

Abstract
Described as an autoimmune collagen vascular disease, the most striking feature of scleroderma may be a systemic vasculopathy. This vasculopathy includes characteristic noninflammatory macrovascular and microvascular changes with dramatic and possibly occlusive formation of a thickened neointima. Scleroderma vessels also have an unusual endothelial phenotype, with loss of normal markers including vascular endothelial (VE)-cadherin. These endothelial cells express type 1 interferon and regulator of G protein signaling 5 (RGS5), two molecules associated with vascular rarefaction. These genes may be important because tissue is hypoxic with high levels of vascular endothelial growth factor (VEGF), especially early in the disease. The combination of VEGF and rarefaction is not necessarily paradoxical. VEGF-mediated angiogenesis creates labile vessels that may not survive unless the vessel acquires a smooth muscle coat. The combination of interferon and RGS5 is consistent with an antiangiogenic phenotype. We offer a hypothesis that places vascular injury at the center of this disease and also suggest possible clinical approaches for arresting and/or reversing the disease.
Introduction
Systemic sclerosis (scleroderma) has two very different subsets: limited scleroderma and diffuse scleroderma. These can be identified by differences in clinical findings and by the temporal relationship to vasospasm.
Limited scleroderma is so named because skin involvement is limited to the hands and face. Limited scleroderma is characterized by calcinosis, Raynaud’s phenomenon, esophageal dysmotility, sclerodactyly, and telangiectasia (CREST syndrome). The most common cause of death in limited scleroderma is pulmonary hypertension or pulmonary fibrosis. Because Raynaud’s phenomenon is a near universal prodrome, there are long-standing suggestions that the initial event in limited scleroderma may affect the vasculature [1]. Even in localized scleroderma, which has fewer systemic features, Raynaud’s progresses to peripheral ischemia with digital ulceration and loss of fingers [2]. Although the vasospasm associated with Raynaud’s phenomenon may be prodromal, it is not itself a sufficient cause of the disease because symptoms of vasospasm can precede diagnosis of limited scleroderma by many years, and the incidence of scleroderma in the population of people with Raynaud’s phenomenon is very low [3•].
Diffuse scleroderma appears with a much wider extent of skin involvement. Patients with diffuse scleroderma are prone to kidney crisis and pulmonary fibrosis. These patients develop Raynaud’s phenomenon within 1 year of onset of symptoms but they tend to not have it as a prodrome. Both limited and diffuse scleroderma patients have ischemic changes in their digits (80%–95% have Raynaud’s phenomenon), some with ulcers, gangrene, and loss of fingers [2]. The ischemia is not limited to digits, although this may be the most clinically obvious site.
Intimal Hyperplasia
Intimal hyperplasia, or more correctly the formation of a neointima, is a common response to arterial injury, and it is a characteristic feature of the arteries in scleroderma. There is an extensive and somewhat contradictory literature concerning neointimal formation. Older studies assumed intima was formed by migration and proliferation of medial smooth muscle cells. However, recent studies suggest that adventitial fibroblasts may be the source of neointimal cells [4•]. One intriguing possibility is that intimal cells may arise from adventitial stem cells under control of the hedgehog signaling pathway [5•]. We use the common term intimal hyperplasia in this article to be consistent with the literature.
The best evidence for intimal hyperplasia in scleroderma is found in a remarkable autopsy series reported by D’Angelo et al. [6] that is summarized in Table 1. The characteristic changes (ie, intimal hyperplasia and fibrosis) are seen in both large muscular arteries and precapillary arterioles. The intimal hyperplasia may be quite spectacular, progressing to what appears to be an occlusive narrowing of the lumen. Skin, gastrointestinal tract, and skeletal muscle findings are most specific for scleroderma because these tissue changes were rarely, if ever, found in nonscleroderma autopsies. Arterial lesions of the kidney and lung are also very prevalent. Overall, these changes affect skin, gastrointestinal tract, lungs, kidneys, and skeletal muscle, and the consequence is death by pulmonary hypertension, heart failure, or kidney crisis [7]. Intimal hyperplasia may be the cause of Raynaud’s phenomenon. There is evidence from animal models that intimal hyperplasia is formed in response to inflammation [7,8], and intimal hyperplasia precedes the vasospasm that closes the ductus arteriosus [9]. Increased vasospasm in intimal hyperplasia might be accounted for by decreased access of endothelial-derived nitric oxide to the media or by vasoactive products of this newly formed tissue [8].
Table 1. Summary of vascular pathology findings*.
| System | Pathology | Scleroderma, % | Control, % | P value |
|---|---|---|---|---|
| Pulmonary | Interstitial fibrosis and arterial thickening | 81 | 22 | N/A |
| Fibrous pleuritis | 28 | 7 | 0.01 | |
| Coronary | Myocardial fibrosis | 81 | 55 | 0.01 |
| Arteriolar concentric intimal thickening | 17 | 2 | 0.01 | |
| Pericarditis fibrous and adhesive | 53 | 14 | 0.001 | |
| Gastrointestinal | Esophageal atrophy and fibrosis | 74 | 0 | 0.001 |
| Lesions of reflux | 40 | 19 | 0.05 | |
| Small intestine atrophy and fibrosis | 48 | 2 | 0.001 | |
| Large intestine atrophy and fibrosis | 39 | 0 | 0.001 | |
| Renal | Artery hyperplasia and bone marrow thickening, fibrinoid necrosis | 58 | 8.9 | 0.001 |
| Skeletal muscle | Atrophy | 41 | 0 | – |
| Round cell myositis | 8 | 0 | – | |
| Arterioles | Noninflammatory intimal proliferation in two or more organ systems | 24 | 0 | 0.01 |
| Inflammatory polyarteritis | 9 | 0 | – |
The systems that are included had atrophy, fibrosis, diffuse arterial and arteriolar thickening, and polyarteritis. Percentages show the number of positive findings.
(Data from D’Angelo et al. [6].)
Endothelial Loss
Characteristic vascular changes in scleroderma also occur at the capillary level. The change most studied in the clinical literature is at the level of the nail beds. Changes in the nailfold capillaries are one of the first signs in scleroderma [10]. Because these changes have only been described by nail bed microscopy, little is known about the mechanism of this change. However, the morphology seen in the scleroderma nail beds closely resembles changes seen when vascular endothelial growth factor (VEGF) is overexpressed locally [11], and nailfold changes appear to be associated with very high levels of circulating VEGF [12].
These abnormalities have led to speculation that endothelial death is a primary and ongoing process in scleroderma. Although there is evidence from in vitro studies that scleroderma serum may contain antiangiogenic or other agents that are toxic to endothelium, apoptotic change or even increased endothelial turnover has not been demonstrated in vivo [13•,14]. Moreover, in our own work, we [15••] have not observed such evidence in studies of skin biopsies. The lack of evidence for apoptosis presents a paradox because we now know that there are marked decreases in the number of capillaries present in both clinically involved and uninvolved skin. These changes are present as early in the disease process as we have studied [15••]. The number of capillaries is decreased, whereas larger vessels appear to persist [15••]. Possible explanations include episodic cell death or detachment of endothelial cells into the circulation. Although we lack clear evidence for either of these, the literature does report an increased number of circulating endothelial cells in scleroderma [16].
Relationship of Vasculopathy to Fibrosis
Although the major clinical manifestations of scleroderma are attributed to fibrosis, the fibrosis is associated with intimal hyperplasia of the arteries supplying those affected organs. This may result in skin fibrosis, pulmonary hypertension, myocardial dysfunction, and renal involvement [17,18]. As a disease model, it is therefore proposed that the intimal changes are causal to organ damage and fibrosis, perhaps by making arteries more likely to undergo vasospasm or by physical restriction of blood flow.
Whatever the mechanism, we do not know whether the vasculopathy precedes fibrosis. Surprisingly, very little is known about the interaction of vascular injury and fibrotic changes. In chronic transplant arteriopathy, both intimal hyperplasia and diffuse fibrosis are observed in an organ-specific way [19], and there is evidence that hypoxia mediates both the angiogenic and the fibrotic signaling pathways [20]. Moreover, an inhibitor of cell migration, fasudil, blocks both intimal hyperplasia and fibrosis in the transplant atherosclerosis model, which is presented in the following text as a potential animal model for scleroderma [19].
Animal Models of Scleroderma Vasculopathy
The most compelling evidence for endothelial injury as an initial event in scleroderma comes from the studies on the University of California, Davis 200 chicken model of genetic progressive fibrosis [21]. The endothelial cells in the skin, lung, kidney, and esophagus of this bird show evidence of apoptosis. In addition, the perivascular spaces develop inflammatory infiltrates, the bird develops anti-endothelial antibodies systemically, and the first signs of fibrosis are observed in the very early stages of disease [22]. The inflammatory infiltrates and signs of apoptotic endothelial cells disappear as the disease progresses, but the birds go on to develop severe fibrotic dermal disease and progressive fibrosis of internal organs, which resembles scleroderma.
Another common model for scleroderma is chronic graft versus host disease. At the clinical level, changes reminiscent of scleroderma have been described in graft versus host disease [23]. This would be an attractive model because one might imagine finding specific endothelial epitopes responsible for the graft effect. Fibrosis is consistently observed in the appropriate mouse strains [24]. However, endothelial death is not obvious in this model, and we have not found such evidence in our own unpublished studies.
Other animal models of scleroderma provide less evidence for an endothelial mechanism. The tight skin murine model of scleroderma is an extremely fibrotic model but it does not show evidence of vascular injury [25]. Other models currently being studied, including bleomycin-induced scleroderma, have not shown evidence for vascular damage, endothelial or otherwise [26,27].
Finally, as already noted, transplant vasculopathy, sometimes mistakenly termed transplant atherosclerosis, has many features in common with scleroderma. This iatrogenic disease occurs in most or all organs months after transplant, and it is not amenable (at least at that time) to immunosuppression. Unlike atherosclerosis, transplant vasculopathy is not marked by lipid accumulation or inflammation, although the syndrome may be exacerbated by the presence of atherosclerosis. Transplant vasculopathy also differs from the extensive inflammatory response associated with the classical autoimmune vasculopathies [19]. Like scleroderma, transplant vasculopathy is a chronic disease characterized by severe intimal hyperplasia. Although less marked, these tissues also show diffuse fibrosis, and fasudil, a Rho kinase inhibitor, blocks both the intimal hyperplasia and fibrotic changes [19].
Role of Endothelial Cell Injury Versus Endothelial Cell Death
Despite in vitro studies [28,29] of endothelial death, if death of endothelial cells does occur in scleroderma, it is an early process not found in biopsies that have been obtained as early as 3 months after diagnosis. Although we do not have evidence for cell death, we have found high levels of interferon (IFN) production in scleroderma. The observation of high levels of IFN in scleroderma skin is consistent with the known antiangiogenic activities of this cytokine [15••].
The mechanism of inhibition of angiogenesis or promotion of capillary loss induced by IFN has not been studied. The absence of evidence for cell death, however, does not rule out other evidence for other forms of endothelial injury. A number of reports have claimed, for example, that scleroderma patients show increased numbers of circulating endothelial cells (either dead cells or precursors) [16,30•]. In addition, systemic evidence from clinical studies for endothelial injury includes elevated levels of endothelin and angiotensin-converting enzyme, among other markers, and a possible role for granzyme B in activating angiogenesis inhibitors [29,31].
Consistent with these systemic markers, our histochemical studies show that the endothelial cells in scleroderma are functionally abnormal. The most characteristic marker of endothelial lineage, vascular endothelial (VE)-cadherin, is lost. Because VE-cadherin is required to form endothelial tubes, we suspect that angiogenesis is not possible for these cells, even in the presence of high levels of VEGF observed in ischemic tissues of scleroderma patients [15••]. Although these scleroderma vessels do not show a prominent inflammatory infiltrate, the remaining endothelial cells do resemble inflammatory endothelium in having a greatly increased cytoplasmic volume, an activated hypochromic nucleus, expression of CD123 (Fig. 1), and expression of inflammatory markers on the cell surface [32,33].
Figure 1.
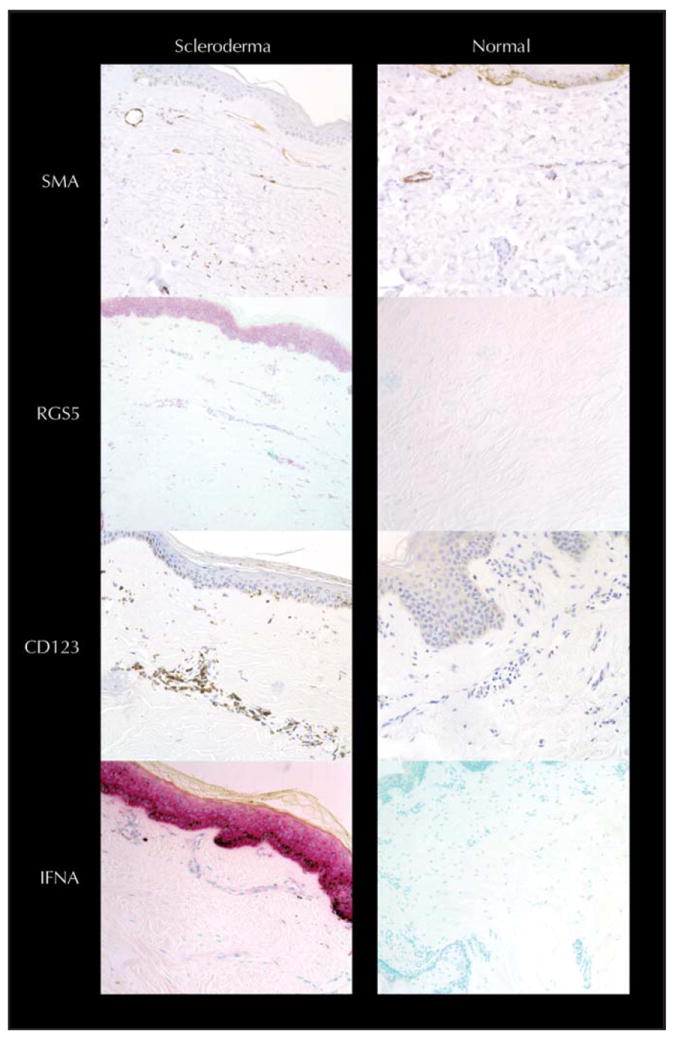
Endothelial phenotype in scleroderma compared with normal control. Immunohistochemistry for smooth muscle actin (SMA) stain (brown Dab stain) in the capillaries of scleroderma smooth muscle actin extends to the very top of the dermal papilla, unlike normal skin where SMA stain ends at the superficial horizontal plexus. SMA staining is also increased in the media of vessels in the reticular dermis (data not shown). SMA staining is present in scattered myofibroblasts present in the dermal collagen matrix of scleroderma. Regulator of G protein signaling 5 (RGS5) in situ expression of RNA is also increased in scleroderma. It is positive in cells (pink color) scattered in the dermal matrix, around blood vessels, and in the keratinocytes of the epidermis. Immunohistochemistry for the inflammatory marker CD123 (brown Dab stain) is increased in scleroderma tissue, especially around the blood vessels and in endothelial cells. In situ expression of interferon α (IFNA) RNA is increased dramatically in scleroderma skin (pink-colored cells) in a variety of cell types.
Role of Interferon
Rarefaction, a decrease in the density of vessels feeding a tissue, is a prominent feature of scleroderma. Even though rarefaction has not been described in other models of diffuse fibrosis, the observations in transplant vasculopathy suggest that rarefaction could itself be one cause of fibrosis. Unfortunately, although rarefaction is well documented in hypertension, congestive heart failure, and in the latter stages of wound healing, there is minimal literature on the mechanisms controlling rarefaction [34].
The data, however, do lead to a tangible hypothesis for scleroderma. As already noted, part of the antiangiogenic phenotype of scleroderma skin is the expression of IFNα, a type 1 IFN (Fig. 1). In our studies, we have shown that both IFNα-1 and INFα-2 RNA are upregulated in the endothelial cells of scleroderma patients. Expression of IFNα is not limited to the endothelium, as many other cells in the scleroderma dermis appear to express IFNα. We also have evidence of signaling induced by IFNα in scleroderma tissues [35••]. The class of type 1 IFN produced appears to be specific. In studies not yet published, we have found that the RNA expression of the other type 1 IFN, IFNβ, is not increased in scleroderma.
Evidence has shown that IFNα and IFNβ have differential effects on angiogenesis and antitumor activity [36]. IFNα appears to have a role in antitumor immune response (sterile inflammation) and tumor recognition, which strongly implicates it in autoimmune response, whereas IFNβ appears to mediate endothelial apoptotic response. It may also be worth considering the possibility that the antitumor activity is directed at the vasculature, given the demonstrated antiangiogenic activity of type 1 IFNs. One example of this is the studies on infantile heman giomas. Not only is the endogenous expression of IFNα inversely correlated with tumor growth, but treatment with exogenous IFNα has been successful on hemangiomas and caused them to regress [37]. Type 1 IFNs are known to inhibit endothelial migration and promote endothelial death [38]. Possible effects of IFN on other vessel wall cells remain to be studied. The cells of interest may be the perivascular cells.
Possible Role of Perivascular Cells
There is abundant evidence that the perivascular cells (smooth muscle, pericytes, and adventitial cells) play a critical role in maintaining vascular integrity. Studies of angiogenesis by Dor et al. [39] and vascular development by Cho et al. [40] have offered a critical perspective on how interactions of endothelial cells with other cell types may determine angiogenesis and stabilization of the newly formed vessels. Development of vascular networks and pruning of these networks are natural events that occur during development and tissue repair or response to injury [41]. Angiogenesis, the formation of new capillaries by branching, is mediated primarily by hypoxia, with the resulting stimulation of production of hypoxia-inducible factor 1α (HIF1α) and VEGF from smooth muscle cells in the vascular wall. During mammalian development, the nascent endothelial tubes are labile until coated with mesenchymal cells that form the smooth muscle or pericytes coats of the final vasculature. Platelet-derived growth factor B plays a critical role in the formation of pericytes along capillary spouts, and it appears that this event correlates with cessation of angiogenic branching. The pericytes express the gene regulator of G protein signaling 5 (RGS5), a smooth muscle gene we have shown to be overexpressed in scleroderma skin and that can identify the afferent vasculature [40].
A possible explanation of the role of RGS5 in scleroderma is indicated by recent studies implicating this gene in stabilization of vessels in cancer. RGS5 is overexpressed in vessels in premalignant lesions, a period of time when angiogenesis is upregulated but where the vasculature, unless coated with pericytes, is poorly organized [42]. RGS5 is also upregulated in wound healing and during ovulation [42]. It is often notably increased in malignancies that are not HIF1α deficient (highly malignant and vascularized astrocytomas and pancreatic islet cells tumors), and it returns to normal expression levels in these tumors after chemotherapy [42].
Although the mechanism of action of RGS5 is unclear, these data from developmental biology and from tumor angiogenesis associate RGS5 with the loss of what Dor et al. [39] have called the “plasticity window,” a time when angiogenic vessels have yet to be stabilized by the recruitment of mural cells. Pericytes also coat the capillaries and venules of mature stable tissues. Once vessels are developed, Dor et al. [39] suggest these pericytes must detach from the vessels to make them more plastic and allow vascular remodeling. Detachment of pericytes from the endothelium, however, destabilizes the tubes, making the vessel vulnerable to pruning. In this theory, VEGF stimulates both branching angiogenesis but also makes the newly formed tubes labile if VEGF is antagonized. The latter effect is believed to be the reason that anti-VEGF antibodies are clinically successful in destroying tumor vasculature. If the VEGF levels fall too low prior to coating and stabilization, the vessels are lost. This process may occur in preeclampsia, as rarefaction of capillaries is mediated though the inhibition of VEGF expression [43•].
In considering the source of perivascular cells or fibroblasts in scleroderma, it is important not to restrict ourselves to smooth muscle. The term adventitial cell is used to describe fibroblasts associated with the connective tissue sheet surrounding the blood vessel. The adventitial cell is traditionally considered to be a perivascular fibrocyte, but this term is not well defined at the level of cell lineage.
The lineage of the adventitial cell may be relevant to the characteristic scarring seen in this disease. Fibroblasts of scleroderma express smooth muscle genes, especially smooth muscle actin, and have been called myofibroblasts. The term myofibroblasts, however, is very descriptive and does not tell us where or how these cells arise. Adventitial cells may well be the source of the myofibroblast because we know that adventitial cells have stem cell properties and can differentiate into smooth muscle [5•,44]. Finally, our studies showed that the scleroderma myofibroblast expresses RGS5, a characteristic marker of arterial smooth muscle and the same gene noted to be characteristic of the pericytes that stabilize newly formed vessels [40]. In addition to expression in myofibroblasts, expression of RGS5 is greatly increased in smooth muscle cell coats of scleroderma vessels (Fig. 1) [16].
Evidence of a specific molecule that might regulate the differentiation of adventitial cells into interstitial cells or intimal cells has come from recent studies of arteriogenesis in the heart. Lavine et al. [45••,46•] have shown that stabilization of blood vessels in adult vascular networks is dependent on the presence of hedgehog signaling, and that the target of this trophic factor is the interstitial cell. Intriguingly, Passman et al. [5•] have shown that the hedgehog responsive cell is an adventitial cell with stem cell properties, whereas Torsney et al. [44] have shown that very similar cells are the source of the neointima in transplant atherosclerosis.
In summary, the outer layer of the vessel wall may be a critical player in the events leading to scleroderma. These cells could be the source of both the myofibroblast and the intimal cell, placing them in the center of the three major components of scleroderma: vasospasm, rarefaction, and fibrosis.
Therapeutic Implications
The final question we need to answer is whether this hypothesis suggests strategies that might intervene in this disfiguring, disabling, and often lethal process. The most obvious example may be the use of fasudil, because this agent has already been shown to have a beneficial effect in transplant vasculopathy [19]. IFN antagonists may also be worthy of consideration [47]. Antagonists to the hedgehog signaling pathway may be worth considering if our hypothesis for adventitial cell holds true.
The most impressive approach, however, may be a return to the underlying immunologic issue. We [48] and others have reported that bone marrow stem cell transplants may arrest scleroderma and, in our data, promote vascular regeneration. Moreover, the abnormal endothelial phenotype is reversible in scleroderma. When we compared the initial baseline biopsies with the 5-year follow-up biopsies, we discovered that the capillaries had grown back in five of the seven cases. Stem cell transplant patients who regained their capillaries also reexpressed VE-cadherin and lost expression of IFNα. Expression of Ki67 and smooth muscle markers, including RGS5, is also lost in the regenerated tissue. The obvious suggestion is that an immunologically active cell mediates the vasculopathy and, when eliminated, scleroderma is at least partially reversible.
Conclusions
We would like to finish this discussion with a unifying hypothesis. Let us hypothesize for a moment that the pathogenesis of scleroderma is initiated by systemic endothelial injury. It is possible that such events occur frequently from exposure to environmental toxins, immune responses to infections, and so forth. In this hypothetical event, many endothelial cells die and their fragmented DNA circulates. We have demonstrated that receptors activated by DNA antibody complexes found in scleroderma serum stimulate the production of IFNα [49••]. This injury would, in our hypothesis, lead to tissue hypoxia and expression of VEGF. Although VEGF expression explains capillary malformations and telangiectasia, VEGF also causes pericytes to detach from vessels. These vulnerable vessels, exposed to type 1 IFNs, are unable to proliferate or migrate, so new vessels never replace the vessels that died in the original injury. Vessels that have thick layers of mural cells are the only ones that survive, and because the vasculature is pruned, increased flow causes the remaining vessels to further remodel towards stability. The paucity of small vessels, however, means that the tissue remains hypoxic. Persistent HIF1α signaling results in expression of transforming growth factor-β, promoting the differentiation of adventitial cells into myofibroblasts, leading to fibrosis [50••]. The mural cells in the vessels continue to proliferate and form a neointima. This increases vessel spasticity, resulting in profound ischemia, digital ulcers, and gangrene.
Taken together, these data suggest that scleroderma has a vasculopathy that is mediated by an initial injurious event that probably causes endothelial cell death and loss of capillaries. This is followed by a noninflammatory reactive fibrosis and formation of a neointima. This results in vessel spasticity that results in more ischemia and vascular injury, thus beginning the cycle again and producing a progressive vasculopathy.
Acknowledgments
Dr. Nash can be contacted at the Fred Hutchinson Cancer Research Center in Seattle, WA.
Footnotes
Disclosures
No potential conflicts of interest relevant to this article were reported.
References and Recommended Reading
Papers of particular interest, published recently, have been highlighted as:
-
•
Of importance
-
••
Of major importance
- 1.LeRoy EC. Systemic sclerosis. A vascular perspective. Rheum Dis Clin North Am. 1996;22:675–694. doi: 10.1016/s0889-857x(05)70295-7. [DOI] [PubMed] [Google Scholar]
- 2.Maricq HR. Capillary abnormalities, Raynaud’s phenomenon, and systemic sclerosis in patients with localized scleroderma. Arch Dermatol. 1992;128:630–632. [PubMed] [Google Scholar]
- 3•.Carpentier PH, Satger B, Poensin D, Maricq HR. Incidence and natural history of Raynaud phenomenon: a long-term follow-up (14 years) of a random sample from the general population. J Vasc Surg. 2006;44:1023–1028. doi: 10.1016/j.jvs.2006.07.037. This article demonstrates that, although Raynaud’s phenomenon does tend to be a prodrome for scleroderma, it is not practical to follow these patients over time. [DOI] [PubMed] [Google Scholar]
- 4•.Iwata H, Sata M. Potential contribution of bone marrow-derived precursors to vascular repair and lesion formation: lessons from animal models of vascular diseases. Front Biosci. 2007;12:4157–4167. doi: 10.2741/2377. This article follows the lineage and contribution of cells in the blood vessel wall. [DOI] [PubMed] [Google Scholar]
- 5•.Passman JN, Dong XR, Wu SP. A Sonic hedgehog signaling domain in the arterial adventitia supports resident sca1+ smooth muscle progenitor cells. Proc Natl Acad Sci U S A. 2008;105:9349–9354. doi: 10.1073/pnas.0711382105. This article and the articles by Carpentier et al. [3•] and Iwata and Sata [4•] provide recent evidence that Sonic hedgehog maintains vasculature in static state and that perturbation of signaling is associated with vascular remodeling. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.D’Angelo WA, Fries JF, Masi AT, Shulman LE. Pathologic observations in systemic sclerosis (scleroderma). A study of fifty-eight autopsy cases and fifty-eight matched controls. Am J Med. 1969;46:428–440. doi: 10.1016/0002-9343(69)90044-8. [DOI] [PubMed] [Google Scholar]
- 7.Yamagishi M, Miyatake K, Tamai J. Intravascular ultrasound detection of atherosclerosis at the site of focal vasospasm in angiographically normal or minimally narrowed coronary segments. J Am Coll Cardiol. 1994;23:352–357. doi: 10.1016/0735-1097(94)90419-7. [DOI] [PubMed] [Google Scholar]
- 8.Suzuki H, Kawai S, Aizawa T. Histological evaluation of coronary plaque in patients with variant angina: relationship between vasospasm and neointimal hyperplasia in primary coronary lesions. J Am Coll Cardiol. 1999;33:198–205. doi: 10.1016/s0735-1097(98)00520-8. [DOI] [PubMed] [Google Scholar]
- 9.Slomp J, van Munsteren JC, Poelmann RE. Formation of intimal cushions in the ductus arteriosus as a model for vascular intimal thickening. An immunohistochemical study of changes in extracellular matrix components. Atherosclerosis. 1992;93:25–39. doi: 10.1016/0021-9150(92)90197-o. [DOI] [PubMed] [Google Scholar]
- 10.Nagy Z, Czirjak L. Nailfold digital capillaroscopy in 447 patients with connective tissue disease and Raynaud’s disease. J Eur Acad Dermatol Venereol. 2004;18:62–68. doi: 10.1111/j.1468-3083.2004.00853.x. [DOI] [PubMed] [Google Scholar]
- 11.Birkenhager R, Schneppe B, Rockl W. Synthesis and physiological activity of heterodimers comprising different splice forms of vascular endothelial growth factor and placenta growth factor. Biochem J. 1996;316:703–707. doi: 10.1042/bj3160703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Distler O, Distler JH, Scheid A. Uncontrolled expression of vascular endothelial growth factor and its receptors leads to insufficient skin angiogenesis in patients with systemic sclerosis. Circ Res. 2004;95:109–116. doi: 10.1161/01.RES.0000134644.89917.96. [DOI] [PubMed] [Google Scholar]
- 13•.Mulligan-Kehoe MJ, Drinane MC, Mollmark J. Antiangiogenic plasma activity in patients with systemic sclerosis. Arthritis Rheum. 2007;56:3448–3458. doi: 10.1002/art.22861. This article provides an example of current research being done to elucidate a mechanism for vascular injury in scleroderma. [DOI] [PubMed] [Google Scholar]
- 14.Jun JB, Kuechle M, Harlan JM, Elkon KB. Fibroblast and endothelial apoptosis in systemic sclerosis. Curr Opin Rheumatol. 2003;15:756–760. doi: 10.1097/00002281-200311000-00012. [DOI] [PubMed] [Google Scholar]
- 15••.Fleming JN, Nash RA, McLeod DO. Capillary regeneration in scleroderma: stem cell therapy reverses phenotype? PLoS ONE. 2008;3:e1452. doi: 10.1371/journal.pone.0001452. This article details the endothelial changes in scleroderma, including the “antiangiogenic phenotype” that endothelial cells in scleroderma appear to have. This article provides a backdrop for the idea that endothelial injury in scleroderma is a very early event and not necessarily an ongoing one. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Del Papa N, Colombo G, Fracchiolla N. Circulating endothelial cells as a marker of ongoing vascular disease in systemic sclerosis. Arthritis Rheum. 2004;50:1296–1304. doi: 10.1002/art.20116. [DOI] [PubMed] [Google Scholar]
- 17.Leroy EC. The vascular defect in scleroderma (systemic sclerosis) Acta Med Scand Suppl. 1987;715:165–167. doi: 10.1111/j.0954-6820.1987.tb09917.x. [DOI] [PubMed] [Google Scholar]
- 18.Maricq HR. Comparison of quantitative and semiquantitative estimates of nailfold capillary abnormalities in scleroderma spectrum disorders. Microvasc Res. 1986;32:271–276. doi: 10.1016/0026-2862(86)90062-2. [DOI] [PubMed] [Google Scholar]
- 19.Libby P, Pober JS. Chronic rejection. Immunity. 2001;14:387–397. doi: 10.1016/s1074-7613(01)00119-4. [DOI] [PubMed] [Google Scholar]
- 20.Corpechot C, Barbu V, Wendum D. Hypoxia-induced VEGF and collagen I expressions are associated with angiogenesis and fibrogenesis in experimental cirrhosis. Hepatology. 2002;35:1010–1021. doi: 10.1053/jhep.2002.32524. [DOI] [PubMed] [Google Scholar]
- 21.Sgonc R, Gruschwitz MS, Dietrich H. Endothelial cell apoptosis is a primary pathogenetic event underlying skin lesions in avian and human scleroderma. J Clin Invest. 1996;98:785–792. doi: 10.1172/JCI118851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nguyen VA, Sgonc R, Dietrich H, Wick G. Endothelial injury in internal organs of University of California at Davis line 200 (UCD 200) chickens, an animal model for systemic sclerosis (scleroderma) J Autoimmun. 2000;14:143–149. doi: 10.1006/jaut.1999.0355. [DOI] [PubMed] [Google Scholar]
- 23.Rouquette-Gally AM, Boyeldieu D, Gluckman E. Autoimmunity in 28 patients after allogeneic bone marrow transplantation: comparison with Sjogren syndrome and scleroderma. Br J Haematol. 1987;66:45–47. doi: 10.1111/j.1365-2141.1987.tb06888.x. [DOI] [PubMed] [Google Scholar]
- 24.Zhang Y, Gilliam AC. Animal models for scleroderma: an update. Curr Rheumatol Rep. 2002;4:150–162. doi: 10.1007/s11926-002-0011-3. [DOI] [PubMed] [Google Scholar]
- 25.Siracusa LD, Christner P, McGrath R. The tight skin (tsk) mutation in the mouse, a model for human fibrotic diseases, is tightly linked to the beta 2-microglobulin (b2m) gene on chromosome 2. Genomics. 1993;17:748–751. doi: 10.1006/geno.1993.1398. [DOI] [PubMed] [Google Scholar]
- 26.Clark SH. Animal models in scleroderma. Curr Rheumatol Rep. 2005;7:150–155. doi: 10.1007/s11926-005-0068-x. [DOI] [PubMed] [Google Scholar]
- 27.Lakos G, Takagawa S, Varga J. Animal models of scleroderma. Methods Mol Med. 2004;102:377–393. doi: 10.1385/1-59259-805-6:377. [DOI] [PubMed] [Google Scholar]
- 28.Ahmed SS, Tan FK, Arnett FC. Induction of apoptosis and fibrillin 1 expression in human dermal endothelial cells by scleroderma sera containing anti-endothelial cell antibodies. Arthritis Rheum. 2006;54:2250–2262. doi: 10.1002/art.21952. [DOI] [PubMed] [Google Scholar]
- 29.Kahaleh MB, Fan PS. Mechanism of serum-mediated endothelial injury in scleroderma: identification of a granular enzyme in scleroderma skin and sera. Clin Immunol Immunopathol. 1997;83:32–40. doi: 10.1006/clin.1996.4322. [DOI] [PubMed] [Google Scholar]
- 30•.Nevskaya T, Bykovskaia S, Lyssuk E. Circulating endothelial progenitor cells in systemic sclerosis: relation to impaired angiogenesis and cardiovascular manifestations. Clin Exp Rheumatol. 2008;26:421–429. This article is a primary example of recent attempts to understand mechanisms related to vascular injury in scleroderma. [PubMed] [Google Scholar]
- 31.Abraham D, Distler O. How does endothelial cell injury start? The role of endothelin in systemic sclerosis. Arthritis Res Ther. 2007;9(Suppl 2):S2. doi: 10.1186/ar2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Anderegg U, Saalbach A, Haustein UF. Chemokine release from activated human dermal microvascular endothelial cells—implications for the pathophysiology of scleroderma? Arch Dermatol Res. 2000;292:341–347. doi: 10.1007/s004030000134. [DOI] [PubMed] [Google Scholar]
- 33.Hebbar M, Lassalle P, Janin A. E-selectin expression in salivary endothelial cells and sera from patients with systemic sclerosis. Role of resident mast cell-derived tumor necrosis factor alpha. Arthritis Rheum. 1995;38:406–412. doi: 10.1002/art.1780380318. [DOI] [PubMed] [Google Scholar]
- 34.Carmeliet P. Angiogenesis in life, disease and medicine. Nature. 2005;438:932–936. doi: 10.1038/nature04478. [DOI] [PubMed] [Google Scholar]
- 35••.Duan H, Fleming J, Pritchard DK. Combined analysis of monocyte and lymphocyte messenger rna expression with serum protein profiles in patients with scleroderma. Arthritis Rheum. 2008;58:1465–1474. doi: 10.1002/art.23451. This article shows the presence of an IFN signaling response present in cells circulating in the blood of scleroderma patients. [DOI] [PubMed] [Google Scholar]
- 36.Rozera C, Carlei D, Lollini PL. Interferon (IFN)-beta gene transfer into ts/a adenocarcinoma cells and comparison with IFN-alpha: differential effects on tumorigenicity and host response. Am J Pathol. 1999;154:1211–1222. doi: 10.1016/s0002-9440(10)65373-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chang E, Boyd A, Nelson CC. Successful treatment of infantile hemangiomas with interferon-alpha-2b. J Pediatr Hematol Oncol. 1997;19:237–244. doi: 10.1097/00043426-199705000-00011. [DOI] [PubMed] [Google Scholar]
- 38.Stout AJ, Gresser I, Thompson WD. Inhibition of wound healing in mice by local interferon alpha/beta injection. Int J Exp Pathol. 1993;74:79–85. [PMC free article] [PubMed] [Google Scholar]
- 39.Dor Y, Porat R, Keshet E. Vascular endothelial growth factor and vascular adjustments to perturbations in oxygen homeostasis. Am J Physiol Cell Physiol. 2001;280:C1367–C1374. doi: 10.1152/ajpcell.2001.280.6.C1367. [DOI] [PubMed] [Google Scholar]
- 40.Cho H, Kozasa T, Bondjers C. Pericyte-specific expression of RGS5: Implications for PDGF and edg receptor signaling during vascular maturation. FASEB J. 2003;17:440–442. doi: 10.1096/fj.02-0340fje. [DOI] [PubMed] [Google Scholar]
- 41.Iruela-Arispe L. Angiogenesis: novel and basic science insights and human therapy—Keystone symposium. IDrugs. 2004;7:111–113. [PubMed] [Google Scholar]
- 42.Berger M, Bergers G, Arnold B. Regulator of G-protein signaling-5 induction in pericytes coincides with active vessel remodeling during neovascularization. Blood. 2005;105:1094–101. doi: 10.1182/blood-2004-06-2315. [DOI] [PubMed] [Google Scholar]
- 43•.Sela S, Itin A, Natanson-Yaron S. A novel human-specific soluble vascular endothelial growth factor receptor 1: cell-type-specific splicing and implications to vascular endothelial growth factor homeostasis and preeclampsia. Circ Res. 2008;102:1566–1574. doi: 10.1161/CIRCRESAHA.108.171504. This article discusses recent evidence that interference with the signaling for VEGF may be involved in the process of rarefaction. [DOI] [PubMed] [Google Scholar]
- 44.Torsney E, Hu Y, Xu Q. Adventitial progenitor cells contribute to arteriosclerosis. Trends Cardiovasc Med. 2005;15:64–68. doi: 10.1016/j.tcm.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 45••.Lavine KJ, Kovacs A, Ornitz DM. Hedgehog signaling is critical for maintenance of the adult coronary vasculature in mice. J Clin Invest. 2008;118:2404–2414. doi: 10.1172/JCI34561. This article both demonstrates how Sonic hedgehog maintains vasculature in a static state and how perturbation of signaling and vascular remodeling are associated. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46•.Lavine KJ, Long F, Choi K. Hedgehog signaling to distinct cell types differentially regulates coronary artery and vein development. Development. 2008;135:3161–3171. doi: 10.1242/dev.019919. This article offers recent evidence regarding the role of Sonic hedgehog in the maintainance of vasculature in a static state and how the perturbation of signaling is associated with vascular remodeling. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sas A, Jones R, Tyor W. Intra-peritoneal injection of polyclonal anti-interferon alpha antibodies cross the blood brain barrier and neutralize interferon alpha. Neurochem Res. 2008;33:2281–2287. doi: 10.1007/s11064-008-9715-8. [DOI] [PubMed] [Google Scholar]
- 48.Nash RA, Bowen JD, McSweeney PA. High-dose immunosuppressive therapy and autologous peripheral blood stem cell transplantation for severe multiple sclerosis. Blood. 2003;102:2364–2372. doi: 10.1182/blood-2002-12-3908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49••.Kim D, Peck A, Santer D. Induction of interferon-alpha by scleroderma sera containing autoantibodies to topoisomerase I: association of higher interferon-alpha activity with lung fibrosis. Arthritis Rheum. 2008;58:2163–2173. doi: 10.1002/art.23486. This article demonstrates that IFNα may participate in the process of fibrosis. [DOI] [PubMed] [Google Scholar]
- 50••.Higgins DF, Kimura K, Iwano M, Haase VH. Hypoxia-inducible factor signaling in the development of tissue fibrosis. Cell Cycle. 2008;7:1128–1132. doi: 10.4161/cc.7.9.5804. This article reviews the effect of chronic hypoxia on tissue. [DOI] [PMC free article] [PubMed] [Google Scholar]