2 protects mice from experimental colitis. J Clin Investig. 2008;118(2):545–59. doi: 10.1172/JCI33145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Petnicki-Ocwieja T, Hrncir T, Liu Y-J, Biswas A, Hudcovic T, et al. Nod2 is required for the regulation of commensal microbiota in the intestine. Proc Natl Acad Sci USA. 2009;106(37):15813–18. doi: 10.1073/pnas.0907722106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Macho Fernandez E, Valenti V, Rockel C, Hermann C, Pot B, et al. Anti-inflammatory capacity of selected lactobacilli in experimental colitis is driven by NOD2-mediated recognition of a specific peptidoglycan-derived muropeptide. Gut. 2011;60(8):1050–59. doi: 10.1136/gut.2010.232918. [DOI] [PubMed] [Google Scholar]
- 133.Cooney R, Baker J, Brain O, Danis B, Pichulik T, et al. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat Med. 2010;16(1):90–97. doi: 10.1038/nm.2069. [DOI] [PubMed] [Google Scholar]
- 134.Penack O, Smith OM, Cunningham-Bussel A, Liu X, Rao U, et al. NOD2 regulates hematopoietic cell function during graft-versus-host disease. J Exp Med. 2009;206(10):2101–10. doi: 10.1084/jem.20090623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Holler E, Rogler G, Brenmoehl J, Hahn J, Herfarth H, et al. Prognostic significance of NOD2/CARD15 variants in HLA-identical sibling hematopoietic stem cell transplantation: effect on long-term outcome is confirmed in 2 independent cohorts and may be modulated by the type of gastrointestinal decontamination. Blood. 2006;107(10):4189–93. doi: 10.1182/blood-2005-09-3741. [DOI] [PubMed] [Google Scholar]
- 136.Holler E, Hahn J, Andreesen R, Rogler G, Brenmoehl J, et al. NOD2/CARD15 polymorphisms in allogeneic stem-cell transplantation from unrelated donors: T depletion matters. J Clin Oncol. 2008;26(2):338–39. doi: 10.1200/JCO.2007.14.1325. [DOI] [PubMed] [Google Scholar]
- 137.Chen M, Wang Y-H, Wang Y, Huang L, Sandoval H, et al. Dendritic cell apoptosis in the maintenance of immune tolerance. Science. 2006;311(5764):1160–64. doi: 10.1126/science.1122545. [DOI] [PubMed] [Google Scholar]
- 138.Stranges PB, Watson J, Cooper CJ, Choisy-Rossi C-M, Stonebraker AC, et al. Elimination of antigen-presenting cells and autoreactive T cells by Fas contributes to prevention of autoimmunity. Immunity. 2007;26(5):629–41. doi: 10.1016/j.immuni.2007.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Park D, Lapteva N, Seethammagari M, Slawin KM, Spencer DM. An essential role for Akt1 in dendritic cell function and tumor immunotherapy. Nat Biotechnol. 2006;24(12):1581–90. doi: 10.1038/nbt1262. [DOI] [PubMed] [Google Scholar]
- 140.Birnberg T, Bar-On L, Sapoznikov A, Caton ML, Cervantes-Barragán L, et al. Lack of conventional dendritic cells is compatible with normal development and T cell homeostasis, but causes myeloid proliferative syndrome. Immunity. 2008;29(6):986–97. doi: 10.1016/j.immuni.2008.10.012. [DOI] [PubMed] [Google Scholar]
- 141.Ohnmacht C, Pullner A, King SBS, Drexler I, Meier S, et al. Constitutive ablation of dendritic cells breaks self-tolerance of CD4 T cells and results in spontaneous fatal autoimmunity. J Exp Med. 2009;206(3):549–59. doi: 10.1084/jem.20082394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Mortier E, Advincula R, Kim L, Chmura S, Barrera J, et al. Macrophage- and dendritic-cell-derived interleukin-15 receptor alpha supports homeostasis of distinct CD8+ T cell subsets. Immunity. 2009;31(5):811–22. doi: 10.1016/j.immuni.2009.09.017. [DOI] [PubMed] [Google Scholar]
- 143.Kang S-J, Liang H-E, Reizis B, Locksley RM. Regulation of hierarchical clustering and activation of innate immune cells by dendritic cells. Immunity. 2008;29(5):819–33. doi: 10.1016/j.immuni.2008.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Marrack P, Kappler J. Positive selection of thymocytes bearing αβ T cell receptors. Curr Opin Immunol. 1997;9(2):250–55. doi: 10.1016/s0952-7915(97)80144-6. [DOI] [PubMed] [Google Scholar]
- 145.Ladi E, Schwickert TA, Chtanova T, Chen Y, Herzmark P, et al. Thymocyte-dendritic cell interactions near sources of CCR7 ligands in the thymic cortex. J Immunol. 2008;181(10):7014–23. doi: 10.4049/jimmunol.181.10.7014. [DOI] [PubMed] [Google Scholar]
- 146.Le Borgne M, Ladi E, Dzhagalov I, Herzmark P, Liao YF, et al. The impact of negative selection on thymocyte migration in the medulla. Nat Immunol. 2009;10(8):823–30. doi: 10.1038/ni.1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Hadeiba H, Lahl K, Edalati A, Oderup C, Habtezion A, et al. Plasmacytoid dendritic cells transport peripheral antigens to the thymus to promote central tolerance. Immunity. 2012;36(3):438–50. doi: 10.1016/j.immuni.2012.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Li J, Park J, Foss D, Goldschneider I. Thymus-homing peripheral dendritic cells constitute two of the three major subsets of dendritic cells in the steady-state thymus. J Exp Med. 2009;206(3):607–22. doi: 10.1084/jem.20082232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Henri S, Vremec D, Kamath A, Waithman J, Williams S, et al. The dendritic cell populations of mouse lymph nodes. J Immunol. 2001;167(2):741–48. doi: 10.4049/jimmunol.167.2.741. [DOI] [PubMed] [Google Scholar]
- 150.Wilson NS, El-Sukkari D, Belz GT, Smith CM, Steptoe RJ, et al. Most lymphoid organ dendritic cell types are phenotypically and functionally immature. Blood. 2003;102(6):2187–94. doi: 10.1182/blood-2003-02-0513. [DOI] [PubMed] [Google Scholar]
- 151.Banchereau J, Pascual V, Palucka AK. Autoimmunity through cytokine-induced dendritic cell activation. Immunity. 2004;20(5):539–50. doi: 10.1016/s1074-7613(04)00108-6. [DOI] [PubMed] [Google Scholar]
- 152.Wan S, Xia C, Morel L. IL-6 produced by dendritic cells from lupus-prone mice inhibits CD4+CD25+ T cell regulatory functions. J Immunol. 2007;178(1):271–79. doi: 10.4049/jimmunol.178.1.271. [DOI] [PubMed] [Google Scholar]
- 153.Ding D, Mehta H, McCune WJ, Kaplan MJ. Aberrant phenotype and function of myeloid dendritic cells in systemic lupus erythematosus. J Immunol. 2006;177(9):5878–89. doi: 10.4049/jimmunol.177.9.5878. [DOI] [PubMed] [Google Scholar]
- 154.Keller AM, Schildknecht A, Xiao Y, van den Broek M, Borst J. Expression of costimulatory ligand CD70 on steady-state dendritic cells breaks CD8+ T cell tolerance and permits effective immunity. Immunity. 2008;29(6):934–46. doi: 10.1016/j.immuni.2008.10.009. [DOI] [PubMed] [Google Scholar]
- 155.Albert ML, Jegathesan M, Darnell RB. Dendritic cell maturation is required for the cross-tolerization of CD8+ T cells. Nat Immunol. 2001;2(11):1010–17. doi: 10.1038/ni722. [DOI] [PubMed] [Google Scholar]
- 156.Merad M, Ginhoux F, Collin M. Origin, homeostasis and function of Langerhans cells and other langerin-expressing dendritic cells. Nat Rev Immunol. 2008;8(12):935–47. doi: 10.1038/nri2455. [DOI] [PubMed] [Google Scholar]
- 157.Merad M, Manz MG. Dendritic cell homeostasis. Blood. 2009;113(15):3418–27. doi: 10.1182/blood-2008-12-180646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Reizis B. Regulation of plasmacytoid dendritic cell development. Curr Opin Immunol. 2010;22(2):206–11. doi: 10.1016/j.coi.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Liu K, Nussenzweig MC. Origin and development of dendritic cells. Immunol Rev. 2010;234(1):45–54. doi: 10.1111/j.0105-2896.2009.00879.x. [DOI] [PubMed] [Google Scholar]
- 160.Shortman K, Naik SH. Steady-state and inflammatory dendritic-cell development. Nat Rev Immunol. 2007;7(1):19–30. doi: 10.1038/nri1996. [DOI] [PubMed] [Google Scholar]
- 161.Varol C, Vallon-Eberhard A, Elinav E, Aychek T, Shapira Y, et al. Intestinal lamina propria dendritic cell subsets have different origin and functions. Immunity. 2009;31(3):502–12. doi: 10.1016/j.immuni.2009.06.025. [DOI] [PubMed] [Google Scholar]
- 162.Cisse B, Caton ML, Lehner M, Maeda T, Scheu S, et al. Transcription factor E2-2 is an essential and specific regulator of plasmacytoid dendritic cell development. Cell. 2008;135(1):37–48. doi: 10.1016/j.cell.2008.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Wu L, D’Amico A, Winkel KD, Suter M, Lo D, Shortman K. RelB is essential for the development of myeloid-related CD8α− dendritic cells but not of lymphoid-related CD8α+ dendritic cells. Immunity. 1998;9(6):839–47. doi: 10.1016/s1074-7613(00)80649-4. [DOI] [PubMed] [Google Scholar]
- 164.Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, et al. Batf3 deficiency reveals a critical role for CD8α+ dendritic cells in cytotoxic T cell immunity. Science. 2008;322(5904):1097–100. doi: 10.1126/science.1164206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Ouaaz F, Arron J, Zheng Y, Choi Y, Beg AA. Dendritic cell development and survival require distinct NF-κB subunits. Immunity. 2002;16(2):257–70. doi: 10.1016/s1074-7613(02)00272-8. [DOI] [PubMed] [Google Scholar]
- 166.Speirs K, Lieberman L, Caamano J, Hunter CA, Scott P. Cutting edge: NF-κB2 is a negative regulator of dendritic cell function. J Immunol. 2004;172(2):752–56. doi: 10.4049/jimmunol.172.2.752. [DOI] [PubMed] [Google Scholar]
- 167.Wang J, Wang X, Hussain S, Zheng Y, Sanjabi S, et al. Distinct roles of different NF-κB subunits in regulating inflammatory and T cell stimulatory gene expression in dendritic cells. J Immunol. 2007;178(11):6777–88. doi: 10.4049/jimmunol.178.11.6777. [DOI] [PubMed] [Google Scholar]
- 168.Macagno A, Napolitani G, Lanzavecchia A, Sallusto F. Duration, combination and timing: the signal integration model of dendritic cell activation. Trends Immunol. 2007;28(5):227–33. doi: 10.1016/j.it.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 169.van der Horst A, Burgering BMT. Stressing the role of FoxO proteins in life span and disease. Nat Rev Mol Cell Biol. 2007;8(6):440–50. doi: 10.1038/nrm2190. [DOI] [PubMed] [Google Scholar]
- 170.Calnan DR, Brunet A. The FoxO code. Oncogene. 2008;27(16):2276–88. doi: 10.1038/onc.2008.21. [DOI] [PubMed] [Google Scholar]
- 171.Dejean AS, Beisner DR, Ch’en IL, Kerdiles YM, Babour A, et al. Transcription factor Foxo3 controls the magnitude of T cell immune responses by modulating the function of dendritic cells. Nat Immunol. 2009;10(5):504–13. doi: 10.1038/ni.1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Fallarino F, Bianchi R, Orabona C, Vacca C, Belladonna ML, et al. CTLA-4-Ig activates forkhead transcription factors and protects dendritic cells from oxidative stress in nonobese diabetic mice. J Exp Med. 2004;200(8):1051–62. doi: 10.1084/jem.20040942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Orabona C, Grohmann U, Belladonna ML, Fallarino F, Vacca C, et al. CD28 induces immunostimulatory signals in dendritic cells via CD80 and CD86. Nat Immunol. 2004;5(11):1134–42. doi: 10.1038/ni1124. [DOI] [PubMed] [Google Scholar]
- 174.Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271(5256):1734–36. doi: 10.1126/science.271.5256.1734. [DOI] [PubMed] [Google Scholar]
- 175.Karandikar NJ, Vanderlugt CL, Walunas TL, Miller SD, Bluestone JA. CTLA-4: a negative regulator of autoimmune disease. J Exp Med. 1996;184(2):783–88. doi: 10.1084/jem.184.2.783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Lin L, Hron JD, Peng SL. Regulation of NF-κB, Th activation, and autoinflammation by the forkhead transcription factor Foxo3a. Immunity. 2004;21(2):203–13. doi: 10.1016/j.immuni.2004.06.016. [DOI] [PubMed] [Google Scholar]
- 177.Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303(5666):2011–15. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- 178.Rosas M, Dijkers PF, Lindemans CL, Lammers J-WJ, Koenderman L, Coffer PJ. IL-5-mediated eosinophil survival requires inhibition of GSK-3 and correlates with β-catenin relocalization. J Leukoc Biol. 2006;80(1):186–95. doi: 10.1189/jlb.1105636. [DOI] [PubMed] [Google Scholar]
- 179.Kastenbauer S, Ziegler-Heitbrock HW. NF-κB1 (p50) is upregulated in lipopolysaccharide tolerance and can block tumor necrosis factor gene expression. Infect Immun. 1999;67(4):1553–59. doi: 10.1128/iai.67.4.1553-1559.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Dissanayake D, Hall H, Berg-Brown N, Elford AR, Hamilton SR, et al. Nuclear factor-κB1 controls the functional maturation of dendritic cells and prevents the activation of autoreactive T cells. Nat Med. 2011;17(12):1663–67. doi: 10.1038/nm.2556. [DOI] [PubMed] [Google Scholar]
- 181.Leung BP, Conacher M, Hunter D, McInnes IB, Liew FY, Brewer JM. A novel dendritic cell-induced model of erosive inflammatory arthritis: distinct roles for dendritic cells in T cell activation and induction of local inflammation. J Immunol. 2002;169(12):7071–77. doi: 10.4049/jimmunol.169.12.7071. [DOI] [PubMed] [Google Scholar]
- 182.Eriksson U, Ricci R, Hunziker L, Kurrer MO, Oudit GY, et al. Dendritic cell-induced autoimmune heart failure requires cooperation between adaptive and innate immunity. Nat Med. 2003;9(12):1484–90. doi: 10.1038/nm960. [DOI] [PubMed] [Google Scholar]
- 183.Georgiev M, Agle LMA, Chu JL, Elkon KB, Ashany D. Mature dendritic cells readily break tolerance in normal mice but do not lead to disease expression. Arthritis Rheum. 2005;52(1):225–38. doi: 10.1002/art.20759. [DOI] [PubMed] [Google Scholar]
- 184.Martin DA, Zhang K, Kenkel J, Hughes G, Clark E, et al. Autoimmunity stimulated by adoptively transferred dendritic cells is initiated by both αβ and γδ T cells but does not require MyD88 signaling. J Immunol. 2007;179(9):5819–28. doi: 10.4049/jimmunol.179.9.5819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Kurts C, Carbone FR, Barnden M, Blanas E, Allison J, et al. CD4+ T cell help impairs CD8+ T cell deletion induced by cross-presentation of self-antigens and favors autoimmunity. J Exp Med. 1997;186(12):2057–62. doi: 10.1084/jem.186.12.2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Hamilton-Williams EE, Lang A, Benke D, Davey GM, Wiesmüller K-H, Kurts C. Cutting edge: TLR ligands are not sufficient to break cross-tolerance to self-antigens. J Immunol. 2005;174(3):1159–63. doi: 10.4049/jimmunol.174.3.1159. [DOI] [PubMed] [Google Scholar]
- 187.O’Keeffe M, Grumont RJ, Hochrein H, Fuchsberger M, Gugasyan R, et al. Distinct roles for the NF-κB1 and c-Rel transcription factors in the differentiation and survival of plasmacytoid and conventional dendritic cells activated by TLR-9 signals. Blood. 2005;106(10):3457–64. doi: 10.1182/blood-2004-12-4965. [DOI] [PubMed] [Google Scholar]
- 188.Artis D, Kane CM, Fiore J, Zaph C, Shapira S, et al. Dendritic cell-intrinsic expression of NF-κB1 is required to promote optimal Th2 cell differentiation. J Immunol. 2005;174(11):7154–59. doi: 10.4049/jimmunol.174.11.7154. [DOI] [PubMed] [Google Scholar]
- 189.Melli K, Friedman RS, Martin AE, Finger EB, Miao G, et al. Amplification of autoimmune response through induction of dendritic cell maturation in inflamed tissues. J Immunol. 2009;182(5):2590–600. doi: 10.4049/jimmunol.0803543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Mercurio F, DiDonato JA, Rosette C, Karin M. p105 and p98 precursor proteins play an active role in NF-κB-mediated signal transduction. Genes Dev. 1993;7(4):705–18. doi: 10.1101/gad.7.4.705. [DOI] [PubMed] [Google Scholar]
- 191.Waterfield MR, Zhang M, Norman LP, Sun SC. NF-κB1/p105 regulates lipopolysaccharide-stimulated MAP kinase signaling by governing the stability and function of the Tpl2 kinase. Mol Cell. 2003;11(3):685–94. doi: 10.1016/s1097-2765(03)00070-4. [DOI] [PubMed] [Google Scholar]
- 192.Saccani S, Pantano S, Natoli G. Modulation of NF-κB activity by exchange of dimers. Mol Cell. 2003;11(6):1563–74. doi: 10.1016/s1097-2765(03)00227-2. [DOI] [PubMed] [Google Scholar]
- 193.Tong X, Yin L, Washington R, Rosenberg DW, Giardina C. The p50-p50 NF-κB complex as a stimulus-specific repressor of gene activation. Mol Cell Biochem. 2004;265(1–2):171–83. doi: 10.1023/b:mcbi.0000044394.66951.4d. [DOI] [PubMed] [Google Scholar]
- 194.Carmody RJ, Ruan Q, Palmer S, Hilliard B, Chen YH. Negative regulation of Toll-like receptor signaling by NF-κB p50 ubiquitination blockade. Science. 2007;317(5838):675–78. doi: 10.1126/science.1142953. [DOI] [PubMed] [Google Scholar]
- 195.Sha WC, Liou HC, Tuomanen EI, Baltimore D. Targeted disruption of the p50 subunit of NF-κB leads to multifocal defects in immune responses. Cell. 1995;80(2):321–30. doi: 10.1016/0092-8674(95)90415-8. [DOI] [PubMed] [Google Scholar]
- 196.Cao S, Zhang X, Edwards JP, Mosser DM. NF-κB1 (p50) homodimers differentially regulate pro-and anti-inflammatory cytokines in macrophages. J Biol Chem. 2006;281(36):26041–50. doi: 10.1074/jbc.M602222200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Driessler F, Venstrom K, Sabat R, Asadullah K, Schottelius AJ. Molecular mechanisms of interleukin-10-mediated inhibition of NF-κB activity: a role for p50. Clin Exp Immunol. 2004;135(1):64–73. doi: 10.1111/j.1365-2249.2004.02342.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Garrett WS, Lord GM, Punit S, Lugo-Villarino G, Mazmanian SK, et al. Communicable ulcerative colitis induced by T-bet deficiency in the innate immune system. Cell. 2007;131(1):33–45. doi: 10.1016/j.cell.2007.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Garrett WS, Punit S, Gallini CA, Michaud M, Zhang D, et al. Colitis-associated colorectal cancer driven by T-bet deficiency in dendritic cells. Cancer Cell. 2009;16(3):208–19. doi: 10.1016/j.ccr.2009.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Garrett WS, Gallini CA, Yatsunenko T, Michaud M, DuBois A, et al. Enterobacteriaceae act in concert with the gut microbiota to induce spontaneous and maternally transmitted colitis. Cell Host Microbe. 2010;8(3):292–300. doi: 10.1016/j.chom.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Uhlig HH, McKenzie BS, Hue S, Thompson C, Joyce-Shaikh B, et al. Differential activity of IL-12 and IL-23 in mucosal and systemic innate immune pathology. Immunity. 2006;25(2):309–18. doi: 10.1016/j.immuni.2006.05.017. [DOI] [PubMed] [Google Scholar]
- 202.Hapfelmeier S, Müller AJ, Stecher B, Kaiser P, Barthel M, et al. Microbe sampling by mucosal dendritic cells is a discrete, MyD88-independent step in δinvG S. Typhimurium colitis. J Exp Med. 2008;205(2):437–50. doi: 10.1084/jem.20070633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Feng T, Wang L, Schoeb TR, Elson CO, Cong Y. Microbiota innate stimulation is a prerequisite for T cell spontaneous proliferation and induction of experimental colitis. J Exp Med. 2010;207(6):1321–32. doi: 10.1084/jem.20092253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Farmer MA, Sundberg JP, Bristol IJ, Churchill GA, Li R, et al. A major quantitative trait locus on chromosome 3 controls colitis severity in IL-10-deficient mice. Proc Natl Acad Sci USA. 2001;98(24):13820–25. doi: 10.1073/pnas.241258698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Ermann J, Garrett WS, Kuchroo J, Rourida K, Glickman JN, et al. Severity of innate immune-mediated colitis is controlled by the cytokine deficiency-induced colitis susceptibility-1 (Cdcs1) locus. Proc Natl Acad Sci USA. 2011;108(17):7137–41. doi: 10.1073/pnas.1104234108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Trombetta ES, Mellman I. Cell biology of antigen processing in vitro and in vivo. Annu Rev Immunol. 2005;23:975–1028. doi: 10.1146/annurev.immunol.22.012703.104538. [DOI] [PubMed] [Google Scholar]
- 207.Faure-André G, Vargas P, Yuseff M-I, Heuzé M, Diaz J, et al. Regulation of dendritic cell migration by CD74, the MHC class II-associated invariant chain. Science. 2008;322(5908):1705–10. doi: 10.1126/science.1159894. [DOI] [PubMed] [Google Scholar]
- 208.Cresswell P. Invariant chain structure and MHC class II function. Cell. 1996;84(4):505–7. doi: 10.1016/s0092-8674(00)81025-9. [DOI] [PubMed] [Google Scholar]
- 209.Lämmermann T, Bader BL, Monkley SJ, Worbs T, Wedlich-Söldner R, et al. Rapid leukocyte migration by integrin-independent flowing and squeezing. Nature. 2008;453(7191):51–55. doi: 10.1038/nature06887. [DOI] [PubMed] [Google Scholar]
- 210.Shin J-S, Ebersold M, Pypaert M, Delamarre L, Hartley A, Mellman I. Surface expression of MHC class II in dendritic cells is controlled by regulated ubiquitination. Nature. 2006;444(7115):115–18. doi: 10.1038/nature05261. [DOI] [PubMed] [Google Scholar]
- 211.Matsuki Y, Ohmura-Hoshino M, Goto E, Aoki M, Mito-Yoshida M, et al. Novel regulation of MHC class II function in B cells. EMBO J. 2007;26(3):846–54. doi: 10.1038/sj.emboj.7601556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Walseng E, Furuta K, Bosch B, Weih KA, Matsuki Y, et al. Ubiquitination regulates MHC class II-peptide complex retention and degradation in dendritic cells. Proc Natl Acad Sci USA. 2010;107(47):20465–70. doi: 10.1073/pnas.1010990107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Ma JK, Platt MY, Eastham-Anderson J, Shin J-S, Mellman I. MHC class II distribution in dendritic cells and B cells is determined by ubiquitin chain length. Proc Natl Acad Sci USA. 2012;109(23):8820–27. doi: 10.1073/pnas.1202977109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Coscoy L, Ganem D. PHD domains and E3 ubiquitin ligases: viruses make the connection. Trends Cell Biol. 2003;13(1):7–12. doi: 10.1016/s0962-8924(02)00005-3. [DOI] [PubMed] [Google Scholar]
- 215.Thibodeau J, Bourgeois-Daigneault M-C, Huppé G, Tremblay J, Aumont A, et al. Interleukin-10- induced MARCH1 mediates intracellular sequestration of MHC class II in monocytes. Eur J Immunol. 2008;38(5):1225–30. doi: 10.1002/eji.200737902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Jabbour M, Campbell EM, Fares H, Lybarger L. Discrete domains of MARCH1 mediate its localization, functional interactions, and posttranscriptional control of expression. J Immunol. 2009;183(10):6500–12. doi: 10.4049/jimmunol.0901521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Ohmura-Hoshino M, Matsuki Y, Mito-Yoshida M, Goto E, Aoki-Kawasumi M, et al. Cutting edge: requirement of MARCH-I-mediated MHC II ubiquitination for the maintenance of conventional dendritic cells. 2009;183(11):6893–97. doi: 10.4049/jimmunol.0902178. [DOI] [PubMed] [Google Scholar]
- 218.Baravalle G, Park H, McSweeney M, Ohmura-Hoshino M, Matsuki Y, et al. Ubiquitination of CD86 is a key mechanism in regulating antigen presentation by dendritic cells. J Immunol. 2011;187(6):2966–73. doi: 10.4049/jimmunol.1101643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Al-Daccak R, Mooney N, Charron D. MHC class II signaling in antigen-presenting cells. Curr Opin Immunol. 2004;16(1):108–13. doi: 10.1016/j.coi.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 220.Muth S, Schütze K, Schild H, Probst HC. Release of dendritic cells from cognate CD4+ T-cell recognition results in impaired peripheral tolerance and fatal cytotoxic T-cell mediated autoimmunity. Proc Natl Acad Sci USA. 2012;109(23):9059–64. doi: 10.1073/pnas.1110620109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Langenkamp A, Messi M, Lanzavecchia A, Sallusto F. Kinetics of dendritic cell activation: impact on priming of TH1, TH2 and nonpolarized T cells. Nat Immunol. 2000;1(4):311–16. doi: 10.1038/79758. [DOI] [PubMed] [Google Scholar]
- 222.Kajino K, Nakamura I, Bamba H, Sawai T, Ogasawara K. Involvement of IL-10 in exhaustion of myeloid dendritic cells and rescue by CD40 stimulation. Immunology. 2007;120(1):28–37. doi: 10.1111/j.1365-2567.2006.02474.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Abdi K, Singh NJ, Matzinger P. Lipopolysaccharide-activated dendritic cells: “exhausted” or alert and waiting? J Immunol. 2012;188(12):5981–89. doi: 10.4049/jimmunol.1102868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Kobayashi K, Hernandez LD, Galán JE, Janeway CA, Medzhitov R, Flavell RA. IRAK-M is a negative regulator of Toll-like receptor signaling. Cell. 2002;110(2):191–202. doi: 10.1016/s0092-8674(02)00827-9. [DOI] [PubMed] [Google Scholar]
- 225.Foster SL, Hargreaves DC, Medzhitov R. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature. 2007;447(7147):972–78. doi: 10.1038/nature05836. [DOI] [PubMed] [Google Scholar]
- 226.Liu X, Zhan Z, Li D, Xu L, Ma F, et al. Intracellular MHC class II molecules promote TLR-triggered innate immune responses by maintaining activation of the kinase Btk. Nat Immunol. 2011;12(5):416–24. doi: 10.1038/ni.2015. [DOI] [PubMed] [Google Scholar]
- 227.Wahl MI, Fluckiger AC, Kato RM, Park H, Witte ON, Rawlings DJ. Phosphorylation of two regulatory tyrosine residues in the activation of Bruton’s tyrosine kinase via alternative receptors. Proc Natl Acad Sci USA. 1997;94(21):11526–33. doi: 10.1073/pnas.94.21.11526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Jefferies CA, Doyle S, Brunner C, Dunne A, Brint E, et al. Bruton’s tyrosine kinase is a Toll/interleukin-1 receptor domain-binding protein that participates in nuclear factor κB activation by Toll-like receptor 4. J Biol Chem. 2003;278(28):26258–64. doi: 10.1074/jbc.M301484200. [DOI] [PubMed] [Google Scholar]
- 229.Josefowicz SZ, Lu L-F, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol. 2012;30:531–64. doi: 10.1146/annurev.immunol.25.022106.141623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Belkaid Y, Tarbell K. Regulatory T cells in the control of host-microorganism interactions. Annu Rev Immunol. 2009;27:551–89. doi: 10.1146/annurev.immunol.021908.132723. [DOI] [PubMed] [Google Scholar]
- 231.Darrasse-Jèze G, Deroubaix S, Mouquet H, Victora GD, Eisenreich T, et al. Feedback control of regulatory T cell homeostasis by dendritic cells in vivo. J Exp Med. 2009;206(9):1853–62. doi: 10.1084/jem.20090746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Bar-On L, Birnberg T, Kim K-W, Jung S. Dendritic cell-restricted CD80/86 deficiency results in peripheral regulatory T-cell reduction but is not associated with lymphocyte hyperactivation. Eur J Immunol. 2010;41:291–98. doi: 10.1002/eji.201041169. [DOI] [PubMed] [Google Scholar]
- 233.Seneschal J, Clark RA, Gehad A, Baecher-Allan CM, Kupper TS. Human epidermal Langerhans cells maintain immune homeostasis in skin by activating skin resident regulatory T cells. Immunity. 2012;36(5):873–84. doi: 10.1016/j.immuni.2012.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Hill JA, Hall JA, Sun C-M, Cai Q, Ghyselinck N, et al. Retinoic acid enhances Foxp3 induction indirectly by relieving inhibition from CD4+CD44hi cells. Immunity. 2008;29(5):758–70. doi: 10.1016/j.immuni.2008.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Hall JA, Grainger JR, Spencer SP, Belkaid Y. The role of retinoic acid in tolerance and immunity. Immunity. 2011;35(1):13–22. doi: 10.1016/j.immuni.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Shull MM, Ormsby I, Kier AB, Pawlowski S, Diebold RJ, et al. Targeted disruption of the mouse transforming growth factor-β1 gene results in multifocal inflammatory disease. Nature. 1992;359(6397):693–99. doi: 10.1038/359693a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Li MO, Wan YY, Sanjabi S, Robertson A-KL, Flavell RA. Transforming growth factor-β regulation of immune responses. Annu Rev Immunol. 2006;24:99–146. doi: 10.1146/annurev.immunol.24.021605.090737. [DOI] [PubMed] [Google Scholar]
- 238.Travis MA, Reizis B, Melton AC, Masteller E, Tang Q, et al. Loss of integrin αvβ8 on dendritic cells causes autoimmunity and colitis in mice. Nature. 2007;449(7160):361–65. doi: 10.1038/nature06110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Coombes JL, Siddiqui KRR, Arancibia-Cárcamo CV, Hall J, Sun C-M, et al. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-β and retinoic acid-dependent mechanism. J Exp Med. 2007;204(8):1757–64. doi: 10.1084/jem.20070590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Jaensson E, Uronen-Hansson H, Pabst O, Eksteen B, Tian J, et al. Small intestinal CD103+ dendritic cells display unique functional properties that are conserved between mice and humans. J Exp Med. 2008;205(9):2139–49. doi: 10.1084/jem.20080414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Worthington JJ, Czajkowska BI, Melton AC, Travis MA. Intestinal dendritic cells specialize to activate transforming growth factor-β and induce Foxp3+ regulatory T cells via integrin αvβ8. Gastroenterology. 2011;141(5):1802–12. doi: 10.1053/j.gastro.2011.06.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Li MO, Sanjabi S, Flavell RA. Transforming growth factor-β controls development, homeostasis, and tolerance of T cells by regulatory T cell-dependent and -independent mechanisms. Immunity. 2006;25(3):455–71. doi: 10.1016/j.immuni.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 243.Zhang N, Bevan MJ. TGF-β signaling to T cells inhibits autoimmunity during lymphopenia-driven proliferation. Nat Immunol. 2012;13(7):667–73. doi: 10.1038/ni.2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Melton AC, Bailey-Bucktrout SL, Travis MA, Fife BT, Bluestone JA, Sheppard D. Expression of αvβ8 integrin on dendritic cells regulates Th17 cell development and experimental autoimmune encephalomyelitis in mice. J Clin Investig. 2010;120(12):4436–44. doi: 10.1172/JCI43786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Mellor AL, Munn DH. IDO expression by dendritic cells: tolerance and tryptophan catabolism. Nat Rev Immunol. 2004;4(10):762–74. doi: 10.1038/nri1457. [DOI] [PubMed] [Google Scholar]
- 246.Grohmann U, Orabona C, Fallarino F, Vacca C, Calcinaro F, et al. CTLA-4-Ig regulates tryptophan catabolism in vivo. Nat Immunol. 2002;3(11):1097–101. doi: 10.1038/ni846. [DOI] [PubMed] [Google Scholar]
- 247.Pallotta MT, Orabona C, Volpi C, Vacca C, Belladonna ML, et al. Indoleamine 2,3-dioxygenase is a signaling protein in long-term tolerance by dendritic cells. Nat Immunol. 2011;12(9):870–78. doi: 10.1038/ni.2077. [DOI] [PubMed] [Google Scholar]
- 248.Mellor AL, Baban B, Chandler P, Marshall B, Jhaver K, et al. Cutting edge: induced indoleamine 2,3 dioxygenase expression in dendritic cell subsets suppresses T cell clonal expansion. J Immunol. 2003;171(4):1652–55. doi: 10.4049/jimmunol.171.4.1652. [DOI] [PubMed] [Google Scholar]
- 249.Manicassamy S, Reizis B, Ravindran R, Nakaya H, Salazar-Gonzalez RM, et al. Activation of β-catenin in dendritic cells regulates immunity versus tolerance in the intestine. Science. 2010;329(5993):849–53. doi: 10.1126/science.1188510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Denning TL, Wang Y-C, Patel SR, Williams IR, Pulendran B. Lamina propria macrophages and dendritic cells differentially induce regulatory and interleukin 17-producing T cell responses. Nat Immunol. 2007;8(10):1086–94. doi: 10.1038/ni1511. [DOI] [PubMed] [Google Scholar]
- 251.Park S-J, Nakagawa T, Kitamura H, Atsumi T, Kamon H, et al. IL-6 regulates in vivo dendritic cell differentiation through STAT3 activation. J Immunol. 2004;173(6):3844–54. doi: 10.4049/jimmunol.173.6.3844. [DOI] [PubMed] [Google Scholar]
- 252.Lunz JG, Specht SM, Murase N, Isse K, Demetris AJ. Gut-derived commensal bacterial products inhibit liver dendritic cell maturation by stimulating hepatic interleukin-6/signal transducer and activator of transcription 3 activity. Hepatology. 2007;46(6):1946–59. doi: 10.1002/hep.21906. [DOI] [PubMed] [Google Scholar]
- 253.Müller M, Fink K, Geisel J, Kahl F, Jilge B, et al. Intestinal colonization of IL-2 deficient mice with non-colitogenic B. vulgatus prevents DC maturation and T-cell polarization. PLoS ONE. 2008;3(6):e2376. doi: 10.1371/journal.pone.0002376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Kitamura H, Kamon H, Sawa S-I, Park S-J, Katunuma N, et al. IL-6-STAT3 controls intracellular MHC class II αβ dimer level through cathepsin S activity in dendritic cells. Immunity. 2005;23(5):491–502. doi: 10.1016/j.immuni.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 255.Murray PJ. The JAK-STAT signaling pathway: input and output integration. J Immunol. 2007;178(5):2623–29. doi: 10.4049/jimmunol.178.5.2623. [DOI] [PubMed] [Google Scholar]
- 256.Kühn R, Löhler J, Rennick D, Rajewsky K, Müller W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 1993;75(2):263–74. doi: 10.1016/0092-8674(93)80068-p. [DOI] [PubMed] [Google Scholar]
- 257.Melillo JA, Song L, Bhagat G, Blazquez AB, Plumlee CR, et al. Dendritic cell (DC)-specific targeting reveals Stat3 as a negative regulator of DC function. J Immunol. 2010;184(5):2638–45. doi: 10.4049/jimmunol.0902960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Alexander WS, Hilton DJ. The role of suppressors of cytokine signaling (SOCS) proteins in regulation of the immune response. Annu Rev Immunol. 2004;22:503–29. doi: 10.1146/annurev.immunol.22.091003.090312. [DOI] [PubMed] [Google Scholar]
- 259.Hanada T, Yoshida H, Kato S, Tanaka K, Masutani K, et al. Suppressor of cytokine signaling-1 is essential for suppressing dendritic cell activation and systemic autoimmunity. Immunity. 2003;19(3):437–50. doi: 10.1016/s1074-7613(03)00240-1. [DOI] [PubMed] [Google Scholar]
- 260.Shen L, Evel-Kabler K, Strube R, Chen S-Y. Silencing of SOCS1 enhances antigen presentation by dendritic cells and antigen-specific anti-tumor immunity. Nat Biotechnol. 2004;22(12):1546–53. doi: 10.1038/nbt1035. [DOI] [PubMed] [Google Scholar]
- 261.Evel-Kabler K, Song X-T, Aldrich M, Huang XF, Chen S-Y. SOCS1 restricts dendritic cells’ ability to break self tolerance and induce antitumor immunity by regulating IL-12 production and signaling. J Clin Investig. 2006;116(1):90–100. doi: 10.1172/JCI26169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Hong B, Ren W, Song X-T, Evel-Kabler K, Chen S-Y, Huang XF. Human suppressor of cytokine signaling 1 controls immunostimulatory activity of monocyte-derived dendritic cells. Cancer Res. 2009;69(20):8076–84. doi: 10.1158/0008-5472.CAN-09-1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Kinjyo I, Hanada T, Inagaki-Ohara K, Mori H, Aki D, et al. SOCS1/JAB is a negative regulator of LPS-induced macrophage activation. Immunity. 2002;17(5):583–91. doi: 10.1016/s1074-7613(02)00446-6. [DOI] [PubMed] [Google Scholar]
- 264.Baetz A, Frey M, Heeg K, Dalpke AH. Suppressor of cytokine signaling (SOCS) proteins indirectly regulate Toll-like receptor signaling in innate immune cells. J Biol Chem. 2004;279(52):54708–15. doi: 10.1074/jbc.M410992200. [DOI] [PubMed] [Google Scholar]
- 265.Mansell A, Smith R, Doyle SL, Gray P, Fenner JE, et al. Suppressor of cytokine signaling 1 negatively regulates Toll-like receptor signaling by mediating Mal degradation. Nat Immunol. 2006;7(2):148–55. doi: 10.1038/ni1299. [DOI] [PubMed] [Google Scholar]
- 266.Morita Y, Naka T, Kawazoe Y, Fujimoto M, Narazaki M, et al. Signals transducers and activators of transcription (STAT)-induced STAT inhibitor-1 (SSI-1)/suppressor of cytokine signaling-1 (SOCS-1) suppresses tumor necrosis factor α-induced cell death in fibroblasts. Proc Natl Acad Sci USA. 2000;97(10):5405–10. doi: 10.1073/pnas.090084797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Tsukada J, Ozaki A, Hanada T, Chinen T, Abe R, et al. The role of suppressor of cytokine signaling 1 as a negative regulator for aberrant expansion of CD8α+ dendritic cell subset. Int Immunol. 2005;17(9):1167–78. doi: 10.1093/intimm/dxh294. [DOI] [PubMed] [Google Scholar]
- 268.Tezuka H, Abe Y, Asano J, Sato T, Liu J, et al. Prominent role for plasmacytoid dendritic cells in mucosal T cell-independent IgA induction. Immunity. 2011;34(2):247–57. doi: 10.1016/j.immuni.2011.02.002. [DOI] [PubMed] [Google Scholar]
- 269.Fuertes MB, Kacha AK, Kline J, Woo S-R, Kranz DM, et al. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8α+ dendritic cells. J Exp Med. 2011;208(10):2005–16. doi: 10.1084/jem.20101159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Seitz HM, Camenisch TD, Lemke G, Earp HS, Matsushima GK. Macrophages and dendritic cells use different Axl/Mertk/Tyro3 receptors in clearance of apoptotic cells. J Immunol. 2007;178(9):5635–42. doi: 10.4049/jimmunol.178.9.5635. [DOI] [PubMed] [Google Scholar]
- 271.Lemke G, Burstyn-Cohen T. TAM receptors and the clearance of apoptotic cells. Ann N Y Acad Sci. 2010;1209:23–29. doi: 10.1111/j.1749-6632.2010.05744.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Rothlin CV, Ghosh S, Zuniga EI, Oldstone MBA, Lemke G. TAM receptors are pleiotropic inhibitors of the innate immune response. Cell. 2007;131(6):1124–36. doi: 10.1016/j.cell.2007.10.034. [DOI] [PubMed] [Google Scholar]
- 273.Sen P, Wallet MA, Yi Z, Huang Y, Henderson M, et al. Apoptotic cells induce Mer tyrosine kinase-dependent blockade of NF-κB activation in dendritic cells. Blood. 2007;109(2):653–60. doi: 10.1182/blood-2006-04-017368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Wallet MA, Sen P, Flores RR, Wang Y, Yi Z, et al. MerTK is required for apoptotic cell-induced T cell tolerance. J Exp Med. 2008;205(1):219–32. doi: 10.1084/jem.20062293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Yi Z, Li L, Matsushima GK, Earp HS, Wang B, Tisch R. A novel role for c-Src and STAT3 in apoptotic cell-mediated MerTK-dependent immunoregulation of dendritic cells. Blood. 2009;114(15):3191–98. doi: 10.1182/blood-2009-03-207522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Lu Q, Lemke G. Homeostatic regulation of the immune system by receptor tyrosine kinases of the Tyro 3 family. Science. 2001;293(5528):306–11. doi: 10.1126/science.1061663. [DOI] [PubMed] [Google Scholar]
- 277.Gohlke PR, Williams JC, Vilen BJ, Dillon SR, Tisch R, Matsushima GK. The receptor tyrosine kinase MerTK regulates dendritic cell production of BAFF. Autoimmunity. 2009;42(3):183–97. doi: 10.1080/08916930802668586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Suh C-H, Hilliard B, Li S, Merrill JT, Cohen PL. TAM receptor ligands in lupus: protein S but not Gas6 levels reflect disease activity in systemic lupus erythematosus. Arthritis Res Ther. 2010;12(4):R146. doi: 10.1186/ar3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Lee EG, Boone DL, Chai S, Libby SL, Chien M, et al. Failure to regulate TNF-induced NF-κB and cell death responses in A20-deficient mice. Science. 2000;289(5488):2350–54. doi: 10.1126/science.289.5488.2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Wertz IE, O’Rourke KM, Zhou H, Eby M, Aravind L, et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-κB signalling. Nature. 2004;430(7000):694–99. doi: 10.1038/nature02794. [DOI] [PubMed] [Google Scholar]
- 281.Boone DL, Turer EE, Lee EG, Ahmad R-C, Wheeler MT, et al. The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nat Immunol. 2004;5(10):1052–60. doi: 10.1038/ni1110. [DOI] [PubMed] [Google Scholar]
- 282.Hitotsumatsu O, Ahmad R-C, Tavares R, Wang M, Philpott D, et al. The ubiquitin-editing enzyme A20 restricts nucleotide-binding oligomerization domain containing 2 triggered signals. Immunity. 2008;28(3):381–90. doi: 10.1016/j.immuni.2008.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Tavares RM, Turer EE, Liu CL, Advincula R, Scapini P, et al. The ubiquitin modifying enzyme A20 restricts B cell survival and prevents autoimmunity. Immunity. 2010;33(2):181–91. doi: 10.1016/j.immuni.2010.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Kool M, van Loo G, Waelput W, De Prijck S, Muskens F, et al. The ubiquitin-editing protein A20 prevents dendritic cell activation, recognition of apoptotic cells, and systemic autoimmunity. Immunity. 2011;35(1):82–96. doi: 10.1016/j.immuni.2011.05.013. [DOI] [PubMed] [Google Scholar]
- 285.Evans PC, Ovaa H, Hamon M, Kilshaw PJ, Hamm S, et al. Zinc-finger protein A20, a regulator of inflammation and cell survival, has de-ubiquitinating activity. Biochem J. 2004;378(Pt. 3):727–34. doi: 10.1042/BJ20031377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Bosanac I, Wertz IE, Pan B, Yu C, Kusam S, et al. Ubiquitin binding to A20 ZnF4 is required for modulation of NF-κB signaling. Mol Cell. 2010;40(4):548–57. doi: 10.1016/j.molcel.2010.10.009. [DOI] [PubMed] [Google Scholar]
- 287.Shembade N, Ma A, Harhaj EW. Inhibition of NF-κB signaling by A20 through disruption of ubiquitin enzyme complexes. Science. 2010;327(5969):1135–39. doi: 10.1126/science.1182364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Skaug B, Chen J, Du F, He J, Ma A, Chen ZJ. Direct, noncatalytic mechanism of IKK inhibition by A20. Mol Cell. 2011;44(4):559–71. doi: 10.1016/j.molcel.2011.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Song HY, Rothe M, Goeddel DV. The tumor necrosis factor-inducible zinc finger protein A20 interacts with TRAF1/TRAF2 and inhibits NF-κB activation. Proc Natl Acad Sci USA. 1996;93(13):6721–25. doi: 10.1073/pnas.93.13.6721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Heyninck K, Van Huffel S, Kreike M, Beyaert R. Yeast two-hybrid screening for proteins interacting with the anti-apoptotic protein A20. Methods Mol Biol. 2004;282:223–41. doi: 10.1385/1-59259-812-9:223. [DOI] [PubMed] [Google Scholar]
- 291.Verstrepen L, Verhelst K, van Loo G, Carpentier I, Ley SC, Beyaert R. Expression, biological activities and mechanisms of action of A20 (TNFAIP3) Biochem Pharmacol. 2010;80(12):2009–20. doi: 10.1016/j.bcp.2010.06.044. [DOI] [PubMed] [Google Scholar]
- 292.Vereecke L, Sze M, Mc Guire C, Rogiers B, Chu Y, et al. Enterocyte-specific A20 deficiency sensitizes to tumor necrosis factor-induced toxicity and experimental colitis. J Exp Med. 2010;207(7):1513–23. doi: 10.1084/jem.20092474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Caton ML, Smith-Raska MR, Reizis B. Notch-RBP-J signaling controls the homeostasis of CD8− dendritic cells in the spleen. J Exp Med. 2007;204(7):1653–64. doi: 10.1084/jem.20062648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Naik SH, Metcalf D, van Nieuwenhuijze A, Wicks I, Wu L, et al. Intrasplenic steady-state dendritic cell precursors that are distinct from monocytes. Nat Immunol. 2006;7(6):663–71. doi: 10.1038/ni1340. [DOI] [PubMed] [Google Scholar]
- 295.Kato M, Sanada M, Kato I, Sato Y, Takita J, et al. Frequent inactivation of A20 in B-cell lymphomas. Nature. 2009;459(7247):712–16. doi: 10.1038/nature07969. [DOI] [PubMed] [Google Scholar]
- 296.Schmitz R, Hansmann M, Bohle V, Martin-Subero J, Hartmann S, et al. TNFAIP3 (A20) is a tumor suppressor gene in Hodgkin lymphoma and primary mediastinal B cell lymphoma. J Exp Med. 2009;206:981–89. doi: 10.1084/jem.20090528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Wu H-J, Ivanov II, Darce J, Hattori K, Shima T, et al. Gut-residing segmented filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity. 2010;32(6):815–27. doi: 10.1016/j.immuni.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature. 2012;482(7384):179–85. doi: 10.1038/nature10809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Turer EE, Tavares RM, Mortier E, Hitotsumatsu O, Advincula R, et al. Homeostatic MyD88-dependent signals cause lethal inflammation in the absence of A20. J Exp Med. 2008;205(2):451–64. doi: 10.1084/jem.20071108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Melis L, Elewaut D. Progress in spondylarthritis. Immunopathogenesis of spondyloarthritis: Which cells drive disease? Arthritis Res Ther. 2009;11(3):233. doi: 10.1186/ar2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Gaston H. Mechanisms of disease: the immunopathogenesis of spondyloarthropathies. Nat Clin Pract Rheumatol. 2006;2(7):383–92. doi: 10.1038/ncprheum0219. [DOI] [PubMed] [Google Scholar]
- 302.Armaka M, Apostolaki M, Jacques P, Kontoyiannis DL, Elewaut D, Kollias G. Mesenchymal cell targeting by TNF as a common pathogenic principle in chronic inflammatory joint and intestinal diseases. J Exp Med. 2008;205(2):331–37. doi: 10.1084/jem.20070906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Kontoyiannis D, Boulougouris G, Manoloukos M, Armaka M, Apostolaki M, et al. Genetic dissection of the cellular pathways and signaling mechanisms in modeled tumor necrosis factor-induced Crohn’s-like inflammatory bowel disease. J Exp Med. 2002;196(12):1563–74. doi: 10.1084/jem.20020281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Keller C, Webb A, Davis J. Cytokines in the seronegative spondyloarthropathies and their modification by TNF blockade: a brief report and literature review. Ann Rheum Dis. 2003;62(12):1128–32. doi: 10.1136/ard.2003.011023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Milia AF, Ibba-Manneschi L, Manetti M, Benelli G, Generini S, et al. Evidence for the prevention of enthesitis in HLA-B27/hβ2m transgenic rats treated with a monoclonal antibody against TNF-α. J Cell Mol Med. 2011;15(2):270–79. doi: 10.1111/j.1582-4934.2009.00984.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Taurog JD. The role of HLA-B27 in spondyloarthritis. J Rheumatol. 2010;37(12):2606–16. doi: 10.3899/jrheum.100889. [DOI] [PubMed] [Google Scholar]