

Abstract
Several successful pathogens have evolved mechanisms to evade host defense, resulting in the establishment of persistent and chronic infections. One such pathogen, Porphyromonas gingivalis, induces chronic low-grade inflammation associated with local inflammatory bone loss and systemic inflammation manifested as atherosclerosis. P. gingivalis expresses an atypical lipopolysaccharide (LPS) structure containing heterogeneous lipid A species, that exhibit Toll-like receptor-4 (TLR4) agonist or antagonist activity, or are non-activating at TLR4. In this study, we utilized a series of P. gingivalis lipid A mutants to demonstrate that antagonistic lipid A structures enable the pathogen to evade TLR4-mediated bactericidal activity in macrophages resulting in systemic inflammation. Production of antagonistic lipid A was associated with the induction of low levels of TLR4-dependent proinflammatory mediators, failed activation of the inflammasome and increased bacterial survival in macrophages. Oral infection of ApoE−/− mice with the P. gingivalis strain expressing antagonistic lipid A resulted in vascular inflammation, macrophage accumulation and atherosclerosis progression. In contrast, a P. gingivalis strain producing exclusively agonistic lipid A augmented levels of proinflammatory mediators and activated the inflammasome in a caspase-11-dependent manner, resulting in host cell lysis and decreased bacterial survival. ApoE−/− mice infected with this strain exhibited diminished vascular inflammation, macrophage accumulation, and atherosclerosis progression. Notably, the ability of P. gingivalis to induce local inflammatory bone loss was independent of lipid A expression, indicative of distinct mechanisms for induction of local versus systemic inflammation by this pathogen. Collectively, our results point to a pivotal role for activation of the non-canonical inflammasome in P. gingivalis infection and demonstrate that P. gingivalis evades immune detection at TLR4 facilitating chronic inflammation in the vasculature. These studies support the emerging concept that pathogen-mediated chronic inflammatory disorders result from specific pathogen-mediated evasion strategies resulting in low-grade chronic inflammation.
Author Summary
Several human pathogens express structurally divergent forms of lipid A, the endotoxic portion of lipopolysaccharide (LPS), as a strategy to evade host innate immune detection and establish persistent infection. Expression of modified lipid A species promotes pathogen evasion of host recognition by Toll-like receptor-4 (TLR4) and the non-canonical inflammasome. The Gram-negative oral anaerobe, Porphyromonas gingivalis, expresses lipid A structures that function as TLR4 agonists or antagonists, or are immunologically inert. It is currently unclear how modulation of P. gingivalis lipid A expression contributes to innate immune recognition, survival, and the ability of the pathogen to induce local and systemic inflammation. In this study, we demonstrate that P. gingivalis expression of antagonist lipid A species results in attenuated production of proinflammatory mediators and evasion of non-canonical inflammasome activation, facilitating bacterial survival in the macrophage. Infection of atherosclerosis-prone ApoE−/− mice with this strain resulted in progression of chronic inflammation in the vasculature. Notably, the ability of P. gingivalis to induce local inflammatory bone loss was independent of lipid A modifications, supporting distinct mechanisms for induction of local versus systemic inflammation. Our work demonstrates that evasion of immune detection at TLR4 contributes to pathogen persistence and facilitates low-grade chronic inflammation.
Introduction
Host recognition of Gram-negative bacteria occurs via detection of LPS expressed on the bacterial membrane by the innate immune receptor, TLR4 [1]. This initial recognition is critical for instructing host immunity and promoting an inflammatory response that eradicates the pathogen from the host [1], [2]. However, a number of Gram-negative organisms have evolved mechanisms to modify their lipid A species, the component of bacterial LPS that directly activates the TLR4 complex, as a strategy to evade immune detection and establish infection [3]. Lipid A is initially synthesized as a β-1′,6-linked disaccharide of glucosamine that is phosphorylated and fatty acylated [4]. An unmodified version of this lipid A structure is typically expressed by E. coli and induces a robust inflammatory response [5]. Modifications to this basic lipid A structure are observed in alterations to acyl chains or terminal phosphate groups [6]. Helicobacter pylori [7], Legionella pneumophila [8], Yersinia pestis [9], and Francisella novicida [10] express underacylated lipid A moieties, in comparison to the canonical LPS expressed by E. coli, and are poorly recognized by TLR4. Yersinia pestis [11] and Francisella tularensis [12] expression of structurally divergent forms of lipid A is highly regulated by local environmental conditions such as temperature, allowing these pathogens to adapt to harsh environmental conditions in the host. It has been postulated that the ability of these pathogens to cause persistent infection and severe disease is partially due to evasion of host immune detection at TLR4 [13].
Recently, it has been revealed that in addition to evasion of TLR4 signaling, lipid A modifications promote evasion of the non-canonical inflammasome by preventing activation of caspase-11 [14]. Activation of the inflammasome is characterized by the production of the proinflammatory mediators IL-1β and IL-18 and is associated with downstream events such as pyroptosis [15]. Due to its role in host innate defense, a number of pathogens have evolved strategies to evade activation of this complex [16]. Pathogen evasion of inflammasome activation has been proposed to serve a dual role: to dampen cytokine production and to prevent host cell death in order to provide an intracellular niche for the pathogen to survive [17]. One pathogen that has successfully adapted to evade the inflammasome is H. pylori, through expression of its tetra-acylated lipid A [14].
The oral pathogen Porphyromonas gingivalis weakly activates TLR4 through expression of a heterogeneous LPS that contains lipid A structures that vary in the number of phosphate groups and the amount and position of lipid A fatty acids [18], [19]. P. gingivalis expresses underacylated lipid A structures that can be penta-acylated forms, conferring TLR4 agonistic activity, or tetra-acylated forms, functioning as TLR4 antagonists, or are non-activating [20], [21]. These structures typically express mono- or di-phosphate terminal groups. Expression of divergent structural moieties by P. gingivalis changes depending on growth phase, temperature, and levels of hemin [22]–[24]. Recently, it has been demonstrated that P. gingivalis also expresses a unique non-phosphorylated, tetra-acylated lipid A that is regulated by levels of hemin [23]. During hemin-deplete conditions, P. gingivalis utilizes endogenous lipid A 1- and 4′-phosphatase activities to express a non-phosphorylated, tetra-acylated lipid A that is immunologically inert at the TLR4 complex, as well as a mono-phosphorylated, penta-acylated lipid A that functions as a weak TLR4 agonist [22], [23], [25]. Under hemin-replete conditions, the activity of 1-phosphatase is suppressed, resulting in the expression of a mono-phosphorylated, tetra-acylated lipid A species that functions as TLR4 antagonists [23]. Expression of these different structural types is believed to allow P. gingivalis to evade immune detection at TLR4 [26].
A hallmark of chronic infection with P. gingivalis is the induction of a local inflammatory response that results in destruction of supporting tissues of the teeth and resorption of alveolar bone [27]–[29]. In addition to inflammation induced at the initial site of infection, P. gingivalis has been associated with systemic diseases such as diabetes, pre-term birth, pancreatic cancer, and cardiovascular disease [30]–[32]. P. gingivalis has been detected in human atherosclerotic lesions and shown to be viable in atherosclerotic tissue [33]–[35]. Studies from our laboratory have validated human studies by demonstrating that oral infection of atherosclerosis-prone ApoE−/− mice with P. gingivalis results in local oral bone loss and systemic inflammation in atherosclerotic lesions [36]. We have demonstrated that P. gingivalis-induced oral inflammatory bone loss and acceleration of systemic inflammation and atherosclerosis is dependent on TLR2 signaling [37], [38]. P. gingivalis engages TLR2 through the expression of several outer membrane components that include lipoprotein, major and minor fimbriae, and phosphorylated dihydroceramides [39]–[41]. The unique ability of P. gingivalis to induce TLR2 signaling and to evade TLR4 signaling has been proposed to enable this organism to cause low-grade persistent infection [42]; however, the expression of multiple P. gingivalis lipid A structures simultaneously has complicated the interpretation of how distinct lipid A moieties contribute to chronic inflammation [22].
To define the role of distinct lipid A species in P. gingivalis evasion of TLR4 signaling, innate immune recognition, survival, and the ability of the pathogen to induce local and systemic chronic inflammation, we constructed genetically modified strains of P. gingivalis that lack either 1- or 4′-phosphatase activity [23]. These resulting strains express lipid A species that are not under genetic regulation and function as TLR4 agonists or TLR4 antagonists. Utilizing these strains, we demonstrate that P. gingivalis expression of antagonist lipid A species results in attenuated production of proinflammatory mediators and evasion of non-canonical inflammasome activation, facilitating bacterial survival in the macrophage. Infection of atherosclerosis-prone ApoE−/− mice with this strain resulted in progression of chronic inflammation in the vasculature. Notably, the ability of P. gingivalis to induce local inflammatory bone loss was independent of lipid A modifications, supporting distinct mechanisms for induction of local versus systemic inflammation. Collectively, these results indicate that expression of P. gingivalis lipid A structures that fail to engage TLR4 or function as TLR4 antagonists enables this pathogen to evade host innate immune detection and induce inflammation at sites distant from infection.
Results
P. gingivalis strain 381 modifies its lipid A structures through expression of endogenous lipid A 1- and 4′-phosphatase activities
MALDI analysis of LPS isolated from P. gingivalis strain 381 revealed an ion cluster at m/z 1368 ( Figure 1A and 1D ). This structure represents the non-phosphorylated and tetra-acylated lipid A species that was predicted to be functionally inert at the TLR4 complex [23]. Additionally, we observed the expression of TLR4 antagonist (m/z 1448) and TLR4 agonist (m/z 1688 and m/z 1768) structures ( Figure 1A and 1E–G ). In order to examine the role of distinct lipid A species on the induction of inflammation, we constructed P. gingivalis strains lacking lipid A 1- and 4′- phosphatase activities in P. gingivalis 381. MALDI analysis of P. gingivalis strain PG1587 381, that lacks 4′-phosphatase activity, revealed TLR4 agonist lipid A structures that centered at m/z 1768 and m/z 1688 ( Figure 1B and 1F–G ). MALDI analysis of P. gingivalis strain PG1773 381, which lacks 1-phosphatase activity, revealed a TLR4 antagonist lipid A mass ion that was predominantly centered at ∼1448 m/z as well as the agonistic lipid A centered at ∼1768 m/z ( Figure 1C, 1E and 1G ).
Figure 1. P. gingivalis strain 381 utilizes endogenous lipid A 1- and 4′- phosphatase activities to modify lipid A species and evade TLR4 activation.

Lipid A isolated from P. gingivalis wild-type strain 381 (A) or lipid A mutant strains PG1587 381 (B) and PG1773 381 (C) were examined by MALDI-TOF MS. Arrows indicate the predominant lipid A species that are expressed for each strain (A–C). The major lipid A structures examined in this study have been identified in P. gingivalis as previously described (D–G) [19].
To confirm the predicted TLR4 activation phenotype of the lipid A expressed by wild-type 381 and the lipid A mutants, we stimulated HEK cells that overexpress mouse TLR4-MD2 with purified LPS from each strain. Notably, the LPS preparations purified from all three strains similarly activated mouse TLR4-MD2 ( Figure 2A ). In contrast, when live bacteria were used to stimulate the HEK cells only strain PG1587 381 resulted in a significant increase in TLR4-dependent NF-κB activation as compared to wild-type 381 and PG1773 381 ( Figure 2B ). These results suggest that the lipid A structures are differentially distributed within the bacterial cell membranes depending upon the strains, and that the relative localization of the specific agonistic and antagonistic lipid A forms to the outer cell membrane determines the respective abilities of the different strains to activate TLR4. The less potent lipid A forms (m/z 1368, 1448, 1688) may be primarily expressed on the bacterial outer membrane whereas the most potent lipid A form (m/z 1768) predominates in the inner membrane where it is initially synthesized prior to processing by phosphatases and deacylase(s).
Figure 2. Lipid A phosphatase activity contributes to the ability of P. gingivalis to evade host innate immune defenses.

HEK cells overexpressing mouse TLR4-MD2 (A–B) or TLR2 (D–E) were stimulated with corresponding LPS concentration purified from each strain (A,D) or whole bacterium (B,E) overnight. Relative NFkB activity indicates inducible firefly luciferase activity over the media control. Wild-type 381 and the lipid A mutants PG1587 381 and PG1773 381 were grown in BHI broth cultures overnight in the presence or absence of polymyxin B (PMB 1, 5, 10, 25, 50 µg/mL). Growth was measured by spectrophotometry (OD660) (C). Wild-type P. gingivalis (solid), PG1587 381 (dotted), and PG1773 381 (dashed). Graphs show the mean ± SEM of triplicate wells and are representative of two independent experiments. (See also Figure S1).
In addition to direct impact on TLR4 activation, modifications in lipid A structure can significantly alter the ability of cationic peptides to kill bacteria. We have previously reported that two different strains of P. gingivalis (33277 and A7436) deficient in PG1587 exhibit the most pronounced sensitivity to polymyxin B as compared to the wild-type and PG1773 strains, consistent with a critical role of the lipid A 4′-phosphate in rendering bacteria susceptible to this drug [23], [43]. Assessment of these mutations in the 381 strains revealed a comparable pattern. Strain PG1587 381 exhibits a pronounced susceptibility to polymyxin B while the P. gingivalis wild-type strain 381 and strain PG1773 381 were relatively more resistant ( Figure 2C ). These data correlate well with the above TLR4 activation data indicating that lipid A structures localized in the outer membrane of the PG1587 381 mutant contain 4′-phosphate (m/z 1688). In contrast, the PG1773 381 strain is the most resistant, indicating the predominance of lipid A lacking 4′-phosphate in the outer membrane (m/z 1448). Wild-type 381 has an intermediate polymyxin B resistance phenotype consistent with an increased presence of lipid A containing 4′-phosphate as compared to strain PG1773 381.
To verify that the mutant 381 strains exhibit phenotypes that are consistent with bacterial surface lipid A modifications rather than modifications of other surface virulence factors, we further assessed the ability of the P. gingivalis wild-type strain and the lipid A mutants to activate TLR2. All three P. gingivalis strains induced a similarly significant increase in NF-κB activation in HEK293 cells over expressing TLR2; however, we observed a slight decrease in the ability of the P. gingivalis wild-type strain 381 to activate TLR2 at lower MOIs ( Figure 2E and data not shown). Stimulation of HEK-TLR2 cells with purified LPS isolated from the P. gingivalis wild-type strain 381 and the lipid A mutants resulted in equivalent activation of TLR2 ( Figure 2D ). These results were expected since P. gingivalis strongly activates TLR2 via expression of fimbriae and lipoproteins [39], [40]. To confirm that modification of lipid A structures in P. gingivalis strains PG1587 381 and PG1773 381 did not alter the expression of other outer membrane components, we examined the major fimbriae protein and activity of the cell-associated cysteine proteases, gingipain R and gingipain K. Similar levels of fimbriae expression were observed in P. gingivalis strains 381, PG1587 381 and PG1773 381 (Figure S1-A-B). We observed a slight decrease in gingipain activity (KGP and RGP) in P. gingivalis strain PG1587 381 as compared to that observed in the wild-type strain (Figure S1-C). We did not observe significant differences in the growth of P. gingivalis strains PG1587 381 and PG1773 381 as compared to the wild-type strain (Figure S1-D and data not shown). Taken together, these results indicate that deletion of PG1587 or PG1773 alters the ability of the whole bacteria to activate TLR4 but does not alter the expression of other outer membrane components or the ability of the pathogen to activate TLR2. Therefore, the use of strains PG1587 381 and PG1773 381 in this study allowed us to assess the immunological consequences of differential lipid A expression by P. gingivalis.
P. gingivalis expression of antagonistic lipid A attenuates induction of TLR4-dependent inflammatory mediators
We examined the ability of the P. gingivalis strains lacking lipid A 1- and 4′-phosphatase activities to induce NF-κB-dependent proinflammatory cytokines in bone marrow-derived macrophages (BMDM). Stimulation of BMDM with P. gingivalis strain PG1587 381 resulted in increased production of KC, IL-6, IL-1β, and IL-1α compared to P. gingivalis strains 381 and PG1773 381 in a dose-dependent manner ( Figure 3B–C ; Figure 4A–B ). In contrast, all three P. gingivalis strains induced significant levels of TNF-α. ( Figure 3A ). The role of TLR2 and TLR4 in the production of these inflammatory cytokines was assessed in BMDM obtained from TLR2- and TLR4-deficient mice. P. gingivalis-induced TNF-α production required both TLR2 and TLR4 signaling; however, TLR2 signaling was more dominant ( Figure 3D ). Additionally, we observed that the ability of P. gingivalis to induce IL-1β and IL-1α was dependent on both TLR2 and TLR4 signaling (Figure S2). In contrast, P. gingivalis-induced expression of KC and IL-6 were primarily dependent on TLR4 signaling ( Figure 3E–F ). We observed that PG1587 381 induced enhanced KC and IL-6 levels in TLR2-deficient macrophages as compared to the P. gingivalis wild-type strain 381, suggesting the increased cytokine production we observed in wild-type BMDM was mediated via TLR4 signaling. Furthermore, we observed comparable KC and IL-6 levels in BMDM deficient in TLR4 following stimulation with all 3 strains of P. gingivalis, suggesting additional signaling through TLR2 in the absence of TLR4 or an inability of lipid A to antagonize production of these cytokines. Overall, these results indicate that expression of antagonistic or inert lipid A attenuates the production of NFκB-dependent proinflammatory mediators in macrophages.
Figure 3. Expression of antagonistic or inert lipid A attenuates the production of NFkB-dependent proinflammatory mediators.

BMDMs from C57BL/6 were stimulated with P. gingivalis wild-type strain 381 (381) or the lipid A mutant strains PG1587 381 (1587) and PG1773 381 (1773) at an MOI of 1 (white) 10 (gray) and 100 (black). Levels of TNFα (A), KC (B), and IL-6 (C) were assayed by ELISA at 24 h. BMDMs from wild-type C57BL/6 mice (white), TLR2-deficient (gray), or TLR4-deficient (black) were stimulated with P. gingivalis wild-type strain 381 or the lipid A mutant strains at an MOI of 100 (D) or MOI of 10 (E–F) and levels of TNFα (D) KC (E) and IL-6 (F) were assessed by ELISA at 24 h (See also Figure S2). Cells treated with E. coli LPS (100 ng/mL) or Pam3CysSk4 (1 µg/mL) for 5 h served as a positive control. Graphs show the mean ± SEM of triplicate wells and are representative of three independent experiments. *p<.05 **p≤.001 ***p<.0001; ANOVA with Bonferroni's posttest.
Figure 4. P. gingivalis evades activation of the inflammasome through expression of modified lipid A species.

C57BL/6 WT BMDM were stimulated with P. gingivalis wild-type strain 381 or the lipid A mutant strains PG1587 381 and PG1773 381 at an MOI of 1 (white), 10 (gray) or 100 (black). Levels of IL-1β (A) and IL-1α (B) were assayed by ELISA at 24 h. LDH release (Promega) was assayed in BMDM stimulated with P. gingivalis wild-type strain 381 or the lipid A mutants at an MOI 100 at 2 h (white), 6 h (gray) and 24 h (black) (C). BMDM from wild-type (white bars) and caspase-11-deficient (black bars) mice were stimulated with P. gingivalis wild-type strain 381 or the lipid A mutants at an MOI of 100 and levels of IL-1β (D) and IL-1α (E) were assessed by ELISA at 24 h. Viable counts (CFU) of internalized P. gingivalis were determined by plating serial dilutions of macrophage lysates on blood agar plates at 2 h (white) and 6 h (gray) (F). Western blot analysis was performed on BMDM cell lysates to assess levels of the proform of IL-1β, pro Caspase-1 and β-actin at 24 h (G). Levels of mature IL-1β and active caspase-1 (p10) were detected in cell supernatants. BMDMs treated with media alone (−) or with E. coli LPS (100 ng/mL) for 5 h and then ATP (5 mM) for 20 m (+) served as the negative and positive control respectively. Graphs depict the mean ± SEM of triplicate wells and are representative of at least three independent experiments. * p<.05 **p≤.001 ***p<.0001; two-tailed unpaired t-tests. (See also Figure S2).
P. gingivalis evades inflammasome activation through expression of lipid A structures that evade TLR4 detection
IL-1β is considered an “alarm” cytokine and has been shown to be critical for host defense against infection [16], [44]. Previous studies have documented that P. gingivalis fails to induce significant levels of IL-1β production in macrophages [45], [46]. Thus, the increased production of IL-1β observed in macrophages stimulated with P. gingivalis strain PG1587 381 was an unexpected finding ( Figure 4A ). IL-1β is first produced as an inactive zymogen, through activation of TLR signaling [47]. We observed that all three P. gingivalis strains induced expression of the proform of IL-1β ( Figure 4G ), which was dependent on TLR2 and TLR4 (data not shown). These results indicated that the attenuated production of IL-1β observed with P. gingivalis strains 381 and PG1773 381 was not due to an obstruction of TLR-mediated synthesis of pro-IL-1β.
These findings led us to investigate the role of P. gingivalis lipid A modifications on inflammasome activation, the second signal required for production of mature and active IL-1β. Stimulation of macrophages with P. gingivalis strain PG1587 381 resulted in activation of the inflammasome, as assessed by detection of the active caspase-1 p10 subunit in cell supernatants by Western blot analysis ( Figure 4G ). We also observed an increase in macrophage cell lysis following stimulation with P. gingivalis strain PG1587 381 ( Figure 4C ), an event that is downstream from inflammasome activation. These results correlated with an inability of PG1587 381 to survive in macrophages. We observed a significant decrease in survival of P. gingivalis strain PG1587 381 in macrophages compared to P. gingivalis strains 381 and strain PG1773 381 ( Figure 4F ).
Caspase-11 has emerged as an important mediator of inflammasome activation in Gram-negative bacterial infections [48], [49]. We thus examined the role of caspase-11 in P. gingivalis-induced IL-1β production. Interestingly, IL-1β production was completely ablated in macrophages deficient in caspase-11 following stimulation with P. gingivalis strain PG1587 381 ( Figure 4D ), while TNFα levels were not altered (data not shown). In agreement with the requirement for caspase-11 in IL-1α production [50], we observed decreased production of IL-1α following stimulation of BMDM obtained from caspase-11-deficient mice with P. gingivalis strain PG1587 381. However, we did not observe significant differences in the levels of IL-1α in BMDM obtained from caspase-11-deficient mice when stimulated with wild-type 381 compared to strain PG1587 381 ( Figure 4E ). These results suggest distinct mechanism(s) for IL-1α production independent of lipid A modifications. Collectively, these findings indicate that expression of modified lipid A species by P. gingivalis facilitates evasion of non-canonical inflammasome activation through a caspase-11-mediated pathway and obstructs downstream events associated with activation of this complex.
Alternative lipid A structures produced by P. gingivalis differentially influence systemic inflammation and local oral inflammation
To determine if the expression of modified lipid A by P. gingivalis is associated with the ability of the organism to promote chronic inflammation in vivo, we utilized a mouse model that mimics chronic P. gingivalis exposure as seen during human infection [51], [52]. Atherosclerosis-prone ApoE−/− mice were orally infected with P. gingivalis strains 381, PG1587 381, and PG1773 381 and chronic inflammation at local (oral bone loss) and systemic (atherosclerosis) sites was evaluated [36].
Plaque accumulation in the innominate artery was examined by magnetic resonance angiogram (MRA) throughout the course of the study to assess progression of site-specific inflammatory atherosclerosis. The innominate artery exhibits a high degree of lesion progression and expresses features of human disease including vessel narrowing and perivascular inflammation [53]. MRA at 8 weeks after oral infection resulted in an increase in the change of luminal area for mice infected with all 3 strains, indicating that the mice were still growing at this age (14–16 weeks of age) ( Figure 5A ). At 16 weeks after oral infection, MRA revealed that oral infection with P. gingivalis strains 381 and strain PG1773 381 resulted in significant luminal narrowing compared to sham-infected controls ( Figure 5A ). In contrast, oral infection with P. gingivalis strain PG1587 381 induced minimal luminal narrowing compared to sham-infected controls ( Figure 5A ). Furthermore, P. gingivalis strains 381 and PG1773 381 induced progressive luminal narrowing from 8 to 16wks compared to strain PG1587 381 and sham-infected controls ( Figure 5A inset).
Figure 5. Expression of immunologically silent or antagonistic lipid A structures exacerbates atherosclerotic plaque progression in the innominate artery.

Innominate arteries of ApoE−/− mice were imaged by MRA at baseline (wk 0) and at 8 and 16 wks after first oral infection. The temporal change in luminal area (mm2) was calculated for individual mice normalized to baseline luminal area (n = 10–12/group) (A). Sham-infected ApoE−/− (orange); 381-infected ApoE−/− (black); PG1587 381-infected ApoE−/− (blue); PG1773 381- infected ApoE−/− (red). Inset - the temporal change in luminal area calculated for individual mice normalized to 8 wk luminal area (n = 10–12/group). *p≤.01 **p<.001 ***p<.0001; two-tailed unpaired t-tests compared to sham-infected and PG1587 381-infected mice. (B) Representative images of the innominate artery with F4/80 staining (macrophages stain brown) and hematoxylin counterstaining for each group at 10× and 40× (n = 3/group) (See also Figure S3).
Luminal narrowing was further validated by histological assessment of the innominate artery in postmortem sections. Hematoxylin and eosin staining of the innominate artery corresponding to the region of MRA analysis revealed an occlusion of the lumen in ApoE−/− mice infected with P. gingivalis strain 381 (Figure S3), in agreement with our previous studies [53]. Oral infection with P. gingivalis strain PG1773 381 resulted in plaque accumulation that was comparable to that observed with P. gingivalis strain 381 (Figure S3). Oral infection with P. gingivalis strain PG1587 381 resulted in thickening of the vessel wall without occlusion of the vasculature (Figure S3). To assess macrophage infiltrate of the atherosclerotic lesions, histological sections of the innominate artery were stained with F4/80. We found oral infection with P. gingivalis strains 381 and PG1773 381 resulted in an increase in macrophage accumulation in the innominate lesions compared to that observed with P. gingivalis strain PG1587 381 and sham-infected controls ( Figure 5B ).
Lipid staining of en face aortas revealed that oral infection with P. gingivalis strains 381 and PG1773 381 accelerated plaque accumulation compared to that observed in sham-infected mice ( Figure 6 ). In contrast, oral infection with P. gingivalis strain PG1587 381 induced minimal plaque accumulation ( Figure 6C ). The inability of P. gingivalis strain PG1587 381 to elicit inflammatory disease pathology was not due to failed activation of immunity, since we observed an induction of the humoral response by P. gingivalis strain PG1587 381 that was comparable to that induced by P. gingivalis strains 381 and strain PG1773 381, as observed by serum levels of IgG1, IgG2b, IgG2c and IgG3 (Figure S4).
Figure 6. Expression of immunologically silent or antagonistic lipid A structures exacerbates atherosclerotic plaque progression in the aorta.
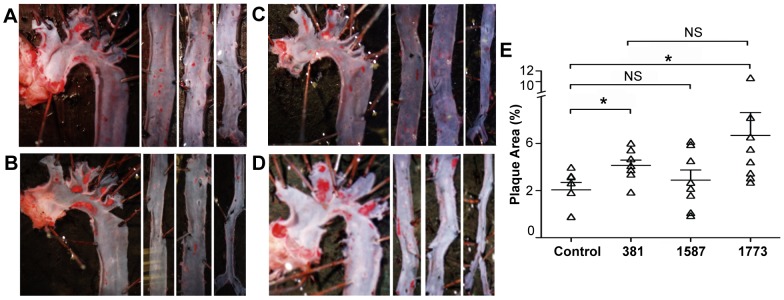
Plaque area was determined from Oil Red O staining for lipids in en face aortic lesions 16 wk after first infection from (A) sham-infected, (B) wild-type 381, (C) PG1587 381, and (D) PG1773 381. (E) Quantification of lipid content within total aorta was calculated using ImageJ software (NIH) (n = 8/group). * p<.05; two-tailed unpaired t-tests. (See also Figure S4).
Assessment of alveolar bone loss in infected mice revealed that P. gingivalis strains 381, PG1587 381, and PG1773 381 all induced oral bone loss at similar levels ( Figure 7 ). These results are consistent with studies demonstrating a predominant role for TLR2 signaling in P. gingivalis-induced oral inflammatory bone loss [54], [55]. Collectively, these results indicate that expression of P. gingivalis lipid A structures that fail to engage TLR4 or function as TLR4 antagonists enables this pathogen to evade host innate immune detection and contributes to inflammation at sites distant from infection.
Figure 7. Expression of immunologically silent, agonistic or antagonistic lipid A does not alter the ability of the pathogen to induce oral bone loss.
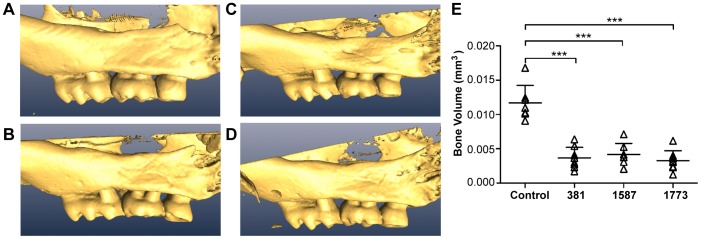
Maxillae were dissected (n = 8/group) 16 wk post initial infection and scanned on a micro CT 40 apparatus. Using AMIRA software, three-dimensional images were generated from micro CT scans. Representative images from (A) sham-infected mouse indicating no bone loss, (B) P. gingivalis wild-type strain 381-induced bone loss, (C) PG1587 381, and (D) PG1773 381. (E) Quantification of bone loss. ***p<.001; two-tailed unpaired t-tests.
Discussion
In this study, we generated genetically defined strains of P. gingivalis expressing TLR4 agonist and antagonist lipid A species to examine the role of TLR4 evasion in P. gingivalis-induced chronic inflammation. Importantly, we determined that expression of TLR4 antagonist lipid A contributes to the ability of P. gingivalis to activate innate immunity and induce inflammation at systemic sites. Notably, induction of local inflammatory bone loss in response to P. gingivalis infection was not dependent on lipid A modifications, indicative of distinct mechanisms for the induction of local versus systemic vascular chronic inflammation. The expression of antagonistic or immunological inert lipid A species was associated with attenuated production of proinflammatory mediators and inflammasome activation, which correlated with increased bacterial survival in macrophages. We conclude that P. gingivalis evades TLR4-mediated bacterial clearance in the host, allowing it to exacerbate vascular inflammation ( Figure 8 ).
Figure 8. P. gingivalis dysregulates host cell immune activation facilitating systemic inflammation.

The host predominantly senses P. gingivalis infection through engagement of TLR2 while the involvement of TLR4-dependent recognition is significantly impaired. Expression of antagonistic or immunologically inert lipid A by P. gingivalis attenuates production of proinflammatory mediators and prevents activation of the inflammasome (potentially through evasion of an unknown sensor) that facilitates intracellular survival. These events allow the pathogen to disseminate and to exacerbate systemic inflammation. In contrast, increased immunostimulatory potential at TLR4, through expression of an agonistic lipid A moiety, results in increased production of proinflammatory mediators, inflammasome activation, and reduced survival of the bacterium in macrophages leading to attenuated systemic inflammation.
We and others have reported that the expression of P. gingivalis lipid A is regulated by growth phase, temperature, and levels of hemin [22]–[24]. The expression of multiple lipid A structures has complicated the interpretation of the host response elicited by P. gingivalis LPS [22]. During growth under hemin-replete conditions, P. gingivalis expresses an antagonistic lipid A, due to the repression of 1-phosphatase activity [23]. The antagonist lipid A expressed by P. gingivalis has been demonstrated to antagonize E. coli LPS binding to TLR4 [56] and to dampen the cytokine response typically induced by other Gram-negative pathogens [45]. The use of P. gingivalis strain PG1773 381 in this study allowed us to specifically define the role of the antagonistic lipid A (typically expressed under hemin-replete conditions) in the induction of chronic inflammation at both local and distant sites from infection. We observed that P. gingivalis strains 381 and PG1773 381 induced comparable levels of inflammatory atherosclerosis suggesting that in vivo P. gingivalis is exposed to a hemin-replete environment and predominantly expresses an antagonistic lipid A that exacerbates chronic systemic inflammation.
Evasion of TLR4 signaling through lipid A modifications has been attributed to the ability of a number of highly pathogenic Gram-negative pathogens to cause disease [3]. For example, F. novicida expresses a tetra-acylated lipid A, with one phosphate group and does not induce a TLR4 response [10]. Y. pestis expresses a tetra-acylated lipid A that functions as a TLR4 antagonist at 37°C, which allows this pathogen to remain undetected in the bloodstream during early stages of infection [9], [11]. Generation of a strain of Y. pestis that expressed a more immunostimulatory LPS conferred a protective immune response against this pathogen [57]. Likewise, Pseudomonas aeruginosa expresses a modified LPS which promotes evasion of TLR4 signaling, favors intracellular survival, and has been postulated to contribute to chronic persistence in Cystic Fibrosis patients [58]. In agreement with these studies, we have shown that P. gingivalis modifies its lipid A structure in order to evade host defenses and establish chronic infection leading to persistent low-grade inflammation in the vasculature. Uniquely, P. gingivalis evasion of host innate immunity at TLR4 results in progression of inflammation at a site that is distant from local infection by gaining access to the vasculature.
A number of reports have proposed that P. gingivalis lipid A modifications are a mechanism for evasion of TLR4 signaling [6], [23], [26]. However, these studies utilized purified P. gingivalis LPS in an in vitro setting. To date, only this study and our recent study in a rabbit model of periodontitis [43] have begun to shed light on the immunological consequences of differential activation of the TLR4 complex by P. gingivalis LPS through the use of live bacteria that express a “locked” lipid A profile that is not responsive to growth conditions. Furthermore, we assessed the host response in the oral cavity and vasculature, physiologically relevant sites of chronic inflammation observed in humans. Notably, the use of purified LPS in our study resulted in discrepancies in TLR4 activation by isolated LPS versus the whole bacterium. These differences observed in the host response may be a reflection of LPS structure and composition on the cell surface, which leave us with new areas of investigation with regards to LPS translocation. Potentially, the less potent lipid A forms (m/z 1368, 1448, 1688) may be primarily expressed on the bacterial outer membrane and, consequently, render wild-type 381 and PG1773 381 unable to activate TLR4. In contrast, strain PG1587 381 is expected to exclusively accumulate lipid A TLR4 agonists (m/z 1688 and possibly m/z 1768) in the outer membrane since the presence of a lipid A 4′-phosphate precludes production of the lipid A antagonist (m/z 1448) or non-activating lipid A (m/z 1368) in this strain.
Overproduction of proinflammatory mediators and dysregulated inflammasome activation has been reported to contribute to inflammatory pathology in a number of chronic diseases [59]. Paradoxically, we observed that increased stimulation of TLR4 by P. gingivalis enhanced production of proinflammatory mediators and activation of the inflammasome, resulting in attenuated systemic inflammation ( Figure 8 ). These results indicate that production of inflammatory mediators is protective against pathogen-mediated chronic inflammation, which is in agreement with our recent results that documented a protective role for TLR4 in P. gingivalis-mediated chronic inflammation at systemic sites [42]. This report, along with our current study, is in contrast to other studies elucidating the role of TLR4 in pathogen-mediated atherosclerosis progression. Infection of ApoE−/− TLR4−/− mice, fed a high-fat diet, with C. pneumoniae resulted in diminished atherosclerosis compared to ApoE−/− infected mice [60]. Additionally, a previous study documented that common mechanisms of signaling via TLR2, TLR4 and MyD88 link stimulation by multiple pathogens and endogenous ligands to atherosclerosis progression [61], [62]. Therapeutic antagonism has been suggested to be beneficial in the treatment of chronic atherosclerosis [63]. However, we have demonstrated TLR4 antagonism exacerbates atherosclerosis progression. Collectively, our study highlights the complexity of chronic inflammatory pathways in diseases like atherosclerosis that are exacerbated by pathogen infection and further elucidate pathogen-specific mechanisms for chronic disease progression.
An important observation from this study was that P. gingivalis failed to activate the inflammasome. It has recently been reported that Gram-negative bacteria utilize a non-canonical pathway for inflammasome activation that is mediated by caspase-11 [48], [49]. Kayagaki et al. [14] have shown that H. pylori, whose tetra-acylated lipid A poorly activates the TLR4 complex, failed to trigger the non-canonical inflammasome. Although TLR4 signaling correlates with non-canonical inflammasome activity, this group reported that activation of the non-canonical inflammasome is independent of TLR4 signaling, further suggesting an unknown sensor in the cytosol that detects modified lipid A ( Figure 8 ). We found that caspase-11 expression was essential for IL-1β production elicited by P. gingivalis strain PG1587 381, pointing to a pivotal role for activation of the non-canonical inflammasome in P. gingivalis infection.
Pathogen evasion of inflammasome activation has been proposed to serve a dual role: to prevent IL-1β release and to circumvent host cell death in order to provide an intracellular niche for the pathogen to survive [17]. Indeed, we observed that the low levels of IL-1β induced by P. gingivalis strains 381 and PG1773 381 correlated with an enhanced ability of these organisms to survive in macrophages. Likewise, the induction of relatively high levels of IL-1β by P. gingivalis strain PG1587 381 correlated with decreased survival of this strain in macrophages. The enhanced survival we observed with wild-type 381 and PG1773 381 are in agreement with our observation that both strains were relatively resistant to killing in the presence of the cationic antimicrobial peptide polymyxin B. In contrast, strain PG1587 381 was unable to survive intracellularly and was rapidly killed. These results suggest that P. gingivalis utilizes multiple mechanisms concurrently to promote its adaptive fitness.
The ability of P. gingivalis to survive intracellularly in the macrophage is intriguing when considering the link between periodontal disease and systemic inflammatory conditions. We propose that P. gingivalis entry and survival into macrophages may be a mechanism for dissemination of the bacterium from the oral cavity to other systemic sites, such as the vasculature. We have recently identified P. gingivalis in blood myeloid dendritic cells of humans with chronic periodontitis, suggesting a role for blood myeloid dendritic cells in harboring and disseminating pathogens from the oral mucosa to atherosclerotic plaques [64]. The mechanism utilized by P. gingivalis for survival in myeloid cells remains elusive. Wang et al. [65] have shown that intracellular survival of P. gingivalis within macrophages is dependent upon TLR2-mediated entry into lipid rafts. Pathogens that hijack lipid rafts do not readily fuse with late endosomes and lysosomes and may potentially fuse to autophagosomes [66], [67]. Whether intracellular trafficking to autophagosomes is the mechanism for P. gingivalis intracellular survival and dissemination to distant sites remains unknown ( Figure 8 ).
An interesting finding from this study was expression of P. gingivalis modified lipid A species did not alter the ability of the organism to induce oral inflammatory bone loss. These results are in agreement with previous studies that show that TLR4 is not needed for the induction of inflammatory bone loss, and it is predominantly mediated via TLR2 signaling [37], [68]. We also recently identified a TLR2- and TNF-dependent macrophage-specific mechanism for P. gingivalis-induced inflammatory bone loss in vivo [38]. In the current study, we observed comparable levels of TNFα were induced in macrophages stimulated with wild-type 381 and the lipid A mutants. In addition to the role of TNFα, it has been recently reported that P. gingivalis is able to induce oral bone loss at very low colonization levels, which triggers changes to the amount and composition of the oral commensal microbiota [69]. We have recently demonstrated in a rabbit model of periodontitis that lipid A phosphatases are required for both colonization of the rabbit and increases in the oral microbial load [43]. Whether this same mechanism for inflammatory bone loss is at play in our study remains to be determined.
It is well established that atherosclerosis progression is due to excessive production of proinflammatory mediators [70]. Although lipid deposition is considered a leading contributor to the inflammation, additional stimuli, such as infectious agents, have been considered as sources for the continuous inflammation [35]. In our study, we observed infection with P. gingivalis induced low levels of proinflammatory mediators but accelerated chronic inflammatory atherosclerosis. Thus, the question arises as to why a pathogen that induces low levels of inflammatory mediators would accelerate chronic inflammatory atherosclerosis. In a recent clinical trial for the inflammasome-mediated disease, Cryopyrin-associated periodic syndrome (CAPS), patients receiving the humanized monoclonal antibody, canakinumab, specific for IL-1β, had a 67% increased risk for infection compared to 25% of patients in the placebo group [71]. This clinical trial supports the results presented in our study that highlight a protective role for activation of innate immunity against low-grade chronic infection. An additional clinical trial was recently launched using canakinumab with the hypothesis that IL-1β inhibition will reduce major cardiovascular events in patients with preexisting coronary artery disease (CAD) [72]. Our results demonstrate that the potential benefit of long-term use of a neutralizing antibody to IL-1β in humans at high risk for atherosclerotic vascular disease must be substantial enough to counter the increased risk of infection [71]. Furthermore, future therapies need to be developed to consider the complexity of inflammatory pathways in chronic inflammation and the role of chronic infection in disease pathology.
Materials and Methods
Ethics statement
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by Boston University's Institutional Animal Care and Use Committee (IACUC) protocol numbers AN15312 and AN14348. Boston University is committed to observing federal policies and regulations and Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) International standards and guidelines for humane care and use of animals. Federal guidelines, the Animal Welfare Act (AWA) and The Guide were followed when carrying out experiments. Procedures involved euthanasia and harvesting of bone marrow macrophages. All efforts were made to minimize discomfort, pain and distress.
Bacteria
Frozen stocks of P. gingivalis wild-type strain 381 and the lipid A mutants (PG1587 381 and PG1773 381) were grown anaerobically at 37°C on blood agar plates (Remel) for 3–5 days [73]. Brain heart infusion broth (Becton-Dickinson Biosciences) supplemented with yeast extract (0.5%; Becton-Dickinson Biosciences), hemin (10 µg/ml; Sigma-Aldrich), and menadione (1 mg/ml; Sigma- Aldrich) was inoculated with plate grown bacteria and cultures grown anaerobically for 16–18 h. P. gingivalis lipid A mutants were grown in the presence of erythromycin (5 µg/mL).
Gene deletions in P. gingivalis strain 381
The genomic nucleotide sequences encoding the putative lipid A 1-phosphatase, PG1773, and the putative lipid A 4′-phosphatase, PG1587, were obtained from searches of the annotated P. gingivalis W83 genome at The Comprehensive Microbial Resource (http://cmr.jcvi.org/tigr-scripts/CMR/CmrHomePage.cgi). Gene deletions were created by introducing an erythromycin resistance cassette (ermF/AM) in place of the coding region for PG1773 and PG1587. Polymerase chain reaction (PCR) amplification of genomic DNA from P. gingivalis 381 was performed using primer sets designed against the W83 sequence to amplify 1000 base-pairs upstream and 1000 base-pairs downstream from the regions adjacent to the PG1773 and PG1587 coding regions, respectively. The amplified 5′ and 3′ flanking regions for PG1773 and PG1587, respectively, were co-ligated with the ermF/AM cassettes respectively into pcDNA3.1(−) to generate the gene disruption plasmids, p1773 5′flank:erm:3′flank and p1587 5′flank:erm:3′flank. P. gingivalis 381 deficient in either PG1587 (PG1587 381) or PG1773 (PG1773 381) was generated by introducing either p1587 5′flank:erm:3′flank or p1773 5′flank:erm:3′flank into P. gingivalis 381 by electroporation in a GenePulser Xcell (BioRad, Hercules, CA). Bacteria were plated on TYHK/agar plates containing the appropriate selective medium, which included erythromycin (5 µg/ml), and incubated anaerobically. One week later, colonies were selected for characterization. Loss of the PG1587 and PG1773 coding sequences were confirmed in all clones by PCR analyses using primers designed to detect the coding sequences in wild-type 381 bacteria.
MALDI-TOF MS analyses
LPS and Lipid A from P. gingivalis 381 and the lipid A mutant strains (PG1587 381 and PG1773 381) were isolated as previously described [23]. For MALDI-TOF MS analyses, lipid A were analyzed in the negative ion mode on an AutoFlex Analyzer (Bruker Daltonics). Data were acquired and processed using Flex Analysis software (Bruker Daltonics) [6], [23].
HEK293 cell TLR activation assays
HEK293 cells were plated in 96-well plates at a density of 4×104 cells per well, and transfected the following day with plasmids bearing firefly luciferase, Renilla luciferase, recombinant murine TLR4 and MD-2 or recombinant murine TLR2 and TLR1 by standard calcium phosphate precipitation. After overnight transfection, the test wells were stimulated in triplicate for 4 hours at 37°C with the indicated doses of LPS isolates or live bacteria. Following stimulation, the transfected HEK293 cells were rinsed with phosphate-buffered saline and lysed with 50 µl of passive lysis buffer (Promega, Madison, WI). Luciferase activity was measured using the Dual Luciferase Assay Reporter System (Promega, Madison, WI). Data are expressed as fold increase of NF-κB-activity which represents the ratio of NF-κB-dependent fire-fly luciferase activity to β-Actin promoter-dependent Renilla luciferase activity.
Polymyxin B sensitivity assays
BHI broth cultures of wild-type 381 and the lipid A mutant strains were started at an optical density of .1 at 660 nm in the presence or absence of increasing concentrations (1, 5, 10, 25, 50 µg/mL) of polymxin B (InvivoGen). After overnight growth under anaerobic conditions, growth was assessed spectrophotometrically at 660 nm [74].
Animals
Male ApoE−/− and C57BL/6 mice were obtained from The Jackson Laboratory (Bar Harbor, ME). C57BL/6 mice deficient in TLR2 and TLR4 were provided by Dr. S. Akira (Osaka University, Osaka, Japan) and bred in house. Mice were maintained under specific-pathogen free conditions and cared for in accordance with the Boston University Institutional Animal Care and Use Committee.
Bone marrow derived macrophage culture and stimulation
Bone marrow derived macrophages (BMDM) from wild-type and knockout mice were cultured in RPMI with 10% fetal bovine serum (Thermo Scientific HyClone Fetal Bovine Serum (U.S.), Defined SH3007003HI Heat inactivated) and 20% L929 supernatants [49] and were allowed to mature into macrophages over 7 days. Cells were seeded into 24-well plates at 2×105 cells/well (ELISA assays) or 6-well plates at 1×106 cells/well (Western blot analysis) and stimulated with bacteria at indicated MOI overnight. Cells stimulated with LPS from E. coli OIII:B4 (InvivoGen) or Pam3CysSk4 (InvivoGen) served as controls.
ELISA
Levels of IL-1β, TNFα, IL-6 (BD Bioscience), IL-1α (eBioscience) and KC (R&D Systems) in cell culture supernatants were analyzed by ELISA.
Immunoblotting
Proteins from cell culture supernatants were precipitated with ethanol at −20°C overnight and resuspended in Laemmli sample buffer. Cellular lysates were collected in RIPA buffer (Thermo Scientific) and samples were prepared in Laemmli sample buffer. Samples were separated by SDS-PAGE, transferred to polyvinyldifluoride membranes, blocked in 5% milk and target proteins were detected using antibodies to IL-1β (H-153) and caspase-1 p10 (M-20) (Santa Cruz Biotechnology). An antibody to β-actin (A1978 Sigma) was used as the loading control.
Antibiotic protection assay
BMDM were stimulated with P. gingivalis or lipid A mutants for 2 h and 6 h. Extracellular nonadherent bacteria were removed by washing with PBS. Adherent bacteria were killed by addition of gentamicin (300 µg/mL) and metronidazole (200 µg/mL) for 1 hr. After PBS wash, BMDM were lysed with HyClone water (Thermo Scientific) for 10 min. Serial dilutions of the lysate were plated on blood agar plates and cultured anaerobically for CFU enumeration [65].
Oral challenge
Mice were fed a normal chow diet (Global 2018; Harlan Teklad, Madison, WI). Six-week old male mice were treated with a 10-day regimen of oral antibiotics to allow for P. gingivalis colonization. Mice were challenged by oral application of vehicle (2% carboxymethylcellulose in PBS) or the P. gingivalis strains (1×109 CFU) at the buccal surface of the maxilla 5 times a week for 3 weeks [36], [37].
In vivo mouse Magnetic Resonance Angiography (MRA) and data analysis
In vivo imaging of the innominate artery was performed using a vertical-bore Bruker 11.7 T Avance spectrometer (Bruker; Billerica, MA) as previously described [53]. Mice were anesthetized with 0.5–2% inhaled isoflurane and placed in a vertical 30 mm probe (Micro 2.5). Respiration was monitored using a small animal monitoring and gating system (SA Instruments, Waukesha, WI). The angiography data was acquired with a fast low-angle shot (FLASH) sequence using the following parameters: slab thickness = 1.5 cm; flip angle = 45°; repetition time = 20 ms; echo time = 2.2 ms; field of view = 1.5×1.5×1.5 cm; matrix = 128×128×128; number of average = 4. The total scan time was ∼25 min. Visualization of the vasculature was achieved by 3D maximum intensity projections (MIP) of angiographic images reconstructed using Paravision. The target cross section of the innominate artery was chosen at 0.3- to 0.5-mm distance below the subclavian bifurcation. Lumen area of the chosen cross section was manually defined and calculated with ImageJ (National Institutes of Health) by two independent observers. The intra-reader reliability was excellent with interclass correlation coefficient values of 0.91.
Immunohistochemistry
Mice were euthanized (n = 3–4/group), perfused with PBS (5 mL) and the aortic arch with heart tissue was embedded in OCT freezing compound. Seven-micrometer serial cryosections were collected every 70 µm in the innominate artery. Immunohistochemistry was performed on cryosections corresponding to greatest plaque accumulation in the innominate artery as previously described [42], [53] using rat anti-mouse F4/80 (no. MCA497R; Serotec, Oxford, U.K.) or isotype controls (no. MCA1125; Serotec). Biotinylated anti-rat (mouse absorbed) IgG was used as secondary Ab (Vector Laboratories, Burlin- game, CA). Nuclei were counter-stained with hematoxylin. Digital micrographs were captured at 10× and 40×.
Atherosclerotic plaque assessment
Aortas were harvested and stained with Oil Red O as described [42]. Digital micrographs were taken, and total area of atherosclerotic plaque was determined using ImageJ (NIH) by a blinded observer.
Microcomputed tomography
Three-dimensional analysis of alveolar bone loss was assessed as previously described [38]. Briefly, cephalons were fixed for 24–48 h in 4% buffered paraformaldehyde and stored at 4°C in 70% ethanol until evaluation by microcomputed tomography (micro-CT). Quantitative three-dimensional analysis of alveolar bone loss in hemi-maxillae was performed using a desktop micro-CT system (μCT 40; Scanco Medical AG, Bassersdorf, Switzerland). Maxillary block biopsies were scanned at a resolution of 12 µm in all three spatial dimensions. Raw images were converted into high-quality dicoms and analyzed using computer software (Amira 5.2.2; Visage Imaging). Residual supporting bone volume was determined for the buccal roots. The apical basis of the measured volume was set mesio-distally parallel to the cemento-enamel junction and bucco-palatinally parallel to the occlusal plane. Results represent residual bone volume (mm3) above the reference plane (180 µm from the cemento-enamel junction).
Statistical analysis
Data were analyzed by two-tailed unpaired Student's t test or ANOVA with Bonferroni's posttest where indicated. A p value of .05 was considered indicative of statistical significance.
Supporting Information
Fimbriae expression, gingipain activity, and growth of P. gingivalis lipid A 1- and 4′ phosphatase mutants. Electron microscopy was performed with P. gingivalis wild-type strain 381, PG1587 381 and PG1773 381 (A). Fimbriae expression was examined by Western blot analysis in whole cell lysates (5×107 CFU) using a monoclonal Ab to major fimbriae (B) [73]. The proteolytic activities of the cell-associated cysteine proteases, gingipain R (RGP) and gingipain K (KGP) for wild-type 381 and the lipid A mutants PG1587 381 and PG1773 381 in whole cultures (lysate) and supernatant fractions were determined by an in vitro gingipain assay [75] (C). Percent proteolytic activity as compared to P. gingivalis wild-type strain 381 gingipain activity for PG1587 381 (dotted) and PG1773 381 (dashed). Bars indicate mean ± SEM from three independent experiments. Brain heart infusion broth cultures of P. gingivalis wild-type (solid), PG1587 381 (dotted), and PG1773 381 (dashed) were inoculated at a starting OD of 0.3. Growth was monitored at indicated time points over 48 h (n = 4) (D).
(TIF)
TLR2 and TLR4 contribute to the production of IL-1β and IL-1α in macrophages stimulated with P. gingivalis wild-type strain 381 and the lipid A mutant strains. BMDMs from wild-type C57BL/6 (white), TLR2-deficient (gray), or TLR4-deficient mice (black) were stimulated with P. gingivalis wild-type strain 381 or the lipid A mutant strains PG1587 381 and PG1773 381 at an MOI of 100 and levels of IL-1β (A) and IL-1α (B) were assessed by ELISA. Bars indicate mean ± SEM for n = 3 sample wells. *p<.05 **p<.01; two-tailed unpaired t-tests.
(TIF)
Plaque accumulation in the innominate artery following oral infection of ApoE−/− mice with P. gingivalis . Representative images of the innominate artery with hematoxylin and eosin staining for each group at 10× and 40× (n = 3/group).
(TIF)
Humoral response following oral infection of ApoE−/− mice with P. gingivalis. P. gingivalis-specific Ab isotypes IgG1 (A), IgG2b (B), IgG2c (C) and IgG3 (D) were measured in serum by ELISA at 16 wk post-infection of ApoE−/− mice with P. gingivalis wild-type strain 381 and the lipid A mutants PG1587 381 and PG1773 381 (n = 10–12 mice/group). * p<.05 **p≤.001; two-tailed unpaired t-tests.
(TIF)
Supporting information on experimental procedures. Describes the experimental procedures utilized for the supplemental data. This includes experimental procedures for Electron Microscopy, Gingipain Assay, ELISA, and Histology.
(DOCX)
Acknowledgments
From Boston University School of Medicine, we acknowledge Dr. Xuemei Zhong and Natalie Bitar (Immunohistochemistry Core), Aaron Brug and Dr. Heidi Schwanz (Magnetic Resonance Imaging), and Dr. Esther Bullitt (Electron Microscopy) for their technical assistance. We thank Dr. Katherine A. Fitzgerald (University of Massachusetts Medical School, Worcester, MA) for providing C57BL/6 mice deficient in caspase-11.
Funding Statement
This work was supported with grants from the NIH NIAID T32AI089673-01A1, NIAID PO1 AI078894-01A1, and NIAID 3RO1DE012768-12S1. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
References
- 1. Akira S, Takeda K, Kaisho T (2001) Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol 2: 675–680 10.1038/90609 [DOI] [PubMed] [Google Scholar]
- 2. Munford RS, Varley AW (2006) Shield as signal: lipopolysaccharides and the evolution of immunity to gram-negative bacteria. PLoS Pathog 2: e67 10.1371/journal.ppat.0020067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Miller SI, Ernst RK, Bader MW (2005) LPS, TLR4 and infectious disease diversity. Nat Rev Micro 3: 36–46 10.1038/nrmicro1068 [DOI] [PubMed] [Google Scholar]
- 4. Needham BD, Trent MS (2013) Fortifying the barrier: the impact of lipid A remodelling on bacterial pathogenesis. Nat Rev Micro 11: 467–481 10.1038/nrmicro3047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Raetz CRH, Whitfield C (2002) Lipopolysaccharide endotoxins. Annu Rev Biochem 71: 635–700 10.1146/annurev.biochem.71.110601.135414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Coats SR, Berezow AB, To TT, Jain S, Bainbridge BW, et al. (2011) The lipid A phosphate position determines differential host Toll-like receptor 4 responses to phylogenetically related symbiotic and pathogenic bacteria. Infection and Immunity 79: 203–210 10.1128/IAI.00937-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cullen TW, Giles DK, Wolf LN, Ecobichon C, Boneca IG, et al. (2011) Helicobacter pylori versus the host: remodeling of the bacterial outer membrane is required for survival in the gastric mucosa. PLoS Pathog 7: e1002454 10.1371/journal.ppat.1002454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Neumeister B, Faigle M, Sommer M, Zähringer U, Stelter F, et al. (1998) Low endotoxic potential of Legionella pneumophila lipopolysaccharide due to failure of interaction with the monocyte lipopolysaccharide receptor CD14. Infection and Immunity 66: 4151–4157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kawahara K, Tsukano H, Watanabe H, Lindner B, Matsuura M (2002) Modification of the structure and activity of lipid A in Yersinia pestis lipopolysaccharide by growth temperature. Infection and Immunity 70: 4092–4098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hajjar AM, Harvey MD, Shaffer SA, Goodlett DR, Sjöstedt A, et al. (2006) Lack of in vitro and in vivo recognition of Francisella tularensis subspecies lipopolysaccharide by Toll-like receptors. Infection and Immunity 74: 6730–6738 10.1128/IAI.00934-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rebeil R, Ernst RK, Jarrett CO, Adams KN, Miller SI, et al. (2006) Characterization of late acyltransferase genes of Yersinia pestis and their role in temperature-dependent lipid A variation. J Bacteriol 188: 1381–1388 10.1128/JB.188.4.1381-1388.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li Y, Powell DA, Shaffer SA, Rasko DA, Pelletier MR, et al. (2012) LPS remodeling is an evolved survival strategy for bacteria. PNAS 109: 8716–8721 10.1073/pnas.1202908109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Maeshima N, Fernandez RC (2013) Recognition of lipid A variants by the TLR4-MD-2 receptor complex. Front Cell Infect Microbiol 3: 3 10.3389/fcimb.2013.00003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, et al. (2013) Noncanonical Inflammasome Activation by Intracellular LPS Independent of TLR4. Science 341: 1246–1249 10.1126/science.1240248 [DOI] [PubMed] [Google Scholar]
- 15. Franchi L, Eigenbrod T, Muñoz-Planillo R, Nuñez G (2009) The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol 10: 241–247 10.1038/ni.1703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Taxman DJ, Huang MT-H, Ting JPY (2010) Inflammasome inhibition as a pathogenic stealth mechanism. Cell Host Microbe 8: 7–11 10.1016/j.chom.2010.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, et al. (2012) Oxidized Mitochondrial DNA Activates the NLRP3 Inflammasome during Apoptosis. Immunity 36: 401–414 10.1016/j.immuni.2012.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bainbridge BW, Coats SR, Darveau RP (2002) Porphyromonas gingivalis lipopolysaccharide displays functionally diverse interactions with the innate host defense system. Ann Periodontol 7: 29–37 10.1902/annals.2002.7.1.29 [DOI] [PubMed] [Google Scholar]
- 19. Kumada H, Haishima Y, Umemoto T, Tanamoto K (1995) Structural study on the free lipid A isolated from lipopolysaccharide of Porphyromonas gingivalis. J Bacteriol 177: 2098–2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Coats SR, Pham T-TT, Bainbridge BW, Reife RA, Darveau RP (2005) MD-2 mediates the ability of tetra-acylated and penta-acylated lipopolysaccharides to antagonize Escherichia coli lipopolysaccharide at the TLR4 signaling complex. J Immunol 175: 4490–4498. [DOI] [PubMed] [Google Scholar]
- 21. Reife RA, Coats SR, Al-Qutub M, Dixon DM, Braham PA, et al. (2006) Porphyromonas gingivalis lipopolysaccharide lipid A heterogeneity: differential activities of tetra- and penta-acylated lipid A structures on E-selectin expression and TLR4 recognition. Cellular Microbiology 8: 857–868 10.1111/j.1462-5822.2005.00672.x [DOI] [PubMed] [Google Scholar]
- 22. Al-Qutub MN, Braham PH, Karimi-Naser LM, Liu X, Genco CA, et al. (2006) Hemin-Dependent Modulation of the Lipid A Structure of Porphyromonas gingivalis Lipopolysaccharide. Infection and Immunity 74: 4474–4485 10.1128/IAI.01924-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Coats SR, Jones JW, Do CT, Braham PH, Bainbridge BW, et al. (2009) Human Toll-like receptor 4 responses to P. gingivalisare regulated by lipid A 1- and 4′-phosphatase activities. Cellular Microbiology 11: 1587–1599 10.1111/j.1462-5822.2009.01349.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Curtis MA, Percival RS, Devine D, Darveau RP, Coats SR, et al. (2011) Temperature-dependent modulation of Porphyromonas gingivalis lipid A structure and interaction with the innate host defenses. Infection and Immunity 79: 1187–1193 10.1128/IAI.00900-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rangarajan M, Aduse-Opoku J, Paramonov N, Hashim A, Bostanci N, et al. (2008) Identification of a second lipopolysaccharide in Porphyromonas gingivalis W50. J Bacteriol 190: 2920–2932 10.1128/JB.01868-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Herath TDK, Darveau RP, Seneviratne CJ, Wang C-Y, Wang Y, et al. (2013) Tetra- and penta-acylated lipid A structures of Porphyromonas gingivalis LPS differentially activate TLR4-mediated NF-κB signal transduction cascade and immuno-inflammatory response in human gingival fibroblasts. PLoS ONE 8: e58496 10.1371/journal.pone.0058496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hayashi C, Gudino CV, Gibson FC III, Genco CA (2010) REVIEW: Pathogen-induced inflammation at sites distant from oral infection: bacterial persistence and induction of cell-specific innate immune inflammatory pathways. Mol Oral Microbiol 25: 305–316 10.1111/j.2041-1014.2010.00582.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Oliver RC, Brown LJ, Löe H (1998) Periodontal diseases in the United States population. J Periodontol 69: 269–278. [DOI] [PubMed] [Google Scholar]
- 29. Pihlstrom BL, Michalowicz BS, Johnson NW (2005) Periodontal diseases. Lancet 366: 1809–1820 10.1016/S0140-6736(05)67728-8 [DOI] [PubMed] [Google Scholar]
- 30. Bohnstedt S, Cullinan MP, Ford PJ, Palmer JE, Leishman SJ, et al. (2010) High antibody levels to P. gingivalis in cardiovascular disease. Journal of Dental Research 89: 938–942 10.1177/0022034510370817 [DOI] [PubMed] [Google Scholar]
- 31. Tonetti MS (2009) Periodontitis and risk for atherosclerosis: an update on intervention trials. J Clin Periodontol 36 (Suppl 10) 15–19 10.1111/j.1600-051X.2009.01417.x [DOI] [PubMed] [Google Scholar]
- 32. Michaud DS, Izard J, Wilhelm-Benartzi CS, You D-H, Grote VA, et al. (2013) Plasma antibodies to oral bacteria and risk of pancreatic cancer in a large European prospective cohort study. Gut 62: 1764–1770 10.1136/gutjnl-2012-303006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Haraszthy VI, Zambon JJ, Trevisan M, Zeid M, Genco RJ (2000) Identification of periodontal pathogens in atheromatous plaques. J Periodontol 71: 1554–1560 10.1902/jop.2000.71.10.1554 [DOI] [PubMed] [Google Scholar]
- 34. Kozarov EV, Dorn BR, Shelburne CE, Dunn WA, Progulske-Fox A (2005) Human atherosclerotic plaque contains viable invasive Actinobacillus actinomycetemcomitans and Porphyromonas gingivalis. Arteriosclerosis, Thrombosis, and Vascular Biology 25: e17–e18 10.1161/01.ATV.0000155018.67835.1a [DOI] [PubMed] [Google Scholar]
- 35. Rosenfeld ME, Campbell LA (2011) Pathogens and atherosclerosis: update on the potential contribution of multiple infectious organisms to the pathogenesis of atherosclerosis. Thromb Haemost 106: 858–867 10.1160/TH11-06-0392 [DOI] [PubMed] [Google Scholar]
- 36. Gibson FC, Hong C, Chou H-H, Yumoto H, Chen J, et al. (2004) Innate immune recognition of invasive bacteria accelerates atherosclerosis in apolipoprotein E-deficient mice. Circulation 109: 2801–2806 10.1161/01.CIR.0000129769.17895.F0 [DOI] [PubMed] [Google Scholar]
- 37. Hayashi C, Madrigal AG, Liu X, Ukai T, Goswami S, et al. (2010) Pathogen-mediated inflammatory atherosclerosis is mediated in part via Toll-like receptor 2-induced inflammatory responses. J Innate Immun 2: 334–343 10.1159/000314686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Papadopoulos G, Weinberg EO, Massari P, Gibson FC, Wetzler LM, et al. (2013) Macrophage-specific TLR2 signaling mediates pathogen-induced TNF-dependent inflammatory oral bone loss. The Journal of Immunology 190: 1148–1157 10.4049/jimmunol.1202511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jain S, Coats SR, Chang AM, Darveau RP (2013) A Novel Class of Lipoprotein Lipase-Sensitive Molecules Mediates Toll-Like Receptor 2 Activation by Porphyromonas gingivalis. Infection and Immunity 81: 1277–1286 10.1128/IAI.01036-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Davey M, Liu X, Ukai T, Jain V, Gudino CV, et al. (2008) Bacterial Fimbriae Stimulate Proinflammatory Activation in the Endothelium through Distinct TLRs. The Journal of Immunology 180: 2187–2195. [DOI] [PubMed] [Google Scholar]
- 41. Nichols FC, Bajrami B, Clark RB, Housley W, Yao X (2012) Free Lipid A Isolated from Porphyromonas gingivalis Lipopolysaccharide Is Contaminated with Phosphorylated Dihydroceramide Lipids: Recovery in Diseased Dental Samples. Infection and Immunity 80: 860–874 10.1128/IAI.06180-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hayashi C, Papadopoulos G, Gudino CV, Weinberg EO, Barth KR, et al. (2012) Protective Role for TLR4 Signaling in Atherosclerosis Progression as Revealed by Infection with a Common Oral Pathogen. The Journal of Immunology 189: 3681–3688 10.4049/jimmunol.1201541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zenobia C, Hasturk H, Nguyen D, Van Dyke TE, Kantarci A, et al. (2014) Porphyromonas gingivalis Lipid A Phosphatase Activity Is Critical for Colonization and Increasing the Commensal Load in the Rabbit Ligature Model. Infection and Immunity 82: 650–659 10.1128/IAI.01136-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tschopp J, Martinon F, Burns K (2003) NALPs: a novel protein family involved in inflammation. Nat Rev Mol Cell Biol 4: 95–104 10.1038/nrm1019 [DOI] [PubMed] [Google Scholar]
- 45. Bostanci N, Allaker RP, Belibasakis GN, Rangarajan M, Curtis MA, et al. (2007) Porphyromonas gingivalis antagonises Campylobacter rectus induced cytokine production by human monocytes. Cytokine 39: 147–156 10.1016/j.cyto.2007.07.002 [DOI] [PubMed] [Google Scholar]
- 46. Taxman DJ, Swanson KV, Broglie PM, Wen H, Holley-Guthrie E, et al. (2012) Porphyromonas gingivalis Mediates Inflammasome Repression in Polymicrobial Cultures through a Novel Mechanism Involving Reduced Endocytosis. Journal of Biological Chemistry 287: 32791–32799 10.1074/jbc.M112.401737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mariathasan S, Monack DM (2007) Inflammasome adaptors and sensors: intracellular regulators of infection and inflammation. Nat Rev Immunol 7: 31–40 10.1038/nri1997 [DOI] [PubMed] [Google Scholar]
- 48. Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, et al. (2011) Non-canonical inflammasome activation targets caspase-11. Nature 479: 117–121 10.1038/nature10558 [DOI] [PubMed] [Google Scholar]
- 49. Rathinam VAK, Vanaja SK, Waggoner L, Sokolovska A, Becker C, et al. (2012) TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by gram-negative bacteria. Cell 150: 606–619 10.1016/j.cell.2012.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Broz P, Monack DM (2013) Noncanonical inflammasomes: caspase-11 activation and effector mechanisms. PLoS Pathog 9: e1003144 10.1371/journal.ppat.1003144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Baker PJ, Evans RT, Roopenian DC (1994) Oral infection with Porphyromonas gingivalis and induced alveolar bone loss in immunocompetent and severe combined immunodeficient mice. Arch Oral Biol 39: 1035–1040. [DOI] [PubMed] [Google Scholar]
- 52. Chou H-H, Yumoto H, Davey M, Takahashi Y, Miyamoto T, et al. (2005) Porphyromonas gingivalis fimbria-dependent activation of inflammatory genes in human aortic endothelial cells. Infection and Immunity 73: 5367–5378 10.1128/IAI.73.9.5367-5378.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hayashi C, Viereck J, Hua N, Phinikaridou A, Madrigal AG, et al. (2011) Porphyromonas gingivalis accelerates inflammatory atherosclerosis in the innominate artery of ApoE deficient mice. Atherosclerosis 215: 52–59 10.1016/j.atherosclerosis.2010.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Burns E, Bachrach G, Shapira L, Nussbaum G (2006) Cutting Edge: TLR2 is required for the innate response to Porphyromonas gingivalis: activation leads to bacterial persistence and TLR2 deficiency attenuates induced alveolar bone resorption. J Immunol 177: 8296–8300. [DOI] [PubMed] [Google Scholar]
- 55. Ukai T, Yumoto H, Gibson FC, Genco CA (2008) Macrophage-Elicited Osteoclastogenesis in Response to Bacterial Stimulation Requires Toll-Like Receptor 2-Dependent Tumor Necrosis Factor-Alpha Production. Infection and Immunity 76: 812–819 10.1128/IAI.01241-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Coats SR, Do CT, Karimi-Naser LM, Braham PH, Darveau RP (2007) Antagonistic lipopolysaccharides block E. coli lipopolysaccharide function at human TLR4 via interaction with the human MD-2 lipopolysaccharide binding site. Cellular Microbiology 9: 1191–1202 10.1111/j.1462-5822.2006.00859.x [DOI] [PubMed] [Google Scholar]
- 57. Montminy SW, Khan N, McGrath S, Walkowicz MJ, Sharp F, et al. (2006) Virulence factors of Yersinia pestis are overcome by a strong lipopolysaccharide response. Nat Immunol 7: 1066–1073 10.1038/ni1386 [DOI] [PubMed] [Google Scholar]
- 58. Cigana C, Curcurù L, Leone MR, Ieranò T, Lorè NI, et al. (2009) Pseudomonas aeruginosa exploits lipid A and muropeptides modification as a strategy to lower innate immunity during cystic fibrosis lung infection. PLoS ONE 4: e8439 10.1371/journal.pone.0008439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Davis BK, Wen H, Ting JPY (2011) The Inflammasome NLRs in Immunity, Inflammation, and Associated Diseases. Annu Rev Immunol 29: 707–735 10.1146/annurev-immunol-031210-101405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Naiki Y, Sorrentino R, Wong MH, Michelsen KS, Shimada K, et al. (2008) TLR/MyD88 and liver X receptor alpha signaling pathways reciprocally control Chlamydia pneumoniae-induced acceleration of atherosclerosis. The Journal of Immunology 181: 7176–7185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Michelsen KS, Wong MH, Shah PK, Zhang W, Yano J, et al. (2004) Lack of Toll-like receptor 4 or myeloid differentiation factor 88 reduces atherosclerosis and alters plaque phenotype in mice deficient in apolipoprotein E. Proc Natl Acad Sci USA 101: 10679–10684 10.1073/pnas.0403249101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Cole JE, Mitra AT, Monaco C (2010) Treating atherosclerosis: the potential of Toll-like receptors as therapeutic targets. Expert Rev Cardiovasc Ther 8: 1619–1635 10.1586/erc.10.149 [DOI] [PubMed] [Google Scholar]
- 63. Leon CG, Tory R, Jia J, Sivak O, Wasan KM (2008) Discovery and development of toll-like receptor 4 (TLR4) antagonists: a new paradigm for treating sepsis and other diseases. Pharm Res 25: 1751–1761 10.1007/s11095-008-9571-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Carrion J, Scisci E, Miles B, Sabino GJ, Zeituni AE, et al. (2012) Microbial carriage state of peripheral blood dendritic cells (DCs) in chronic periodontitis influences DC differentiation, atherogenic potential. The Journal of Immunology 189: 3178–3187 10.4049/jimmunol.1201053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wang M, Shakhatreh M-AK, James D, Liang S, Nishiyama S-I, et al. (2007) Fimbrial proteins of porphyromonas gingivalis mediate in vivo virulence and exploit TLR2 and complement receptor 3 to persist in macrophages. J Immunol 179: 2349–2358. [DOI] [PubMed] [Google Scholar]
- 66. Amer AO, Byrne BG, Swanson MS (2005) Macrophages rapidly transfer pathogens from lipid raft vacuoles to autophagosomes. Autophagy 1: 53–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Simons K, Gruenberg J (2000) Jamming the endosomal system: lipid rafts and lysosomal storage disease. Trends Cell Biol 10: 459–462. [DOI] [PubMed] [Google Scholar]
- 68. Gibson FC III, Genco CA (2007) Porphyromonas gingivalis Mediated Periodontal Disease and Atheroscerlosis: Disparate Disease with Commonalities in Pathogenesis Through TLRs. Current Pharmaceutical Design 13: 3665–3675. [DOI] [PubMed] [Google Scholar]
- 69. Hajishengallis G, Liang S, Payne MA, Hashim A, Jotwani R, et al. (2011) Low-abundance biofilm species orchestrates inflammatory periodontal disease through the commensal microbiota and complement. Cell Host Microbe 10: 497–506 10.1016/j.chom.2011.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Dinarello CA (2011) A clinical perspective of IL-1β as the gatekeeper of inflammation. Eur J Immunol 41: 1203–1217 10.1002/eji.201141550 [DOI] [PubMed] [Google Scholar]
- 71. Tabas I, Glass CK (2013) Anti-inflammatory therapy in chronic disease: challenges and opportunities. Science 339: 166–172 10.1126/science.1230720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Ridker PM, Thuren T, Zalewski A, Libby P (2011) Interleukin-1β inhibition and the prevention of recurrent cardiovascular events: rationale and design of the Canakinumab Anti-inflammatory Thrombosis Outcomes Study (CANTOS). Am Heart J 162: 597–605 10.1016/j.ahj.2011.06.012 [DOI] [PubMed] [Google Scholar]
- 73. Takahashi Y, Davey M, Yumoto H, Gibson FC III, Genco CA (2006) Fimbria-dependent activation of pro-inflammatory molecules in Porphyromonas gingivalis infected human aortic endothelial cells. Cellular Microbiology 8: 738–757 10.1111/j.1462-5822.2005.00661.x [DOI] [PubMed] [Google Scholar]
- 74. Coats SR, To TT, Jain S, Braham PH, Darveau RP (2009) Porphyromonas gingivalis resistance to polymyxin B is determined by the lipid A 4′-phosphatase, PGN_0524. Int J Oral Sci 1: 126–135 10.4248/IJOS.09062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Madrigal AG, Barth K, Papadopoulos G, Genco CA (2012) Pathogen-Mediated Proteolysis of the Cell Death Regulator RIPK1 and the Host Defense Modulator RIPK2 in Human Aortic Endothelial Cells. PLoS Pathog 8: e1002723 10.1371/journal.ppat.1002723.g011 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fimbriae expression, gingipain activity, and growth of P. gingivalis lipid A 1- and 4′ phosphatase mutants. Electron microscopy was performed with P. gingivalis wild-type strain 381, PG1587 381 and PG1773 381 (A). Fimbriae expression was examined by Western blot analysis in whole cell lysates (5×107 CFU) using a monoclonal Ab to major fimbriae (B) [73]. The proteolytic activities of the cell-associated cysteine proteases, gingipain R (RGP) and gingipain K (KGP) for wild-type 381 and the lipid A mutants PG1587 381 and PG1773 381 in whole cultures (lysate) and supernatant fractions were determined by an in vitro gingipain assay [75] (C). Percent proteolytic activity as compared to P. gingivalis wild-type strain 381 gingipain activity for PG1587 381 (dotted) and PG1773 381 (dashed). Bars indicate mean ± SEM from three independent experiments. Brain heart infusion broth cultures of P. gingivalis wild-type (solid), PG1587 381 (dotted), and PG1773 381 (dashed) were inoculated at a starting OD of 0.3. Growth was monitored at indicated time points over 48 h (n = 4) (D).
(TIF)
TLR2 and TLR4 contribute to the production of IL-1β and IL-1α in macrophages stimulated with P. gingivalis wild-type strain 381 and the lipid A mutant strains. BMDMs from wild-type C57BL/6 (white), TLR2-deficient (gray), or TLR4-deficient mice (black) were stimulated with P. gingivalis wild-type strain 381 or the lipid A mutant strains PG1587 381 and PG1773 381 at an MOI of 100 and levels of IL-1β (A) and IL-1α (B) were assessed by ELISA. Bars indicate mean ± SEM for n = 3 sample wells. *p<.05 **p<.01; two-tailed unpaired t-tests.
(TIF)
Plaque accumulation in the innominate artery following oral infection of ApoE−/− mice with P. gingivalis . Representative images of the innominate artery with hematoxylin and eosin staining for each group at 10× and 40× (n = 3/group).
(TIF)
Humoral response following oral infection of ApoE−/− mice with P. gingivalis. P. gingivalis-specific Ab isotypes IgG1 (A), IgG2b (B), IgG2c (C) and IgG3 (D) were measured in serum by ELISA at 16 wk post-infection of ApoE−/− mice with P. gingivalis wild-type strain 381 and the lipid A mutants PG1587 381 and PG1773 381 (n = 10–12 mice/group). * p<.05 **p≤.001; two-tailed unpaired t-tests.
(TIF)
Supporting information on experimental procedures. Describes the experimental procedures utilized for the supplemental data. This includes experimental procedures for Electron Microscopy, Gingipain Assay, ELISA, and Histology.
(DOCX)